3-Phenyl-4-aroyl-5-isoxazolonate complexes of Tb3+ as promising
艾沙康唑硫酸盐说明书

艾沙康唑硫酸盐;Isavuconazonium sulfate产品编号:MB5282质量标准:>95%,BR包装规格:20MG;100MG;产品形式:solid基本信息简介:艾沙康唑硫酸盐是艾沙康唑的前体药物,进入体内后代谢为艾沙康唑后,发挥抗真菌的作用机制。
别名:N-methyl-[2-[[[1-[1-[(2R,3R)-3-[4-(4-cyanophenyl)-2-thiazolyl]-2-(2,5-difluorophenyl)-2-hydroxybutyl]-4H-1,2,4-triazolium-4-yl]ethoxy]carbonyl]methylamino]-3-pyridinyl]methyl ester, glycine, monosulfate物理性状及指标:外观:………………白色至类白色固体溶解性:……………Soluble in DMSO;Water Insoluble含量: (95)储存条件:-20℃,避光防潮密闭干燥生物活性2015年3月6日,美国FDA优先审批批准日本安斯泰来的抗真菌新药艾沙康唑硫酸盐(Isavuconazonium sulfate),以商品名Cresemba上市。
用于治疗侵入性曲霉病和毛霉菌病,这两种真菌感染多发于血癌患者中。
艾沙康唑硫酸盐是艾沙康唑的前体药物,进入体内后代谢为艾沙康唑后,发挥抗真菌的作用机制。
艾沙康唑硫酸盐有口服和注射剂,艾沙康唑较辉瑞的伏立康唑(Vfend)更安全有效,病患死亡率更低。
艾沙呋唑是唑类抗真菌药艾沙呋唑(ISA)的水溶性前药形式。
口服伊沙呋唑铵可提高对唑敏感和耐药的烟曲霉感染大鼠模型在0.25-512毫克/千克/天剂量范围内(伊沙当量=0.12-245.8毫克/千克/天)的存活率。
以40-60 mg/kg的Isa当量剂量给药时,Isavuconazonium可减轻真菌负荷和机体介导的肺损伤,并提高实验性侵袭性肺曲霉病兔模型的存活率。
药物月桂酰阿立哌唑(Aripiprazole lauroxil)合成检索总结报告
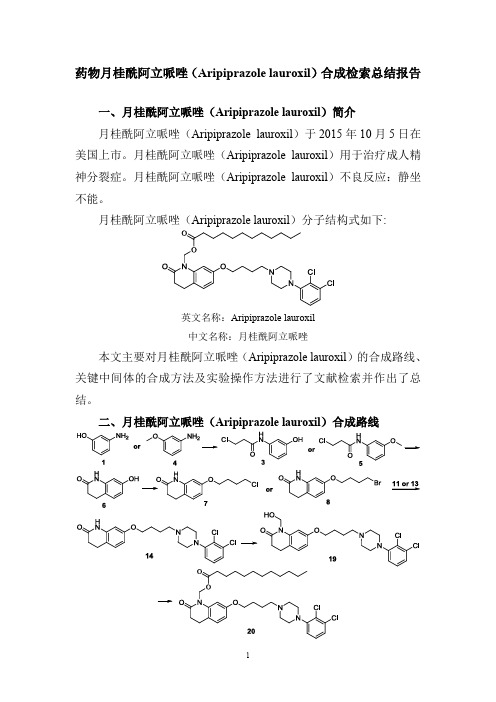
药物月桂酰阿立哌唑(Aripiprazole lauroxil)合成检索总结报告
一、月桂酰阿立哌唑(Aripiprazole lauroxil)简介
月桂酰阿立哌唑(Aripiprazole lauroxil)于2015年10月5日在美国上市。
月桂酰阿立哌唑(Aripiprazole lauroxil)用于治疗成人精神分裂症。
月桂酰阿立哌唑(Aripiprazole lauroxil)不良反应:静坐不能。
月桂酰阿立哌唑(Aripiprazole lauroxil)分子结构式如下:
英文名称:Aripiprazole lauroxil
中文名称:月桂酰阿立哌唑
本文主要对月桂酰阿立哌唑(Aripiprazole lauroxil)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、月桂酰阿立哌唑(Aripiprazole lauroxil)合成路线
三、月桂酰阿立哌唑(Aripiprazole lauroxil)合成检索总结报告(一) 月桂酰阿立哌唑(Aripiprazole lauroxil)中间体3的合成
(二) 月桂酰阿立哌唑(Aripiprazole lauroxil)中间体5的合成
(三)
月桂酰阿立哌唑(Aripiprazole lauroxil )中间体6的合成方法一
(四)月桂酰阿立哌唑(Aripiprazole lauroxil)中间体6的合成方法二
(五) 月桂酰阿立哌唑(Aripiprazole lauroxil)中间体7的合成。
对氯苯氧异丁酸(安妥明)的合成

对氯苯氧异丁酸(安妥明)的合成[适用对象]药物制剂专业[实验学时] 20学时一、实验目的掌握安妥明合成中缩合反应原理及产品精制操作方法。
了解和掌握成盐方法,原理以及基本操作。
二、实验原理1、缩合反应原理:2、成铝盐反应式3、成钙盐反应式三、仪器设备1、主要仪器:100ml三口烧瓶 1个调速搅拌器 1个、100℃温度计 1个球形冷凝管 1个100ml抽滤瓶 1个自动电热套 1个100ml烧杯 1个 250V调压器 1个吸滤瓶 1个布氏漏斗 1个表面皿 1个 b型熔点测定管1个铁架台 1个2、仪器与实验装置图回流装置熔点测试装置四、相关知识点抽滤装置本课程知识点综合:安妥明;【英文名】Clofibrate【类别】调节血脂及抗动脉硬化药【别名】安妥明;对氯苯氧异丁酸乙酯;冠心平;降脂乙酯;氯苯丁酯;祛脂乙酯;心血安,氯贝丁酯【外文名】Clofibrate【性状】无色或黄色的澄清油状液体,有特臭、味辛辣而甜;遇光色变深,几不溶水。
【药理作用】能抑制胆固醇和甘油三酯的合成。
促进胆固醇和排泄,降低甘油三酯较降低胆固醇作用显著。
还有减低血液粘度,降低血浆纤维蛋白原含量,有抗血栓作用。
【适应症】主要用于高甘油三酯血症,尤其适用于Ⅲ、Ⅳ型高脂蛋白血症,还用于动脉粥样硬化。
氯贝丁酯能降低血小板的粘附作用,抑制血小板聚集,使过高的纤维蛋白浓度降低至正常,因而减少血栓形成,可单独应用或与抗凝剂合用于缺血性心脏病人。
多课程知识点综合:缩合反应 condensation reaction两个或两个以上有机分子相互作用后以共价键结合成一个大分子,并常伴有失去小分子(如水、氯化氢、醇等)的反应。
在多官能团化合物的分子内部发生的类似反应则称为分子内缩合反应。
缩合反应在有机化学,尤其是有机合成中应用很广。
①羟醛缩合反应。
为醛、酮或羧酸衍生物等羰基化合物在羰基旁形成新的碳-碳键,从而把两个分子结合起来的反应。
这些反应通常在酸或碱的催化作用下进行。
世卫组织-三类致癌物

81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123
1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987 1987
1-氨基-2-甲基蒽醌 喹诺酮(五氯硝基苯) 二乙酰基氨基甲苯 苏丹红4号 颜料黄AB,苯偶氮-2-萘胺 苏丹红III N-亚硝基二苯胺 肼苯哒嗪 1-萘基硫脲(ANTU) 2,4,6-三甲基苯胺 青霉酸 3,3′-二甲氧基-4,4′-联苯二异 3,3'-Dimethoxybenzidine-4,4'-diisocyanate 氰酸酯 4-Nitrobiphenyl 4-硝基联苯 2,4-Xylidine 2,4-二甲基苯胺 2,5-Diaminotoluene 2,5-二氨基甲苯 2,5-Xylidine 2,5二甲基苯胺 Eugenol 丁子香酚 Disulfiram 双硫仑 1,2-Diamino-4-nitrobenzene 1,2-二氨基-4-硝基苯 5-Nitro-ortho-anisidine 5- 邻氨基苯甲醚 N-Methyl-N,4-dinitrosoaniline N-甲基-N-4-亚硝基苯胺 Chloropropham 氯苯胺灵 Dinitrosopentamethylenetetramine 发泡剂H meta-Cresidine 3-氨基对甲苯甲醚 Phenicarbazide 1-苯基氨基脲 Azobenzene 偶氮苯 para-Anisidine 对甲氧基苯胺 para-Phenylenediamine 对苯二胺 Succinic anhydride 丁二酸酐 meta-Phenylenediamine 间苯二胺 Dicofol 三氯杀螨醇 2-Aminoanthraquinone 2-氨基蒽醌 Anthranilic acid 邻氨基苯甲酸 4-Amino-2-nitrophenol 2-硝基-4-氨基苯酚 Isosafrole 异黄樟素 2-Amino-5-nitrothiazole 2-氨基-5-硝基噻唑 Propham 苯胺灵 Maleic hydrazide 马来酰肼 Sulfafurazole (Sulfisoxazole) 磺胺异恶唑 Butylated hydroxytoluene (BHT) 丁羟甲苯 Blue VRS 酸性蓝1,食品篮3 Oxyphenbutazone 羟基保泰松 Yellow OB 邻甲苯偶氮-2-萘胺 Captan 环己烯亚胺,克菌丹 1-Naphthylamine α-萘胺
阿司咪唑

合成方法
合成方法
化合物(I)和碘甲烷在乙醇中回流8h,环合得到化合物(Ⅱ)。再水解脱去酯基,得到化合物(Ⅲ)。用对甲氧 基苯乙基溴进行N-烷基化,得化合物(Ⅳ)。再用对氟苄基溴烷基化,得阿司咪唑。
1. 1-[(4-氟苯基)甲基]-苯并咪唑-2-(3H)-酮的制备
在反应瓶中加入2-羟基苯并咪唑5.0g(37.3mmol)和NaH 1.6g(53mmol)(NaH含量大约为80%,浸入矿物油中) 的DMF 100ml的悬浮液.加毕.在60ºC.(最好有N2保护)搅拌反应1h.再加入4-氟苄基氯(FBC)5.4g(37mmol),加热 ( 6 0 ºC ) 搅 拌 反 应 5 . 5 h . 冷 却 至 室 温 后 加 入 冰 水 7 0 0 m l , 用 二 氯 甲 烷 ( 5 0 0 m l × 2 ) 提 取 . 有 机 层 用 食 盐 水 洗 . 无 水 N a 2 S O 4 干燥.过滤.滤液减压浓缩.剩余物用石油醚析晶.得1-[(4-氟苯基)甲基]-苯并咪唑-2-(3H)-酮固体8.0g,为无色 结 晶 m p 1 7 8 ~ 1 7 9 ºC , 收 率 8 8 % .
