引物+++保护碱基列表--百度文库
引物保护碱基列表--百度文库

11月13日引物合成的详解4.需要什么级别的引物?答:引物常用的纯化方式C18脱盐,OPC纯化,PAGE纯化,HPLC纯化。
根据实验需要,确定订购引物的纯度级别。
应用引物长度要求纯度级别要求一般PCR扩增<45 base OPC一般PCR扩增>45 base PAGE诊断PCR扩增< 40base OPC, PAGEDNA测序20base左右OPC亚克隆,点突变等根据实验要求定OPC, PAGE,HPLC根据实验要求定PAGE基因构建(全基因合成)反义核酸根据实验要求定PAGEPAGE, HPLC修饰引物根据实验要求定8.如何计算引物的浓度?答:引物保存在高浓度的状况下比较稳定。
引物一般配制成10-50pmol/ul。
一般情况下,建议将引物的浓度配制成50pmol/ul,加水的体积(微升)按下列方式计算:V (微升)= OD数*(乘)33 *(乘)*(乘)20000 / (除) 引物的分子量。
引物的分子量可以从合成报告单上获得。
如果需要配制成其他浓度,按上述公式换算。
注意:1 OD260= 33 ug/ml.9.如何计算引物的Tm值?答:引物设计软件都可以给出Tm,与引物长度、碱基组成、引物使用缓冲的离子强度有关。
长度为25mer以下的引物,Tm计算公式为:Tm = 4℃(G + C)+ 2℃(A + T)对于更长的寡聚核苷酸,Tm计算公式为:Tm = 81.5 + 16.6 x Log10[Na+] + 0.41 (%GC) – 600/size公式中,Size = 引物长度。
11.如何溶解引物?答:干燥后的引物质地非常疏松,开盖前最好离心一下,或管垂直向上在桌面上敲敲,将引物粉末收集到管底。
根据计算出的体积加入去离子无菌水或10mM Tris pH7.5缓冲液,室温放置几分钟,振荡助溶,离心将溶液收集到管底。
溶解引物用的水一般不要用蒸馏水,因为有些蒸馏水的pH值比较低(pH4-5),引物在这种条件下不稳定。
保护碱基表全集文档

保护碱基表全集文档(可以直接使用,可编辑实用优质文档,欢迎下载)
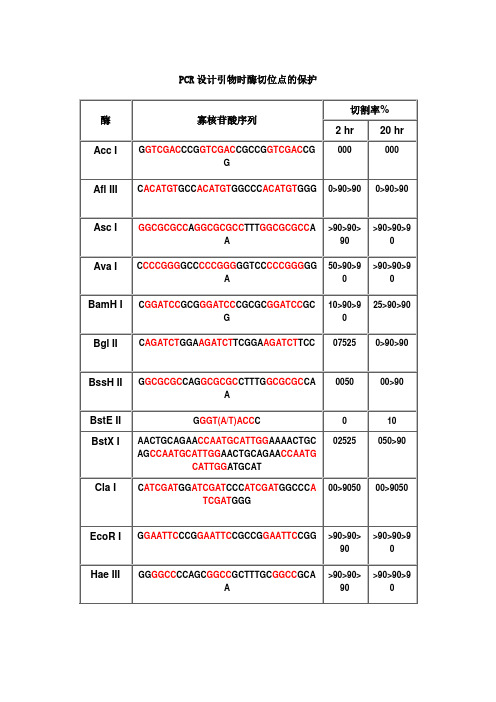
不同内切酶对识别位点以外最少保护碱基数目的要求PCR设计引物时酶切位点的保护碱基表1
PCR设计引物时酶切位点的保护碱基表2
在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
添加保护碱基,需要考虑两个因素:一是碱基数目,一是碱基种类。
添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的,见附表。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
保护碱基列表
PCR设计引物时酶切位点的保护
注释:
1.如果要加在序列的5‘端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3‘端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。
2.切割率:正确识别并酶切的效率
3。
加保护碱基时最好选用切割率高时加的相应碱基。
引物设计保护碱基列表
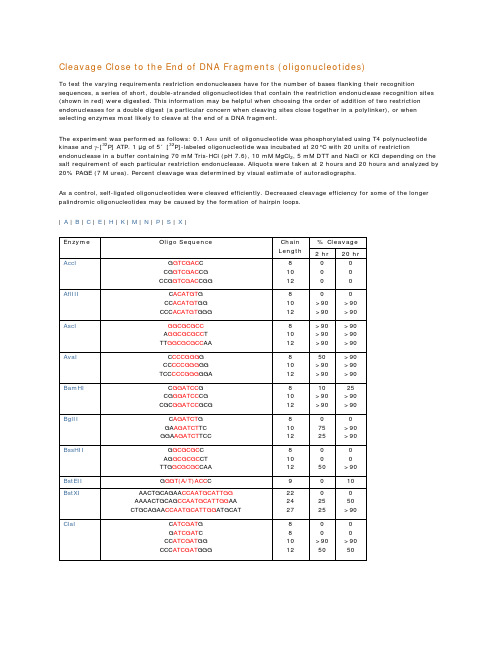
Cleavage Close to the End of DNA Fragments (oligonucleotides)To test the varying requirements restriction endonucleases have for the number of bases flanking their recognition sequences, a series of short, double-stranded oligonucleotides that contain the restriction endonuclease recognition sites (shown in red) were digested. This information may be helpful when choosing the order of addition of two restriction