MPX12
飞思卡尔传感器

飞思卡尔传感器飞思卡尔传感器motorola/freescale氣壓傳感器MPX10D MPX10DP MPX10GP MPX10GS MPXV10GC6U MPXV10GC7U MPX12D MPX12DP MPX12GP MPX2010D MPX2010DP MPX2010GP MPXT2010G7U MPX2050D MPX2050DP MPX2050GP MPX2050GVP MPX2050GS MPX2050GSX MPX2050GVSX MPX2053D MPX2053DP MPX2053GP MPX2100A MPX2100DP MPX2100AP MPX2100AS MPX2100ASX MPX2102A MPX2102D MPX2102DP MPX2102AP MPX2102GP MPX2102GVP MPX2200A MPX2200D MPX2200DP MPX2200AP MPX2200GP MPX2200A MPX2200D MPX2200DP MP2200AP MPX2200GP MPX21002A MPX2102D MPX2102DP MPX2102AP MPX2102GP MPX2102GVP MPX2200A MPX2200D MPX2200DP MPX2200AP MPX2200GP MPX2201GP MPX2300DT1 MPX4080D MPX4100A MPXA4100A6U MPX4100AP MPXA4100A6U MPX4100AS MPX4101A MPXA4101AC6U MPX4105A MPX4115A MPXA4115A6U MPX4115AP MPXA4115AC6U MPX4115AS MPX4200A MPX4200AP MPX4200SA MPX4200ASX MPX4250A MPX4250AP MPX4250A6U MPXA4250A6T1 MPXA4250AC6U MPXA4250AC6T1 MPX4250D MPX4250GP MPX4250DP MPX5010D MPX5010DP MPX5010GP MPX5010GS MPX5010GSX MPXV5010G6U MPXV5010G7U MPXV5010GC6U/T1 MPXV5010GC7U MPX5050D MPX5050DP MPX5050GP MPX5100A MPX5100D MPX5100DP MPX5100AP MPX5100GP MPX5100GVP MPX5100AS MPX5100GS MPX5100GVS MPX5100ASX MPX5100GSX MPX5100GVSX MPX53D MPX53GP MPX5500D MPX5500DP MPX5700D MPX5700A MPX5700DP MPX5700GP MPX5700AP MPX5700GS MPX5700AS MPX5999D飞思卡尔传感器MPXA6115A6U MPXA6115AC6U MPXA6115A MPXA6115A6T1 MPXA6115AC6T1 MPXAZ4100A6U MPXAZ4100A MPXAZ4100A6T1 MPXAZ4100AC6U MPXAZ4100AC6T1 MPXAZ4115A6U MPXZ4115A MPXAZ4115A6T1 MPXAX4115AC6U MPXAZ4115AC6T1 MPXC2011DT1 MPXM2010D MPXM2010DT1 MPXM2010GS MPXM2010GST1 MPXM2053D MPXM2053DT1 MPXM2053GS MPXM2053GST1 MPXV4006GC6U MPXV4006G6U MPXV4115V6U MPXV4115V6T1 MPXV4115VC6U MPXV5004GC6U/T1 MPXV5004G6U/T1 MPXV5004GC7U MPXV5004G7U MPXY8010 MPXY8020 MPXY8030 MPXY8040 MMA6200xxQ 