治疗措施
阿司咪唑中毒的治疗要点为: 1.大量摄入者予洗胃,后灌服活性炭和导泻。 2.对心肌抑制和Q-T间期延长者予5%碳酸氢钠250ml静注可能有效。 3.对症、支持治疗。
专家点评
专家点评
阿司咪阿司咪唑自1983年上市以来,在许多国家得到了广泛应用。国外研究显示阿司咪唑治疗荨麻疹的总有 效率为74%。国内的一项多中心双盲安慰剂对照试验表明阿司咪唑对急性荨麻疹的总有效率为82.9%,对慢性荨麻 疹的总有效率为86.0%,均显著高于安慰剂,主要不良反应为嗜睡、倦怠、口干等,连续用药3个月的患者中,半 数有食欲及体重增加。阿司咪唑的心脏毒性虽然发生率较低,但由于后果严重,已限制了它的应用。阿司咪唑为 强效和长效的H1受体拮抗剂,无中枢镇静和抗毒蕈碱样作用。代谢产物去甲阿司咪唑仍有抗胆胺作用。长期服用 可增进食欲和增加体重,服用过量可引起心脏Q-T间期延长和室性心律失常。适用于各种原因引起过敏性疾病。
盐酸莫西沙星氯化钠注射液说明书

AVELOX®(moxifloxacin hydrochloride) TabletsAVELOX® I.V.(moxifloxacin hydrochloride in sodium chloride injection)XXXXXXX, R.X02/08 To reduce the development of drug-resistant bacteria and maintain the effectiveness of AVELOX® and other antibacterial drugs, AVELOX should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria.DESCRIPTIONAVELOX (moxifloxacin hydrochloride) is a synthetic broad spectrum antibacterial agent and is available as AVELOX Tablets for oral administration and as AVELOX I.V. for intravenous administration. Moxifloxacin, a fluoroquinolone, is available as the monohydrochloride salt of 1-cyclopropyl-7-[(S,S)-2,8-diazabicyclo[4.3.0]non-8-yl]-6-fluoro-8-methoxy-1,4-dihydro-4-oxo-3 quinoline carboxylic acid. It is a slightly yellow to yellow crystalline substance with a molecular weight of 437.9. Its empirical formula is C21H24FN3O4 *HCl and its chemical structure is as follows:AVELOX Tablets are available as film-coated tablets containing moxifloxacin hydrochloride (equivalent to 400 mg moxifloxacin). The inactive ingredients are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, magnesium stearate, hypromellose, titanium dioxide, polyethylene glycol and ferric oxide.AVELOX I.V. is available in ready-to-use 250 mL latex-free flexibags as a sterile, preservative free, 0.8% sodium chloride aqueous solution of moxifloxacin hydrochloride (containing 400 mg moxifloxacin) with pH ranging from 4.1 to 4.6. The appearance of the intravenous solution is yellow. The color does not affect, nor is it indicative of, product stability. The inactive ingredients are sodium chloride, USP, Water for Injection, USP, and may include hydrochloric acid and/or sodium hydroxide for pH adjustment.CLINICAL PHARMACOLOGYAbsorptionMoxifloxacin, given as an oral tablet, is well absorbed from the gastrointestinal tract. The absolute bioavailability of moxifloxacin is approximately 90 percent. Co-administration with a high fat meal (i.e., 500 calories from fat) does not affect the absorption of moxifloxacin. Consumption of 1 cup of yogurt with moxifloxacin does not significantly affect the extent or rate of systemic absorption (AUC).The mean (± SD) C max and AUC values following single and multiple doses of 400 mg moxifloxacin given orally are summarized below.C max (mg/L)AUC(mg•h/L)Half-life(hr)Single Dose OralHealthy (n = 372) 3.1 ± 1.036.1 ± 9.111.5 - 15.6* Multiple Dose OralHealthy young male/female (n = 15) 4.5 ± 0.548.0 ± 2.712.7 ± 1.9 Healthy elderly male (n = 8) 3.8 ± 0.351.8 ± 6.7Healthy elderly female (n = 8) 4.6 ± 0.654.6 ± 6.7Healthy young male (n = 8) 3.6 ± 0.548.2 ± 9.0Healthy young female (n = 9) 4.2 ± 0.549.3 ± 9.5* Range of means from different studiesThe mean (± SD) C max and AUC values following single and multiple doses of 400 mg moxifloxacin given by 1 hour I.V. infusion are summarized below.C max (mg/L)AUC(mg•h/L)Half-life(hr)Single Dose I.V.Healthy young male/female (n = 56) 3.9 ± 0.939.3 ± 8.68.2 - 15.4* Patients (n = 118)Male (n = 64) 4.4 ± 3.7Female (n = 54) 4.5 ± 2.0< 65 years (n = 58) 4.6 ± 4.2≥ 65 years (n = 60) 4.3 ± 1.3Multiple Dose I.V.Healthy young male (n = 8) 4.2 ± 0.838.0 ± 4.714.8 ± 2.2 Healthy elderly (n =12; 8 male, 4 female) 6.1 ± 1.348.2 ± 0.910.1 ± 1.6 Patients** (n = 107)Male (n = 58) 4.2 ± 2.6Female (n = 49) 4.6 ± 1.5< 65 years (n = 52) 4.1 ± 1.4≥ 65 years (n = 55) 4.7 ± 2.7* Range of means from different studies** Expected C max (concentration obtained around the time of the end of the infusion)Plasma concentrations increase proportionately with dose up to the highest dose tested (1200 mg single oral dose). The mean (± SD) elimination half-life from plasma is 12 ± 1.3 hours; steady-state is achieved after at least three days with a 400 mg once daily regimen.Mean Steady-State Plasma Concentrations of Moxifloxacin Obtained With Once Daily Dosing of 400 mg Either Orally (n=10) or by I.V. Infusion (n=12)DistributionMoxifloxacin is approximately 30-50% bound to serum proteins, independent of drug concentration. The volume of distribution of moxifloxacin ranges from 1.7 to 2.7 L/kg. Moxifloxacin is widely distributed throughout the body, with tissue concentrations often exceeding plasma concentrations. Moxifloxacin has been detected in the saliva, nasal and bronchial secretions, mucosa of the sinuses, skin blister fluid, subcutaneous tissue, skeletal muscle, and abdominal tissues and fluids following oral or intravenous administration of 400 mg. Moxifloxacin concentrations measured post-dose in various tissues and fluids following a 400 mg oral or I.V. dose are summarized in the following table. The rates of elimination of moxifloxacin from tissues generally parallel the elimination from plasma.Moxifloxacin Concentrations (mean ± SD) in Tissuesand the Corresponding Plasma Concentrations After a Single 400 mg Oral orIntravenous Dose §Tissue or Fluid NPlasmaConcentration(µg/mL)Tissue or FluidConcentration(µg/mL or µg/g)TissuePlasmaRatio:RespiratoryAlveolar Macrophages5 3.3± 0.761.8± 27.321.2 ± 10.0 Bronchial Mucosa8 3.3± 0.7 5.5± 1.3 1.7 ± 0.3 Epithelial Lining Fluid5 3.3± 0.724.4± 14.78.7 ± 6.1 SinusMaxillary Sinus Mucosa4 3.7± 1.1†7.6± 1.7 2.0 ± 0.3 Anterior Ethmoid Mucosa3 3.7± 1.1†8.8± 4.3 2.2 ± 0.6 Nasal Polyps4 3.7± 1.1†9.8± 4.5 2.6 ± 0.6 Skin, MusculoskeletalBlister Fluid5 3.0± 0.5‡ 2.6± 0.9 0.9 ± 0.2 Subcutaneous Tissue6 2.3± 0.4#0.9± 0.3* 0.4 ± 0.6 Skeletal Muscle6 2.3± 0.4#0.9± 0.2* 0.4 ± 0.1 Intra-AbdominalAbdominal tissue 8 2.9± 0.5 7.6 ± 2.0 2.7 ± 0.8 Abdominal exudate 10 2.3±0.5 3.5 ±1.2 1.6 ± 0.7 Abscess fluid 6 2.7± 0.7 2.3 ±1.5 0.8±0.4§all moxifloxacin concentrations were measured 3 hours after a single 400 mg dose, except the abdominal tissue and exudate concentrations which were measured at 2 hours post-dose and the sinus concentrations which were measured 3 hours post-dose after 5 days of dosing.† N = 5‡N = 7#N = 12* Reflects only non-protein bound concentrations of drug.MetabolismApproximately 52% of an oral or intravenous dose of moxifloxacin is metabolized via glucuronide and sulfate conjugation. The cytochrome P450 system is not involved in moxifloxacin metabolism, and is not affected by moxifloxacin. The sulfate conjugate (M1) accounts for approximately 38% of the dose, and is eliminated primarily in the feces. Approximately 14% of an oral or intravenous dose is converted to a glucuronide conjugate (M2), which is excreted exclusively in the urine. Peak plasma concentrations of M2 are approximately 40% those of the parent drug, while plasma concentrations of M1 are generally less than 10% those of moxifloxacin.In vitro studies with cytochrome (CYP) P450 enzymes indicate that moxifloxacin does not inhibit CYP3A4, CYP2D6, CYP2C9, CYP2C19, or CYP1A2, suggesting that moxifloxacin is unlikely to alter the pharmacokinetics of drugs metabolized by these enzymes.ExcretionApproximately 45% of an oral or intravenous dose of moxifloxacin is excreted as unchanged drug (~20% in urine and ~25% in feces). A total of 96% ± 4% of an oral dose is excreted as either unchanged drug or known metabolites. The mean (± SD) apparent total body clearance and renal clearance are 12 ± 2.0 L/hr and 2.6 ± 0.5 L/hr, respectively.Special PopulationsGeriatricFollowing oral administration of 400 mg moxifloxacin for 10 days in 16 elderly (8 male; 8 female) and 17 young (8 male; 9 female) healthy volunteers, there were no age-related changes in moxifloxacin pharmacokinetics. In 16 healthy male volunteers (8 young; 8 elderly) given a single 200 mg dose of oral moxifloxacin, the extent of systemic exposure (AUC and C max)was not statistically different between young and elderly males and elimination half-life was unchanged. No dosage adjustment is necessary based on age. In large phase III studies, the concentrations around the time of the end of the infusion in elderly patients following intravenous infusion of 400 mg were similar to those observed in young patients.PediatricThe pharmacokinetics of moxifloxacin in pediatric subjects have not been studied.GenderFollowing oral administration of 400 mg moxifloxacin daily for 10 days to 23 healthy males (19-75 years) and 24 healthy females (19-70 years), the mean AUC and C max were 8% and 16% higher, respectively, in females compared to males. There are no significant differences in moxifloxacin pharmacokinetics between male and female subjects when differences in body weight are taken into consideration.A 400 mg single dose study was conducted in 18 young males and females. The comparison of moxifloxacin pharmacokinetics in this study (9 young females and 9 young males) showed no differences in AUC or C max due to gender. Dosage adjustments based on gender are not necessary. RaceSteady-state moxifloxacin pharmacokinetics in male Japanese subjects were similar to those determined in Caucasians, with a mean C max of 4.1 µg/mL, an AUC24 of 47 µg•h/mL, and an elimination half-life of 14 