endonucleases for a double digest (a particular concern when cleaving sites close together in a polylinker), or when selecting enzymes most likely to cleave at the end of a DNA fragment.The experiment was performed as follows: 0.1 A260 unit of oligonucleotide was phosphorylated using T4 polynucleotide kinase and γ-[32P] ATP. 1 µg of 5´ [32P]-labeled oligonucleotide was incubated at 20°C with 20 units of restriction endonuclease in a buffer containing 70 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT and NaCl or KCl depending on the salt requirement of each particular restriction endonuclease. Aliquots were taken at 2 hours and 20 hours and analyzed by 20% PAGE (7 M urea). Percent cleavage was determined by visual estimate of autoradiographs.As a control, self-ligated oligonucleotides were cleaved efficiently. Decreased cleavage efficiency for some of the longer palindromic oligonucleotides may be caused by the formation of hairpin loops.| A | B | C | E | H | K | M | N | P | S | X |。
引物设计保护碱基列表上课讲义

引物设计保护碱基列
表
在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
添加保护碱基,需要考虑两个因素:一是碱基数目,一是碱基种类。
添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的,见附表。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
保护碱基列表。
保护碱基列表

0
0
75
>90
25
>90
0
0
0
0
50
>90
0
10
0
0
25
50
25
>90
0
0
0
0
>90
>90
50
50
>90
>90
>90
>90
>90
>90
>90
>90
>90
>90
>90
>90
2 精心整理,用心做精品
用心整理的精品 word 文档,下载即可编辑!!
Hind III
CAAGCTTG CCAAGCTTGG CCCAAGCTTGGG
GGAATTCC CGGAATTCCG CCGGAATTCCGG
GGGGCCCC AGCGGCCGCT TTGCGGCCGCAA
切割率%
2 hr
0 0 0
20 hr
0 0 0
0
0
>90
>90
>90
>90
>90
>90
>90
>90
>90
>90
50
>90
>90
>90
>90
>90
10
25
>90
>90
>90
>90
CACATGTG CCACATGTGG CCCACATGTGGG
GGCGCGCC AGGCGCGCCT TTGGCGCGCCAA
保护碱基列表
PCR设计引物时酶切位点的保护注释:1.如果要加在序列的5‘端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3‘端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。
2.切割率:正确识别并酶切的效率3。