低重力加速度(low-g)傳感器MPXA6115A 高溫精確集成壓力傳感器MPXAZ6115A 耐抗高溫壓力傳感器MPXH6115A 高溫精確集成壓力傳感器MP3H6115A 高溫精確集成壓力傳感器 MPXHZ6115A 媒體耐抗的高溫精確集成壓力傳感器MPXH6250A 集成壓力傳感器 MPXH6300A 壓力傳感器 MPXH6400A 集成壓力傳感器等汽車用壓力傳感器飞思卡尔传感器图片飞思卡尔传感器技术参数:参数符号最小典型最大单位压力范围Pop 10 100 100 Kpa供电电压Vs 10 16 Vdc供电电流Io 6.0 mAdc满程输出Vfss 38.5 40 41.5 mV零位偏差电压Voff -1.0 0 1.0 mV灵敏度ΔV/ΔP 0.4 mV/Kpa线性度-0.25 0.25 %Vfs压力迟滞±0.25 %Vfs温度迟滞(-40~125℃)±0.5% %Vfs满程温度系数TCVFss -1.0 1.0 %Vfs零位温度系数TCVoff -1.0 1.0 mV输入阻抗Zin 1000 2500 Ω输出阻抗Zout 1400 3000 Ω响应时间TR 1.0 Ms稳定度±0.5 %Vfss最大压力 4 FS破坏压力7 FS工作温度TA -40~125 ℃以上内容技术参数以《OIML60号国际建议》92年版为基础,最新具体变化可查看《JJG669—12 Freescale广州南创传感器事业部检定规程》。
04.1 [BE]-MPX
![04.1 [BE]-MPX](https://img.taocdn.com/s3/m/f301ea2127d3240c8447efbc.png)
悬 挂 控 制
距
总 成
离 警 告
AFS ECU)
(大灯摆动
ECU 总成)
坐 椅 安 全 带 控 制
大 灯 水 平 测 量 电
大 灯 摆 动 电 机
机
ECU*11 ECU
*12
ECU*4
LH LH
5
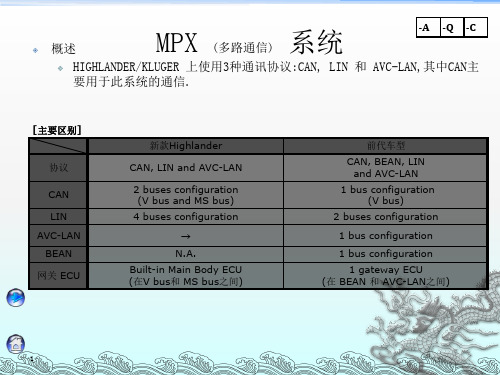
车型概述
发动机
底盘
MPX-MOST
MOST通信 --特性参数
车身
车身电器
协议 应用
MOST 音频视频
AVC-LAN + AVC-LAN
底盘
车身
元件位置
电源管理控制ECU
右侧接线盒
车身电器
转向锁执行器总成 转向锁ECU 主ECU(多路通信网络车身ECU) 左侧接线盒
22
车型概述
发动机
MPX系统-CAN
元件位置
底盘
车身
车身电器
大灯摆动控制ECU
大灯水平控制ECU
23
车型概述
发动机
MPX系统-CAN
元件位置
右侧车门通讯ECU
底盘
电动窗马 达
CANH, CANL
主/从 (单主) 星型
LIN
车型概述
发动机
底盘
MPX-MOST
MOST 通信 • 数据通信线和唤醒线
车身
车身电器
ACC
主设备(ECU) 显示器及导航模块显示屏
唤醒信号
从设备(ECU) 信号接收
MI+ MI-
信息唤醒电路 信息传达电路
WUO MO+ MO-
WUI MI+ MI-
信息唤醒电路 信息传达电路
ECU (副)
MPX

物理地址
这个号码是用作识别每一种部件(例如. 多媒体显 示, Navigation ECU,等)也能确定主要的部件
Logical Address
多媒体显示 这个号码是用作每一种功能(例如:功能按 钮,command 开关,调谐器等)Slave部件将这个号码 发送给主要的部件
Audio Head Unit 物理地址 190 (Audio Head Unit) 25 (Command 开关)
Gateway ECU
Mirror ECUs (D and P)
Power Source Control ECU Rain 感应器
Tilt and Telescopic ECU Double Lock ECU
EPS ECU J/C No. 2 Skid Control ECU*
2
Certification ECU
逻辑地址
29 (Speaker Beep)
60 (Radio Tuner), etc.