hours, following 400 mg p.o. daily.Renal InsufficiencyThe pharmacokinetic parameters of moxifloxacin are not significantly altered in mild, moderate, severe, or end-stage renal disease. No dosage adjustment is necessary in patients with renal impairment, including those patients requiring hemodialysis (HD) or continuous ambulatory peritoneal dialysis (CAPD).In a single oral dose study of 24 patients with varying degrees of renal function from normal to severely impaired, the mean peak concentrations (C max) of moxifloxacin were reduced by 21% and 28% in the patients with moderate (CL CR ≥30 and ≤ 60 mL/min) and severe (CL CR < 30 mL/min) renal impairment, respectively. The mean systemic exposure (AUC) in these patients was increased by 13%. In the moderate and severe renally impaired patients, the mean AUC for the sulfate conjugate (M1) increased by 1.7-fold (ranging up to 2.8-fold) and mean AUC andC max for the glucuronide conjugate (M2) increased by 2.8-fold (ranging up to 4.8-fold) and1.4-fold (ranging up to2.5-fold), respectively.The pharmacokinetics of single dose and multiple dose moxifloxacin were studied in patients with CL CR < 20 mL/min on either hemodialysis or continuous ambulatory peritoneal dialysis (8 HD, 8 CAPD). Following a single 400 mg oral dose, the AUC of moxifloxacin in these HD and CAPD patients did not vary significantly from the AUC generally found in healthy volunteers. C max values of moxifloxacin were reduced by about 45% and 33% in HD and CAPD patients, respectively, compared to healthy, historical controls. The exposure (AUC) to the sulfate conjugate (M1) increased by 1.4- to 1.5-fold in these patients. The mean AUC of the glucuronide conjugate (M2) increased by a factor of 7.5, whereas the mean C max values of the glucuronide conjugate (M2) increased by a factor of 2.5 to 3, compared to healthy subjects. The sulfate and the glucuronide conjugates of moxifloxacin are not microbiologically active, and the clinical implication of increased exposure to these metabolites in patients with renal disease including those undergoing HD and CAPD has not been studied.Oral administration of 400 mg QD moxifloxacin for 7 days to patients on HD or CAPD produced mean systemic exposure (AUC ss) to moxifloxacin similar to that generally seen in healthy volunteers. Steady-state C max values were about 22% lower in HD patients but were comparable between CAPD patients and healthy volunteers. Both HD and CAPD removed only small amounts of moxifloxacin from the body (approximately 9% by HD, and 3% by CAPD). HD and CAPD also removed about 4% and 2% of the glucuronide metabolite (M2), respectively. Hepatic InsufficiencyIn 400 mg single oral dose studies in 6 patients with mild (Child Pugh Class A), and 10 patients with moderate (Child Pugh Class B), hepatic insufficiency, moxifloxacin mean systemic exposure (AUC) was 78% and 102%, respectively, of 18 healthy controls and mean peak concentration (C max)was 79% and 84% of controls.The mean AUC of the sulfate conjugate of moxifloxacin (M1) increased by 3.9-fold (ranging up to 5.9-fold) and 5.7-fold (ranging up to 8.0-fold) in the mild and moderate groups, respectively. The mean C max of M1 increased by approximately 3-fold in both groups (ranging up to 4.7- and 3.9-fold). The mean AUC of the glucuronide conjugate of moxifloxacin (M2) increased by 1.5-fold (ranging up to 2.5-fold) in both groups. The mean C max of M2 increased by 1.6- and 1.3-fold (ranging up to 2.7- and 2.1-fold), respectively. The clinical significance of increased exposure to the sulfate and glucuronide conjugates has not been studied. No dosage adjustment is recommended for mild or moderate hepatic insufficiency (Child Pugh Classes A and B). The pharmacokinetics of moxifloxacin in severe hepatic insufficiency (Child Pugh Class C) have not been studied. (See DOSAGE AND ADMINISTRATION.)Photosensitivity PotentialA study of the skin response to ultraviolet (UVA and UVB) and visible radiation conducted in 32 healthy volunteers (8 per group) demonstrated that moxifloxacin does not show phototoxicity in comparison to placebo. The minimum erythematous dose (MED) was measured before and after treatment with moxifloxacin (200 mg or 400 mg once daily), lomefloxacin (400 mg once daily), or placebo. In this study, the MED measured for both doses of moxifloxacin were not significantly different from placebo, while lomefloxacin significantly lowered the MED. (See PRECAUTIONS, Information for Patients.)It is difficult to ascribe relative photosensitivity/phototoxicity among various fluoroquinolones during actual patient use because other factors play a role in determining a subject’s susceptibility to this adverse event such as: a patient’s skin pigmentation, frequency and duration of sun and artificial ultraviolet light (UV) exposure, wearing of sunscreen andprotective clothing, the use of other concomitant drugs and the dosage and duration of fluoroquinolone therapy (See ADVERSE REACTIONS and ADVERSEREACTIONS/Post-Marketing Adverse Event Reports).Drug-drug InteractionsThe potential for pharmacokinetic drug interactions between moxifloxacin and itraconazole, theophylline, warfarin, digoxin, atenolol, probenecid, morphine, oral contraceptives, ranitidine, glyburide, calcium, iron, and antacids has been evaluated. There was no clinically significant effect of moxifloxacin on itraconazole, theophylline, warfarin, digoxin, atenolol, oral contraceptives, or glyburide kinetics. Itraconazole, theophylline, warfarin, digoxin, probenecid, morphine, ranitidine, and calcium did not significantly affect the pharmacokinetics of moxifloxacin. These results and the data from in vitro studies suggest that moxifloxacin is unlikely to significantly alter the metabolic clearance of drugs metabolized by CYP3A4, CYP2D6, CYP2C9, CYP2C19, or CYP1A2 enzymes.As with all other quinolones, iron and antacids significantly reduced bioavailability of moxifloxacin.Itraconazole:In a study involving 11 healthy volunteers, there was no significant effect of itraconazole (200 mg once daily for 9 days), a potent inhibitor of cytochrome P4503A4, on the pharmacokinetics of moxifloxacin (a single 400 mg dose given on the 7th day of itraconazole dosing). In addition, moxifloxacin was shown not to affect the pharmacokinetics of itraconazole. Theophylline:No significant effect of moxifloxacin (200 mg every twelve hours for 3 days) on the pharmacokinetics of theophylline (400 mg every twelve hours for 3 days) was detected in a study involving 12 healthy volunteers. In addition, theophylline was not shown to affect the pharmacokinetics of moxifloxacin. The effect of co-administration of a 400 mg dose of moxifloxacin with theophylline has not been studied, but it is not expected to be clinically significant based on in vitro metabolic data showing that moxifloxacin does not inhibit the CYP1A2 isoenzyme.Warfarin:No significant effect of moxifloxacin (400 mg once daily for eight days) on the pharmacokinetics of R- and S-warfarin (25 mg single dose of warfarin sodium on the fifth day) was detected in a study involving 24 healthy volunteers. No significant change in prothrombin time was observed. (See PRECAUTIONS, Drug Interactions.)Digoxin:No significant effect of moxifloxacin (400 mg once daily for two days) on digoxin (0.6 mg as a single dose) AUC was detected in a study involving 12 healthy volunteers. The mean digoxin C max increased by about 50% during the distribution phase of digoxin. This transient increase in digoxin C max is not viewed to be clinically significant. Moxifloxacin pharmacokinetics were similar in the presence or absence of digoxin. No dosage adjustment for moxifloxacin or digoxin is required when these drugs are administered concomitantly. Atenolol:In a crossover study involving 24 healthy volunteers (12 male; 12 female), the mean atenolol AUC following a single oral dose of 50 mg atenolol with placebo was similar to that observed when atenolol was given concomitantly with a single 400 mg oral dose of moxifloxacin. The mean C max of single dose atenolol decreased by about 10% following co-administration with a single dose of moxifloxacin.Morphine:No significant effect of morphine sulfate (a single 10 mg intramuscular dose) on the mean AUC and C max of moxifloxacin (400 mg single dose) was observed in a study of 20 healthy male and female volunteers.Oral Contraceptives:A placebo-controlled study in 29 healthy female subjects showed that moxifloxacin 400 mg daily for 7 days did not interfere with the hormonal suppression of oral contraception with 0.15 mg levonorgestrel/0.03 mg ethinylestradiol (as measured by serum progesterone, FSH, estradiol, and LH), or with the pharmacokinetics of the administered contraceptive agents.Probenecid:Probenecid (500 mg twice daily for two days) did not alter the renal clearance and total amount of moxifloxacin (400 mg single dose) excreted renally in a study of 12 healthy volunteers.Ranitidine:No significant effect of ranitidine (150 mg twice daily for three days as pretreatment) on the pharmacokinetics of moxifloxacin (400 mg single dose) was detected in a study involving 10 healthy volunteers.Antidiabetic agents:In diabetics, glyburide (2.5 mg once daily for two weeks pretreatment and for five days concurrently) mean AUC and C max were 12% and 