Sertaconazole_nitrate_DataSheet_MedChemExpress
处方常用拉丁文缩写与中文对照表

处方常用拉丁文及药物缩写中文对照表《执业药师必备手册》一、药物拉丁缩写:二、处方缩写:五、制剂用法:八、医用药品别名:1(丁胺卡那霉素——阿米卡星)2;(醋酸泼泥松——强的松)3(头孢哌酮——先锋必)4(头孢塞肟钠——先锋7号)5(头孢唑林钠——先锋5号)6(头孢曲松钠——菌必治)7(苄星青霉素——长效青霉素)8(大观霉素——淋必治)9(利巴韦林——病毒唑)10(吗啉双呱——病毒灵)11(葡醛内酯——肝泰乐)12(百炎净——复方磺胺甲恶唑-SMZ)13(诺氟沙星——氟哌酸)14(呋喃妥因——呋喃坦定)15(呋喃唑酮——痢特灵)16(甲硝唑——灭滴灵)17(阿昔洛韦——无坏鸟甘)18(庆大霉素普鲁卡因——胃炎灵)19(庆大霉素碳酸必——肠炎宁)20(呋噻米——速尿)21(心律平——普罗帕酮)22(异博定——盐酸维拉帕米)23(硝酸异山梨酯片——消心痛)24(脑复新——盐酸吡硫醇)25(脑脉宁——盐酸托哌酮片)26(曲安奈德——康A——康尼克通)27(心的安——盐酸普萘洛尔)28(脑复康——吡拉西坦)29(硫酸软骨素——康的宁)30(肝安——15AA)(肾安——9AA)31(沙丁胺醇——硫酸舒喘宁)32(必嗽平——溴已新)33(咳必清——枸椽酸喷托维林片)34(脑溢嗪——盐酸桂利嗪)35(盐酸二氧嗪片——咳克敏)36(妇康片——炔诺酮片)37(化痰片——羧甲司坦片)38(维尔新——维生素E烟酸酯胶囊)39(螺内酯——安体舒通)40(西咪替丁——甲青咪瓜)41(胃舒平——氢氧化铝)42(甲疏咪唑——他巴唑)43(肾上腺色腙素——安洛血)44(扑尔敏——马来酸氯苯那敏)45(盐酸异丙嗪——非那根)46(碳酸氢钠——小苏打)47(706代血浆——羟乙基淀粉40氯化钠针)48(低份子右旋糖——右旋糖酐40葡萄糖针)49(酚磺乙胺——止血敏)50(罗通定——颅痛定)51(维生素B2——核黄素)52(维生素C——抗坏血酸)53(A TP——三磷酸腺苷酸)54(GM——庆大霉素)55(潘生丁——双嘧达莫)56(扑炎痛——贝诺酯)57(消炎痛——吲哚美辛)58(扑热息痛——对乙酰胺基酚)59(止血芳酸——氨甲苯酸)60(强力霉素——多西坏素)61(癣敌——硝酸溢康唑软膏)62(治癣必妥——联苯苄唑乳膏)63(维脑路通——曲克芦丁)64(氢氯噻嗪——双克片)65(黄体酮——醋酸甲羟孕酮)66(阿司匹林——乙酰水杨酸)67(吡罗昔康——炎痛喜康)68(盐酸黄莲素——盐酸小檗碱)69(双氯灭痛——双氯芬酸酯)70(强筋松——苯丙氨酯)71(酚酞片——果异片)72(甲氯普胺——胃复安)73(溴丙胺太林片——普鲁苯辛)74(牙痛水——樟脑水合氯醛酊)75(654-2——消旋山莨菪碱片)76(心脉宁——复方毛冬青氯贝酸铝)77(脉通——复方亚油酸乙酯胶丸)78(心痛定——硝苯地平)79(毛花洋地黄苷丙——西地兰)80(苯磺酸阿曲库铵——卡肌宁)81(杜冷丁——哌替啶)82(氨伽黄敏胶囊——速效伤风胶囊)83(乳酶生——表飞明(鸣))84(异烟肼——雷米封)85(卡托普利——克普定)86头孢曲松钠(菌必治)87头孢噻吩(先锋I) 88头孢氨苄(先锋IV)89头孢拉定(先锋VI) 90强力霉素(多西环素)91艾司唑仑(安定)92他巴唑(甲巯咪唑)93甲氧氯普胺(胃复安)94 复方丹参(香丹)95氨苄青霉素(氨苄西林)96氟桂利嗪(西比定)97复方氢氧化铝(胃舒平)98奥美拉唑(洛赛克)99琥乙红霉素(利君沙)100吲哚美辛(消炎痛)101硝酸异山梨酯(消心痛)102硝苯地平(心痛定)103普萘洛尔(心得安)104硫酸新霉素滴眼液(的确当)105硝苯地平缓释片(圣通平)106诺氟沙星(氟哌酸)107复方新诺明(百炎净)108沙丁胺醇(舒喘灵)109水飞蓟宾葡甲胺片(西利复安)110曲安奈德(肤疾灵)111酒石酸美托洛尔(倍他乐克)112维拉帕米(异搏定)113桂利嗪(脑益嗪)114普罗帕酮(心律平)115格列齐特(达美康)116格列本脲(优降糖)117格列吡嗪(美吡达)118曲咪新乳膏(皮康霜)119林旦软膏(疥得治)120酮康他唑(皮康王)。
艾塞那肽结构式

艾塞那肽结构式1. 艾塞那肽的概述艾塞那肽(Acesulfame potassium)是一种高强度甜味剂,也被称为甜蜜素K。
它是一种无卡路里的人造甜味剂,可以用于替代食品中的糖分。
艾塞那肽在食品和饮料工业中被广泛使用,以提供甜味而不增加热量。
2. 艾塞那肽的化学结构艾塞那肽的化学名称为6-methyl-1,2,3-oxathiazin-4(3H)-one 2,2-dioxide potassium salt。
它的分子式为C4H4KNO4S,相对分子质量为201.24 g/mol。
3. 艾塞那肽的制备方法艾塞那肽通常通过化学合成方法制备。
以下是一种常见的制备方法:1.首先,将氯乙酰乙酸酯与氯乙醇反应生成氯乙基乙酸酯。
2.将氯乙基乙酸酯与氨基酮反应生成氯乙基乙酸氨基酮。
3.将氯乙基乙酸氨基酮与亚硫酸钾反应生成艾塞那肽。
4.最后,通过过滤和结晶等步骤得到艾塞那肽结晶体。
4. 艾塞那肽的甜味特性艾塞那肽是一种高强度甜味剂,其甜度约为蔗糖的200倍。
它可以快速溶解在水中,并且在不同温度下都能保持稳定的甜味。
虽然艾塞那肽具有甜味,但它没有提供热量。
这使得它成为许多低热量和无糖产品的理想替代品。
此外,艾塞那肽不会引起龋齿,因为它不能被口腔中的细菌代谢。
5. 艾塞那肽的应用领域艾塞那肽广泛应用于食品和饮料工业中。
以下是一些常见的应用领域:•饮料:艾塞那肽常被用于制作无糖饮料,如无糖碳酸饮料、无糖果汁等。
•烘焙食品:艾塞那肽可用于制作无糖蛋糕、无糖饼干等烘焙食品。
•奶制品:艾塞那肽可用于制作无糖乳制品,如无糖冰淇淋、无糖酸奶等。
•调味品:艾塞那肽可以用于制作低热量的甜味调味品,如低热量果酱、低热量沙拉酱等。
6. 艾塞那肽的安全性评估艾塞那肽经过多次严格的安全性评估,并被许多国家和地区批准用于食品和饮料中。
以下是一些关于艾塞那肽安全性的重要信息:•毒性:大量科学研究表明,艾塞那肽在推荐剂量下对人体没有毒性。
•代谢和排泄:艾塞那肽在人体内不被代谢,大部分通过尿液排出。
药用辅料中英文对照