25
MPX 系统 of Recent Years
多总线传输 –近几年来,因为通讯系统数据量的增加和扩大,由CAN/BEAN等组 成的MPX系统被采用
AFS
ECU
TPWS
ECU
Meter
ECU
Sliding
Roof ECU
可变的结构 (min. 0 byte to max. 8 byte) Cyclic Redundancy 检查:提供一个错误检测的代码 与CRC的的报告向匹配 结构结束
6
0 - 64 15 2 7
15
MPX基本原理
CAN – 错误检测 • 与BEAN一样, CAN通过CRC检查发送的数据 • 如果是 “not good”: 在检测到错误以后,一个错误的信息会 立即产生并发送到各ECU里去
TEF6621中文资料

(USN)检测可控制立体声解调分离度 。 场强(LEVEL)、多径(WAM)和噪声 (USN)检测可控制高切断(HCC)。 场强(LEVEL)、多径(WAM)和噪声 (USN)检测可控制软静音。
典型应用
车载收音系统。
描述
2.6 表9 中频计数结果对应表
IFCN 4
0 0 0 0 0
IFCN 3
0 0 0 0 0
IFCN 2
0 0 0 0 1
1
1
1
1
1
1
IFCN 1 IFCN 0
0
0
0
1
1
0
1
1
0
0
。。。。。。
1
0
1
1
计数频率差
Байду номын сангаас
FM
AM
0KHz~5KHz
0KHZ~0.5KHz
5KHz~10KHz 0.5KHZ~1KHz
--
60
--
dB
--
0.5
0.8
%
45
60
--
dB
--
150
--
KHz
ftune(step)
调谐频率步距
欧洲/美国/日本模式
--
50
--
KHz
fIFc(res)
中频计数分辨率
--
5
--
KHz
Vo
L、R输出幅度
3h: OUTA=0
80
120
175
mV
AM部分
fRF Vi(sens)
AM射频频率
1.8寸彩屏 mpx ( mvs )播放器 操作说明

1.8寸彩屏mpx(mvs)播放器操作说明使用要点:1.本版本最好不要在W98下使用。
2.Win 2000 与PC联机后,不能直接脱离USB线。
一定要使用弹出方式安全删除硬件。
3.如出现Dos Error 显示时,需要您在本机的菜单中进行格式化。
4.使用FM必须要插耳机,否则不能收音。
5.充电说明:充电时最好把电源开关关掉,此时屏会不亮,最大的保护了OLED的寿命!如果想要边充电边听音乐则必须打开电源开关!本机为多媒体播放器(视频多媒体/MP3/FM/电子相册/图片浏览/游戏)。
采用26万色 160X128 点阵TFT彩屏,颜色亮丽;菜单界面美观大方。
本机可以对MPEG1/2的2/3层的所有压缩率以及取样率信号进行实时解码。
通过内置的麦克风,还可以进行SP(WAV)/LP(ASF)语音录音。
除了支持当前流行的多种音频文件格式外支持最新定义的视频多媒体文件,与传统的MP3文件不同,视频多媒体文件是包含有MP3的音频流和视频流的一种新的多媒体文件格式,通过本mpx(mvs)播放器,用户感受音频和视频于一体的全新感觉,是性价比最高的多媒体播放器。
MVS用户软件可以把Windows Media文件(*.wmv、*.wma、*.asf)和Mpeg文件 (*.mpg、*.mpeg)和Quick Time文件(*.mov、*.qt)和AVI文件(*.avi)压缩成本产品支持的视频格式(*.MVS)。
本机具有图片浏览,只需把图片做成BMP格式,大小最好为128X128 即可,如果超过128X128则可以按、上下移动或按、进行左右移动;最大为2048X2048;用户可根据自己喜好制作开机画面和待机屏保;ID3电脑软件编辑器支持编辑歌曲的ID3及歌词。
关于MVS视频文件说明MVS多媒体文件是由音频流和运动图像构成的多媒体文件,音频流的格式选用了当今流行的 MP3 音频编码格式。
目前可以把 DVD、VCD、Windows Media、Mpeg、Quick Time 、AVI 文件压缩成MVS文件,本机播放电影时可达到20祯/秒所以每分钟的MVS文件大小是约8M。
汽车中的MPX多路通信系统
仪表 多信息显示 (变换和设定 [adjustment])
多信息显示可以通过 “DISP” 开关来变换和设定
-A -Q -C
标准显示
设定显示
时钟设定
:设 置
其他巡航信息
显示设定
显示设定
: 回到标 准显示
空调显示
返回
无操作 (10 秒或更长)
: 按 (1秒内.)