21% lower, respectively, when taken with moxifloxacin (400 mg once daily for five days) in comparison to placebo. Nonetheless, blood glucose levels were decreased slightly in patients taking glyburide and moxifloxacin in comparison to those taking glyburide alone, suggesting no interference by moxifloxacin on the activity of glyburide. These interaction results are not viewed as clinically significant. Calcium:Twelve healthy volunteers were administered concomitant moxifloxacin (single 400 mg dose) and calcium (single dose of 500 mg Ca++ dietary supplement) followed by an additional two doses of calcium 12 and 24 hours after moxifloxacin administration. Calcium had no significant effect on the mean AUC of moxifloxacin. The mean C max was slightly reduced and the time to maximum plasma concentration was prolonged when moxifloxacin was given with calcium compared to when moxifloxacin was given alone (2.5 hours versus 0.9 hours). These differences are not considered to be clinically significant.Antacids:When moxifloxacin (single 400 mg tablet dose) was administered two hours before, concomitantly, or 4 hours after an aluminum/magnesium-containing antacid (900 mg aluminum hydroxide and 600 mg magnesium hydroxide as a single oral dose) to 12 healthy volunteers there was a 26%, 60% and 23% reduction in the mean AUC of moxifloxacin, respectively. Moxifloxacin should be taken at least 4 hours before or 8 hours after antacids containing magnesium or aluminum, as well as sucralfate, metal cations such as iron, and multivitamin preparations with zinc, or VIDEX® (didanosine) chewable/ buffered tablets or the pediatric powder for oral solution. (See PRECAUTIONS, Drug Interactions and DOSAGE AND ADMINISTRATION.) Iron:When moxifloxacin tablets were administered concomitantly with iron (ferrous sulfate 100 mg once daily for two days), the mean AUC and C max of moxifloxacin was reduced by 39% and 59%, respectively. Moxifloxacin should only be taken more than 4 hours before or 8 hours after iron products. (See PRECAUTIONS, Drug Interactions and DOSAGE AND ADMINISTRATION.) Electrocardiogram:Prolongation of the QT interval in the ECG has been observed in some patients receiving moxifloxacin. Following oral dosing with 400 mg of moxifloxacin the mean (± SD) change in QTc from the pre-dose value at the time of maximum drug concentration was 6 msec (± 26) (n = 787). Following a course of daily intravenous dosing (400 mg; 1 hour infusion each day) the mean change in QTc from the Day 1 pre-dose value was 9 msec (± 24) on Day 1 (n = 69) and 3 msec (± 29) on Day 3 (n = 290). (See WARNINGS.)There is limited information available on the potential for a pharmacodynamic interaction in humans between moxifloxacin and other drugs that prolong the QTc interval of the electrocardiogram. Sotalol, a Class III antiarrhythmic, has been shown to further increase the QTc interval when combined with high doses of intravenous (I.V.) moxifloxacin in dogs. Therefore, moxifloxacin should be avoided with Class IA and Class III antiarrhythmics. (See ANIMAL PHARMACOLOGY, WARNINGS,and PRECAUTIONS.)MICROBIOLOGYMoxifloxacin has in vitro activity against a wide range of Gram-positive and Gram-negative microorganisms. The bactericidal action of moxifloxacin results from inhibition of the topoisomerase II (DNA gyrase) and topoisomerase IV required for bacterial DNA replication, transcription, repair, and recombination. It appears that the C8-methoxy moiety contributes to enhanced activity and lower selection of resistant mutants of Gram-positive bacteria compared to the C8-H moiety. The presence of the bulky bicycloamine substituent at the C-7 position prevents active efflux, associated with the NorA or pmrA genes seen in certain Gram-positive bacteria. The mechanism of action for quinolones, including moxifloxacin, is different from that of macrolides, beta-lactams, aminoglycosides, or tetracyclines; therefore, microorganisms resistant to these classes of drugs may be susceptible to moxifloxacin and other quinolones. There is no known cross-resistance between moxifloxacin and other classes of antimicrobials.In vitro resistance to moxifloxacin develops slowly via multiple-step mutations. Resistance to moxifloxacin occurs in vitro at a general frequency of between 1.8 x 10–9 to < 1 x 10–11 for Gram-positive bacteria.Cross-resistance has been observed between moxifloxacin and other fluoroquinolones against Gram-negative bacteria. Gram-positive bacteria resistant to other fluoroquinolones may, however, still be susceptible to moxifloxacin.Moxifloxacin has been shown to be active against most strains of the following microorganisms, both in vitro and in clinical infections as described in the INDICATIONS AND USAGE section. Aerobic Gram-positive microorganismsEnterococcus faecalis (many strains are only moderately susceptible)Staphylococcus aureus (methicillin-susceptible strains only)Streptococcus anginosusStreptococcus constellatusStreptococcus pneumoniae (including multi-drug resistant strains [MDRSP]*) Streptococcus pyogenes* MDRSP, Multi-drug resistant Streptococcus pneumoniae includes isolates previously known as PRSP (Penicillin-resistant S. pneumoniae), and are strains resistant to two or more of the following antibiotics: penicillin (MIC ≥ 2 μg/mL), 2nd generation cephalosporins (e.g., cefuroxime), macrolides, tetracyclines, and trimethoprim/sulfamethoxazole.Aerobic Gram-negative microorganismsEnterobacter cloacaeEscherichia coliHaemophilus influenzaeHaemophilus parainfluenzaeKlebsiella pneumoniaeMoraxella catarrhalisProteus mirabilisAnaerobic microorganismsBacteroides fragilisBacteroides thetaiotaomicronClostridium perfringensPeptostreptococcus speciesOther microorganismsChlamydia pneumoniaeMycoplasma pneumoniaeThe following in vitro data are available, but their clinical significance is unknown.Moxifloxacin exhibits in vitro minimum inhibitory concentrations (MICs) of 2 µg/mL or lessagainst most (≥ 90%) strains of the following microorganisms; however, the safety andeffectiveness of moxifloxacin in treating clinical infections due to these microorganisms have notbeen established in adequate and well-controlled clinical trials.Aerobic Gram-positive microorganismsStaphylococcus epidermidis (methicillin-susceptible strains only)Streptococcus agalactiaeStreptococcus viridans groupAerobic Gram-negative microorganismsCitrobacter freundiiKlebsiella oxytocaLegionella pneumophilaAnaerobic microorganismsFusobacterium speciesPrevotella speciesSusceptibility TestsDilution Techniques:Quantitative methods are used to determine antimicrobial minimuminhibitory concentrations (MICs). These MICs provide estimates of the susceptibility of bacteriato antimicrobial compounds. The MICs should be determined using a standardized procedure.Standardized procedures are based on a dilution method1 (broth or agar) or equivalent withstandardized inoculum concentrations and standardized concentrations of moxifloxacin powder.The MIC values should be interpreted according to the following criteria:For testing Enterobacteriaceae and methicillin-susceptible Staphylococcus aureus:MIC (µg/mL)Interpretation≤2.0 Susceptible(S)4.0Intermediate(I)≥8.0Resistant(R)For testing Haemophilus influenzae and Haemophilus parainfluenzae a:MIC (µg/mL) Interpretation(S) ≤1.0 Susceptiblea This interpretive standard is applicable only to broth microdilution susceptibility tests withHaemophilus influenzae and Haemophilus parainfluenzae using Haemophilus Test Medium1.The current absence of data on resistant strains precludes defining any results other than“Susceptible”. Strains yielding MIC results suggestive of a “nonsusceptible” category should besubmitted to a reference laboratory for further testing.For testing Streptococcus species including Streptococcus pneumoniae b and Enterococcus faecalis:MIC (µg/mL)Interpretation≤1.0 Susceptible(S)2.0Intermediate(I)≥ 4.0Resistant(R)b These interpretive standards are applicable only to broth microdilution susceptibility tests using cation-adjusted Mueller-Hinton broth with 2 - 5% lysed horse blood.A report of “Susceptible” indicates that the pathogen is likely to be inhibited if the antimicrobial compound in the blood reaches the concentrations usually achievable. A report of “Intermediate” indicates that the result should be considered equivocal, and, if the microorganism is not fully susceptible to alternative, clinically feasible drugs, the test should be repeated. This category implies possible clinical applicability in body sites where the drug is physiologically concentrated or in situations where a high dosage of drug can be used. This category also provides a buffer zone which prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of “Resistant” indicates that the pathogen is not likely to be inhibited if the antimicrobial compound in the blood reaches the concentrations usually achievable; other therapy should be selected.Standardized susceptibility test procedures require the use of laboratory control microorganisms to control the technical aspects of the laboratory procedures. Standard moxifloxacin powder should provide the following MIC values:Microorganism MIC (µg/mL)Enterococcus faecalis ATCC 292120.06- 0.5Escherichia coli ATCC 259220.008- 0.06Haemophilus influenzae ATCC 49247c0.008- 0.03Staphylococcus aureus ATCC 292130.015- 0.06Streptococcus pneumoniae ATCC 49619d0.06- 0.25c This quality control range is applicable to only H. influenzae ATCC 49247 tested by a broth microdilution procedure using Haemophilus Test Medium (HTM)1.d This quality control range is applicable to only S. pneumoniae ATCC 49619 tested by a broth microdilution procedure using cation-adjusted Mueller-Hinton broth with 2 - 5% lysed horse blood.Diffusion Techniques:Quantitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. One such standardized procedure2 requires the use of standardized inoculum concentrations. This procedure uses paper disks impregnated with 5-µg moxifloxacin to test the susceptibility of microorganisms to moxifloxacin.Reports from the laboratory providing results of the standard single-disk susceptibility test with a 5-µg moxifloxacin disk should be interpreted according to the following criteria:。