加保护碱基时最好选用切割率高时加的相应碱基。
加盟支持内容列表华夏帝王列表中国皇帝(君王)包括正统朝代和少数民族建立的政权,还有一些政变、夺权所建立的政权,再加上农民起义建立的政权,中国皇帝共有1000多位呢!附:南越、东越、闽越、东瓯、匈奴、突厥、回纥(回鹘)、吐蕃、高昌、于阗、柔然、吐谷浑、渤海国(大震)、南诏(大蒙、大礼、大封民)、大长和、大天兴、大义宁、大理国(前理汉武帝刘彻、后理)、大中、东夏(大真)(以上不包括十六国时期和五代十国时期的少数民族政权)其中云南列朝自世隆以下【南诏(大蒙、大礼、大封民)、大长和、大天兴、大义宁、大理国(前理、后理)、大中】和东夏(大真)的君主称皇帝;南越(吕后时)、于阗(五代时)的君主一度称皇帝;南越、东越、闽越、东瓯、高昌、于阗、吐谷浑、渤海国(大震)作为中原王朝的藩属国,君主称王;匈奴的君主称单于;回纥(回鹘)、柔然的君主称可汗;吐蕃的君主称赞普。
十六国时期:汉赵、后赵、成汉、前燕、后燕、南燕、前秦、后秦、胡夏9个政权称皇帝,后凉、北燕2个政权称天王,前凉、西秦、南凉、北凉、西凉5个半独立政权称王。
其中:汉赵刘渊304年—308年称汉王;后赵石勒319年—328年称赵王,328年—330年称天王,石虎335年—349年称天王;成汉李雄304年—306年称成都王;前燕慕容皝337年—348年、慕容儁348年—352年称燕王;后燕慕容垂384年—386年称燕王,慕容盛400年—401年、慕容熙401年—407年称天王;南燕慕容德398年—400年称燕王;前秦苻健351年—352年、苻坚357年—385年称天王;后秦姚苌384年—386年称万年秦王,姚兴399年—416年称天王;胡夏赫连勃勃407年—418年称天王。
保护碱基列表

25
90
AAGGAAAAAAGCGGCCGCAAAAGGAAAA
25
>90
NsiI
TGCATGCATGCA
10
>90
CCAATGCATTGGTTCTGCAGTT
>90
>90
PacI
TTAATTAA GTTAATTAAC CCTTAATTAAGG
0
0
0
25
0
>90
PmeI
GTTTAAAC GGTTTAAACC GGGTTTAAACCC AGCTTTGTTTAAACGGCGCGCCGG
GGGT(A/T)ACCC
AACTGCAGAACCAATGCATTGG AAAACTGCAGCCAATGCATTGGAA CTGCAGAACCAATGCATTGGATGCAT
CATCGATG GATCGATC CCATCGATGG CCCATCGATGGG
GGAATTCC CGGAATTCCG CCGGAATTCCGG
GACGCGTC CGACGCGTCG
0
0
25
50
NcoI
CCCATGGG CATGCCATGGCATG
0
0
50
75
NdeI
CCATATGG CCCATATGGG CGCCATATGGCG GGGTTTCATATGAAACCC GGAATTCCATATGGAATTCC GGGAATTCCATATGGAATTCCC
0
0
>90
>90
75
>90
75
>90
XhoI
CCTCGAGG CCCTCGAGGG CCGCTCGAGCGG
保护碱基列表
010>90>900
Pvu I
GCGATCGCATCGATCGATCGACGATCGTCG
0100
02510
Sac I
GGAGCTCC
10
10
Sac II
CCCGCGGGGGACCGCGGTCC
050
0>90
Sal I
GTCAAAAGGCCATAGCGGCCGCGTCGACGTCTTGGCCATAGCGGCCGCGGGTCGACGCGTCGGCCATAGCGGCCGCGACGCGTCGACGAA
.
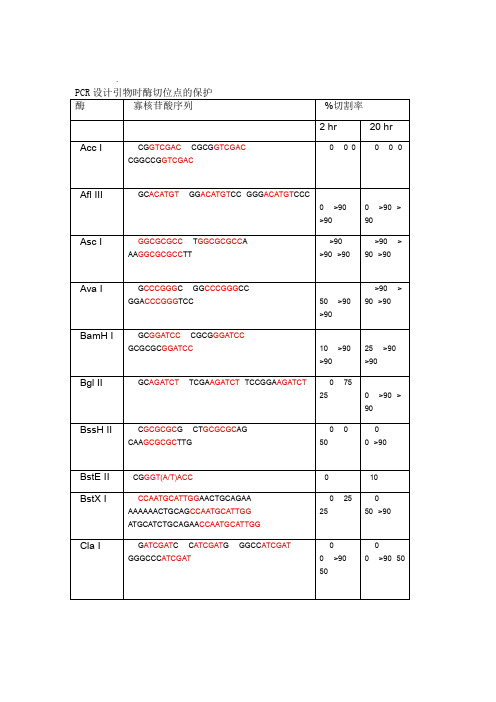
PCR设计引物时酶切位点的保护
酶
寡核苷酸序列
%切割率
2 hr
20 hr
Acc I
CGGTCGACCGCGGTCGACCGGCCGGTCGAC
000
000
Afl III
GCACATGTGGACATGTCCGGGACATGTCCC
0>90>90
0>90>90
Asc I
GGCGCGCCTGGCGCGCCAAAGGCGCGCCTT
0>907575
0>90>90>90
Xho I
GCCTCGAGGGCCCTCGAG
010
025
3 / 4
.