药用辅料中英文对照1 阿拉伯胶(Acacia)2 乙酰舒泛钾(Acesulfame Potassium)3 冰醋酸(Acetic Acid,Glacial)4 乙酰枸橼酸三丁酯(AcetyltributylCitrate)5 乙酰枸橼酸三乙酯(AcetyltriethylCitrate)6 人血白蛋白(Albumin)7 乙醇(Alcohol)8 海藻酸(Alginic Acid)9 脂肪族聚酯(Aliphatic Polyesters)10 阿力糖(Alitame)11 杏仁油(Almond Oil)12 维生素E(Alpha Tocopherol)13 氨溶液(Ammonia Solution)14 维生素C(Ascorbic Acid)15 棕榈酸维生素C酯(Ascorbyl Palmitate)16 阿司帕坦(Aspartame)17 绿坡缕石(Attapulgite)18 皂土(Bentonite)19 苯扎氯铵(Benzalkonium Chloride)20 苄索氯铵(Benzethonium Chloride)21 苯甲酸(Benzoic Acid)22 苯甲醇(Benzyl Alcohol)23 苯甲酸苄酯(Benzyl Benzoate)24 溴硝丙二醇(Bronopol)25 丁羟茴醚(Butylated Hydroxyanisole)26 丁羟甲苯(Butylated Hydroxytoluene)27 羟苯丁酯(Butylparaben)28 碳酸钙(Calcium Carbonate)29 无水磷酸氢钙(Calcium Phosphate,Dibasic Anhydrous)30 磷酸氢钙二水合物(Calcium Phosphate,Dibasic Dihydrate)31 磷酸钙(Calcium Phosphate,Tribasic)32 硬脂酸钙(Calcium Stearate)33 硫酸钙(Calcium Sulfate)34 低芥酸菜籽油(Canola Oil)35 卡波姆(Carbomer)36 二氧化碳(Carbon Dioxide)37 羧甲纤维素钙(Carboxymethylcellulose Calcium)38 羧甲纤维素钠(Carboxymethylcellulose Sodium)39 角叉菜胶(Carrageenan)40 蓖麻油(Castor Oil)41 氢化蓖麻油(Castor Oil,Hydro-genated)42 微晶纤维素(Cellulose,Microcr ystalline)43 粉状纤维素(Cellulose,Powdered)44 微粉硅胶微晶纤维素(Cellulose, Silicified Microcrystalline)45 醋酸纤维素(Cellulose Acetate)46 纤维醋法酯(Cellulose Acetate Phthalate)47 角豆胶(Ceratonia)48 十八十六醇(Cetostearyl Alcohol)49 西曲溴铵(Cetrimide)50 十六醇(Cetyl Alcohol)51 壳聚糖(Chitosan)52 氯己定(Chlorhexidine)53 三氯叔丁醇(Chlorobutanol)54 氯甲酚(Chlorocresol)55 一氯二氟乙烷(Chlorodifluoroe-thane)56 氟里昂(Chlorofluorocabons)57 对氯间二甲酚(Chloroxylenol)58 胆固醇(Cholesterol)59 枸橼酸(Citric Acid Monohydrate)60 胶态二氧化硅(微粉硅胶)(Colloidal Silicon Dioxide)61 着色剂(Coloring Agents)62 玉米油(Corn Oil)63 棉籽油(Cottonseed Oil)64 甲酚(Cresol)65 交联羧甲纤维素钠(Croscarmellose Sodium)66 交联聚维酮(Crospovidone)67 环糊精(Cyclodextrins)68 环甲基硅酮(Cyclomethicone)69 苯甲地那铵(Denatonium Benzoate)70 葡萄糖结合剂(Dextrates)71 糊精(Dextrin)72 葡萄糖(Dextrose)73 邻苯二甲酸二丁酯(Dibutyl Phthalate)74 癸二酸二丁酯(Dibutyl Sebacate)75 二乙醇胺(Diethanolamine)76 邻苯二甲酸二乙酯(Diethyl Phthalate)77 二氟乙烷(Difluoroethane)78 二甲硅油(Dimethicone)79 二甲醚(Dimethyl Ether)80 邻苯二甲酸二甲酯(Dimethyl Phthalate)81 二甲亚砜(Dimethyl Sulfoxide)82 多库酯钠(Docusate Sodium)83 依地酸(乙二胺四乙酸)(Edetic Acid)84 乙酸乙酯(Ethyl Acetate)85 乙基麦芽酚(Ethyl Maltol)86 油酸乙酯(Ethyl Oleate)87 乙基香草醛(Ethyl Vanillin)88 乙基纤维素(Ethylcellulose)89 硬脂酸棕榈酸乙二醇酯(Ethylene Glycol Palmitostearate)90 羟苯乙酯(Ethylparaben)91 果糖(Fructose)92 富马酸(Fumaric Acid)93 明胶(Gelatin)94 液体葡萄糖(Glucose,Liquid)95 甘油(Glycerin)96 山萮酸甘油酯(Glyceryl Behenate)97 单油酸甘油酯(Glyceryl Monooleate)98 单硬脂酸甘油酯(Glyceryl Monostearate)99 硬脂酸棕榈酸甘油酯(Glyceryl Palmitostearate) 100 四氢呋喃聚乙二醇醚(Glycofurol)101 瓜耳胶(Guar Gum)102 七氟丙烷(HFC)(Heptafluoro-propane)103 海克西定(Hexetidine)104 烷烃类(HC) (Hydrocarbons)105 盐酸(Hydrochloric Acid)106 羟乙纤维素(Hydroxyethyl Cellulose)107 羟乙甲纤维素(Hydroxyethylmethyl Cellulose) 108 羟丙纤维素(Hydroxypropyl Cellulose)109 低取代羟丙纤维素(Hydroxypropyl Cellulose,Low-substituted)110 羟丙甲纤维素(Hypromellose)111 羟丙甲纤维素酞酸酯(Hypromellose Phthalate)112 咪唑烷脲(Imidurea)113 异丙醇(Isopropyl Alcohol)114 肉豆蔻酸异丙酯(Isopropyl Myristate)115 棕榈酸异丙酯(Isopropyl Palmitate)116 白陶土(Kaolin)117 乳酸(Lactic Acid)118 拉克替醇(Lactitol)119 乳糖(Lactose)120 羊毛脂(Lanolin)121 含水羊毛脂(Lanolin,Hydrous)122 羊毛醇(Lanolin Alcohols)123 卵磷脂(Lecithin)124 硅酸镁铝(Magnesium Aluminum Silicate) 125 碳酸镁(Magnesium Carbonate)126 氧化镁(Magnesium Oxide)127 硅酸镁(Magnesium Silicate)128 硬脂酸镁(Magnesium Stearate)129 三硅酸镁(Magnesium Trisilicate)130 苹果酸(Malic Acid)131 麦芽糖醇(Maltitol)132 麦芽糖醇溶液(Maltitol Solution)133 麦芽糖糊精(Maltodextrin)134 麦芽酚(Maltol)135 麦芽糖(Maltose)136 甘露醇(Mannitol)137 中链脂肪酸甘油三酯(Medium-chain Triglycerides) 138 葡甲胺(Meglumine)139 薄荷脑(Menthol)140 甲基纤维素(Methylcellulose)141 羟苯甲酯(Methylparaben)142 液体石蜡(Mineral Oil)143 轻质液体石蜡(Mineral Oil,Light)144 液体石蜡羊毛醇(Mineral Oil and Lanolin Alcohols) 145 单乙醇胺(Monoethanolamine)146 谷氨酸一钠(Monosodium Glutamate)147 硫代甘油(Monothioglycerol)148 氮(Nitrogen)149 一氧化二氮(Nitrous Oxide)150 油酸(Oleic Acid)151 橄榄油(Olive Oil)152 石蜡(Paraffin)153 花生油(Peanut Oil)154 凡士林(Petrolatum)155 凡士林羊毛醇(Petrolatum and Lanolin Alcohols) 156 苯酚(Phenol)157 苯氧乙醇(Phenoxyethanol)158 苯乙醇(Phenylethyl Alcohol)159 醋酸苯汞(Phenylmercuric Acetate)160 硼酸苯汞(Phenylmercuric Borate)161 硝酸苯汞(Phenylmercuric Nitrate)162 磷酸(Phosphoric Acid)163 波拉克林钾(Polacrilin Potassium)164 泊洛沙姆(Poloxamer)165 葡聚糖(Polydextrose)166 聚乙二醇(Polyethylene Glycol)167 聚氧乙烯(Polyethylene Oxide)168 聚(甲基)丙烯酸树脂(Polymethacr-ylates)169 聚氧乙烯烷基醚(Polyoxyethylene Alkyl Ethers)170 聚氧乙烯蓖麻油衍生物(Polyoxyeth-ylene Castor Oil Derivatives)171 聚山梨酯(Polyoxyethylene Sorbitan Fatty Acid Esters) 172 硬脂酸聚氧乙烯酯(Polyoxyethylene Stearates)173 聚醋酸乙烯酞酸酯(Polyvinyl Acetate Phthalate)174 聚乙烯醇(Polyvinyl Alcohol)175 苯甲酸钾(Potassium Benzoate)176 碳酸氢钾(Potassium Bicarbonate)177 氯化钾(Potassium Chloride)178 枸橼酸钾(Potassium Citrate)179 氢氧化钾(Potassium Hydroxide)180 焦亚硫酸钾(Potassium Metabisulfite)181 山梨酸钾(Potassium Sorbate)182 聚维酮(Povidone)183 丙酸(Propionic Acid)184 没食子酸丙酯(Propyl Gallate)185 碳酸丙烯酯(Propylene Carbonate)186 丙二醇(Propylene Glycol)187 海藻酸丙二醇酯(Propylene Glycol Alginate)188 羟苯丙酯(Propylparaben)189 糖精(Saccharin)190 糖精钠(Saccharin Sodium)191 芝麻油(Sesame Oil)192 虫胶(Shellac)193 二氧化硅二甲硅油(Simethicone)194 海藻酸钠(Sodium Alginate)195 抗坏血酸钠(Sodium Ascorbate)196 苯甲酸钠(Sodium Benzoate)197 碳酸氢钠(Sodium Bicarbonate)198 氯化钠(Sodium Chloride)199 枸橼酸钠二水合物(Sodium Citrate Dihydrate)200 环拉酸钠(Sodium Cyclamate)201 氢氧化钠(Sodium Hydroxide)202 月桂硫酸钠(十二烷基硫酸钠)(Sodium Lauryl Sulfate)203 焦亚硫酸钠(偏亚硫酸钠)(Sodium Metabisulfite)204 磷酸氢二钠(Sodium Phosphate,Dibasic)205 磷酸二氢钠(Sodium Phosphate ,Monobasic)206 丙酸钠(Sodium Propionate)207 羧甲淀粉钠(Sodium Starch Glycolate)208 硬脂富马酸钠(Sodium Stearyl Fumarate)209 山梨酸(Sorbic Acid)210 山梨坦酯Sorbitan Esters(Sorbitan Fatty Acid Esters) 211 山梨醇(Sorbitol)212 大豆油(Soybean Oil)213 淀粉(Starch)214 预胶化淀粉(Starch,Pregelatinized)215 灭菌玉米淀粉(Starch,Sterilizable Maize)216 硬脂酸(Stearic Acid)217 硬脂醇(Stearyl Alcohol)218 羟糖氯(Sucralose)219 蔗糖(Sucrose)220 可压性蔗糖(Sugar,Compressible)221 蔗糖粉(Sugar,Confectioner’s)222 蔗糖球形颗粒(Sugar Spheres)223 硫酸(Sulfuric Acid)224 葵花籽油(Sunflower Oil)225 氢化植物油(硬脂)栓剂基质(Sup-pository Bases,Hard Fat)226 滑石粉(Talc)227 酒石酸(Tartaric Acid)228 四氟乙烷(HFC)(Tetrafluoroe-thane)229 硫柳汞(Thimerosal)230 二氧化钛(Titanium Dioxide)231 西黄蓍胶(Tragacanth)232 海藻糖(Trehalose)233 三醋汀(Triacetin)234 枸橼酸三丁酯(Tributyl Citrate)235 三乙醇胺(Triethanolamine)236 枸橼酸三乙酯(Triethyl Citrate)237 香草醛(Vanillin)238 氢化植物油(Vegetable Oil,Hydrogenated) 239 水(Water)240 阴离子乳化蜡(Wax,Anionic Emulsifying) 241 巴西棕榈蜡(Wax,Carnauba)242 十六醇酯蜡(Wax,Cetyl Esters)243 微晶蜡(Wax,Microcrystalline)244 非离子乳化蜡(聚西托醇乳化蜡)(Wax,Nonionic Emulsifying)245 白蜡(Wax,White)246 黄蜡(Wax,Yellow)247 黄原酸胶(Xanthan Gum)248 木糖醇(Xylitol)796249 玉米朊(玉米蛋白)(Zein) 250 硬脂酸锌(Zinc Stearate)。
常用蛋白酶切割位点
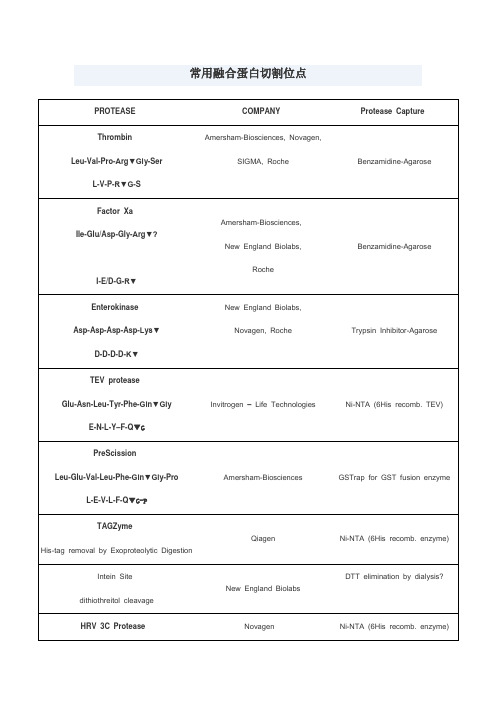
4.溴化氰处理,专一性的切割甲硫氨酸羧基端的肽键。
SIGMA, Roche
Benzamidine-Agarose
Factor Xa
Ile-Glu/Asp-Gly-Arg▼?
I-E/D-G-R▼
Amersham-Biosciences,
New England Biolabs,
Roche
Benzamidine-Agarose
Enterokinase
Asp-Asp-Asp-Asp-Lys▼
羧肽酶
羧肽酶B可以切割C端的Lys或Arg;羧肽酶A可以切割C端除了Lys、Arg、Pro的氨基酸,但如果倒数第二个氨基酸为Pro两种羧肽酶均不能作用
1.胰蛋白酶属肽链内切酶,能把多肽链中Lys和Arg残基中的羧基侧切断。
2.胰凝乳蛋白酶(亦称糜蛋白酶)属肽链内切酶,主要切断多肽链中的芳香族氨基酸(Phe、Trp、Tyr)残基的羧基一侧。
LifeSensors
Ni-NTA (6His recomb. enzyme)
Kex-2
-Arg-X-Lys/Arg-Arg▼
Invitrogen – Life Technologies,
Ni-NTA (6His recomb. enzyme)
KEX2对arg的专一性高,要求最重要。
Arg前为lys效率最高,不切-Arg-lys,Pro影响KEX2切割
Ni-NTA (6His recomb. TEV)
PreScission
Leu-Glu-Val-Leu-Phe-Gln▼Gly-Pro
艾塞那肽FDA说明书

HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use BYETTA safely and effectively. See full prescribing information for BYETTA.BYETTA® (exenatide) InjectionInitial U.S. Approval: 2005--------------------------RECENT MAJOR CHANGES------------------------Indications and Usage 10/2009Monotherapy and Combination Therapy (1.1)Important Limitations of Use 10/2009History of Pancreatitis (1.2)Warnings and Precautions 10/2009Pancreatitis (5.1)Renal Impairment (5.3)Macrovascular Outcomes (5.7)---------------------------INDICATIONS AND USAGE--------------------------- BYETTA is a glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.Important Limitations of Use• BYETTA is not a substitute for insulin. BYETTA should not be used in patients with type 1 diabetes or for the treatment of diabetic ketoacidosis(1.2).• The concurrent use of BYETTA with insulin has not been studied and cannot be recommended (1.2).• BYETTA has not been studied in patients with a history of pancreatitis.Consider other antidiabetic therapies in patients with a history ofpancreatitis (1.2).-------------------------DOSAGE AND ADMINISTRATION------------------- • Inject subcutaneously within 60 minutes prior to morning and evening meals (or before the two main meals of the day, approximately 6 hours or more apart) (2.1).• Initiate at 5 mcg per dose twice daily; increase to 10 mcg twice daily after 1 month based on clinical response (2.1).------------------------DOSAGE FORMS AND STRENGTHS------------------- BYETTA is supplied as 250 mcg/mL exenatide in:• 5 mcg per dose, 60 doses, 1.2 mL prefilled pen• 10 mcg per dose, 60 doses, 2.4 mL prefilled pen-----------------------------CONTRAINDICATIONS------------------------------ • History of severe hypersensitivity to exenatide or any product components(4.1).-----------------------WARNINGS AND PRECAUTIONS----------------------- • Pancreatitis: Postmarketing reports, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis. Discontinue BYETTA promptly.FULL PRESCRIBING INFORMATION: CONTENTS*1 INDICATIONS AND USAGE1.1 Type 2 Diabetes Mellitus1.2 Important Limitations of Use2 DOSAGE AND ADMINISTRATION2.1 Recommended Dosing3 DOSAGE FORMS AND STRENGTHS4 CONTRAINDICATIONS4.1 Hypersensitivity5 WARNINGS AND PRECAUTIONS5.1 Acute Pancreatitis5.2 Hypoglycemia5.3 Renal Impairment5.4 Gastrointestinal Disease5.5 Immunogenicity5.6 Hypersensitivity5.7 Macrovascular Outcomes6 ADVERSE REACTIONS6.1 Clinical Trial Experience6.2 Post-Marketing Experience7 DRUG INTERACTIONS7.1 Orally Administered Drugs7.2 Warfarin8 USE IN SPECIFIC POPULATIONS8.1 Pregnancy8.3 Nursing MothersBYETTA should not be restarted. Consider other antidiabetic therapies in patients with a history of pancreatitis (5.1).• Hypoglycemia: Increased risk when BYETTA is used in combination witha sulfonylurea. Consider reducing the sulfonylurea dose (5.2).• Renal Impairment: Postmarketing reports, sometimes requiring hemodialysis and kidney transplantation. BYETTA should not be used in patients with severe renal impairment or end-stage renal disease and should be used with caution in patients with renal transplantation. Caution should be applied when initiating BYETTA or escalating the dose of BYETTA in patients with moderate renal failure (5.3).• Severe Gastrointestinal Disease: Use of BYETTA is not recommended in patients with severe gastrointestinal disease (e.g., gastroparesis) (5.4). • Hypersensitivity: Postmarketing reports of hypersensitivity reactions (e.g.anaphylaxis and angioedema). The patient should discontinue BYETTA and other suspect medications and promptly seek medical advice (5.6). • There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with BYETTA or any other antidiabetic drug(5.7).-----------------------------ADVERSE REACTIONS------------------------------- • Most common (≥5%) and occurring more frequently than placebo in clinical trials: nausea, hypoglycemia, vomiting, diarrhea, feeling jittery, dizziness, headache, dyspepsia. Nausea usually decreases over time (5.2;6).• Postmarketing reports of increased international normalized ratio (INR) with concomitant use of warfarin, sometimes with bleeding (6.2).To report SUSPECTED ADVERSE REACTIONS contact Amylin Pharmaceuticals, Inc. and Eli Lilly and Company at 1-800-868-1190 and or FDA at 1-800-FDA-1088 or /medwatch ------------------------------DRUG INTERACTIONS------------------------------ • Warfarin: Postmarketing reports of increased INR sometimes associated with bleeding. Monitor INR frequently until stable upon initiation oralteration of BYETTA therapy (7.2).-------------------------USE IN SPECIFIC POPULATIONS--------------------- • Pregnancy: Based on animal data, BYETTA may cause fetal harm.BYETTA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. To report drug exposure duringpregnancy call 1-800-633-9081 (8.1).• Nursing Mothers: Caution should be exercised when BYETTA is administered to a nursing woman (8.3).See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.Revised: 10/20098.4 Pediatric Use8.5 Geriatric Use8.6 Renal Impairment8.7 Hepatic Impairment10 OVERDOSAGE11 DESCRIPTION12 CLINICAL PHARMACOLOGY12.1 Mechanism of Action12.2 Pharmacodynamics12.3 Pharmacokinetics13 NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, Mutagenesis, Impairment ofFertility13.3 Reproductive and Developmental Toxicology14 CLINICAL STUDIES14.1 Monotherapy14.2 Combination Therapy16 HOW SUPPLIED/STORAGE AND HANDLING16.1 How Supplied16.2 Storage and Handling17 PATIENT COUNSELING INFORMATION* Sections or subsections omitted from the full prescribing information are not listed.FULL PRESCRIBING INFORMATION1 INDICATIONS AND USAGE1.1 Type 2 Diabetes MellitusBYETTA is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.1.2 Important Limitations of UseBYETTA is not a substitute for insulin. BYETTA should not be used in patients with type 1 diabetes or for the treatment of diabetic ketoacidosis, as it would not be effective in these settings.The concurrent use of BYETTA with insulin has not been studied and cannot be recommended.Based on postmarketing data BYETTA has been associated with acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis. BYETTA has not been studied in patients with a history of pancreatitis. It is unknown whether patients with a history of pancreatitis are at increased risk for pancreatitis while using BYETTA. Other antidiabetic therapies should be considered in patients with a history of pancreatitis.2 DOSAGE AND ADMINISTRATION2.1 Recommended DosingBYETTA should be initiated at 5 mcg administered twice daily at any time within the 60-minute period before the morning and evening meals (or before the two main meals of the day, approximately 6 hours or more apart). BYETTA should not be administered after a meal. Based on clinical response, the dose of BYETTA can be increased to 10 mcg twice daily after 1 month of therapy. Initiation with 5 mcg reduces the incidence and severity of gastrointestinal side effects. Each dose should be administered as a subcutaneous (SC) injection in the thigh, abdomen, or upper arm. No data are available on the safety or efficacy of intravenous or intramuscular injection of BYETTA.Use BYETTA only if it is clear, colorless and contains no particles.3 DOSAGE FORMS AND STRENGTHSBYETTA is supplied as a sterile solution for subcutaneous injection containing 250 mcg/mL exenatide in the following packages:• 5 mcg per dose, 60 doses, 1.2 mL prefilled pen• 10 mcg per dose, 60 doses, 2.4 mL prefilled peni p i a 4 CONTRAINDICATIONS4.1 HypersensitivityBYETTA is contraindicated in patients with prior severe hypersensitivity reactions to exenatide or to any of the product components.5 WARNINGS AND PRECAUTIONS5.1 Acute PancreatitisBased on postmarketing data BYETTA has been associated with acute pancreatitis,ncluding fatal and non-fatal hemorrhagic or necrotizing pancreatitis. After initiation ofBYETTA, and after dose increases, observe patients carefully for signs and symptoms ofancreatitis (including persistent severe abdominal pain, sometimes radiating to the back, which may or may not be accompanied by vomiting). If pancreatitis is suspected,BYETTA should promptly be discontinued and appropriate management should benitiated. If pancreatitis is confirmed, BYETTA should not be restarted. Considerntidiabetic therapies other than BYETTA in patients with a history of pancreatitis.5.2 HypoglycemiaThe risk of hypoglycemia is increased when BYETTA is used in combination with asulfonylurea (hypoglycemia can also occur when other antidiabetic agents are used incombination with a sulfonylurea). Therefore, patients receiving BYETTA and a sulfonylureamay require a lower dose of the sulfonylurea to reduce the risk of hypoglycemia. It is alsopossible that the use of BYETTA with other glucose-independent insulin secretagogues (e.g.meglitinides) could increase the risk of hypoglycemia.For additional information on glucose dependent effects see Mechanism of Action (12.1).5.3 Renal ImpairmentBYETTA should not be used in patients with severe renal impairment (creatinine clearance< 30 mL/min) or end-stage renal disease and should be used with caution in patients with renaltransplantation [see Use in Specific Populations (8.6)]. In patients with end-stage renal disease receiving dialysis, single doses of BYETTA 5 mcg were not well-tolerated due to gastrointestinal side effects. Because BYETTA may induce nausea and vomiting with transient hypovolemia,treatment may worsen renal function. Caution should be applied when initiating or escalatingdoses of BYETTA from 5 mcg to 10 mcg in patients with moderate renal impairment (creatinine clearance 30 to 50 mL/min).There have been postmarketing reports of altered renal function, including increased serumcreatinine, renal impairment, worsened chronic renal failure and acute renal failure, sometimesrequiring hemodialysis or kidney transplantation. Some of these events occurred in patientsreceiving one or more pharmacologic agents known to affect renal function or hydration status, such as angiotensin converting enzyme inhibitors, nonsteroidal anti-inflammatory drugs, or diuretics. Some events occurred in patients who had been experiencing nausea, vomiting, or diarrhea, with or without dehydration. Reversibility of altered renal function has been observed in many cases with supportive treatment and discontinuation of potentially causative agents, including BYETTA. Exenatide has not been found to be directly nephrotoxic in preclinical or clinical studies.5.4 Gastrointestinal DiseaseBYETTA has not been studied in patients with severe gastrointestinal disease, including gastroparesis. Because BYETTA is commonly associated with gastrointestinal adverse reactions, including nausea, vomiting, and diarrhea, the use of BYETTA is not recommended in patients with severe gastrointestinal disease.5.5 ImmunogenicityPatients may develop antibodies to exenatide following treatment with BYETTA, consistent with the potentially immunogenic properties of protein and peptide pharmaceuticals. In a small proportion of patients, the formation of antibodies to exenatide at high titers could result in failure to achieve adequate improvement in glycemic control. If there is worsening glycemic control or failure to achieve targeted glycemic control, alternative antidiabetic therapy should be considered [see Adverse Reactions (6.1)].5.6 HypersensitivityThere have been postmarketing reports of serious hypersensitivity reactions (e.g. anaphylaxis and angioedema) in patients treated with BYETTA. If a hypersensitivity reaction occurs, the patient should discontinue BYETTA and other suspect medications and promptly seek medical advice [see Adverse Reactions (6.2)].5.7 Macrovascular OutcomesThere have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with BYETTA or any other antidiabetic drug.6 ADVERSE REACTIONS6.1 Clinical Trial ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.HypoglycemiaTable 1 summarizes the incidence and rate of hypoglycemia with BYETTA in five placebo-controlled clinical trials.Table 1: Incidence (%) and Rate of Hypoglycemia When BYETTA was Used as Monotherapy or WithConcomitant Antidiabetic Therapy in Five Placebo-Controlled Clinical Trials*BYETTAPlacebo twice daily 5 mcg twice daily 10 mcg twice dailyMonotherapy (24 Weeks)77 77 78N%1.3% 5.2% 3.8%OverallRate0.03 0.21 0.52 (episodes/patientyear)% Severe 0.0% 0.0% 0.0%With Metformin (30 Weeks)N 113 110 1135.3% 4.5% 5.3%Overall%Rate0.12 0.13 0.12 (episodes/patientyear)% Severe 0.0% 0.0% 0.0%With a Sulfonylurea (30 Weeks)N 123 125 129 % Overall 3.3% 14.4% 35.7%Rate0.07 0.64 1.61 (episodes/patientyear)% Severe 0.0% 0.0% 0.0%With Metformin and a Sulfonylurea (30 Weeks)N 247 245 241 % Overall 12.6% 19.2% 27.8%Rate0.58 0.78 1.71 (episodes/patientyear)% Severe 0.0% 0.4% 0.0%With a Thiazolidinedione (16 Weeks)N 112 Dose not studied 121% Overall 7.1% Dose not studied 10.7%Rate0.56 Dose not studied 0.98(episodes/patientyears)% Severe 0.0% Dose not studied 0.0%* For the 30-week trials, a hypoglycemia episode was recorded if the patient reported symptoms consistent with hypoglycemia and was recorded as severe if the subject required the assistance of another person to treat theevent. For the other trials, a hypoglycemic episode was recorded if a patient reported signs or symptoms ofhypoglycemia or had a blood glucose value consistent with hypoglycemia regardless of associated symptoms ortreatment and was recorded as severe if the subject required the assistance of another person to treat the event.The requirement for assistance had to be accompanied by a blood glucose measurement of <50 mg/dL orprompt recovery after administration of oral carbohydrate.N = The number of Intent-to-Treat subjects in each treatment group.Immunogenicitypatients had low titer antibodies to exenatide at 30 weeks. For this group, the level of glycemic control (hemoglobin A1c [HbA1c]) was generally comparable to that observed in those without antibody titers. An additional 6% of patients had higher titer antibodies at 30 weeks. In about half of this 6% (3% of the total patients given BYETTA in the 30-week controlled studies), the glycemic response to BYETTA was attenuated; the remainder had a glycemic response comparable to that of patients without antibodies.In the 16-week trial of BYETTA add-on to thiazolidinediones, with or without metformin, 9% of patients had higher titer antibodies at 16 weeks. In the 24-week trial of BYETTA used as monotherapy, 3% of patients had higher titer antibodies at 24 weeks. Compared with patients who did not develop antibodies to BYETTA, on average the glycemic response in patients with higher titer antibodies was attenuated [see Warnings and Precautions (5.5)].Other Adverse ReactionsMonotherapyFor the 24-week placebo-controlled study of BYETTA used as a monotherapy, Table 2 summarizes adverse reactions (excluding hypoglycemia) occurring with an incidence ≥2% and occurring more frequently in BYETTA-treated patients compared with placebo-treated patients. Table 2: Treatment-Emergent Adverse Reactions ≥2% Incidence With BYETTA Used as Monotherapy (Excluding Hypoglycemia)*Monotherapy Placebo BIDN = 77%All BYETTA BIDN = 155%Nausea 0 8Vomiting 0 4Dyspepsia 0 3 * In a 24-week placebo-controlled trial.BID = twice daily.Adverse reactions reported in ≥1.0 to <2.0% of patients receiving BYETTA and reported more frequently than with placebo included decreased appetite, diarrhea, and dizziness. The most frequently reported adverse reaction associated with BYETTA, nausea, occurred in a dose-dependent fashion.Two of the 155 patients treated with BYETTA withdrew due to adverse reactions of headache and nausea. No placebo-treated patients withdrew due to adverse reactions.Combination TherapyAdd-on to metformin and/or sulfonylureaadverse reactions (excluding hypoglycemia) with an incidence ≥2% and occurring more frequently in BYETTA-treated patients compared with placebo-treated patients [see Warnings and Precautions (5.2)] are summarized in Table 3.Table 3: Treatment-Emergent Adverse Reactions ≥2% Incidence and Greater Incidence With BYETTA Treatment Used With Metformin and/or a Sulfonylurea (Excluding Hypoglycemia)*Placebo BID N = 483% All BYETTA BIDN = 963%Nausea 18 44Vomiting 4 13Diarrhea 6 13 Feeling Jittery 4 9Dizziness 6 9Headache 6 9Dyspepsia 3 6Asthenia 2 4 Gastroesophageal RefluxDisease1 3Hyperhidrosis 1 3 * In three 30-week placebo-controlled clinical trials.BID = twice daily.Adverse reactions reported in ≥1.0 to <2.0% of patients receiving BYETTA and reported morefrequently than with placebo included decreased appetite. Nausea was the most frequentlyreported adverse reaction and occurred in a dose-dependent fashion. With continued therapy, the frequency and severity decreased over time in most of the patients who initially experiencednausea. Patients in the long-term uncontrolled open-label extension studies at 52 weeks reportedno new types of adverse reactions than those observed in the 30-week controlled trials.The most common adverse reactions leading to withdrawal for BYETTA-treated patients werenausea (3% of patients) and vomiting (1%). For placebo-treated patients, <1% withdrew due tonausea and none due to vomiting.Add-on to thiazolidinedione with or without metforminFor the 16-week placebo-controlled study of BYETTA add-on to a thiazolidinedione, with orwithout metformin, Table 4 summarizes the adverse reactions (excluding hypoglycemia) with an incidence of ≥2% and occurring more frequently in BYETTA-treated patients compared withplacebo-treated patients.Table 4: Treatment-Emergent Adverse Reactions ≥2% Incidence With BYETTA Used With a Thiazolidinedione, With or Without Metformin (Excluding Hypoglycemia)*With a TZD or TZD/MET PlaceboN = 112%All BYETTA BIDN = 121%Nausea 15 40Vomiting 1 13Dyspepsia 1 7Diarrhea 3 6 GastroesophagealReflux Disease0 3* In a 16-week placebo-controlled clinical trial.BID = twice daily.Adverse reactions reported in ≥1.0 to <2.0% of patients receiving BYETTA and reported more frequently than with placebo included decreased appetite. Chills (n = 4) and injection-sitereactions (n = 2) occurred only in BYETTA-treated patients. The two patients who reported an injection-site reaction had high titers of antibodies to exenatide. Two serious adverse events(chest pain and chronic hypersensitivity pneumonitis) were reported in the BYETTA arm. Noserious adverse events were reported in the placebo arm.The most common adverse reactions leading to withdrawal for BYETTA-treated patients werenausea (9%) and vomiting (5%). For placebo-treated patients, <1% withdrew due to nausea.6.2 Post-Marketing ExperienceThe following additional adverse reactions have been reported during post-approval use of BYETTA. Because these events are reported voluntarily from a population of uncertain size, itis generally not possible to reliably estimate their frequency or establish a causal relationship todrug exposure.Allergy/Hypersensitivity: injection-site reactions, generalized pruritus and/or urticaria, macularor papular rash, angioedema, anaphylactic reaction [see Warnings and Precautions (5.6)].Drug Interactions: International normalized ratio (INR) increased with concomitant warfarin use sometimes associated with bleeding [see Drug Interactions (7.2)].Gastrointestinal: nausea, vomiting, and/or diarrhea resulting in dehydration; abdominaldistension, abdominal pain, eructation, constipation, flatulence, acute pancreatitis, hemorrhagicand necrotizing pancreatitis sometimes resulting in death [see Limitations of Use (1.2) andWarnings and Precautions (5.1)].Neurologic: dysgeusia; somnolenceRenal and Urinary Disorders: altered renal function, including increased serum creatinine, renal impairment, worsened chronic renal failure or acute renal failure (sometimes requiringhemodialysis), kidney transplant and kidney transplant dysfunction [see Warnings and Precautions (5.3)].7 DRUG INTERACTIONS7.1 Orally Administered DrugsThe effect of BYETTA to slow gastric emptying can reduce the extent and rate of absorption of orally administered drugs. BYETTA should be used with caution in patients receiving oral medications that have narrow therapeutic index or require rapid gastrointestinal absorption [see Adverse Reactions (6.2)]. For oral medications that are dependent on threshold concentrations for efficacy, such as contraceptives and antibiotics, patients should be advised to take those drugs at least 1 hour before BYETTA injection. If such drugs are to be administered with food, patients should be advised to take them with a meal or snack when BYETTA is not administered [see Clinical Pharmacology (12.3)].7.2 WarfarinThere are postmarketing reports of increased INR sometimes associated with bleeding, with concomitant use of warfarin and BYETTA [see Adverse Reactions (6.2)]. In a drug interaction study, BYETTA did not have a significant effect on INR [see Clinical Pharmacology (12.3)]. In patients taking warfarin, prothrombin time should be monitored more frequently after initiation or alteration of BYETTA therapy. Once a stable prothrombin time has been documented, prothrombin times can be monitored at the intervals usually recommended for patients on warfarin.8 USE IN SPECIFIC POPULATIONS8.1 PregnancyPregnancy Category CThere are no adequate and well-controlled studies of BYETTA use in pregnant women. In animal studies, exenatide caused cleft palate, irregular skeletal ossification and an increased number of neonatal deaths. BYETTA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.Female mice given SC doses of 6, 68, or 760 mcg/kg/day beginning 2 weeks prior to and throughout mating until gestation day 7 had no adverse fetal effects. At the maximal dose,760 mcg/kg/day, systemic exposures were up to 390 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC [see Nonclinical Toxicology (13.3)].In developmental toxicity studies, pregnant animals received exenatide subcutaneously during organogenesis. Specifically, fetuses from pregnant rabbits given SC doses of 0.2, 2, 22, 156, or260 mcg/kg/day from gestation day 6 through 18 experienced irregular skeletal ossifications from exposures 12 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC. Moreover, fetuses from pregnant mice given SC doses of 6, 68, 460, or 760 mcg/kg/day from gestation day 6 through 15 demonstrated reduced fetal and neonatal growth, cleft palate and skeletal effects at systemic exposure 3 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC [see Nonclinical Toxicology (13.3)].Lactating mice given SC doses of 6, 68, or 760 mcg/kg/day from gestation day 6 through lactation day 20 (weaning), experienced an increased number of neonatal deaths. Deaths were observed on postpartum days 2-4 in dams given 6 mcg/kg/day, a systemic exposure 3 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC [see Nonclinical Toxicology (13.3)].Pregnancy RegistryAmylin Pharmaceuticals, Inc. maintains a Pregnancy Registry to monitor pregnancy outcomes of women exposed to exenatide during pregnancy. Physicians are encouraged to register patients by calling 1-800-633-9081.8.3 Nursing MothersIt is not known whether exenatide is excreted in human milk. However, exenatide is present at low concentrations (less than or equal to 2.5% of the concentration in maternal plasma following subcutaneous dosing) in the milk of lactating mice. Many drugs are excreted in human milk and because of the potential for clinically significant adverse reactions in nursing infants from exenatide, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account these potential risks against the glycemic benefits to the lactating woman. Caution should be exercised when BYETTA is administered to a nursing woman.8.4 Pediatric UseSafety and effectiveness of BYETTA have not been established in pediatric patients.8.5 Geriatric UsePopulation pharmacokinetic analysis of patients ranging from 22 to 73 years of age suggests that age does not influence the pharmacokinetic properties of exenatide [see Clinical Pharmacology (12.3)]. BYETTA was studied in 282 patients 65 years of age or older and in 16 patients75 years of age or older. No differences in safety or effectiveness were observed between these patients and younger patients. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection in the elderly based on renal function.8.6 Renal ImpairmentBYETTA is not recommended for use in patients with end-stage renal disease or severe renal impairment (creatinine clearance < 30 mL/min) and should be used with caution in patients with renal transplantation. No dosage adjustment of BYETTA is required in patients with mild renal impairment (creatinine clearance 50 to 80 mL/min). Caution should be applied when initiating or escalating doses of BYETTA from 5 mcg to 10 mcg in patients with moderate renal impairment (creatinine clearance 30 to 50 mL/min) [see Clinical Pharmacology (12.3)].8.7 Hepatic ImpairmentNo pharmacokinetic study has been performed in patients with a diagnosis of acute or chronic hepatic impairment. Because exenatide is cleared primarily by the kidney, hepatic dysfunction is not expected to affect blood concentrations of exenatide [see Clinical Pharmacology (12.3)].10 OVERDOSAGEIn a clinical study of BYETTA, three patients with type 2 diabetes each experienced a single overdose of 100 mcg SC (10 times the maximum recommended dose). Effects of the overdoses included severe nausea, severe vomiting, and rapidly declining blood glucose concentrations. One of the three patients experienced severe hypoglycemia requiring parenteral glucose administration. The three patients recovered without complication. In the event of overdose, appropriate supportive treatment should be initiated according to the patient’s clinical signs and symptoms.11 DESCRIPTIONBYETTA (exenatide) is a synthetic peptide that was originally identified in the lizard Heloderma suspectum. Exenatide differs in chemical structure and pharmacological action from insulin, sulfonylureas (including D-phenylalanine derivatives and meglitinides), biguanides, thiazolidinediones, alpha-glucosidase inhibitors, amylinomimetics and dipeptidyl peptidase-4 inhibitors.Exenatide is a 39-amino acid peptide amide. Exenatide has the empirical formulaC184H282N50O60S and molecular weight of 4186.6 Daltons. The amino acid sequence for exenatide is shown below.H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu -Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-NH2BYETTA is supplied for SC injection as a sterile, preserved isotonic solution in a glass cartridge that has been assembled in a pen-injector (pen). Each milliliter (mL) contains 250 micrograms (mcg) synthetic exenatide, 2.2 mg metacresol as an antimicrobial preservative, mannitol as a tonicity-adjusting agent, and glacial acetic acid and sodium acetate trihydrate in water for。
注射用头孢洛林酯说明书(美国,英文)