沙奎那韦

谢谢观看
联合治疗可以使AIDS合并症或垂危状态的危险性减少53%,死亡率减少72%。这与治疗18个月后AIDS合并症或 死亡率由29.4%降至16.0%是相符的;同样,单纯死亡率由8.6%降至4.1%。在3个治疗组中,平均疗程为11~13个 月,平均随访时间是17个月。
该研究中,所有治疗组CD4细胞基线计数平均为156~176/立方毫米。16周后(DAVG16),沙奎那韦联合ddc 治疗组CD4细胞增加26/立方毫米,血浆病毒载量减少0.6log10RNA拷贝/毫升。16周时,CD4细胞平均值增加47/ 立方毫米。12周时,血浆病毒载量平均值降低0.7log10RNA拷贝/毫升。
与核苷类似物(齐多夫定等)不同,沙奎那韦直接作用于病毒靶酶,不需经代谢激活,对静止细胞也有潜在 作用。在10-10摩尔/升浓度下,沙奎那韦对淋巴母细胞株和单核细胞株以及被实验室病毒株或临床分离的HIV-1 感染的淋巴细胞和单核细胞的起始培养有作用。
实验室细胞培养结果显示,沙奎那韦在与其他逆转录酶抑制剂(包括AZT(齐多夫定)、ddc(扎西他滨)、 ddI(去羟肌苷)进行两联或三联治疗HIV-1感染时,有附加的协同抗病毒作用,但毒性并不增加 。
机体对静注沙奎那韦6、36、72毫克后清除率很高,为1.14升/小时/千克(CV12%),略高于肝血流,并为常 数。体内存留时间平均为7小时。
适应症
沙奎那韦可与其他抗逆转录病毒药物联合使用治疗成人HIV-1感染 。
用法与用量
1
标准剂量
2
剂量调整
3
不良反应
4
禁忌
5
药物相互作用
成人及16岁以上儿童:推荐方案是与核苷类似物联合用药,餐后2小时内服用沙奎那韦600毫克,每天3次。 联合使用的抗逆转录病毒药物的剂量参考处方手册。与其他蛋白酶抑制剂合用时,沙奎那韦应减量(见【药物相 互作用】)。与其他蛋白酶抑制剂一样,强烈推荐按医嘱服药。
1例艾沙康唑致患者全血细胞减少的病例分析

1例艾沙康唑致患者全血细胞减少的病例分析刘晓平1*,林小鲁1,李剑芳2 #(1.广州开发区医院药学部,广州 510730;2.中山大学孙逸仙纪念医院药学部,广州 510120)中图分类号 R969.3;R978.5文献标志码 A 文章编号 1001-0408(2024)07-0881-05DOI 10.6039/j.issn.1001-0408.2024.07.20摘要目的正确识别和应对艾沙康唑引起的全血细胞减少的不良反应,为该药的安全使用提供参考。
方法临床药师通过参与1例严重感染合并肾功能不全的患者使用艾沙康唑后出现全血细胞减少的病例分析,筛查患者住院期间所用药物,并结合药物的半衰期和相关文献,评估了该不良反应与艾沙康唑的关系及可能的发生机制。
结果与结论患者全血细胞减少与艾沙康唑的关系评估为“可能相关”。
使用艾沙康唑时应提高警惕,避免同时联用相同机制或有潜在相互作用的药物。
如疗程大于2周或用药前血液系统异常或合并肝肾功能损害或需要联合使用相同机制的药物者,可考虑增加血常规监测频率。
关键词艾沙康唑;全血细胞减少;病例分析;药物不良反应Case analysis of a patient with isavuconazonium-caused pancytopeniaLIU Xiaoping1,LIN Xiaolu1,LI Jianfang2(1. Dept. of Pharmacy,Guangzhou Development District Hospital,Guangzhou 510730,China;2. Dept. of Pharmacy,Sun Yat-sen Memorial Hospital,Sun Yat-sen University,Guangzhou 510120, China)ABSTRACT OBJECTIVE To correctly identify and deal with the adverse drug reaction as pancytopenia caused by isavuconazonium and to provide reference for the safe use of isavuconazonium. METHODS Clinical pharmacists analyzed a case of severe infection and renal insufficiency who experienced pancytopenia after using isavuconazonium. Clinical pharmacists screened the drugs used during hospitalization and evaluated the relationship between this adverse drug reaction and isavuconazonium,as well as the possible mechanisms,based on the half-life of the drugs and relevant literature. RESULTS &CONCLUSIONS The relationship between pancytopenia and isavuconazonium was assessed as “possibly related”. When using isavuconazonium, attention should be paid to avoiding the combination of drugs with the same mechanism or potential interaction. For patients who have a course of treatment for more than 2weeks,have hematological abnormalities or complicated with liver and renal insufficiency,or should use it combined with other drug with same mechanism,it may be considered to increase the frequency of blood routine monitoring.KEYWORDS isavuconazonium; pancytopenia; case analysis; adverse drug reaction硫酸艾沙康唑是三唑类抗真菌药物艾沙康唑(一种14-α-去甲基化酶抑制剂)的水溶性前药,可被血浆中的酯酶(主要是丁基胆碱酯酶)快速水解成活性成分艾沙康唑和非活性裂解产物[1]。
锂离子电池电解液添加剂物性数据

1,3-丙烷磺酸内酯 (1,3-PS)
1,3-Propane sultone;
精品资料
______________________________________________________________________________________________________________
______________________________________________________________________________________________________________
化学名称
别名 英文名称 CAS 号
分子式
锂离子电池电解液添加剂物性数据
环己基苯(CHB)
CAS 号 分子式
4427-96-7 C5H6O3
分子结构
分子量 熔点/沸点/闪点
114.10
?/237℃/733mmHg/96.6
密度(g/mL at 25℃) 粘度(40℃) 折光率 外观 特性
1.188
1.45 无色液体
见附注
1073-05-8 C3H6O4S
1-Phenyl-2-acetone
2、添加了 MMDS 的电池具有很好的高温循 环性能。适用于动力电池,特别是锰酸锂做 正极材料的动力电池,MMDS 能防止高温下 熔出的 Mn 吸附在负极表面,抑制了阻抗上 升,有效提高了循环周期特性,可以大大增 加其循环寿命。
变色。还可用作底涂剂。
包装材料为 PE
精品资料
______________________________________________________________________________________________________________
氢溴酸达非那新

适应证
氢溴酸达非那新用于膀胱过度刺激引起的尿频、尿急、尿失禁。
禁忌证
1.对氢溴酸达非那新及其中成分过敏者禁用。 2.尿潴留、胃潴留及未控制的闭角型青光眼患者禁用。 3.重度肝功能损害患者不推荐使用。
注意事项
1.由于尿潴留的可能,有明显膀胱尿道阻塞症状的患者使用时应谨慎。 2.氢溴酸达非那新具有抗胆碱作用,能降低胃肠道动力,胃肠道阻塞性疾病患者有胃潴留的可能,使用时应 谨慎。严重便秘、溃疡性结肠炎和重症肌无力患者慎用。 3.已控制的闭角型青光眼患者慎用。 4.氢溴酸达非那新生殖毒性分级为C,只有当对母体的益处高于对胎儿的危险时方可用于孕妇。 5.氢溴酸达非那新可经大鼠乳汁分泌,尚不知氢溴酸达非那新是否经人乳汁分泌,哺乳期妇女应慎用。
用法用量
口服,推荐剂量为7.5mg,1次/d,整片服下,不得嚼碎、掰开或压碎,可单服或与食物同服。根据个人临床 反应,剂量可增至15mg。中度肝功能损伤患者及与CYP3A4抑制剂(如酮康唑、伊曲康唑、利托那韦、奈非那韦、 克拉霉素、奈法唑酮)同服时,剂量不得超过7.5mg。
药物相互作用
1.氢溴酸达非那新主要经CYP2D6和CYP3A4代谢,CYP3A4抑制剂(酮康唑、伊曲康唑、利托那韦、奈非那韦、 克拉霉素、奈法唑酮)可使氢溴酸达非那新代谢减少,日剂量不应超过7.5mg。
尿失禁治疗药物是一个潜力巨大但尚未完全开发的市场,临床特征均是在24h内需要小便数不少于十次。据 世界卫生组织(WHO)有关人员估计,全球约有10%~15%中年人(50岁以下)和40%~70%老年人不同程度地受到此病 困扰。膀胱过动症一般没有神经源性损伤或疾病,可由膀胱的快速充盈、体位改变、甚至行走、咳嗽诱发。估计 全世界约有4~5亿名尿失禁患者,女性的发生率为男性的2倍。男性的发生率随着年龄的增长而升高,是一种常 见和令人痛苦的疾病。(另有一组数据估计世界7个主要国家受影响的人群达1.54亿,其中0.73亿人被分类为明 显尿失禁症。)。
三氟乙酸脱苄基O-debenzylation_of_ortho-substituted_phenols_with_trifluoroacetic_acid

Mild,efficient and rapid O-debenzylation of ortho -substituted phenols with trifluoroacetic acidSteven Fletcher *,Patrick T.Gunning *Department of Chemistry,University of Toronto,Mississauga,ON L5L 1C6,Canadaa r t i c l e i n f o Article history:Received 21May 2008Revised 2June 2008Accepted 4June 2008Available online 10June 2008a b s t r a c tThe mild and efficient deblocking of aryl benzyl ethers with TFA is reported.Cleavage was fastest with ortho -electron-withdrawing groups on the phenolic ring,which we have attributed to a proton chelation effect,furnishing the deprotected phenols in excellent yields.The corresponding para -methoxybenzyl,allyl and iso -propyl ethers were also cleanly removed under these conditions.In addition,the selective aryl benzyl ether debenzylation in the presence of benzyl ester,Cbz carbamate and Boc carbamate func-tionalities was also observed.Crown Copyright Ó2008Published by Elsevier Ltd.All rights reserved.Phosphotyrosines feature in the design of inhibitors of several protein targets,including protein tyrosine phosphatase 1B (PTP1B).1However,these moieties suffer from hydrolytic lability to cellular phosphatases and poor cell penetration due to the asso-ciated dianionic charge.1To address these issues,salicylic acid derivatives (and closely-related analogues)have become popular mimetics of phosphotyrosine in small molecule inhibitors.1–5Turk-son et al.have recently reported on NSC74859(1),a potent,sali-cylic acid-based inhibitor of the oncogenic protein Stat3.6As part of our structure–activity relationship (SAR)studies on NSC74859(1),we sought to debenzylate both the phenol ether and benzoate ester in 2without reducing the aryl-bromide bond,a common undesired side reaction that occurs with hydrogen gas and Pd/C catalyst.7O -Benzyl-protected phenols are known to undergo debenzyla-tion with trifluoroacetic acid (TFA)8by an initial protonation of the weakly basic phenol oxygen,although additives such as strongorganic acids (e.g.,trifluoromethanesulfonic acid 9)or a large excess of nucleophilic scavenger (e.g.,thioanisole,which accelerates the reaction by a ‘push–pull’mechanism 10)are typically required.Re-cent work by Ploypradith et al.describes the mild deprotection of aromatic ethers with sub-stoichiometric para -toluenesulfonic acid on solid support.11In a special case,O -benzyl-protected ortho -nitrophenol was cleaved rapidly (<5min)with neat TFA,12which we considered was due to the ability of the substrate to chelate a proton since the structurally-similar ortho -hydroxybenzoates (salicylates)are well-known to chelate copper ions and iron ions.We reasoned that 2(and indeed 3)may similarly undergo acceler-ated debenzylation with TFA.In fact,as shown in Scheme 1,treat-ment of 2(or 3)with a 1:1mixture of TFA/toluene led to rapid debenzylation (5min for 2;1h for 3)in 91%yield for 2(or 85%yield for 3).In this Letter,we will explore the structural require-ments of the phenol component that increase the lability of the O -benzyl phenol ether bond in the presence of TFA.In addition,0040-4039/$-see front matter Crown Copyright Ó2008Published by Elsevier Ltd.All rights reserved.