7510CTCGAGCGGCCG
Xma I
GCCCCGGGGGCCCCCGGGGGGCCCCCCGGGGGGATCCCCCCGGG
02550>90
075>90>90
注释:'端加55‘端,就在酶切位点识别碱基序列(红色)的1.如果要加在序列的‘端加保护碱基,就在酶切位点识上相应的碱基(黑色),相同如果要在3 '端加上相应的碱基(黑色)。别碱基序列(红色)的3 2.切割率:正确识别并酶切的效率3。加保护碱基时最好选用切割率高时加的相应碱基。4 / 4
保护碱基列表

10
25
75
75
SmaI
CCCGGG CCCCGGGG CCCCCGGGGG TCCCCCGGGGGA
0
10
0
10
10
50
>90
>90
SpeI
GACTAGTC GGACTAGTCC CGGACTAGTCCG CTAGACTAGTCTAG
10
>90
10
>90
0
50
0
50
SphI
GGCATGCC CATGCATGCATG ACATGCATGCATGT
PCR 设计引物时酶切位点的保护
精心整理
酶
寡核苷酸序列
AccI AflIII AscI AvaI BamHI BglII BssHII BstEII BstXI ClaI
GGTCGACC CGGTCGACCG CCGGTCGACCGG
CACATGTG CCACATGTGG CCCACATGTGGG
GGCGCGCC AGGCGCGCCT TTGGCGCGCCAA
ቤተ መጻሕፍቲ ባይዱ
10
SacII
GCCGCGGC TCCCCGCGGGGA
0
0
50
>90
SalI
GTCGACGTCAAAAGGCCATAGCGGCCGC GCGTCGACGTCTTGGCCATAGCGGCCGCGG
0 10
0 50
ACGCGTCGACGTCGGCCATAGCGGCCGCG
10
75
GAA
ScaI
GAGTACTC AAAAGTACTTTT
0
0
10
10
AACTGCAGAACCAATGCATTGG
引物设计保护碱基列表

在分子克隆试验中,有时我们会在待扩增目标基因片段两端加上特定酶切位点,用于后续酶切和连接反应。
因为直接暴露在末端酶切位点不轻易直接被限制性核酸内切酶切开,所以在设计PCR引物时,人为在酶切位点序列5‘端外侧添加额外碱基序列,即保护碱基,用来提升未来酶切时活性。
添加保护碱基,需要考虑两个原因:一是碱基数目,一是碱基种类。
添加保护碱基时,最关心应该是保护碱基数目,而不是种类。
什么样酶切位点,添加多个保护碱基,是有数据能够参考,见附表。
添加什么保护碱基,假如严格点,是依据两条引物Tm值和各引物碱基分布及GC含量。
假如某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
保护碱基列表。
保护碱基列表

Sph I Stu I Xba I
Xho I
GCTGCAGC TGCACTGCAGTGCA AACTGCAGAACCAATGCATTGG AAAACTGCAGCCAATGCATTGGAA CTGCAGAACCAATGCATTGGATGCAT
CCGATCGG ATCGATCGAT TCGCGATCGCGA
GACTAGTC GGACTAGTCC CGGACTAGTCCG CTAGACTAGTCTAG
GGCATGCC CATGCATGCATG ACATGCATGCATGT
AAGGCCTT GAAGGCCTTC AAAAGGCCTTTT
CTCTAGAG GCTCTAGAGC TGCTCTAGAGCA CTAGTCTAGACTAG
10 >90
0 0 0
0 0 0 75
〉90 〉90 〉90
0 0 75
0 >90 〉90
0 50
75
0 0 0 0 〉90 〉90
0 25 50
0 10 10 90 〉90
〉90 >90
0 25 〉90
0 25 50 〉90
Pst I
Pvu I Sac I Sac II Sal I
Sca I Sma I
TTAATTAA GTTAATTAAC CCTTAATTAAGG
GTTTAAAC GGTTTAAACC GGGTTTAAACCC AGCTTTGTTTAAACGGCGCGCCGG
〉90 >90 〉90
0 0 10
0 〉90 >90
0 25
0 50
0 0 0 0 75 75
0 10 10
0 10 10 25 25
保护碱基列表

10
75
GAA
ScaI