9
: 按住 (1秒或更长)
: 设定 : 选择项目 : 返回标准显示画面
DRL) • H-LP LL Relay (w/
DRL) • H-LP RH, LH Relay
[IFL2]
• PBD Fuse • ALT Fuse • EPS Fuse • ST Fuse 发动机室继电器盒
Mechanical Relay • EFI MAIN Relay • A/F Relay • S-HORN Relay • HORN Relay • C/OPN Relay • H-LP RL Relay (w/o
(6)
(17)
注册快速语音
—
(6)
注册组
—
(20)
1自5 动音量设定
: 可以 —: 不可以 ( ): 最大输入容量 *: Only for –A models
系统图
蓝牙免提 系统
Overhead Module • 麦克风
多功能显示或音响机头组件*
“音量” 开关
组合仪表
• 车速
“语音” 开 关
DRL) • H-LP LL Relay (w/o
维修要点 IFL 能投过观察窗免拆检查是否熔断 (电力分配)
熔断部分
正常
熔断
5提示: PD 和 IFL1/2 为不可分解零件. 如果PD总成发生故障,则必
PTX-LCD 系列激励器说明书

PTX-LCD系列激励器说明书目 录1 概述 (3)1.1 综述 (3)1.2 激励器外观图 (3)1.3 激励器内部布局 (3)1.4 激励器原理框图 (4)1.5 技术指标 (5)2 操作 (8)2.1 面板布局及各功能键功能说明 (8)2.2 操作方法 (14)3 内部模块调整 (28)3.1 电源模块 (28)3.2 音频母板 (29)3.3 音频输入 (30)3.4 编码板 (32)3.5 锁相/驱动板及VCO板 (33)3.6 功率放大模块 (34)3.7 面板卡板 (35)3.8 CPU板 (37)3.9 遥测板(选件) (38)3.10 数字音频接口(AUDINP-DIG,选件) (38)3.11 基于DSP的数/模音频接口(TRDSP) (41)4 日常维护 (42)5 故障分析与维修 (43)5.1 液晶屏“白屏”或“全黑” (43)5.2 无音频或音频幅度低 (43)5.3 过载 (43)5.4 本振失锁 (43)1 概述1.1 综述PTX-LCD系列激励器采用19英寸标准机箱封装,频率调节灵活方便。
其频率范围涵盖87.5~108MHz,可10kHz步进设置。
如用户需要,亦可定制为其他频段。
按其最大输出功率,PTX-LCD系列可分为三种机型,PTX30-LCD为30W机,PTX60-LCD为60W机,PTX100-LCD为100W机,额定负载50Ω。
输出功率在0至标称值间任意可调。
现设备配置的机型为PTX30-LCD为30W机。
PTX-LCD可预置为立体声型或单声道/MPX型,单声道/MPX型可用于单声道信号或经外部编码器处理后的立体声信号的广播。
激励器的用户接口由液晶显示屏和一个旋钮(脉冲编码器)组成,操作简单明了。
借助此接口,使用者可参看所有的工作参数,并轻松调节可设置项如输出功率和工作频率。
激励器提供各种连接口,可使其集成到复合系统中。
它易于与外部单元如放大器、转换系统及同轴继电器等交流信息并提供控制,亦可与其它调制器组成网络。
串口模块MP3模块使用说明书

MPx 系列高保真放音模块根据存储介质的不同可分为 TF 卡版本(简称 T 版)、SD 卡版本(简称 S 版)和 FLASH 版本(简称 F 版)。其中 T 版体积小巧,节省空间;S 版具有最高性能价格比;F 版 专为震动场合设计,功能可靠。另有低成本 SPI 版本正在开发中,敬请期待。
MPx 系列高保真放音模块的推荐使用电压为 6V-24V,最低可低至 5V,最高可高达 30V,满足大 多数场合的需要。
MPx 系列高保真放音模块直接使用标准的内存卡存储音乐,通过电脑录音编辑后即可直接通过 非常廉价的读写卡器写入内存卡中,不使用昂贵且通用性差的专业编程器,在满足灵活性的前提下 最大限度为用户节省成本。
MPx 系列高保真放音模块使用非常简便,配合本公司提供的免费技术支持和驱动程序,可最大 限度的缩短用户的开发时间。
MPx 系列高保真放音模块有非常灵活的工作模式可供选择使用:MP3 模式、循环直放模式、普通 直放模式、并行模式和串行模式。
MPx 系列高保真放音模块可是以最简便的方式使用在各种各样需要高保真音响的场合:商场等 需要背景音的公共场合、各种游乐设备音响系统、学校等各种定时播放音乐的场合……
二、MP38x 系列放音模块选型表及全部照片
型号 MP380 MP383 MP385 MP386 MP381
型号 MP380
MP383
MP385
MP386
MP381
主要特点 存储介质
基本型 可扩展型 SD 卡座横置 可加功放型
Lexicon MPX Native Reverb插件商品说明书

ቤተ መጻሕፍቲ ባይዱ
2 9 MPX Native Reverb
Installation
Installing the MPX Plug-In
1. Install the MPX Plug-In on your computer. 2. Connect your iLok2 to an available USB port. 3. If you haven't already, go to and download/install the required
4 9 MPX Native Reverb
on the store button also allows you to manage existing presets. The store window displays existing presets. Click on an existing preset to either replace or delete it.