*Tel.:+19058285354;fax:+19058285425(P.T.G.).E-mail addresses:steven.fletcher@utoronto.ca (S.Fletcher),patrick.gunning@utoronto.ca (P.T.Gunning).Tetrahedron Letters 49(2008)4817–4819Contents lists available at ScienceDirectTetrahedron Lettersj o ur na l h om e pa ge :w w w.e ls e v ie r.c o m/lo c at e/t et l e twe will explore the selectivity of this mild debenzylation tech-nique with respect to other aromatic ethers and examine the sta-bility of other benzyl-based protecting groups to these reaction conditions.A series of 12O -benzyl-protected phenols was prepared by standard procedures in near quantitative yields.Each of these ethers was then deprotected with a 1:1mixture of TFA/toluene;our observations are summarized in Table 1.In certain cases,O ?C benzyl migration (Friedel–Crafts reaction)by-products (610%)were occasionally inseparable from the product by silica gel flash column chromatography.Thus,several benzyl cation cap-tors were investigated for their abilities to improve yields and puri-ties of the debenzylation reactions.Three to ten equivalents of p -cresol,anisole and triethylsilane were employed,but these exerted little effects on reducing by-product formation.Conversely,we dis-covered that including the more nucleophilic scavenger thioanisole as an additive to the co-solvent toluene typically,after silica gel flash column chromatography,furnished products in P 95%puri-ties (and higher yields),as judged by 1H NMR.Nevertheless,we envisaged any Friedel–Crafts impurities would be more readily separable on slightly more complex aryl benzyl ethers,as we ob-served with the substrates shown in Scheme 1and Tables 3and 4(>99%purities (1H NMR)in each case).Whilst likely leading to even higher yields and purities,large excesses of thioanisole (50equiv)are also known to accelerate TFA-mediated debenzyla-tion.10However,in our hands just 3equiv of thioanisole had little effect on the rate of debenzylation,allowing us to attribute the deprotection rates solely to the structure of the phenol.Electron-rich phenols are good scavengers of benzyl cations,13and since preliminary experiments with electron-rich phenols generated complex mixtures of Friedel–Crafts by-products under these deb-enzylation conditions,we chose to investigate only electron-poor phenols in this study.O -Benzyl-protected phenols with p -ortho -electron-withdraw-ing groups (6a ,6b ,6d ,6f )were swiftly (several in less than 3h cf.24h for unsubstituted phenol 6l )and cleanly debenzylated,with less than 5%of the undesired C-benzylated phenol by-prod-ucts.In contrast,meta -and para -electron-withdrawing groups slo-wed down the debenzylation (e.g.,entries 6g and 6h ),relative to the control compound 6l ,which itself could only be obtained in moderate purity by this method.The r -withdrawing (and p -donating)bromophenols 6i –k were insufficiently deactivated to benzyl cation scavenging and were contaminated with several by-products.Importantly,n -butyl benzyl ether 8was unaffected by TFA under the reaction conditions,indicating this procedure is selective for aryl benzyl ethers.In addition,the results in Table 1suggest that this procedure is suitable only for phenols substituted with p -electron-withdrawing groups.Since the debenzylation mechanism with TFA proceeds via an initial protonation of the phenol ether oxygen,the more available the ether oxygen lone pairs are,the faster the reaction will be.Hence,the slower reaction times for the phenols bearing meta -and para -electron-withdrawing groups make sense,although this is not true for the ortho -functionalized aryl benzyl ethers.As hypothesized for the bis-benzyl salicylate derivative 2earlier,we considered these ortho -substituted phenols were capable of chelat-ing the acidic hydrogen atom from TFA which therein facilitated the acid-mediated debenzylation via a six-membered cyclic inter-mediate,as proposed in Scheme 2.A similar chelation intermediate has been put forward by Baldwin and Haraldsson to account for the Lewis acid MgBr 2-mediated debenzylation of aromatic benzyl ethers ortho to an aldehyde group.14Accordingly,to test this hypothesis we expanded this series of ortho -substituted aryl benzyl ethers,and the results from their deb-enzylation reactions with TFA are summarized in Table 2.These substrates have been listed in order of increasing approximateTable 1TFA-mediated debenzylation of O -benzyl-protected phenols aTFAtolueneOBnROHR67Substrate RTime (h)b Yield c (%)6a o -CO 2Me,m d -NHAc 5min 936b o -CO 2Me 5min 946c p -CO 2Me 36e 63(85f )6d o -CO 2Bn 5min 936e p -CO 2Bn 36e 58(79f )6f o -NO 23976g m -NO 236e 75(98f )6h p -NO 236e 66(98f )6i o -Br 16—g 6j m -Br 30—g 6k p -Br 36—g 6lH 24—gn -BuOBn (8)—24No reactionaThe reaction was carried out with 6(0.5mmol)in a 1:1mixture of TFA/toluene (5ml)at rt,with 3equiv of thioanisole.bTime taken for all starting material to be consumed.cIsolated yield after silica gel flash column chromatography.dmeta to phenol oxygen AND para to ester.eReaction was slow and incomplete after 3days.fYield based on recovered starting material.gComplex mixture of products.Table 2TFA-mediated debenzylation of O -benzyl-protected,ortho -substituted phenols aTFA tolueneOBnOH67RRSubstrate R p K aH b Time c (h)Yield d (%)Relative rate 6m CO 2NH 2À2248316n CHO À7 3.594e 6.96o CO 2H À8191246b CO 2Me À8.55min 942886d CO 2Bn À8.55min 932886p CN À10>4851(95f )—6f NO 2À1239786i Br —16—g 1.56lH—24—g1aThe reaction was carried out with 6(0.5mmol)in a 1:1mixture of TFA/toluene (5ml)at rt,with 3equiv of thioanisole.bApproximate p K aH of conjugate acid of R group.15cTime taken for all starting material to be consumed.dIsolated yield after silica gel flash column chromatography.eIncluding thioanisole in the deprotection of 6n led to further by-products,thus no scavenger was used and compound 7n could be obtained in only 90%purity.fYield based on recovered starting material.gComplex mixture of products.4818S.Fletcher,P.T.Gunning /Tetrahedron Letters 49(2008)4817–4819acidity of the conjugate acid (decreasing p K aH )of the ortho -elec-tron-withdrawing substituent.15There appears to be an optimal p K aH of around À8.5,that is exhibited by carboxylic esters,which lead to the fastest rate of debenzylation with TFA.In an approxi-mate bell-shaped distribution of reaction rate versus ortho -substi-tuent p K aH —that was interrupted only by ortho -cyanophenol 6p —protonatable groups with p K aH ’s <À8.5or >À8.5were less effective at accelerating the TFA-mediated debenzylation.These data concur with our chelation hypothesis:groups that are too ba-sic bind more strongly to the TFA proton making it less available for sharing with,and ultimately releasing to,the phenol ether oxygen;groups that are weakly basic do not bind the TFA proton as well,leading to reduced chelation and hence less rate enhancement.The anomalous result for ortho -cyanophenol 6p was anticipated since this compound was selected as a negative control.Phenol 6p is geometrically incapable of chelating a proton,because the lin-ear,sp -hybridized nitrile functionality directs its basic nitrogen atom (p K aH %À10)away from the phenol oxygen.As predicted,there was no rate enhancement for the TFA-mediated debenzyla-tion of 6p relative to phenol 6l .In fact,6p was only slowly deben-zylated,at a rate that was comparable with the m -nitro and p -nitro derivatives 6g and 6h ,respectively.We next wanted to investigate the selectivity for the deprotec-tion of the benzyl group over other phenol protecting groups.Accordingly,the benzyl group in salicylate derivative 9a was varied with para -methoxybenzyl (PMB;9b ),methyl (9c ),allyl (9d )and iso -propyl (i -Pr;9e ).These substrates were then debenzylated with a 1:1mixture of TFA/toluene;our findings are reported in Table 3.Any impurities this time were minor and readily separable from the products,eliminating the need for the additive thioanisole.The relative rates at which these protecting groups were removed was para -methoxybenzyl >benzyl >allyl >iso -propyl )methyl,which reflects the stability of the carbocations.These data suggest that in salicylates such as 9,the benzyl phenol protecting group (R =Bn)can be removed with TFA in the presence of the corres-ponding allyl,iso -propyl and methyl ethers.Finally,we explored the selectivity of this mild debenzylation technique over other benzyl-based protecting groups,as shown in Table 4.As the results demonstrate,it was possible to deblock the O -benzyl ether in the presence of a benzyl ester (6d )and in the presence of a benzyl carbamate (11b ),thereby increasing the orthogonality of O -benzyl phenol ethers of salicylate derivatives.Interestingly,it was even possible to cleave the benzyl group in 11c with TFA in the presence of an N -Boc-protected aniline.In summary,we have presented the mild,efficient and rapid deblocking of ortho -substituted aryl benzyl ethers with TFA.Deb-enzylation was fastest when the ortho group was a carboxylic ester,which we have attributed to a proton chelation effect.Other ortho groups that accelerated the TFA-mediated debenzylation included carboxylic acid,aldehyde and nitro.In addition,we have shown that in such ortho -functionalized phenols,benzyl could be removed in the presence of the corresponding iso -propyl,allyl and methyl ethers.Moreover,the benzyl ether could be selectively cleaved in the presence of benzyl ester,Cbz carbamate and Boc carbamate functionalities.AcknowledgementsThe authors gratefully acknowledge financial support for this work from the Canadian Foundation of Innovation and the Univer-sity of Toronto (Connaught Foundation).References and notes1.Zhang,S.;Zhang,Z.-Y.Drug Discov.Today 2007,12,373–381.2.(a)Pei,Z.;Li,X.;Liu,G.;Abad-Zapatero,C.;Lubben,T.;Zhang,T.;Ballaron,S.J.;Hutchins,C.W.;Trevillyana,J.M.;Jirouseka,M.R.Bioorg.Med.Chem.Lett.2003,13,3129–3132;(b)Xin,Z.;Liu,G.;Abad-Zapatero,C.;Pei,Z.;Szczepankiewicz,B.G.;Li,X.;Zhang,T.;Hutchins,C.W.;Hajduk,P.J.;Ballaron,S.J.;Stashko,M.A.;Lubben,T.H.;Trevillyana,J.M.;Jirouseka,M.R.Bioorg.Med.Chem.Lett.2003,13,3947–3950.3.Tautz,L.;Bruckner,S.;Sareth,S.;Alonso,A.;Bogetz,J.;Bottini,N.;Pellecchia,M.;Mustelin,T.J.Biol.Chem.2005,280,9400–9408.4.Shrestha,S.;Bhattarai,B.R.;Chang,K.J.;Leea,K.