GAGTACTC AAAAGTACTTTT
10
25
75
75
SmaI
CCCGGG CCCCGGGG CCCCCGGGGG TCCCCCGGGGGA
0
10
0
10
10
50
>90
>90
SpeI
GACTAGTC GGACTAGTCC CGGACTAGTCCG CTAGACTAGTCTAG
10
>90
10
>90
0
50
50
SphI
GGCATGCC CATGCATGCATG ACATGCATGCATGT
0
0
0
25
10
50
StuI
AAGGCCTT GAAGGCCTTC AAAAGGCCTTTT
>90
>90
>90
>90
>90
>90
XbaI
CTCTAGAG GCTCTAGAGC TGCTCTAGAGCA CTAGTCTAGACTAG
0
0
>90
>90
75
>90
75
>90
XhoI
CCTCGAGG CCCTCGAGGG CCGCTCGAGCGG
0
0
10
25
10
75
仅供个人学习参考
XmaI
CCCCGGGG CCCCCGGGGG CCCCCCGGGGGG TCCCCCCGGGGGGA
0
0
25
75
50
>90
保护碱基列表

25
90
AAGGAAAAAAGCGGCCGCAAAAGGAAAA
25
>90
Nsi I
TGCATGCATGCA CCAATGCATTGGTTCTGCAGTT
10
>90
>90
>90
Pac I
TTAATTAA GTTAATTAAC CCTTAATTAAGG
0
0
0
25
0
>90
Pme I
GTTTAAAC GGTTTAAACC GGGTTTAAACCC AGCTTTGTTTAAACGGCGCGCCGG
10
75
GAA
Sca I
GAGTACTC AAAAGTACTTTT
10
25
75
75
Sma I
CCCGGG CCCCGGGG CCCCCGGGGG TCCCCCGGGGGA
0
10
0
10
10
50
>90
>90
Spe I
GACTAGTC GGACTAGTCC CGGACTAGTCCG CTAGACTAGTCTAG
GGGT(A/T)ACCC
AACTGCAGAACCAATGCATTGG AAAACTGCAGCCAATGCATTGGAA CTGCAGAACCAATGCATTGGATGCAT
CATCGATG GATCGATC CCATCGATGG CCCATCGATGGG
GGAATTCC CGGAATTCCG CCGGAATTCCGG
Hae III
GGGGCCCC AGCGGCCGCT TTGCGGCCGCAA
>90
>90
保护碱基列表
GGGTACCC GGGGTACCCC CGGGGTACCCCG
GACGCGTC CGACGCGTCG
50
>90
0
10
0
0
25
50
25
>90
0
0
0
0
>90
>90
50
50
>90
>90
>90
>90
>90
>90
>90
>90
>90
>90
>90
>90
酶 Acc I Afl III Asc I Ava I BamH I Bgl II BssH II
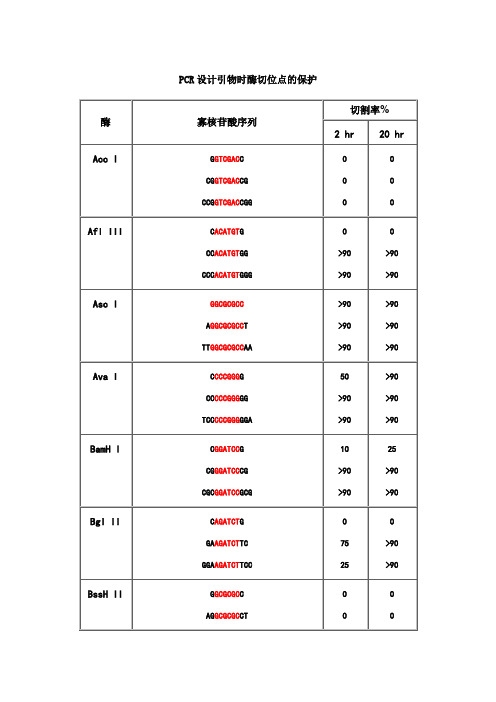
PCR 设计引物时酶切位点的保护
寡核苷酸序列
GGTCGACC CGGTCGACCG CCGGTCGACCGG
切割率% 2 hr 20 hr
0
0
0
0
0
0
CACATGTG CCACATGTGG CCCACATGTGGG
75
>90
0
0
10
10
>90
>90
>90
>90
0
0
0
0
10
25
0
10
10
10
0
0
50
>90
0
0
10
50
10
75
10
25
75
75
0
10
0
10
10
50
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
11月13日引物合成的详解4.需要什么级别的引物?答:引物常用的纯化方式C18脱盐,OPC纯化,PAGE纯化,HPLC纯化。
根据实验需要,确定订购引物的纯度级别。
应用引物长度要求纯度级别要求一般PCR扩增<45 base OPC一般PCR扩增>45 base PAGE诊断PCR扩增< 40base OPC, PAGEDNA测序20base左右OPC亚克隆,点突变等根据实验要求定OPC, PAGE,HPLC根据实验要求定PAGE基因构建(全基因合成)反义核酸根据实验要求定PAGEPAGE, HPLC修饰引物根据实验要求定8.如何计算引物的浓度?答:引物保存在高浓度的状况下比较稳定。