1 23 4 5 678 91011 12 1314151617 18192021 22 23242554HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use TEFLARO safely and effectively. See full prescribing information for TEFLARO®.TEFLARO® (ceftaroline fosamil) injection for intravenous (IV) use Initial U.S. Approval: 2010To reduce the development of drug-resistant bacteria and maintain theeffectiveness of Teflaro and other antibacterial drugs, Teflaro should be used onlyto treat infections that are proven or strongly suspected to be caused by bacteria.-----------------------RECENT MAJOR CHANGES------------------------------------ Dosage and Administration (2.3) XX/2012 --------------------------INDICATIONS AND USAGE--------------------------------Teflaro ® is a cephalosporin antibacterial indicated for the treatment of the following infections caused by designated susceptible bacteria:• Acute bacterial skin and skin structure infections (ABSSSI)(1.1) • Community-acquired bacterial pneumonia (CABP) (1.2) ------------------------DOSAGE AND ADMINISTRATION-------------------------• 600 mg every 12 hours by IV infusion administered over 1 hour in adults ≥ 18 years of age (2.1) • Dosage adjustment in patients with renal impairment (2.2)Estimated CreatinineClearance # (mL/min) Teflaro Dosage Regimen > 50 No dosage adjustment necessary > 30 to ≤ 50 400 mg IV (over 1 hour) every 12 hours ≥ 15 to ≤ 30 300 mg IV (over 1 hour) every 12 hours End-stage renal disease (ESRD), including hemodialysis200 mg IV (over 1 hour) every 12 hours#As calculated using the Cockcroft-Gault formula -----------------------DOSAGE FORMS AND STRENGTHS -----------------------600 mg or 400 mg of sterile Teflaro powder in single-use 20 mL vials. (3) 26--------------------------CONTRAINDICATIONS---------------------------- 27 ∙Known serious hypersensitivity to ceftaroline or other members of28 the cephalosporin class. (4)29 -----------------------WARNINGS AND PRECAUTIONS----------------- 30 ∙Serious hypersensitivity (anaphylactic) reactions have been31 reported with beta-lactam antibiotics, including ceftaroline. 32 Exercise caution in patients with known hypersensitivity to beta33 lactam antibiotics. (5.1) 34 ∙Clostridium difficile -associated diarrhea (CDAD) has been35 reported with nearly all systemic antibacterial agents, including36 Teflaro. Evaluate if diarrhea occurs. (5.2)37 ∙Direct Coombs’ test seroconversion has been reported with38 Teflaro. If anemia develops during or after therapy, a diagnostic 39 workup for drug-induced hemolytic anemia should be performed 40 and consideration given to discontinuation of Teflaro. (5.3) 41 -----------------------------ADVERSE REACTIONS------------------------42 The most common adverse reactions occurring in >2 % of patients are 43 diarrhea, nausea, and rash. (6.3) 44 45 To report SUSPECTED ADVERSE REACTIONS, contact Forest46 Pharmaceuticals, Inc., at 1-800-678-1605 or FDA at 1-800-FDA47 1088 or /medwatch. 48 ---------------------------USE IN SPECIFIC POPULATIONS-------------49 ∙Dosage adjustment is required in patients with moderate or severe50 renal impairment and in ESRD patients, including patients on51 hemodialysis.(2.2, 12.3) 52 53See 17 for PATIENT COUNSELING INFORMATIONRevised:XX/2012 5556 FULL PRESCRIBING INFORMATION: CONTENTS* 84 8.4 Pediatric Use85 8.5 Geriatric Use 57 1 INDICATIONS AND USAGE86 8.6 Patients with Renal Impairment58 1.1 Acute Bacterial Skin and Skin Structure 87 10 OVERDOSAGE59 Infections 88 11 DESCRIPTION 60 1.2 Community-Acquired Bacterial Pneumonia 89 12 CLINICAL PHARMACOLOGY61 1.3 Usage90 12.1 Mechanism of Action62 2 DOSAGE ANDADMINISTRATION 91 12.2 Pharmacodynamics 63 2.1 Recommended Dosage 92 12.3 Pharmacokinetics64 2.2 Patients with Renal Impairment 93 12.4 Microbiology65 2.3 Preparation of Solutions 94 13 NONCLINICAL TOXICOLOGY66 3 DOSAGE FORMS AND STRENGTHS 95 13.1 Carcinogenesis,Mutagenesis, Impairment of 67 4 CONTRAINDICATIONS 96 Fertility 68 5 WARNINGS AND PRECAUTIONS 97 14 CLINICAL TRIALS69 5.1 Hypersensitivity Reactions 98 14.1 Acute Bacterial Skin and Skin Structure70 5.2 Clostridium difficile -associated Diarrhea 99 Infections71 5.3 Direct Coombs’ Test Seroconversion 100 14.2 Community-Acquired Bacterial Pneumonia72 5.4 Development of Drug-Resistant Bacteria 101 15 REFERENCES 73 6 ADVERSE REACTIONS 102 16 HOW SUPPLIED/STORAGE AND HANDLING 74 6.1 Adverse Reactions from Clinical Trials 103 17 PATIENT COUNSELING INFORMATION 75 6.2 Serious Adverse Events and Adverse 10476 Events Leading to Discontinuation 77 6.3 Most Common Adverse Reactions 10578 6.4 Other Adverse Reactions Observed During 106 *Sections or subsections omitted from the full prescribing information79 Clinical Trials of Teflaro107 are not listed.80 7 DRUGINTERACTIONS 81 8 USE IN SPECIFIC POPULATIONS82 8.1 Pregnancy83 8.3 Nursing MothersPage 1 of 13108 FULL PRESCRIBING INFORMATION 109 1. INDICATIONS AND USAGE110 Teflaro® (ceftaroline fosamil) is indicated for the treatment of patients with the following infections caused by susceptible isolates of the designated 111 microorganisms. 112 1.1Acute Bacterial Skin and Skin Structure Infections113 Teflaro is indicated for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible isolates of the following Gram114 positive and Gram-negative microorganisms:Staphylococcus aureus (including methicillin-susceptible and -resistant isolates), Streptococcus pyogenes ,115 Streptococcus agalactiae , Escherichia coli , Klebsiella pneumoniae, and Klebsiella oxytoca. 116 1.2Community-Acquired Bacterial Pneumonia117 Teflaro is indicated for the treatment of community-acquired bacterial pneumonia (CABP) caused by susceptible isolates of the following Gram-positive118 and Gram-negative microorganisms: Streptococcus pneumoniae (including cases with concurrent bacteremia),Staphylococcus aureus (methicillin119 susceptible isolates only), Haemophilus influenzae, Klebsiella pneumoniae, Klebsiella oxytoca, and Escherichia coli. 120 1.3 Usage121 To reduce the development of drug-resistant bacteria and maintain the effectiveness of Teflaro and other antibacterial drugs, Teflaro should be used to 122 treat only ABSSSI or CABP that are proven or strongly suspected to be caused by susceptible bacteria. Appropriate specimens for microbiological123 examination should be obtained in order to isolate and identify the causative pathogens and to determine their susceptibility to ceftaroline. When culture124 and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local125 epidemiology and susceptibility patterns may contribute to the empiric selection of therapy. 126 2. DOSAGE AND ADMINISTRATION 127 2.1Recommended Dosage128 The recommended dosage of Teflaro is 600 mg administered every 12 hours by intravenous (IV) infusion over 1 hour in patients ≥ 18 years of age. The129 duration of therapy should be guided by the severity and site of infection and the patient’s clinical and bacteriological progress. 130 The recommended dosage and administration by infection is described in Table 1.131Table 1: Dosage of Teflaro by InfectionInfection Dosage FrequencyInfusion Time(hours)RecommendedDuration ofTotal Antimicrobial TreatmentAcute Bacterial Skin and Skin Structure Infection(ABSSSI) 600 mg Every 12 hours 1 5-14 days Community-Acquired Bacterial Pneumonia (CABP)600 mg Every 12 hours 1 5-7 days 132133 2.2 Patients with Renal Impairment 134Table 2: Dosage of Teflaro in Patients with Renal Impairment 135 136 137 138 139 140 141Estimated CrCl a (mL/min) Recommended Dosage Regimen for Teflaro> 50No dosage adjustment necessary > 30 to ≤ 50 400 mg IV (over 1 hour) every 12 hours ≥ 15 to ≤ 30 300 mg IV (over 1 hour) every 12 hours End-stage renal disease,including hemodialysis b 200 mg IV (over 1 hour) every 12 hours c ab End-stage renal disease is defined as CrCl < 15 mL/min.cTeflaro is hemodialyzable; thus Teflaro should be administered after hemodialysis on hemodialysis days.2.3 Preparation of SolutionsAseptic technique must be followed in preparing the infusion solution. The contents of Teflaro vial should be constituted with 20 mL Sterile Water forInjection, USP; or 0.9% of sodium chloride injection (normal saline); or 5% of dextrose injection; or lactated ringer’s injection . The preparation of Teflaro solutions is summarized in Table 3.143 Table 3: Preparation of Teflaro for Intravenous UseDosage Strength(mg) Volume of Diluent To BeAdded(mL)Approximate Ceftarolinefosamil Concentration(mg/mL)Amount to Be Withdrawn400 20 20 Total Volume600 20 30 Total Volume144145 The constituted solution must be further diluted in 250 mL before infusion. Use the same diluent for this further dilution, unless sterile water for 146 injection was used earlier. If sterile water for injection was used earlier, then appropriate infusion solutions include: 0.9% Sodium Chloride 147 Injection, USP (normal saline); 5% Dextrose Injection, USP; 2.5% Dextrose Injection, USP, and 0.45% Sodium Chloride Injection, USP; or Lactated 148 Ringer’s Injection, USP. The resulting solution should be administered over approximately 1 hour.149 Constitution time is less than 2 minutes. Mix gently to constitute and check to see that the contents have dissolved completely. Parenteral drug products 150 should be inspected visually for particulate matter prior to administration.151 The color of Teflaro infusion solutions ranges from clear, light to dark yellow depending on the concentration and storage conditions. When stored as 152 recommended, the product potency is not affected.153 Studies have shown that the constituted solution in the infusion bag should be used within 6 hours when stored at room temperature or within 24 hours 154 when stored under refrigeration at 2 to 8º C (36 to 46º F).155 The compatibility of Teflaro with other drugs has not been established. Teflaro should not be mixed with or physically added to solutions containing other 156 drugs.157 3. DOSAGE FORMS AND STRENGTHS158 Teflaro is supplied in single-use, clear glass vials containing either 600 mg or 400 mg of sterile ceftaroline fosamil powder.159 4. CONTRAINDICATIONS160 Teflaro is contraindicated in patients with known serious hypersensitivity to ceftaroline or other members of the cephalosporin class. Anaphylaxis and 161 anaphylactoid reactions have been reported with ceftaroline.162 5. WARNINGS AND PRECAUTIONS163 5.1 Hypersensitivity Reactions164 Serious and occasionally fatal hypersensitivity (anaphylactic) reactions and serious skin reactions have been reported in patients receiving beta-lactam 165 antibacterials. Before therapy with Teflaro is instituted, careful inquiry about previous hypersensitivity reactions to other cephalosporins, penicillins, or 166 carbapenems should be made. If this product is to be given to a penicillin- or other beta-lactam-allergic patient, caution should be exercised because cross 167 sensitivity among beta-lactam antibacterial agents has been clearly established.168 If an allergic reaction to Teflaro occurs, the drug should be discontinued. Serious acute hypersensitivity (anaphylactic) reactions require emergency 169 treatment with epinephrine and other emergency measures, that may include airway management, oxygen, intravenous fluids, antihistamines, 170 corticosteroids, and vasopressors as clinically indicated.171 5.2 Clostridium difficile-associated Diarrhea172 Clostridium difficile-associated diarrhea (CDAD) has been reported for nearly all systemic antibacterial agents, including Teflaro, and may range in 173 severity from mild diarrhea to fatal colitis.174 Treatment with antibacterial agents alters the normal flora of the colon and may permit overgrowth of C. difficile.175 C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin-producing strains of C. difficile cause increased 176 morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all 177 patients who present with diarrhea following antibiotic use. Careful medical history is necessary because CDAD has been reported to occur more than 2 178 months after the administration of antibacterial agents.179 If CDAD is suspected or confirmed, antibacterials not directed against C. difficile should be discontinued, if possible. Appropriate fluid and electrolyte 180 management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated [see Adverse 181 Reactions (6.3)].182 5.3 Direct Coombs’ Test Seroconversion183 Seroconversion from a negative to a positive direct Coombs’ test result occurred in 120/1114 (10.8%) of patients receiving Teflaro and 49/1116 (4.4%) of 184 patients receiving comparator drugs in the four pooled Phase 3 trials.185 In the pooled Phase 3 CABP trials, 51/520 (9.8%) of Teflaro-treated patients compared to 24/534 (4.5%) of ceftriaxone-treated patients seroconverted 186 from a negative to a positive direct Coombs’ test result. No adverse reactions representing hemolytic anemia were reported in any treatment group.187 If anemia develops during or after treatment with Teflaro, drug-induced hemolytic anemia should be considered. Diagnostic studies including a direct 188 Coombs’ test, should be performed. If drug-induced hemolytic anemia is suspected, discontinuation of Teflaro should be considered and supportive care 189 should be administered to the patient (i.e. transfusion) if clinically indicated.191192 5.4 Development of Drug-Resistant Bacteria193 Prescribing Teflaro in the absence of a proven or strongly suspected bacterial infection is unlikely to provide benefit to the patient and increases the risk 194 of the development of drug-resistant bacteria.195 6. ADVERSEREACTIONS196 The following serious events are described in greater detail in the Warnings and Precautions section197 ∙Hypersensitivity reactions [see Warnings and Precautions (5.1)]198 ∙Clostridium difficile-associated diarrhea [see Warnings and Precautions (5.2)]199 ∙Direct Coombs’ test seroconversion [see Warnings and Precautions (5.3)]200 6.1 Adverse Reactions from Clinical Trials201 Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be compared 202 directly to rates from clinical trials of another drug and may not reflect rates observed in practice.203 Teflaro was evaluated in four controlled comparative Phase 3 clinical trials (two in ABSSSI and two in CABP) which included 1300 adult patients treated 204 with Teflaro (600 mg administered by IV over 1 hour every 12h) and 1297 patients treated with comparator (vancomycin plus aztreonam or ceftriaxone) 205 for a treatment period up to 21 days. The median age of patients treated with Teflaro was 54 years, ranging between 18 and 99 years old. Patients treated 206 with Teflaro were predominantly male (63%) and Caucasian (82%).207 6.2 Serious Adverse Events and Adverse Events Leading to Discontinuation208 In the four pooled Phase 3 clinical trials, serious adverse events occurred in 98/1300 (7.5%) of patients receiving Teflaro and 100/1297 (7.7%) of patients 209 receiving comparator drugs. The most common SAEs in both the Teflaro and comparator treatment groups were in the respiratory and infection system 210 organ classes (SOC). Treatment discontinuation due to adverse events occurred in 35/1300 (2.7%) of patients receiving Teflaro and 48/1297 (3.7%) of 211 patients receiving comparator drugs with the most common adverse events leading to discontinuation being hypersensitivity for both treatment groups at a 212 rate of 0.3% in the Teflaro group and 0.5% in comparator group.213 6.3 Most Common Adverse Reactions214 No adverse reactions occurred in greater than 5% of patients receiving Teflaro. The most common adverse reactions occurring in > 2% of patients 215 receiving Teflaro in the pooled phase 3 clinical trials were diarrhea, nausea, and rash.216 Table 4 lists adverse reactions occurring in ≥ 2% of patients receiving Teflaro in the pooled Phase 3 clinical trials.217 Table 4: Adverse Reactions Occurring in ≥ 2% of Patients Receiving Teflaro in the Pooled Phase 3 Clinical TrialsSystem Organ Class/ Preferred TermPooled Phase 3 Clinical Trials(four trials, two in ABSSSI and two in CABP)Teflaro(N=1300)Pooled Comparators a(N=1297) Gastrointestinal disordersDiarrhea 5 % 3 %Nausea 4 % 4 %Constipation 2 % 2 %Vomiting 2 % 2 %InvestigationsIncreased transaminases 2% 3 %Metabolism and nutrition disordersHypokalemia 2 % 3 %Skin and subcutaneous tissue disordersRash 3%2%Vascular disordersPhlebitis 2%1% 218 a219 IV every 24h in the Phase 3 CABP trials.220221222 6.4 Other Adverse Reactions Observed During Clinical Trials of Teflaro223 Following is a list of additional adverse reactions reported by the 1740 patients who received Teflaro in any clinical trial with incidences less than 2%. 224 Events are categorized by System Organ Class.225 Blood and lymphatic system disorders - Anemia, Eosinophilia, Neutropenia, Thrombocytopenia226 Cardiac disorders - Bradycardia, Palpitations227 Gastrointestinal disorders -Abdominal pain228 General disorders and administration site conditions - Pyrexia229 Hepatobiliary disorders - Hepatitis230 Immune system disorders - Hypersensitivity, Anaphylaxis231 Infections and infestations -Clostridium difficile colitis232 Metabolism and nutrition disorders - Hyperglycemia, Hyperkalemia233 Nervous system disorders -Dizziness, Convulsion234 Renal and urinary disorders - Renal failure235 Skin and subcutaneous tissue disorders - Urticaria236 7. DRUGINTERACTIONS237 No clinical drug-drug interaction studies have been conducted with Teflaro. There is minimal potential for drug-drug interactions between Teflaro and 238 CYP450 substrates, inhibitors, or inducers; drugs known to undergo active renal secretion; and drugs that may alter renal blood flow [see Clinical 239 Pharmacology (12.3)].240 8. USE IN SPECIFIC POPULATIONS241 8.1 Pregnancy242 Category B243 Developmental toxicity studies performed with ceftaroline fosamil in rats at IV doses up to 300 mg/kg demonstrated no maternal toxicity and no effects 244 on the fetus. A separate toxicokinetic study showed that ceftaroline exposure in rats (based on AUC) at this dose level was approximately 8 times the 245 exposure in humans given 600 mg every 12 hours. There were no drug-induced malformations in the offspring of rabbits given IV doses of 25, 50, and 246 100 mg/kg, despite maternal toxicity. Signs of maternal toxicity appeared secondary to the sensitivity of the rabbit gastrointestinal system to broad247 spectrum antibacterials and included changes in fecal output in all groups and dose-related reductions in body weight gain and food consumption at > 50 248 mg/kg; these were associated with an increase in spontaneous abortion at 50 and 100 mg/kg. The highest dose was also associated with maternal 249 moribundity and mortality. An increased incidence of a common rabbit skeletal variation, angulated hyoid alae, was also observed at the maternally toxic 250 doses of 50 and 100 mg/kg. A separate toxicokinetic study showed that ceftaroline exposure in rabbits (based on AUC) was approximately 0.8 times the 251 exposure in humans given 600 mg every 12 hours at 25 mg/kg and 1.5 times the human exposure at 50 mg/kg.252 Ceftaroline fosamil did not affect the postnatal development or reproductive performance of the offspring of rats given IV doses up to 450 mg/kg/day. 253 Results from a toxicokinetic study conducted in pregnant rats with doses up to 300 mg/kg suggest that exposure was ≥ 8 times the exposure in humans 254 given 600 mg every 12 hours.255 There are no adequate and well-controlled trials in pregnant women. Teflaro should be used during pregnancy only if the potential benefit justifies the 256 potential risk to the fetus.257 8.3 Nursing Mothers258 It is not known whether ceftaroline is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when Teflaro 259 is administered to a nursing woman.260 8.4 Pediatric Use261 Safety and effectiveness in pediatric patients have not been established.262 8.5 Geriatric Use263 Of the 1300 patients treated with Teflaro in the Phase 3 ABSSSI and CABP trials, 397 (30.5%) were ≥65 years of age. The clinical cure rates in the 264 Teflaro group (Clinically Evaluable [CE] Population) were similar in patients ≥65 years of age compared with patients < 65 years of age in both the 265 ABSSSI and CABP trials.266 The adverse event profiles in patients ≥ 65 years of age and in patients < 65 years of age were similar. The percentage of patients in the Teflaro group who 267 had at least one adverse event was 52.4% in patients ≥ 65 years of age and 42.8% in patients < 65 years of age for the two indications combined.268 Ceftaroline is excreted primarily by the kidney, and the risk of adverse reactions may be greater in patients with impaired renal function. Because elderly 269 patients are more likely to have decreased renal function, care should be taken in dose selection in this age group and it may be useful to monitor renal 270 function. Elderly subjects had greater ceftaroline exposure relative to non-elderly subjects when administered the same single dose of Teflaro. However, 271 higher exposure in elderly subjects was mainly attributed to age-related changes in renal function. Dosage adjustment for elderly patients should be based 272 on renal function [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].275 8.6 Patients with Renal Impairment276 Dosage adjustment is required in patients with moderate (CrCl > 30 to ≤50 mL/min) or severe (CrCl ≥ 15 to ≤30 mL/min) renal impairment and in 277 patients with end-stage renal disease (ESRD – defined as CrCl < 15 mL/min), including patients on hemodialysis (HD) [see Dosage and Administration 278 (2.2) and Clinical Pharmacology (12.3)].279 10. OVERDOSAGE280 In the event of overdose, Teflaro should be discontinued and general supportive treatment given.281 Ceftaroline can be removed by hemodialysis. In subjects with ESRD administered 400 mg of Teflaro, the mean total recovery of ceftaroline in the 282 dialysate following a 4-hour hemodialysis session started 4 hours after dosing was 76.5 mg (21.6% of the dose). However, no information is available on 283 the use of hemodialysis to treat overdosage [see Clinical Pharmacology (12.3)].284 11. DESCRIPTION285 Teflaro is a sterile, semi-synthetic, broad-spectrum, prodrug antibacterial of cephalosporin class of beta-lactams (β-lactams). Chemically, the prodrug, 286 ceftaroline fosamil monoacetate monohydrate is (6R,7R)-7-{(2Z)-2-(ethoxyimino)-2-[5-(phosphonoamino)-1,2,4-thiadiazol-3-yl]acetamido}-3-{[4-(1287 methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl}-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate monoacetate monohydrate. Its molecular 288 weight is 762.75. The empirical formula is C22H21N8O8PS4.C2H4O2.H2O.289 Figure 1: Chemical structure of ceftaroline fosamil290291292293294295296297298299300 301 302 Teflaro vials contain either 600 mg or 400 mg of anhydrous ceftaroline fosamil. The powder for injection is formulated from ceftaroline fosamil monoacetate monohydrate, a pale yellowish-white to light yellow sterile powder. All references to ceftaroline activity are expressed in terms of the prodrug, ceftaroline fosamil. The powder is constituted for IV injection [see Dosage and Administration (2.3)].303 Each vial of Teflaro contains ceftaroline fosamil and L-arginine, which results in a constituted solution at pH 4.8 to 6.5. 304 12. CLINICAL PHARMACOLOGY305 Ceftaroline fosamil is the water-soluble prodrug of the bioactive ceftaroline [see Clinical Pharmacology (12.3)]. 306 12.1 Mechanism of Action307 Ceftaroline is an antibacterial drug [see Clinical Pharmacology (12.4)].308 12.2 Pharmacodynamics309 310 311 As with other beta-lactam antimicrobial agents, the time that unbound plasma concentration of ceftaroline exceeds the minimum inhibitory concentration (MIC) of the infecting organism has been shown to best correlate with efficacy in a neutropenic murine thigh infection model with S. aureus and S. pneumoniae.312 313 314 Exposure-response analysis of Phase 2/3 ABSSSI trials supports the recommended dosage regimen of Teflaro 600 mg every 12 hours by IV infusion over 1 hour. For Phase 3 CABP trials, an exposure-response relationship could not be identified due to the limited range of ceftaroline exposures in the majority of patients.315 Cardiac Electrophysiology316 317 318 In a randomized, positive- and placebo-controlled crossover thorough QTc study, 54 healthy subjects were each administered a single dose of Teflaro 1500 mg, placebo, and a positive control by IV infusion over 1 hour. At the 1500 mg dose of Teflaro, no significant effect on QTc interval was detected at peak plasma concentration or at any other time.319 12.3 Pharmacokinetics320 321 322 The mean pharmacokinetic parameters of ceftaroline in healthy adults (n=6) with normal renal function after single and multiple 1-hour IV infusions of 600 mg ceftaroline fosamil administered every 12 hours are summarized in Table 5. Pharmacokinetic parameters were similar for single and multiple dose administration.323326 Table 5: Mean (Standard Deviation) Pharmacokinetic Parameters of Ceftaroline IV in Healthy AdultsParameter Single 600 mg Dose Administered asa 1-Hour Infusion(n=6)Multiple 600 mg Doses Administered Every12 Hours as 1-Hour Infusions for 14Days(n=6)C max (mcg/mL) 19.0 (0.71) 21.3 (4.10)T max (h)a 1.00 (0.92-1.25) 0.92 (0.92-1.08)AUC (mcg h/mL) b 56.8 (9.31) 56.3 (8.90)T1/2 (h) 1.60 (0.38) 2.66 (0.40)CL (L/h) 9.58 (1.85) 9.60 (1.40)a Reported as median (range)b AUC0-∞,for single-dose administration; AUC0-tau, for multiple-dose administration; C max, maximum observed concentration; T max, time of C max; AUC0-∞, area under concentration-time curve from time 0 to infinity; AUC0-tau, area under concentration-time curveover dosing interval (0-12 hours); T1/2, terminal elimination half-life; CL, plasma clearance327328 The C max and AUC of ceftaroline increase approximately in proportion to dose within the single dose range of 50 to 1000 mg. No appreciable 329 accumulation of ceftaroline is observed following multiple IV infusions of 600 mg administered every 12 hours for up to 14 days in healthy adults with 330 normal renal function.331 Distribution332 The average binding of ceftaroline to human plasma proteins is approximately 20% and decreases slightly with increasing concentrations over 1-50 333 mcg/mL (14.5-28.0%). The median (range) steady-state volume of distribution of ceftaroline in healthy adult males (n=6) following a single 600 mg IV 334 dose of radiolabeled ceftaroline fosamil was 20.3 L (18.3-21.6 L), similar to extracellular fluid volume.335 Metabolism336 Ceftaroline fosamil is converted into bioactive ceftaroline in plasma by a phosphatase enzyme and concentrations of the prodrug are measurable in plasma 337 primarily during IV infusion. Hydrolysis of the beta-lactam ring of ceftaroline occurs to form the microbiologically inactive, open-ring metabolite 338 ceftaroline M-1. The mean (SD) plasma ceftaroline M-1 to ceftaroline AUC0-∞ ratio following a single 600 mg IV infusion of ceftaroline fosamil in 339 healthy adults (n=6) with normal renal function is 28% (3.1%).340 When incubated with pooled human liver microsomes, ceftaroline was metabolically stable (< 12% metabolic turnover), indicating that ceftaroline is not a 341 substrate for hepatic CYP450 enzymes.342 Excretion343 Ceftaroline and its metabolites are primarily eliminated by the kidneys. Following administration of a single 600 mg IV dose of radiolabeled ceftaroline 344 fosamil to healthy male adults (n=6), approximately 88% of radioactivity was recovered in urine and 6% in feces within 48 hours. Of the radioactivity 345 recovered in urine approximately 64% was excreted as ceftaroline and approximately 2% as ceftaroline M-1. The mean (SD) renal clearance of ceftaroline 346 was 5.56 (0.20) L/h, suggesting that ceftaroline is predominantly eliminated by glomerular filtration.347 Specific Populations348 Renal Impairment349 Following administration of a single 600 mg IV dose of Teflaro, the geometric mean AUC0-∞ of ceftaroline in subjects with mild (CrCl > 50 to ≤ 80 350 mL/min, n=6) or moderate (CrCl > 30 to ≤50 mL/min, n=6) renal impairment was 19% and 52% higher, respectively, compared to healthy subjects with 351 normal renal function (CrCl > 80 mL/min, n=6). Following administration of a single 400 mg IV dose of Teflaro, the geometric mean AUC0-∞ of 352 ceftaroline in subjects with severe (CrCl ≥ 15 to ≤30 mL/min, n=6) renal impairment was 115% higher compared to healthy subjects with normal renal 353 function (CrCl > 80 mL/min, n=6). Dosage adjustment is recommended in patients with moderate and severe renal impairment [see Dosage and 354 Administration (2.2)].355 A single 400 mg dose of Teflaro was administered to subjects with ESRD (n=6) either 4 hours prior to or 1 hour after hemodialysis (HD). The geometric 356 mean ceftaroline AUC0-∞ following the post-HD infusion was 167% higher compared to healthy subjects with normal renal function (CrCl > 80 mL/min, 357 n=6). The mean recovery of ceftaroline in the dialysate following a 4-hour HD session was 76.5 mg, or 21.6% of the administered dose. Dosage 358 adjustment is recommended in patients with ESRD (defined as CrCL < 15 mL/min), including patients on HD [see Dosage and Administration (2.2)]. 359 Hepatic Impairment360 The pharmacokinetics of ceftaroline in patients with hepatic impairment have not been established. As ceftaroline does not appear to undergo significant 361 hepatic metabolism, the systemic clearance of ceftaroline is not expected to be significantly affected by hepatic impairment.362 Geriatric Patients363 Following administration of a single 600 mg IV dose of Teflaro to healthy elderly subjects (≥65 years of age, n=16), the geometric mean AUC0-∞ of 364 ceftaroline was ~33% higher compared to healthy young adult subjects (18-45 years of age, n=16). The difference in AUC0-∞ was mainly attributable to。
迷迭香酸