6. Real Time Visualization – Real Time Visualization can be turned on and off by clicking anywhere in the Real Time Visualization window. When off, the plug-in name is displayed – this conserves CPU power. When on, the Spectral Intensity Analyzer is displayed, providing real-time visual feedback of the effect signal's amplitude (Y axis) over the frequency spectrum (X Axis).
MPX-X2

http://
MPX/MKP-X2(MP2)
Metallized Polypropylene Film Capacitors,Class X2
金属化聚丙烯膜抑制电源电磁干扰用电容器
MPX/MKP-X2产品编码说明: ■ 15位数产品代码如下:
1
2
3
4
5
6
7
8
Plastic Capacitors
引线加工 代码
常规 L0=15±1 L0=4.5±0.5 L0=3±0.5 电子引线
0
4
3
V
L0=25±1
铜线
其他
2
A
Q
Note: 使用涉及到和极限参数有关问题请与
联系。
Page2
http://
MPX/MKP-X2(MP2)
Metallized Polypropylene Film Capacitors,Class X2
气候类别 -40---+110℃/56d 额定电压 275vac/300vac 容量范围 0.0022 F---4.7 F
塑壳型号 脚距 mm
引线直径 mm dv/dt(v/ s)
C类 10 0.6 500
D类
E类
15
22.5
0.6/0.8
300
100
F类 G类 27.5 31.5 0.8
100 100
第 9位数
产品塑壳型号代码 C类壳=C D类壳=D E类壳=E F类壳=F G类壳=G T类壳=T
第 10位数
内部识别码
第 11位数
塑壳颜色代码“-” 灰色+SRD=R 黄色+SRD=S 黑色+SRD=T
Supermicro X12系列主板内存配置指南说明书

S upermicro X12系列主板的内存配置基于第三代i ntel® X eon®可扩展处理器用户指南版本1.0cThe information in this user’s manual has been carefully reviewed and is believed to be accurate. The vendor assumes no responsibility for any inaccuracies that may be contained in this document, and makes no commitment to update or to keep current the information in this manual, or to notify any person or organization of the updates. Please Note: For the most up-to-date version of this manual, please see our website at .Super Micro Computer, Inc. ("Supermicro") reserves the right to make changes to the product described in this manual at any time and without notice. This product, including software and documentation, is the property of Supermicro and/ or its licensors, and is supplied only under a license. Any use or reproduction of this product is not allowed, except as expressly permitted by the terms of said license.IN NO EVENT WILL SUPER MICRO COMPUTER, INC. BE LIABLE FOR DIRECT, INDIRECT, SPECIAL, INCIDENTAL, SPECULATIVE OR CONSEQUENTIAL DAMAGES ARISING FROM THE USE OR INABILITY TO USE THIS PRODUCT OR DOCUMENTATION, EVEN IF ADVISED OF THE POSSIBILITY OF SUCH DAMAGES. IN PARTICULAR, SUPER MICRO COMPUTER, INC. SHALL NOT HAVE LIABILITY FOR ANY HARDWARE, SOFTWARE, OR DATA STORED OR USED WITH THE PRODUCT, INCLUDING THE COSTS OF REPAIRING, REPLACING, INTEGRATING, INSTALLING OR RECOVERING SUCH HARDWARE, SOFTWARE, OR DATA.Any disputes arising between manufacturer and customer shall be governed by the laws of Santa Clara County in the State of California, USA. The State of California, County of Santa Clara shall be the exclusive venue for the resolution of any such disputes. Supermicro's total liability for all claims will not exceed the price