引物一般配制成10-50pmol/ul。
一般情况下,建议将引物的浓度配制成50pmol/ul,加水的体积(微升)按下列方式计算:V (微升)= OD数*(乘)33 *(乘)*(乘)20000 / (除) 引物的分子量。
引物的分子量可以从合成报告单上获得。
如果需要配制成其他浓度,按上述公式换算。
注意:1 OD260= 33 ug/ml.9.如何计算引物的Tm值?答:引物设计软件都可以给出Tm,与引物长度、碱基组成、引物使用缓冲的离子强度有关。
长度为25mer以下的引物,Tm计算公式为:Tm = 4℃(G + C)+ 2℃(A + T)对于更长的寡聚核苷酸,Tm计算公式为:Tm = 81.5 + 16.6 x Log10[Na+] + 0.41 (%GC) – 600/size公式中,Size = 引物长度。
11.如何溶解引物?答:干燥后的引物质地非常疏松,开盖前最好离心一下,或管垂直向上在桌面上敲敲,将引物粉末收集到管底。
根据计算出的体积加入去离子无菌水或10mM Tris pH7.5缓冲液,室温放置几分钟,振荡助溶,离心将溶液收集到管底。
溶解引物用的水一般不要用蒸馏水,因为有些蒸馏水的pH值比较低(pH4-5),引物在这种条件下不稳定。
12.如何保存引物?答:引物合成后,经过一系列处理和纯化步骤,旋转干燥而成片状物质。
引物在溶解前,室温状态下可以长期保存。
溶解后的引物-20度可以长期保存。
如果对实验的重复性要求较高,合成的OD数较大,建议分装,避免反复冻融。
修饰荧光引物需要避光保存。
13.合成的引物5’端是否有磷酸化答:合成的引物5’为羟基,没有磷酸基团。
如果需要您可以用多核苷酸激酶进行5′端磷酸化,或者要求引物合成公司合成时直接在5′或3′端进行磷酸化,需要另外收费。
14.引物片段退火后不能连接到载体上是什么问题?连接反应需要引物的5’磷酸基团。
如果需要将合成的引物退火直接连接相应的载体上,引物需要磷酸化。
磷酸化的产物如果还不能连接载体上,需要检查载体的酶切效果,需要改善引物退火的条件。
SiRNA分子具有特殊的对称结构,退火的难度较大,退火时需要提高退火温度。
16.长链引物为什么出错的几率非常高?答:引物合成时,每一步反应效率都不能达到100%,产生碱基插入、缺失、置换突变的因素客观条件都有一直存在。
引物链越长,突变的频率累加起来就越高。
研究人员总希望合成的引物万无一失,这种心情可以理解。
但是犹如PCR扩增,不可能绝对保证扩增产物中没有突变,引物合成也不可能保证100%正确。
要知道,引物合成中发生错误(非人为因素)的频率,比任何高保真高温聚合酶PCR扩增过程所产生的频率都要高。
做引物合成,长链引物合成,您要有引物中部分引物可能有突变的思想准备。
19. TaqMan 探针设计的基本原则是什么?答:下列原则供您参考。
◆TaqMan 探针位置尽可能靠近扩增引物(扩增产物50-150bp),但不能与引物重叠。
◆长度一般为18-40mer 。
◆G-C含量控制在40-80%左右。
◆避免连续相同碱基的出现,特别是要避免GGGG或更多G出现。
◆在引物的5’端避免使用G。
◆选用比较多的碱基C。
◆退火温度Tm控制在 68-70C左右。
有用的荧光染料参数colorsName吸收波长发射波长6-FAM (6-carboxy-fluorescein)494nm518nm GreenTET (5-tetrachloro-fluorescein)521nm 538nm OrangeHEX (5-hexachloro-fluorescein)535nm 553nm PinkTAMRA(tetramethyl-6-carboxyrhodamine)560nm582nm RoseROX (6-carboxy-x-rhodamine)587nm607nm RedCy3 (Indodicarbocyanine)552nm570nm RedCy5(Indodicarbocyanine)643nm667nm Violet20.Primer设计的基本原则是什么?答:引物设计的下列原则供您参考。
◆引物长度一般在18-35mer。
◆G-C含量控制在40-60%左右。
◆避免近3’端有酶切位点或发夹结构。
◆如果可能避免在3’端最后5个碱基有2个以上的G或C。