-H.;Choa,H.Bioorg.Med.Chem.Lett.2007,17,2760–2764.5.Liljebris,C.;Larsen,S.D.;Ogg,D.;Palazuk,B.J.;Bleasdale,J.E.J.Med.Chem.2002,45,1785–1798.6.Siddiquee,K.;Zhang,S.;Guida,W.C.;Blaskovich,M.A.;Greedy,B.;Lawrence,H.R.;Yip,M.L.R.;Jove,R.;Laughlin,M.M.;Lawrence,N.J.;Sebti,S.M.;Turkson,J.Proc.Natl.Acad.Sci.U.S.A.2007,104,7391–7396.7.Pandey,P.N.;Purkayastha,M.L.Synthesis 1982,876–878.8.(a)Greene,T.W.;Wuts,P.G.M.Protective Groups in Organic Synthesis ,3rd ed.;John Wiley &Sons:New York,1999;(b)Kocienski,P.J.Protecting Groups ,3rd ed.;Georg Thieme:Stuttgart,Germany,2003.9.Kiso,Y.;Isawa,H.;Kitagawa,K.;Akita,T.Chem.Pharm.Bull.1978,26,2562–2564.10.Kiso,Y.;Ukawa,K.;Nakamura,S.;Ito,K.;Akita,T.Chem.Pharm.Bull.1980,28,673–676.11.Ploypradith,P.;Cheryklin,P.;Niyomtham,N.;Bertoni,D.R.;Ruchirawat,.Lett.2007,9,2637–2640.12.Marsh,J.P.,Jr.;Goodman,.Chem.1965,30,2491–2492.13.(a)Eberle,A.N.J.Chem.Soc.,Perkin Trans.11986,361–367;(b)Bodanszky,M.;Tolle,J.C.;Deshmane,S.S.;Bodanszky,A.Int.J.Pept.Protein Res.1978,12,57–68.14.Haraldsson,G.G.;Baldwin,J.E.Tetrahedron 1997,53,215–224.15.(a)Ionization Constants of Organic Acids in Solution ;Serjeant,E.P.,Dempsey,B.,Eds.IUPAC Chemical Data Series No.23;Pergamon Press:Oxford,UK,1979;(b)see also:/labs/evans/pdf/evans_pKa_table.pdf .Table 3TFA-mediated deprotection of O-blocked phenol ether derivatives of methyl 4-acetamidosalicylate aTFAtolueneNHAcNHAcORO OMeOH OMeO 910Substrate R Time b (h)Yield c (%)9a Bn 5min 919b PMB 2min 909c Me 480d 9d Allyl 20919ei -Pr3692aThe reaction was carried out with 9(0.5mmol)in a 1:1mixture of TFA/toluene (5ml)at rt.bTime taken for all starting material to be consumed.cIsolated yield after silica gel flash column chromatography.dOnly starting material remained after 48h,at which point the reaction was aborted.Table 4Selectivity investigation into the TFA-mediated debenzylation of aryl benzyl ethers aTFA tolueneOBnOH2Bn2Bn1112RRSubstrate R Yield b (%)6d c H 9311a NHAc 9211b NHCbz 9311c dNHBoc54aThe reaction was carried out with 11(0.5mmol)in a 1:1mixture of TFA/toluene (5ml)at rt for 5min,then all solvents were evaporated.bIsolated yield after silica gel flash column chromatography.cFor compound 6d ,3equiv of thioanisole were also used.dAfter 5min,the reaction mixture was diluted with CH 2Cl 2and then immedi-ately neutralized with 1M NaOH.The organic layer was then separated and evaporated.S.Fletcher,P.T.Gunning /Tetrahedron Letters 49(2008)4817–48194819。
世界卫生组织国际癌症研究机构三类致癌物清单
36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80
1-Amino-2-methylanthraquinone Quintozene (Pentachloronitrobenzene) Diacetylaminoazotoluene Scarlet Red Yellow AB Sudan III N-Nitrosodiphenylamine Hydralazine 1-Naphthylthiourea (ANTU) 2,4,6-Trimethylaniline Penicillic acid
N-Phenyl-2-naphthylamine 2,4,5-Trimethylaniline Shikimic acid Nithiazide para-Dimethylaminoazobenzenediazo sodium sulfonate Vinblastine sulfate Methyl selenac Sodium diethyldithiocarbamate 8-Hydroxyquinoline Patulin para-Aminobenzoic acid Dulcin para-Nitrosodiphenylamine Phenelzine sulfate Coronene Methyl parathion Evans blue Zectran Diazomethane Ethylene sulfide Retrorsine Seneciphylline 2,4'-Diphenyldiamine Methyl red Acridine orange Chlorobenzilate Citrinin Kaempferol CI Acid Orange 20 Angelicin plus ultraviolet A radiation Chrysoidine Ethionamide Tris(1-aziridinyl)phosphine oxide Mannomustine dihydrochloride Semicarbazide hydrochloride Methyl carbamate 9-Nitroanthracene 2,6-Dichloro-para-phenylenediamine n-Propyl carbamate Dihydroxymethylfuratrizine (see also Panfuran S) Aziridyl benzoquinone Nitrovin Dimethoxane
硫代爱地那非分子量

硫代爱地那非分子量
硫代爱地那非(Sildenafil)是一种口服的治疗男性勃起功能障碍的药物,其分子式为C22H30N6O4S,分子量为474.58 g/mol。
它属于磷酸二酯酶-5抑制剂(PDE5抑制剂)的一种。
硫代爱地那非的化学名为
1-[[3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4 -ethoxyphenyl]sulfonyl]-4-methylpiperazine,它的英文名称为 sildenafil,常见的商标包括 Viagra、Revatio等。
硫代爱地那非的作用是通过抑制磷酸二酯酶-5 (PDE5) 酶来扩张血管,增加血流量,改善勃起功能障碍。
它通常在性刺激下才能发挥作用,因为它不会直接导致勃起。
硫代爱地那非通常是以25mg、50mg和100mg剂量的药丸服用,建议在性行为前1小时内服用。
但是,硫代爱地那非的不良反应包括头痛、面部潮红、腹泻、视觉障碍甚至听力障碍等,应该遵循医生的建议使用。
总之,硫代爱地那非的出现,为治疗男性勃起功能障碍提供了新的选择,但在使用药物前仍需慎重,尤其是需要遵循医生的建议和注意药物的不良反应。
泮托拉唑钠的相关中间体
结构式:
产品名称:5-(二氟甲氧基)-2-{[(4-氯-3-甲氧基-2-吡4-Chloro-3-methoxy-2-pyridinyl)methy l]thio-1 H-benz im idazo le [CAS]:[368890-20-4] 结构式:
结构式:
产品名称:5-(二氟甲氧基)-2-{[(3,4-二甲氧基-2-吡啶基)甲基]硫}-1H-苯并咪唑 5-Difluoromethoxy-2-[(3,4-dimethoxy-2-pyridinyl)methy l]thio-1 H-benz im idazo le [CAS]:[102625-64-9]
产品名称:4-氯-3-甲氧基-2-甲基吡啶 N-氧化物 4-Chloro-3-methoxy-2-methylpyridine N-oxide [CAS]:[122307-41-9] 结构式:
产品名称:2-羟甲基-3,4-二甲氧基吡啶 2-Hydroxymethyl-3,4-dimethoxypyr id ine [CAS]:[72830-08-1] 结构式:
中文名:4-二氟甲氧基苯胺 4-difluoromethoxyaniline CAS:[22236-10-8 ] 分子式:C7 H7F2NO 分子量:159.133 结构式:
中文名:3-二氟甲氧基苯胺 3-Difluoromethoxy aniline CAS :[22236-08-4] 结构式:
产品名称: 4-氯-3-甲氧基-2-氯甲基吡啶盐酸盐 4-Chloro-2-chloromethyl-3-methoxypyridine,Hydrochloride Salt 结构式:
产品名称: 4-二氟甲氧基-乙酰苯胺 4-Difluoromethoxy-acetanilid N-(4-二氟甲氧基苯基)-乙酰胺 N-(4-Difluoromethoxyphenyl)acetamide
26 阿昔洛韦、异丙啶、辛那卡塞特、植物二酮、间苯三酚、卡比多巴、倍他司汀、乐卡地平
中文名称
英文名称
CAS
规格
用途
结构式
阿昔洛韦杂质31
Acyclovir Impurity 31
2190509-40-9
10mg
25mg
50mg
100mg
更大规格请咨询
项目报批
纯度高于98%
异丙啶杂质7
Picaridin Impurity 7
25mg
50mg
100mg
更大规格请咨询Βιβλιοθήκη 项目报批纯度高于98%
乐卡地平杂质37
Lercanidipine Impurity 37
624-58-8
10mg
25mg
50mg
100mg
更大规格请咨询
项目报批
纯度高于98%
有Kou各q:类2标8准5品3对1照1品7和7杂5质2
10mg
25mg
50mg
100mg
更大规格请咨询
项目报批
纯度高于98%
卡比多巴杂质18
Carbidopa Impurity 18
85933-19-3
10mg
25mg
50mg
100mg
更大规格请咨询
项目报批
纯度高于98%
倍他司汀杂质7
Betahistine Impurity 7
88796-99-0
10mg
6214-01-3
10mg
25mg
50mg
100mg
更大规格请咨询
项目报批
纯度高于98%
辛那卡塞特杂质82
Cinacalcet Impurity 82
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
3-Phenyl-4-aroyl-5-isoxazolonate complexes of Tb 3+as promisinglight-conversion molecular devicesS.Biju a ,M.L.P.Reddya,*,Ricardo O.FreirebaChemical Sciences and Technology Division,Regional Research Laboratory (CSIR),Thiruvananthapuram 695019,IndiabDepartmento de Quimica Fundamental –UFPE,50670-901,Recife,PE,BrazilReceived 6November 2006;accepted 12December 2006Available online 21December 2006AbstractNew tris(3-phenyl-4-aroyl-5-isoxazolonate)terbium(III)complexes have been synthesized and characterized by various spectroscopic techniques.Due to an efficient energy transfer from the heterocyclic b -diketonate ligand to the central Tb 3+,these complexes show a strong emission corresponding to Tb 3+5D 4–7F J (J =6,5,4,3)transitions,with 5D 4–7F 5(545nm)green emission as the most prominent one.The overall quantum yields and luminescent lifetimes of these complexes were found to be promising as compared to previously reported terbium-1-phenyl-3-methyl-4-acyl-5-pyrazolonate complexes.Ó2006Elsevier B.V.All rights reserved.Keywords:Tb 3+;3-Phenyl-4-aroyl-5-isoxazolonate complexes;Crystal structure;LuminescenceRecently,luminescent lanthanide complexes have attr-acted much attention because of their academic interest and potential application in a wide variety of photonic applications,such as planar waveguide amplifiers,plastic lasers,light emitting diodes and luminescent probes [1,2].The two most useful lanthanides,Eu 3+and Tb 3+,have unusual spectroscopic characteristics,including millisecond lifetime,very sharp emission bands,and large Stokes shifts [2].Generally,the Eu 3+and Tb 3+ions show very weak absorption in the visible region of the spectrum and often require the application of strongly absorbing ‘‘antennae’’for light harvesting to obtain efficient photoluminescence.Among them,b -diketone ligand is one kind of important antennae for the Eu 3+and Tb 3+ions [3].Although the use of tris(8-hydroxyquinoline)aluminum (ALQ)as a green-emitting material has been investigated extensively,terbium complexes are still of great interest in electrolumi-nescent devices because they offer several distinct advanta-ges such as 100%quantum efficiency theoretically,extremely sharp emission bands and the ability to modifythe ligand without affecting the emission characteristics of the central metal ion.Previous reports showed efficient green electroluminescence from terbium complexes containing 1-phenyl-3-methyl-4-isobutyl-5-pyrazolone or 1-phenyl-3-methyl-4-(2-ethylbutyryl)-5-pyrazolone and tri-phenylphosphine oxide [4,5].However,another novel class of heterocyclic b -diketones such as 3-phenyl-4-aroyl-5-iso-xazolones has not been utilized for the preparation of ter-bium luminescent materials even though they are well known as complexing agents for lanthanides [6,7].In this paper,new tris(heterocyclic b -diketonato)terbium(III)complexes,Tb(PBI)3(H 2O)2(1)and Tb(TPI)3(H 2O)2(2)(where HPBI =3-phenyl-4-benzoyl-5-isoxazolone and HTPI =3-phenyl-4-(4-toluoyl)-5-isoxazolone)have been synthesized and characterized by elemental analyses,Fourier transform infrared,high resolution mass spectrom-etry,thermogravimetric analysis and photoluminescence spectroscopy.The synthesis procedures of terbium complexes are shown in Scheme 1(Supplementary data ).The elemental and HRMS analyses of the complexes 1and 2show that Tb 3+ion has reacted with HPBI and HTPI in a metal-to-ligand mole ratio of 1:3.The IR spectrum of the complexes1387-7003/$-see front matter Ó2006Elsevier B.V.All rights reserved.doi:10.1016/j.inoche.2006.12.008*Corresponding