迷迭香酸迷迭香酸<英文名>:Rosmarinic acid <化学式>:C18H16O8<分子量>:360.33<结构式>:O CO2HO OHOHHOR E<来源>:迷迭香酸广泛存在于各类植物中,尤以唇型科和紫草科植物中含量较高。
<基本性质>:<物理性质>:为浅黄色粉末,易溶于水及乙醇水溶液,难溶于氯仿,不溶于油脂、无水乙醇。
稳定性较好,适宜在酸性及低温条件下保存,使用。
<化学性质>:抗氧化性<生理活性>:具有极强清除体内自由基的活性和抗氧化作用。
<作用机理>:与不饱和脂肪酸竞争性地与脂质过氧基结合,以终止脂质过氧化的连锁反应,降低脂质过氧化速率,而迷迭香酸被氧化为醌式;迷迭香酸可抑制中性粒细胞呼吸爆发及通过减少细胞内钙离子浓度而抑制溶酶体的释放。
迷迭香酸的抗氧化作用与其结构有关,邻二酚羟基是清除自由基活性的物质基础,而且C3位的共轭双键具有增效作用。
抗炎活性德国Nattermann公司于1991年把它作为抗炎、镇痛、解毒药物投放市场。
迷迭香酸对肾炎有一定的抑制作用。
抗炎机理可能是:1.与抑制花生四烯酸代谢的5-脂氧化酶( 5-LOX)有关;2.对补体依赖性PGL2 的合成产生抑制作用,干扰不同途径C3转移酶的活性;3.抗氧化和消除自由基作用;4.抑制肥大细胞中组胺的释放。
抗菌活性国内外的研究证明,迷迭香酸有许多医药功能,对金黄色葡萄球菌、大肠杆菌等具有较强的抑制作用。
另外发现,迷迭香酸对植物病原真菌也有抑制效果:其中对番茄灰霉病菌、芒果灰斑病菌、柑橘青霉和梨黑斑病菌抑制作用较强,在研究中还发现迷迭香酸具有很强的热稳定性和耐低温贮藏性;该结果和已有的研究文献说明,迷迭香酸具有广谱的抗微生物活性,深入研究迷迭香酸的生物活性将有力地促进其在医药、农药等方面的开发应用。
抗菌作用机理:1.显著增加细菌细胞膜的通透性,加速糖类和蛋白质的渗漏,使细胞代谢发生紊乱,进而影响细菌蛋白质代谢;2.还可以通过抑制DNA聚合酶的活性而影响DNA的复制,来发挥其抑菌作用。
卫喜康说明书