◆如果可能避免在3’端最后1个碱基为A。
◆避免连续相同碱基的出现,特别是要避免GGGG或更多G出现。
◆退火温度Tm控制在 58-60C左右。
◆如果是设计点突变引物,突变点应尽可能在引物的中间。
21.为什么引物的OD260/OD280小于1.5 ?答:引物应该全是DNA,但是OD260/OD280的比值为什么那么低,怎么会有蛋白质污染?引物化学合成,哪里有机会污染到蛋白质?需要指出的是OD260/OD280的比值不能用来衡量引物的纯度。
OD260/OD280的比值过低一般是由于引物中C/T 的含量比较高所致。
下表是一个20mer 同聚体引物的OD260/OD280的比值,清楚表明OD260/OD280的比值与引物的碱基组成密切相关。
A260/280 ratios of Crude 20-mer Oligos of Differing Base CompositionsBase Composition A260/2805-AAAAAAAAAAAAAAAAAAAA-3 2.505-GGGGGGGGGGGGGGGGGGGG-3 1.855-CCCCCCCCCCCCCCCCCCCC-3 1.155-TTTTTTTTTTTTTTTTTTTT-3 1.145-AAAAAGGGGGTTTTTCCCCC-3 1.6622.同样的OD用PAGE检测,EB染色为什么深浅不一?答:通常可以用EB染色的方法来判断双链DNA的量(如质粒DNA),因为EB可以嵌合到双链DNA中。
而合成的单链DNA,由于碱基组成不同,形成二级结构的可能性不同,EB的染色程度也会有差异,比如Oligo(dT)等不形成二级结构,EB染色效果就非常差。
所以不要用EB染色的方法来定量,而用紫外分光光度计检测。
同样道理,用EB染色来照片不适合所有引物。
23.引物不纯会有什么后果?答:引物不纯可能会导致:1)非特异性扩增;2)无法用预先设计在引物5′端酶切位点的酶切开,特别是没有保护碱基的引物;3) 用于测序出现双峰或乱峰。
解决办法重新合成或重新纯化。
24.已经溶解的引物,为什么原先使用正常,而过一段时间再使用就不好了?答:如果溶解引物的水PH过低或污染了菌或核酸酶,会使引物降解。
使用时没有充分解冻混合,液体不均匀也可能会造成引物加入量不准确。
建议分装引物,避免反复冻溶。
建议使用10mM Tris pH7.5缓冲液溶解引物,因为有些蒸馏水的pH值比较低(pH4-5), 引物在这种条件下不稳定。
还有一种可能性是引物没有问题,而是PCR使用材料特别是模板的质量与先前使用的不完全一致。
25.PCR扩增不出就引物有问题吗?答:基本不是。
当今发展出各色各样的PCR扩增技术,各色各样的高温聚合酶,就是来解决PCR扩增中遇到的扩不出、扩增效率低的问题。
如巢式PCR就是扩增那些拷贝数很低的基因片段。
有些重复片段的扩增, GC含量高的片段,非要采用特殊扩增手段才能扩增出了。
引物扩增不出,主要是下列两种情况比较常见(1) RT-PCR。
请注意,很多基因通过常规RT –PCR方法是很难扩增出来的。
RT- PCR成功的关键在于RT的反应的RNA质量和目标基因在特定组织和细胞中含量。
(2)从基因组中扩增。
一般情况下,基因在基因组中都是单拷贝,基因组作为模板需要严格控制用量。
基因组DNA过高,会影响反应体系中的Mg和pH。
16:14 | 添加评论 | 固定链接 | 写入日志保护碱基列表AccI G GTCGAC CCG GTCGAC CGCCG GTCGAC CGG81012AflIII C ACATGT GCC ACATGT GGCCC ACATGT GGG81012>90>90>90>90AscI GGCGCGCCA GGCGCGCC TTT GGCGCGCC AA81012>90>90>90>90>90>90AvaI C CCCGGG GCC CCCGGG GGTCC CCCGGG GGA8101250>90>90>90>90>90BamHI C GGATCC GCG GGATCC CGCGC GGATCC GCG8101210>90>9025>90>90BglII C AGATCT GGA AGATCT TCGGA AGATCT TCC810127525>90>90BssHII G GCGCGC CAG GCGCGC CTTTG GCGCGC CAA8101250>90BstEII G GGT(A/T)ACC