paid for the hardware product.FCC Statement: This equipment has been tested and found to comply with the limits for a Class A digital device pursuant to Part 15 of the FCC Rules. These limits are designed to provide reasonable protection against harmful interference when the equipment is operated in an industrial environment. This equipment generates, uses, and can radiate radio frequency energy and, if not installed and used in accordance with the manufacturer’s instruction manual, may cause harmful interference with radio communications. Operation of this equipment in a residential area is likely to cause harmful interference, in which case you will be required to correct the interference at your own expense.California Best Management Practices Regulations for Perchlorate Materials: This Perchlorate warning applies only to products containing CR (Manganese Dioxide) Lithium coin cells. “Perchlorate Material-special handling may apply. See /hazardouswaste/perchlorate”.The products sold by Supermicro are not intended for and will not be used in life support systems, medical equipment, nuclear facilities or systems, aircraft, aircraft devices, aircraft/emergency communication devices or other critical systems whose failure to perform be reasonably expected to result in significant injury or loss of life or catastrophic property damage. Accordingly, Supermicro disclaims any and all liability, and should buyer use or sell such products for use in such ultra-hazardous applications, it does so entirely at its own risk. Furthermore, buyer agrees to fully indemnify, defend and hold Supermicro harmless for and against any and all claims, demands, actions, litigation, and proceedings of any kind arising out of or related to such ultra-hazardous use or sale.Manual Revision 1.0cRelease Date: May 21, 2021Unless you request and receive written permission from Super Micro Computer, Inc., you may not copy any part of this document. Information in this document is subject to change without notice. Other products and companies referred to herein are trademarks or registered trademarks of their respective companies or mark holders.Copyright © 2021 by Super Micro Computer, Inc.All rights reserved.Printed in the United States of AmericaDIMM安装准则X12系列主板的内存支持本文档简明介绍了如何正确配置和安装X12系列主板,这些主板使用速度为3200/2933/2666的3DS LRDIMM/LRDIMM/3DS RDIMM/RDIMM DDR4(288针)ECC REG内存模组。
佳能 LBP-1210激光打印机 说明书

EN60825-1:1994 CLASS 1 LASER PRODUCT LASER KLASSE 1 APPAREIL A RAYONNEMENT LASER DE CLASSE 1 APPARECCHIO LASER DI CLASSE 1 PRODUCTO LASER DE CLASE 1 APARELHO A LASER DE CLASSE 1 220-240 V
• • • • 30%
xi
•
0˚C-35˚C (32˚F-95˚F).