核准日期: 2009年11月09日 修改日期:2012年09月17日2014年05月29日2015年12月18日 2018年12月10日 2020年09月09日 2022年03月01日琥珀酸索利那新片说明书请仔细阅读说明书并在医师指导下使用【药品名称】通用名称: 琥珀酸索利那新片 商品名称: 卫喜康(V esicare ) 英文名称: Solifenacin Succinate Tablets 汉语拼音: Huposuansuolinaxin Pian 【成份】化学名称:琥珀酸索利那新,(3R )-1-氮杂双环[2.2.2]辛-3-基 (1S )-1-苯基-3,4-二氢异喹啉-2(1H )-羧酸酯单琥珀酸盐化学结构式:分子式:C 23H 26N 2O 2·C 4H 6O 4 分子量:480.55辅料:乳糖单水合物、玉米淀粉、羟丙甲纤维素、硬脂酸镁、欧巴代黄色03F12967 【性状】浅黄色薄膜衣片,刻有公司标识和“150”字样。
【适应症】OO NOOOH OH•用于膀胱过度活动症患者伴有的尿失禁和/或尿频、尿急症状的治疗。
【规格】5mg【用法用量】本品的推荐剂量为每日一次,每次一片(5mg),必要时可增至每日一次,每次两片(10mg)。
本品必须整片用水送服,餐前或餐后均可服用。
肾功能障碍患者轻、中度肾功能障碍患者(肌酐清除率>30 ml/min)用药剂量不需要调整。
严重肾功能障碍患者(肌酐清除率≤30 ml/min)应谨慎用药,剂量不超过每日5 mg。
肝功能障碍患者轻度肝功能障碍患者用药剂量不需要调整。
中度肝功能障碍(Child-Pugh评分7至9分)患者应谨慎用药,剂量不超过每日一次5 mg。
强力的细胞色素P450 3A4抑制剂与酮康唑或治疗剂量的其它强力CYP3A4抑制剂例如利托那韦、奈非那韦和伊曲康唑同时用药时,本品的最大剂量不超过5mg。
【不良反应】由于索利那新的药理作用,本品可能引起抗胆碱副作用,通常为轻、中度,其发生频率与剂量有关。
曲安奈德 - Drug Future药物在线 首页

谱 仪 , 记 录 色 谱 图 ; 另 取 曲 安 奈 德 对 照 品 约 1 5 . 6 m g ,精密称 定,置 25ml 量瓶中,加甲醇溶解并稀释至刻度,摇匀,作为曲 安 奈 德 对 照 品 溶 液 ; 另 取 硝 酸 益 康 唑 对 照 品 约 1 2 . 5mg ,精密 称 定 , 置 2 5 m l 量 瓶 中 , 精 密 加 曲 安 奈 德 对 照 品 溶 液 2 m l ,加四 氢 呋 喃 2 m l ,用甲醇稀释至刻度,摇勾,同法测定,按外标法以
曲安奈德益康唑乳奮
沉,振摇后成均匀的乳白色混悬液。 【鉴别】( 1) 在含量测定项下记录的色谱图中,供试品溶 液主峰的保留时间应与对照品溶液主峰的保留时间一致。 (2) 取本品适量(约相当于曲安奈德 50mg) ,用乙醚提取 2 次,每次 10ml ,弃去乙醚液,分取水层,滤过,残渣用少量水洗 涤后,置105°C干燥1小时。取干燥残渣用无水乙醇溶解并稀 释制成每lml中约含12jxg的溶液,照紫外-可见分光光度法 (附录IVA)测定,在239nm的波长处有最大吸收。 (3) 取 本 品 适 量 ( 约 相 当 于 曲 安 奈 德 4 0 m g ) , 加 水 5 m r 混 匀,加乙醚 10ml ,振摇提取后,取水层,水浴蒸干,残渣经减压 干燥,依法测定。本品的红外光吸收图谱应与对照的图谱(光 谱集 603 图)一致。 【 检 查 】 p H 值 应 为 5 . 0 ~ 7 . 5( 附录 YI H:)。 有 关 物 质 取 本 品 , 摇 匀 , 精 密 量 取 5 m l ,置 200ml 量瓶 中,加流动相适量,振摇使曲安奈德溶解,用流动相稀释至刻 度,摇匀,滤过,取续滤液作为供试品溶液。照曲安奈德有关 物质项下的方法试验。供试品溶液色谱图中如有杂质峰,单 个 杂 质 峰 面 积 不 得 大 于 对 照 溶 液 主 峰 面 积 的 0 . 5倍(0. 5 % ) , 各杂质峰面积的和不得大于对照溶液主峰面积的1.5倍 (1.5%)。供试品溶液色谱图中任何小于对照溶液主峰面积 0.01倍的峰可忽略不计。 k度取本品,充分振摇,取1滴,照粒度测定法(附录 K E第一法)检査3个视野,颗粒均应小于15pm,其中5pm
阿司咪唑