C 9 0 10BstXI AACTGCAGAA CCAATGCATTGGAAAACTGCAG CCAATGCATTGG AACTGCAGAA CCAATGCATTGG ATGCAT 222427252550>90ClaI C ATCGAT GG ATCGAT CCC ATCGAT GGCCC ATCGAT GGG881012>9050>9050EcoRI G GAATTC CCG GAATTC CGCCG GAATTC CGG81012>90>90>90>90>90>90HaeIII GG GGCC CCAGC GGCC GCTTTGC GGCC GCAA81012>90>90>90>90>90>90HindIII C AAGCTT GCC AAGCTT GGCCC AAGCTT GGG810121075KpnI G GGTACC CGG GGTACC CCCGG GGTACC CCG81012>90>90>90>90MluI G ACGCGT CCG ACGCGT CG8102550NcoI C CCATGG GCATG CCATGG CATG8145075NdeI C CATATG GCC CATATG GGCGC CATATG GCGGGGTTT CATATG AAACCCGGAATTC CATATG GAATTCCGGGAATTC CATATG GAATTCCC810121820227575>90>90NheI G GCTAGC CCG GCTAGC CGCTA GCTAGC TAG8101210102550NotI TT GCGGCCGC AAATTT GCGGCCGC TTTAAAATAT GCGGCCGC TATAAAATAAGAAT GCGGCCGC TAAACTATAAGGAAAAAA GCGGCCGC AAAAGGAAAA 121620242810102525101090>90NsiI TGC ATGCAT GCACCA ATGCAT TGGTTCTGCAGTT 122210>90>90>90PacI TTAATTAAG TTAATTAA CCC TTAATTAA GG8101225>90PmeI GTTTAAACG GTTTAAAC CGG GTTTAAAC CCAGCTTT GTTTAAAC GGCGCGCCGG8101224752550>90PstI G CTGCAG CTGCA CTGCAG TGCAAA CTGCAG AACCAATGCATTGGAAAA CTGCAG CCAATGCATTGGAACTGCAG AACCAATGCATTGGATGCAT81422242610>90>9010>90>90PvuI C CGATCG GAT CGATCG ATTCG CGATCG CGA81012102510SacI C GAGCTC G 8 10 10SacII G CCGCGG CTCC CCGCGG GGA81250>90SalI GTCGAC GTCAAAAGGCCATAGCGGCCGCGC GTCGAC GTCTTGGCCATAGCGGCCGCGGACGC GTCGAC GTCGGCCATAGCGGCCGCGGAA 28303210105075ScaI G AGTACT CAAA AGTACT TTT81210752575SmaI CCCGGGC CCCGGG GCC CCCGGG GG681010101050TCC CCCGGG GGA 12 >90 >90SpeI G ACTAGT CGG ACTAGT CCCGG ACTAGT CCGCTAG ACTAGT CTAG81012141010>90>905050SphI G GCATGC CCAT GCATGC ATGACAT GCATGC ATGT81214102550StuI A AGGCCT TGA AGGCCT TCAAA AGGCCT TTT81012>90>90>90>90>90>90XbaI C TCTAGA GGC TCTAGA GCTGC TCTAGA GCACTAG TCTAGA CTAG8101214>907575>90>90>90XhoI C CTCGAG GCC CTCGAG GGCCG CTCGAG CGG8101210102575XmaI C CCCGGG GCC CCCGGG GGCCC CCCGGG GGGTCCC CCCGGG GGGA81012142550>9075>90>90。