author.Tel.:+914712515360;fax:+91471249712.E-mail address:mlpreddy@ (M.L.P.Reddy)./locate/inocheInorganic Chemistry Communications 10(2007)393–3961and2shows a broad absorption in the region3000–3500cmÀ1,indicating the presence of solvent molecules in the complex.The existence of solvent molecules in lan-thanide complexes with heterocyclic b-diketones such as 1-phenyl-3-methyl-4-acylpyrazolones and3-phenyl-4-aroyl-5-isoxazolones is well documented[8,9].The car-bonyl stretching frequency of HPBI(1699cmÀ1)and HTPI (1705cmÀ1)has been shifted to lower wave numbers in complexes1and2(1641cmÀ1in1and1639cmÀ1in2), indicating the involvement of carbonyl oxygen in the complex formation with Tb3+ion.It is clear from the ther-mogravimetric analysis data that the complexes1and2 undergo a mass loss of4%(calcd.:3.6%for1and3.5% for2)up to200°C,which corresponds to the removal of coordinated water molecules.Further,decomposition takes place in the region200–800°C for both the complexes.The structure of the complex Tb(PBI)3(EtOH) (H2O)Æ0.25(H2O)was characterized by single crystal X-ray crystallography and it is interesting to note that one of the water molecules in complex1is replaced by an eth-anol molecule from the medium(ethanol/CH2Cl2)during crystallization process.One water molecule is present in the lattice with0.25occupancy,i.e.,one full water mole-cule in the unit cell.Details of crystal data and data collec-tion parameters are given in Table1in Supplementary Material.The asymmetric unit of Tb(PBI)3(EtOH)(H2O)Æ0.25(H2O)is shown in Fig.1.The central Tb3+ion is coordinated with eight oxygen atoms,six of which from the three bidentate HPBI ligands,one is from water mole-cule and another from an ethanol molecule.The coordina-tion geometry of the metal centre is best described as a bicapped triagonal prism.The average bond length between the terbium ion and the isoxazolone oxygen atoms is2.36A˚,which is slightly shorter than that of ter-bium and water oxygen atom(2.38A˚)and also than that of terbium and ethanol oxygen atom(2.44A˚).This may be due to the result of the negative charge of the isoxazo-lone oxygen,which could be more strongly coordinated to the terbium ion due to electrostatic effects.Similar behav-iour has been reported elsewhere in the X-ray single-crystal structure of Tb(PMPP)3Æ2H2O(where PMPP=1-phenyl-3-methyl-4-propionyl-5-pyrazolone)[12],here the average bond length between the terbium ion and the PMPP oxy-gen atoms is2.34A˚and that of terbium and water oxygen atom is2.45A˚.The coordination geometries of the com-plexes Tb(PBI)3(EtOH)(H2O)and Tb(TPI)3(EtOH)(H2O) were calculated using Sparkle/AM1model[10,11].The optimized structures,selected bond lengths and bond angles for complexes are given in Supplementary data. The average bond length between the terbium ion and the isoxazolone oxygen atoms(2.39A˚)obtained from sparkle model is also in good agreement with the crystal data.Fig.1.The asymmetric unit of compound Tb(PBI)3(C2H5OH)(H2O)Æ0.25(H2O). 394S.Biju et al./Inorganic Chemistry Communications10(2007)393–396The excitation spectra of the complexes1and2moni-tored around the peak of the intense5D4!7F5transition of the Tb3+ion,exhibits a broad band between250and 450nm(k max=370nm)which can be assigned to p–p* electron transition of the ligands[8].A small peak at 490nm observed as a result of f–f absorption transition (7F6!5D4)of Tb3+ion.This transition is much weaker than the absorption of organic ligands,which proves that luminescence sensitization via excitation of the ligand,is more effective than direct excitation of the Tb3+ion.The room-temperature normalized emission spectra of Tb3+ complexes1and2(Fig.2)shows characteristic emission bands of Tb3+(k ex=370nm)centered at490,545,585 and620nm,resulting from the deactivation of the5D4 excited state to the corresponding ground state7F J (J=6,5,4,3)of the Tb3+ion.The strongest emission is centered on545nm,which corresponds to the hypersensi-tive transition of5D4!7F5[13,14].The broad emission peaks obtained may be due to the greater non-homogeneityfor Tb3+local coordination site due to the presence of water molecules.The overall quantum yields(U overall)of terbium com-plexes(1and2)were measured at room-temperature using the technique for powdered samples described by Bril and De Jager-Veenis[15].The overall quantum yields of com-plexes1and2were calculated as11%and22%,respec-tively,and were found to be promising as compared to the recently reported Tb(PMIP)3(H2O)2(where PMIP =1-phenyl-3-methyl-4-isobutaryl-5-pyrazolone;U overall= 29.7·10À3%)[16]and terbium-1-phenyl-3-[G-3]-4-pheny-lacetyl-5-pyrazolonates(G stands for poly aryl ether;U overall =2.26%)[17].Among complexes1and2,the later shows better quantum yields due to the presence of electron-releasing group(–CH3)on the benzoyl moiety.The luminescence lifetimes(s)were also investigated for terbium complexes and found to be400and472l s,respec-tively for1and2(Fig.3).The measured luminescent decays of complexes could be described by mono-exponen-tial kinetics,which suggests that only one species exists in the excited state of these complexes.In combination with the data for the overall quantum yields,the data for the luminescence lifetimes show that longer the luminescent lifetimes,higher the quantum yields of the complexes.The singlet and triplet energy levels of HPBI and HTPI were estimated by referring to the wavelengths of UV–Vis absorbance edges(HPBI and HTPI are365and360nm) and the lower wavelength emission edges(HPBI and HTPI are450and442nm)of the corresponding phosphorescence spectra of the complexes Gd(PBI)3(H2O)2and Gd(TPI)3-(H2O)2(Supplementary data).The triplet energy level of HTPI(22620cmÀ1)was found to be moderately higher than HPBI(22220cmÀ1),may be due to the presence of electron-donating group(–CH3)on the benzoyl moiety of the HPBI system.Generally,the sensitization pathway in luminescent terbium complexes consists of the excitation of the ligands from the ground state to their excited singlet states,and subsequently through the intersystem crossing of the ligands to their triplet states,following the energy transfer from the triplet state of the ligand to the central ion.In this process,the4f electrons of the Tb3+ion are excited to the5D J manifold from the ground state,finally the Tb3+ion emits when the4f electrons undergo a transi-tion from the excited state of5D4to the ground state.It has been noticed that the energy gaps D E(1pp*–3pp*) between the1pp*and3pp*levels are5180and5160cmÀ1 for HPBI and HTPI,respectively.According to Rein-houdt’s empirical rule[18]that the intersystem crossing process will be effective when D E(1pp*–3pp*)is at least 5000cmÀ1,thus the intersystem crossing processes are effective for all the ligands.According to Latva’s empirical rule[19],an optimal ligand-to-metal energy transfer pro-cess for Tb3+is when D E(3pp*–5D4)>2000cmÀ1.It can be concluded that the transfer process is effective from HTPI to Tb3+and that HTPI is a suitable as a sensitizer for Tb3+(D E(3pp*–5D4)>2220cmÀ1).On the other hand,S.Biju et al./Inorganic Chemistry Communications10(2007)393–396395the3pp*state(22220cmÀ1)of HPBI is so close to 5D4(20400cmÀ1)of Tb3+,giving D E(3pp*–5D4)=1820 cmÀ1,which is too low to prevent the back-energy transfer from the Tb3+excited state to the triplet state of HPBI.AcknowledgementsThe authors acknowledge thefinancial support from Defence Research and Development Organization and University Grants commission,New Delhi,India.The Bra-zilian author acknowledges CNPq and Instituto do Milenio de Materiais Complexos,forfinancial support.Appendix A.Supplementary dataSupplementary data associated with this article can be found,in the online version,at doi:10.1016/j.inoche. 2006.12.008.References[1]J.Kido,Y.Okamoto,Chem.Rev.102(2002)2357.[2]J.C.G.Bunzli,C.Piguet,Chem.Soc.Rev.34(2005)1048.[3]G.F.de Sa,O.L.Malta,C.de Mello Donega,A.M.Simas,R.L.Longo,P.A.Santa-Cruz,E.F.da Silva Jr.,Coord.Chem.Rev.196 (2000)165.[4]H.Xin,M.Shi,X.C.Gao,Y.Y.Huang,Z.L.Gong,D.B.Nie,H.Cao,Z.Q.Bian,F.Y.Li,C.H.Huang,J.Phys.Chem.B108(2004) 10796.[5]H.Xin,F.Y.Li,M.shi,Z.Q.Bian,C.H.Huang,J.Am.Chem.Soc.125(2003)7166.[6]L.Gan,L.Xu, C.Luo, C.Huang,S.Umetani,M.Matsui,Polyhedron13(1994)3167.[7]R.Pavithran,M.L.P.Reddy,Radiochim.Acta92(2004)31.[8]R.Pavithran,N.S.Saleesh Kumar,S.Biju,M.L.P.Reddy,S.AlvesJr.,R.O.Freire,Inorg.Chem.45(2006)2184.[9]C.Pettinari,F.Marchetti,A.Cingolann,A.Drozdov,I.Timokhin,S.I.Troyanov,V.Tsaryuk,V.Zolin,Inorg.Chim.Acta357(2004) 4181.[10]R.O.Freire,G.B.Rocha,A.M.Simas,Inorg.Chem.44(2005)3299.[11]G.B.Rocha,R.O.Freire,N.B.da Costa Jr.,G.F.de Sa,A.M.Simas,Inorg.Chem.4(2004)2346.[12]D.Zhou,Q.Li,C.H.Huang,G.Yao,S.Umetani,M.Matsui,L.Ying,A.Yu,X.Zhao,Polyhedron16(1997)381.[13]A.Dias,S.Viswanathan,Dalton Trans.34(2006)4093.[14]Z-Q.Zhang,Q.Shen,Y.Zhang,Y-M.Yao,J.Lin,Inorg.Chem.Commun.7(2004)305.[15]A.Bril,A.W.De Jager-Veenis,J.Electrochem.Soc.123(1976)396.[16]D.Zhang,M.Shi,Z.Liu,F.Li,T.Yi,C.Huang,Eur.J.Inorg.Chem.11(2006)2277.[17]L.Shen,M.Shi,F.Li,D.Zhang,X.Li,E.Shi,T.Yi,Inorg.Chem.45(2006)6188.[18]F.J.Steemers,W.Verboom,D.N.Reinhoudt,E.B.Vander Tol,J.W.Verhoeven,J.Am.Chem.Soc.117(1995)9408.[19]tva,H.Takalo,V.M.Mukkala,C.Matachescu,J.C.Rodri-guez-Ubis,J.Kanakare,J.Lumin.75(1997)149.396S.Biju et al./Inorganic Chemistry Communications10(2007)393–396。