合成方法
合成方法
化合物(I)和碘甲烷在乙醇中回流8h,环合得到化合物(Ⅱ)。再水解脱去酯基,得到化合物(Ⅲ)。用对甲氧 基苯乙基溴进行N-烷基化,得化合物(Ⅳ)。再用对氟苄基溴烷基化,得阿司咪唑。
1. 1-[(4-氟苯基)甲基]-苯并咪唑-2-(3H)-酮的制备
在反应瓶中加入2-羟基苯并咪唑5.0g(37.3mmol)和NaH 1.6g(53mmol)(NaH含量大约为80%,浸入矿物油中) 的DMF 100ml的悬浮液.加毕.在60ºC.(最好有N2保护)搅拌反应1h.再加入4-氟苄基氯(FBC)5.4g(37mmol),加热 ( 6 0 ºC ) 搅 拌 反 应 5 . 5 h . 冷 却 至 室 温 后 加 入 冰 水 7 0 0 m l , 用 二 氯 甲 烷 ( 5 0 0 m l × 2 ) 提 取 . 有 机 层 用 食 盐 水 洗 . 无 水 N a 2 S O 4 干燥.过滤.滤液减压浓缩.剩余物用石油醚析晶.得1-[(4-氟苯基)甲基]-苯并咪唑-2-(3H)-酮固体8.0g,为无色 结 晶 m p 1 7 8 ~ 1 7 9 ºC , 收 率 8 8 % .
治疗措施
阿司咪唑中毒的治疗要点为: 1.大量摄入者予洗胃,后灌服活性炭和导泻。 2.对心肌抑制和Q-T间期延长者予5%碳酸氢钠250ml静注可能有效。 3.对症、支持治疗。
专家点评
专家点评
阿司咪阿司咪唑自1983年上市以来,在许多国家得到了广泛应用。国外研究显示阿司咪唑治疗荨麻疹的总有 效率为74%。国内的一项多中心双盲安慰剂对照试验表明阿司咪唑对急性荨麻疹的总有效率为82.9%,对慢性荨麻 疹的总有效率为86.0%,均显著高于安慰剂,主要不良反应为嗜睡、倦怠、口干等,连续用药3个月的患者中,半 数有食欲及体重增加。阿司咪唑的心脏毒性虽然发生率较低,但由于后果严重,已限制了它的应用。阿司咪唑为 强效和长效的H1受体拮抗剂,无中枢镇静和抗毒蕈碱样作用。代谢产物去甲阿司咪唑仍有抗胆胺作用。长期服用 可增进食欲和增加体重,服用过量可引起心脏Q-T间期延长和室性心律失常。适用于各种原因引起过敏性疾病。
肝病专业英语词汇

3α-羟类固醇脱氢酶(Y' 蛋白) γ -谷氨酰转移酶 γ-氨基丁酸 甲胎蛋白 人兽共患病 异种肝移植 脂肪性纤维瘤,黄色瘤 黄嘌呤氧化酶 黄斑瘤 全球移植中心名录 窗口期 肝豆状核变性 肥达反应 外斐反应 韦克斯勒成人智力测验 呕吐 视觉诱发电位 病毒学应答 病毒复制 病毒性肝炎 静脉-静脉转流 VOD 肝小静脉闭塞病 静脉-动脉转流 血管活性肽 静脉曲张 胆管消失综合征 疫苗 熊去氧胆酸 尿胆素原 尿胆素 二磷酸尿苷异构酶 尿素生成 鸟氨酸循环,尿素循环 上消化道出血 粗纤维调节素 肝未分化肉瘤 充盈不足学说 非结合高胆红素血症 游离胆红素,非结合胆红素 超声(波)检查法
短潜伏期肝炎 移动性浊音 腹水白蛋白浓度梯度 血清肝炎 血清诊断 血清胆红素 血清白蛋白 血清学应答 乙肝血清学检查 血清转换 败血症相关胆汁淤积 正链,有义链 镇静剂 次级胆酸 海蓝组织细胞增多症 硬化疗法 硬化性胆管炎 日本血吸虫病 湄公血吸虫 曼氏血吸虫 日本血吸虫 间插血吸虫 埃及血吸虫 血吸虫 瘢痕形成期 粗面内质网 滚环机制(环状DNA复制的机理) RNA干扰 核酶 核糖体 胆固醇逆向转运
伤寒 Ⅳ型胶原 Ⅲ型前胶原 甲肝和乙肝疫苗 抑癌基因 肿瘤坏死因子 肝结核 滋养体 甘油三酯 颠换效应 经颈静脉肝内门腔分流 转换 输血传染的病毒 输血性肝炎 转化生长因子
transcatheter arterial chemoembolization 肝动脉化疗栓塞 trans-activation toxic hepatitis total cholesterin total bilirubin tomor necrosis factor tocopherol TNF-related apoptosis inducing ligand tissue inhibitor of metalloproteinase thymosin Thymopolypeptides for Injection thromboxane the core promoter element tentative diagnosis tension of muscle tenderness Telbivudine taurocholic acid taurochenodeoxycholate acid systemic inflammatory response syndrome syncytial giant-cell hepatitis sustained virus response sustained response 反式激活 中毒性肝炎 总胆固醇 总胆红素 肿瘤坏死因子 生育酚,维生素E 肿瘤坏死因子相关凋亡诱导配体 基质金属蛋白酶组织抑制物 胸腺肽 胸腺肽 血栓素 核心启动子元件,启动子核心元件 暂时的(假定的)诊断,试验性诊断 肌张力 压痛 LdT 牛(磺)胆酸 牛磺鹅(去氧)胆酸盐,牛磺鹅(脱氧)胆酸盐 全身炎症反应综合症 融合巨细胞性肝炎 持续病毒应答 持久应答
西那卡塞片剂药品说明书(英文)-日本

1
Revised: January 2010 (version 5)
Standard Commodity Classification No. of Japan
873999 - DRUG FOR TREATMENT OF SECONDARY HYPERPARATHYROIDISM (CALCIUM RECEPTOR AGONIST)
Approval No. Date of listing in the NHI reimbursement price
December 2007 January 2008
March 2004
Expiration date Specified on the outer package.
Date of initial marketing in Japan International birth date
1. Composition Brand name Active ingredients (per tablet) Regpara Tablets Regpara Tablets 25 mg 75 mg Cinacalcet Cinacalcet hydrochloride hydrochloride 27.55 mg (including 82.65 mg (including 75 mg of cinacalcet) 25 mg of cinacalcet) Pregelatinized starch, Pregelatinized starch, Crystalline cellulose, Crystalline cellulose, Povidone, Povidone, Crospovidone, Crospovidone, Magnesium stearate, Light anhydrous Lactose hydrate, silicic acid, Hypromellose, Talc, Titanium oxide, Magnesium stearate, Triacetin, Lactose hydrate, Macrogol 400, Hypromellose, Yellow ferric oxide Titanium oxide, Triacetin, Macrogol 400, Yellow ferric oxide, Blue No.2 aluminum lake Light-yellow, Light-green to lightfilm-coated tablets yellowish green, film-coated tablets
Contents-日本药典目录英文版

CONTENTSPreface (i)The Japanese Pharmacopoeia,Sixteenth Edition (1)General Notices (1)General Rules for Crude Drugs (5)General Rules for Preparations (7)General Tests,Processes and Apparatus (25)1.Chemical Methods1.01Alcohol Number Determination (25)1.02Ammonium Limit Test (27)1.03Chloride Limit Test (28)1.04Flame Coloration Test (28)1.05Mineral Oil Test (28)1.06Oxygen Flask Combustion Method (28)1.07Heavy Metals Limit Test (29)1.08Nitrogen Determination(Semimicro-Kjeldahl Method) (30)1.09Qualitative Tests (31)1.10Iron Limit Test (37)1.11Arsenic Limit Test (37)1.12Methanol Test (39)1.13Fats and Fatty Oils Test (39)1.14Sulfate Limit Test (41)1.15Readily Carbonizable Substances Test (41)2.Physical MethodsChromatography2.01Liquid Chromatography (42)2.02Gas Chromatography (45)2.03Thin-layer Chromatography (47)2.04Amino Acid Analysis of Proteins (47)Spectroscopic Methods2.21Nuclear Magnetic ResonanceSpectroscopy (48)2.22Fluorometry (50)2.23Atomic AbsorptionSpectrophotometry (51)2.24Ultraviolet-visible Spectrophotometry (52)2.25Infrared Spectrophotometry (53)Other Physical Methods2.41Loss on Drying Test (55)2.42Congealing Point Determination (55)2.43Loss on Ignition Test (56)2.44Residue on Ignition Test (56)2.45Refractive Index Determination (56)2.46Residual Solvents Test (57)2.47Osmolarity Determination (57)2.48Water Determination(Karl FischerMethod) (58)2.49Optical Rotation Determination (61)2.50Endpoint Detection Methods inTitrimetry (62)2.51Conductivity Measurement (63)2.52Thermal Analysis (65)2.53Viscosity Determination (67)2.54pH Determination (69)2.55Vitamin A Assay (71)2.56Determination of Specific Gravity andDensity (72)2.57Boiling Point and Distilling RangeTest (74)2.58X-Ray Powder Diffraction Method (75)2.59Test for Total Organic Carbon (79)2.60Melting Point Determination (80)3.Powder Property Determinations3.01Determination of Bulk and TappedDensities (82)3.02Specific Surface Area by GasAdsorption (84)3.03Powder Particle DensityDetermination (86)3.04Particle Size Determination (87)4.Biological Tests/Biochemical Tests/Microbial Tests4.01Bacterial Endotoxins Test (92)4.02Microbial Assay for Antibiotics (96)4.03Digestion Test (100)4.04Pyrogen Test (103)4.05Microbial Limit Test (103)4.06Sterility Test (114)5.Tests for Crude Drugs5.01Crude Drugs Test (117)5.02Microbial Limit Test for Crude Drugs (120)6.Tests for Preparations6.01Test for Metal Particles in OphthalmicOintments (126)6.02Uniformity of Dosage Units (127)6.03Particle Size Distribution Test forPreparations (129)6.04Test for Acid-neutralizing Capacity ofGastrointestinal Medicines (129)6.05Test for Extractable Volume ofParenteral Preparations (130)6.06Foreign Insoluble Matter Test forInjections (131)6.07Insoluble Particulate Matter Test forInjections (131)6.08Insoluble Particulate Matter Test forOphthalmic Solutions (134)6.09Disintegration Test (135)6.10Dissolution Test (137)JP XVI Contents6.11Foreign Insoluble Matter Test forOphthalmic Solutions (141)7.Tests for Containers and Packing Materials7.01Test for Glass Containers for Injections..1417.02Test Methods for Plastic Containers (142)7.03Test for Rubber Closure for AqueousInfusions (148)8.Other Methods8.01Sterilization and Aseptic Manipulation (149)9.Reference Standards;Standard Solutions;Reagents,Test Solutions;MeasuringInstruments,Appliances,etc.Reference Standards9.01Reference Standards (150)Standard Solutions9.21Standard Solutions for VolumetricAnalysis (153)9.22Standard Solutions (164)9.23Matching Fluids for Color (166)Reagents,Test Solutions,etc.9.41Reagents,Test Solutions (167)9.42Solid Supports/Column Packings forChromatography (306)9.43Filter Papers,Filters for filtration,Test Papers,Crucibles,etc (308)9.44Standard Particles,etc (308)Measuring Instruments and Appliances,Thermometers,etc.9.61Optical Filters for Wavelength andTransmission Rate Calibration (309)9.62Measuring Instruments,Appliances (309)9.63Thermometers (310)Official Monographs (313)Crude Drugs (1593)Infrared Reference Spectra.....................1775–1961 Ultraviolet-visible Reference Spectra.........1965–2131General InformationG1Physics and ChemistryGuideline for Residual Solvents and Models for the Residual Solvents Test (2135)Inductively Coupled Plasma Atomic Emission Spectrometry (2136)Near Infrared Spectrometry (2141)pH Test for Gastrointestinal Medicine (2144)System Suitability (2145)Test for Trace Amounts of Aluminum inTrans Parenteral Nutrition(TPN)Solutions (2146)Validation of Analytical Procedures (2148)G2Solid-state PropertiesLaser Diffraction Measurement ofParticle Size (2151)Powder Fineness (2154)Powder Flow (2155)Solid and Particle Densities (2158)G3Biotechnological/Biological Products Amino Acid Analysis (2159)Basic Requirements for Viral Safety ofBiotechnological/Biological Productslisted in Japanese Pharmacopoeia (2166)Capillary Electrophoresis (2179)Isoelectric Focusing (2184)Mass Spectrometry of Peptides andProteins (2186)Mycoplasma Testing for Cell Substrates used for the Production of Biotechnological/Biological Products (2188)Peptide Mapping (2191)Qualification of Animals as Origin ofAnimal-derived Medicinal Productsprovided in the General Notices ofJapanese Pharmacopoeia and OtherStandards (2194)SDS-Polyacrylamide Gel Electrophoresis (2196)Total Protein Assay (2201)G4MicroorganismsDecision of Limit for BacterialEndotoxins (2205)Disinfection and Sterilization Methods (2205)Media Fill Test(Process Simulation) (2206)Microbial Attributes of Non-sterilePharmaceutical Products (2209)Microbiological Evaluation of Processing Areas for Sterile PharmaceuticalProducts (2211)Preservatives-Effectiveness Tests (2215)Rapid Counting of Microbes usingFluorescent Staining (2217)Rapid Identification of MicroorganismsBased on Molecular Biological Method (2220)Sterility Assurance for Terminally Sterilized Pharmaceutical Products (2221)Terminal Sterilization and SterilizationIndicators (2225)G5Crude DrugsAristolochic Acid (2227)Purity Tests on Crude Drugs Using Genetic Information (2228)On the Scientific Names of Crude DrugsListed in the JP (2231)G6Drug FormulationTablet Friability Test (2244)G7Containers and PackagePlastic Containers for PharmaceuticalJP XVI ContentsProducts (2244)G8WaterQuality Control of Water for PharmaceuticalUse (2246)Water to be used in the Tests of Drugs (2253)G9OthersInternational Harmonization Implementedin the Japanese Pharmacopoeia SixteenthEdition (2253)AppendixAtomic Weight Table(2010) (2287)Standard Atomic Weights2010 (2288)Index (2291)Index in Latin name (2307)Index in Japanese (2309)PREFACEThe15th Edition of the Japanese Pharmacopoeia (JP)was promulgated by Ministerial Notification No. 285of the Ministry of Health,Labour and Welfare (MHLW)on March31,2006.In July2006,the Committee on JP established the basic principles for the preparation of the JP16th Edi-tion,setting out the roles and characteristics of the JP, the definite measures for the revision,and the date of the revision.At the Committee,the five basic principles of JP, which we refer to as the``five pillars'',were estab-lished as follows:1)Including all drugs which are im-portant from the viewpoint of health care and medical treatment;2)Making qualitative improvement by in-troducing the latest science and technology;3)Pro-moting internationalization;4)Making prompt partial revision as necessary and facilitating smooth adminis-trative operation;and5)Ensuring transparency regarding the revision,and disseminating the JP to the public.It was agreed that the Committee on JP should make efforts,on the basis of these principles,to en-sure that the JP is used more effectively in the fields of health care and medical treatment by taking appropri-ate measurements,including getting the understanding and cooperation of other parties concerned.It was agreed that the JP should provide an official standard,being required to assure the quality of medi-cines in Japan in response to the progress of science and technology and medical demands at the time.It should define the standards for specifications,as well as the methods of testing to assure overall quality of all drugs in principle,and it should have a role in clarifying the criteria for quality assurance of drugs that are recognized to be essential for public health and medical treatment.The JP has been prepared with the aid of the knowledge and experience of many professionals in the pharmaceutical field.Therefore,the JP should have the characteristics of an official standard,which might be widely used by all parties concerned.It should provide information and understanding about the quality of drugs to the public,and it should be conducive to smooth and effective regulatory control of the quality of drugs,as well as promoting and maintaining international consistency and harmoniza-tion of technical requirements.It was also agreed that JP articles should cover drugs,which are important from the viewpoint of health care and medical treatment,clinical results and frequency of use,as soon as possible after they reach the market.The target date for the publication of JP16th Edi-tion(the Japanese edition)was set as April2011.JP Expert Committees are organized with the fol-lowing panels:Panel on the Principles of Revisions; Sub-committee on the Principles of Revisions;Panel on Medicinal Chemicals;Panel on Antibiotics;Panel on Biologicals;Panel on Crude Drugs;Panel on Phar-maceutical Excipients;Panel on Physico-Chemical Methods;Panel on Preparations;Panel on Physical Methods;Panel on Biological Tests;Panel on Nomen-clature;Panel on International Harmonization;Panel on Pharmaceutical Water;and Panel on Reference Standards.Furthermore,working groups are estab-lished under the Panel on Physico-Chemical Methods, Panel on Preparations and Panel on Biological Tests to expedite discussion on revision drafts.In the Committee on JP,Takao Hayakawa took the role of chairman from July2003to December2010, and Mitsuru Hashida from January2011to March 2011.In addition to the regular revision every five years in line with the basic principles for the preparation of the JP it was agreed that partial revision should be done as necessary to take account of recent progress of science and in the interests of international harmonization.In accordance with the above principles,the panels initiated deliberations on selection of articles,and on revisions for General Notices,General Rules for Crude Drugs,General Rules for Preparations,General Tests, Monographs and so on.Draft revisions covering subjects in General Notices, General Rules for Crude Drugs,General Rules for Preparations,General Tests and Monographs,for which discussions were finished between September 2005and March2007,were prepared for a supplement to the JP15.They were examined by the Committee on JP in April2007,followed by the Pharmaceutical Affairs and Food Sanitation Council(PAFSC)in June 2007,and then submitted to the Minister of Health, Labour and Welfare.The supplement was named ``Supplement I to the JP15th Edition'',promulgated on September28,2007by Ministerial Notification No. 316of MHLW,and became effective on October1,i。
艾塞那肽注射液说明书

核准日期:年月日艾塞那肽注射液说明书请仔细阅读说明书并在医师指导下使用对艾塞那肽及其它成份过敏者禁用【药物名称】通用名称:艾塞那肽注射液英文名称:Exenatide Injection汉语拼音:Aisainatai Zhusheye【成份】本品主要成份为艾塞那肽。
化学结构式:分子式:C184H282N50O60S分子量:4186.6辅料:甘露醇、醋酸钠三水合物、间甲酚(2.00-2.4mg/ml)、冰醋酸、注射用水。
【性状】本品为无色澄明液体【适应症】本品用于改善2型糖尿病患者的血糖控制,适用于单用二甲双胍、磺酰脲类,以及二甲双胍合用磺酰脲类,血糖仍控制不佳的患者。
【规格】(1)5μg剂量刻度注射笔:0.25 mg/ml,1.2 ml/支,单次注射药量5μg,内含60次注射的药量。
(2)10μg剂量刻度注射笔:0.25 mg/ml,2.4 ml/支,单次注射药量10μg,内含60次注射的药量。
【用法用量】本品的起始剂量为每次5微克(μg),每日2次,在早餐和晚餐前60分钟内(或每天的2顿主餐前;给药间隔大约6小时或更长)皮下注射。
不应在餐后注射本品。
根据临床应答,在治疗1个月后剂量可增加至10微克。
每日2次。
每次给药应在大腿、腹部或上臂皮下注射给药。
本品推荐用于接受二甲双胍、一种磺酰脲类、二甲双胍合用一种磺酰脲类治疗,血糖仍控制不佳的2型糖尿病患者。
在二甲双胍治疗的基础上加用本品时,可继续使用二甲双胍的目前剂量,因为合用本品发生低血糖而需要进行调整二甲双胍剂量的可能性较低。
在磺酰脲类治疗基础上加用本品,应该考虑降低磺酰脲类的剂量,以降低低血糖发生的风险。
(参照【注意事项】,低血糖)本品为无色澄明液体,当溶液有颗粒、浑浊或变色时不得使用。
过有效期后不得使用。
尚无本品静脉或肌肉注射的安全性和有效性资料。
注射笔使用指南注射笔的详细操作图示和注射指导请参见本品包装盒中所附“注射笔使用手册”。
重要提示使用本注射笔前请仔细阅读“注射笔使用手册”,如果不完全按照使用手册操作可能会出现剂量错误、注射笔损坏或者发生感染。
鲑降钙素鼻喷剂-详细说明书与重点

鞋降钙素鼻喷剂英文名称:Calcitonin (Salmon) Nasal Spray【成分】活性成份:鞋鱼降钙素(32个氨基酸单链组成),化学结构式:【性状】本品在耐压容器中的药液为无色的澄明液体,撒压阀门,药液即呈雾状喷出。
【适应症】- 由于骨质溶解或骨质减少引起的骨痛- 其他药物治疗无效的骨质疏松症患者:- 早期和晚期的绝经后骨质疏松症;- 老年性骨质疏松症;- 继发性骨质疏松症,例如继发于皮质激素治疗或制动。
为防止进行性骨量丢失,在使用本品的同时应根据个体需要给予适量的钙和维生素Do- 其他药物治疗无效或不适于接受其他药物治疗的Paget氏骨病(变形性骨炎),特别是伴有下列情况的患者:- 骨痛;- 神经并发症;- 骨转换增加,表现为血清碱性磷酸酶升高和尿羟脯氨酸排泌增加;- 骨损害范围进行性扩大;- 不完全或反复骨折。
- 由下列情况引起的高血钙症:- 继发于乳腺癌、肺癌、肾癌、骨髓瘤和其它恶性疾病的肿瘤性骨溶解;- 甲状旁腺机能亢进、制动或维生素D中毒;- 慢性高血钙症需作长期治疗的患者,应持续到对其基本疾病的特殊治疗见效为止。
- 神经营养不良症(痛性神经营养不良或sudeck氏病):-由不同病因和诱因所致,诸如创伤后骨质疏松症、反射性神经营养不良、肩臂综合征、外周神经受伤所致的灼痛、药物引起的神经营养不良。
【规格】2ml : 4400IU.本品的活性成份为鞋鱼降钙素。
他的生物活性以国际单位(IU)表示,本品每喷200IU. 每瓶至少含供14喷的使用剂量。
【用法用量】给药装置使用说明:在首次使用鼻喷瓶前,按压驱动装置3次,以启动喷药泵(直到鼻喷瓶颈边缺口的计数窗显示绿色)。
无论何时若喷药嘴阻塞,请用力按压驱动装置以排除阻塞。
但千万不要用针或其它尖锐的物体来排除阻塞,因为这样可能会损坏喷药装置。
1.取下瓶盖。
2,初次使用,如图所示手持喷鼻瓶,用力按压瓶帽,至出现‘咔嗒“声,然后放松,重复操作3次,瓶帽缺口显示绿色,鼻喷瓶已准备好可使用了。
琥珀酸曲格列汀片(Zafatek)日本说明书译稿

Zafatek®日本说明书译文【禁忌】(以下患者禁用)(1)严重酮症、糖尿病昏迷或昏迷前期、1型糖尿病患者[由于输液、胰岛素快速校正高血糖是必要的,故不适合使用本品。
](2)严重感染、手术前后、严重创伤患者[由于需要注射胰岛素控制血糖,故不适合使用本品。
](3)严重肾功能障碍患者或需透析的晚期肾衰竭患者[由于本品主要经肾脏排泄,排泄延迟可能导致本品的血药浓度上升。
](见【药代动力学】)(4)对本品的成份有过敏史的患者【组成·性状】Zafatek ® 100mg Zafatek ® 50mg1片中的有效成份琥珀酸曲格列汀133mg(曲格列汀100mg)琥珀酸曲格列汀66.5mg(曲格列汀50mg)剂型两面有凹痕的薄膜衣片薄膜衣片颜色淡红色淡橙色验证码D389 D388形状上面下面侧面长直径(mm) 11.0 8.2 短直径(mm) 5.6 4.7 厚度(mm) 约3.8 约3.1 质量(mg) 约187 约93添加剂:D-甘露醇、微晶纤维素、羧甲基纤维素钠、羟丙纤维素、硬脂富马酸钠、羟丙甲纤维素、聚乙二醇6000、氧化钛、氧化铁(以上两个规格均含有),黄色氧化铁(仅50mg含有)。
【功能主治】2型糖尿病【用法·用量】通常成人每周一次口服曲格列汀100mg。
【使用上的注意】1. 慎重给药(以下患者慎用)下列患者或状态:(1)中度肾功能损害患者(见<用法用量相关使用上的注意>、【药代动力学】)(2)正在使用磺脲类药物或胰岛素制剂的患者[与其他DPP-4抑制剂并用时有严重低血糖的报道](见「重要注意事项」、「相互作用」、「严重不良反应」)(3)脑垂体功能不全或肾上腺功能不全[有引发低血糖的风险](4)营养不良、饥饿、饮食不规律、进食量不足或虚弱状态[有引发低血糖的风险] (5)激烈肌肉运动[有引发低血糖的风险](6)饮酒过量者[有引发低血糖的风险]2. 重要的注意事项(1)应注意,由于本品与其他糖尿病药物并用时有引发低血糖的风险,因此在与这些药物并用时需对患者详细说明低血糖症状及其处理方法。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Inhibitors, Agonists, Screening Libraries
Data Sheet
BIOLOGICAL ACTIVITY:
Sertaconazole nitrate is a topical broad–spectrum antifungal that is developed to provide an additional agent for the treatment of superficial cutaneous and mucosal infections.
Target: Antifungal
Sertaconazole nitrate reduces the release of cytokines from activated lymphocytes and mitigated inflammation in animal models of irritant contact dermatitis and neurogenic inflammation in a dose–dependent fashion. Sertaconazole nitrate is found to inhibit the proliferation of stimulated human lymphocytes with IC50 of 4 μg/mL [1]. Sertaconazole nitrate inhibits ergosterol synthesis by
blockade of the P450–dependent enzyme pathway that catalyzes the methylation of lanosterol to ergosterol, thus inhibits fungal cell growth. Sertaconazole nitrate binds directly to nonsterol lipids in the membrane, which interferes with the regulation of the permeability of fungal cell membranes, thus induces fungal cell death [2].
The mean ear weight of Tetradecanoyl phorbol acetate (TPA)–challenged murine treated with sertaconazole nitrate (1%) is 7.23 mg compared with 14.7 mg for controls, indicating a statistically significant reduction in irritant dermatitis. Sertaconazole nitrate 1% elicits a significant reduction in Resiniferatoxin–induced ear edema when compared with controls in CD–1 mice. Topical treatment with sertaconazole nitrate 1% significantly inhibits contact hypersensitivity and decreases the content of the pro–inflammatory cytokines TNFα, IL–2, and IFNγ in oxazolone exposed murine skin [1]. Clinical trials with sertaconazole nitrate cream 2% show efficacy in the treatment of superficial cutaneous fungal infections [2]. Sertaconazole reduces inflammation via inducing PGE2 production and the COX–2 inhibitor blocks sertaconazole from exerting its anti–inflammatory effects in a mouse model of TPA–induced ear edema [3].References:
[1]. Liebel, F., et al., Anti–inflammatory and anti–itch activity of sertaconazole nitrate. Arch Dermatol Res, 2006. 298(4): p. 191–9.
[2]. Pfaller, M.A. and D.A. Sutton, Review of in vitro activity of sertaconazole nitrate in the treatment of superficial fungal infections. Diagn Microbiol Infect Dis, 2006. 56(2): p. 147–52.
[3]. Sur, R., et al., Anti–inflammatory activity of sertaconazole nitrate is mediated via activation of a p38–COX–2–PGE2 pathway. J Invest Dermatol, 2008.128(2): p. 336–44.
Product Name:
Sertaconazole (nitrate)Cat. No.:
HY-B0736A CAS No.:
99592-39-9Molecular Formula:
C 20H 16Cl 3N 3O 4S Molecular Weight:
500.78Target:
Fungal Pathway:
Anti–infection Solubility:
10 mM in DMSO
Caution: Product has not been fully validated for medical applications. For research use only.
Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@ Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。