•
35-85% RH
EP-.................................................................................................... 1 ............................................................................................ 2 ........................................................................................ 4 ........................................................................................... 4 ........................................................................................... 4 ........................................................................... 5 ................................................................................................ 7 ................................................................................................ 8 ........................................................................................... 9 ....................................................................................... 9 ............................................................................................. 10 ............................................................................................. 10 ......................................................................................... 11 .............................................................................................. 12 .............................................................................................. 13 ...................................................................................... 14 ................................................................................. 14 ................................................................................. 15 .................................................................................. 16 ......................................................................... 19 ...................................................................................... 24 Canon Advanced Printing Technology .................................... 27 CAPT .................................................................................. 28 Windows 95/98/Me .............................................................................. 28 Plug and Play ....................................................... 28 ......................................................................... 32 Windows NT 4.0/2000/XP ................................................................... 36 Windows 2000 Plug and Play ............................. 36 Windows XP Plug and Play ................................ 39 Windows NT 4.0 ............................................ 42 Windows 2000 ................................................ 46 Windows XP ................................................... 52 LBP-1210 ..................................................... 58 ..................................................................................... 58 Windows 95/98/Me ........................................ 59 ............................................ 60 Windows NT 4.0 Windows 2000 ................................................ 60 ................................................... 61 Windows XP .......................................................................................... 62 ...................................... 62 LBP-1210
西门子 S7-1200 功能安全手册 - 设备手册说明书

SIMATICS7S7-1200 功能安全手册设备手册Siemens AGDigital IndustriesⓅ 10/2022 本公司保留更改的权利 Copyright © Siemens AG 2022. 保留所有权利法律资讯警告提示系统为了您的人身安全以及避免财产损失,必须注意本手册中的提示。
人身安全的提示用一个警告三角表示,仅与财产损失有关的提示不带警告三角。
警告提示根据危险等级由高到低如下表示。
危险表示如果不采取相应的小心措施,将会导致死亡或者严重的人身伤害。
警告表示如果不采取相应的小心措施,可能导致死亡或者严重的人身伤害。
小心表示如果不采取相应的小心措施,可能导致轻微的人身伤害。
注意表示如果不采取相应的小心措施,可能导致财产损失。
当出现多个危险等级的情况下,每次总是使用最高等级的警告提示。
如果在某个警告提示中带有警告可能导致人身伤害的警告三角,则可能在该警告提示中另外还附带有可能导致财产损失的警告。
合格的专业人员本文件所属的产品/系统只允许由符合各项工作要求的合格人员进行操作。
其操作必须遵照各自附带的文件说明,特别是其中的安全及警告提示。
由于具备相关培训及经验,合格人员可以察觉本产品/系统的风险,并避免可能的危险。
按规定使用 Siemens 产品请注意下列说明:警告Siemens 产品只允许用于目录和相关技术文件中规定的使用情况。
如果要使用其他公司的产品和组件,必须得到 Siemens 推荐和允许。
正确的运输、储存、组装、装配、安装、调试、操作和维护是产品安全、正常运行的前提。
必须保证允许的环境条件。
必须注意相关文件中的提示。
商标所有带有标记符号 ® 的都是 Siemens AG 的注册商标。
本印刷品中的其他符号可能是一些其他商标。
若第三方出于自身目的使用这些商标,将侵害其所有者的权利。
责任免除我们已对印刷品中所述内容与硬件和软件的一致性作过检查。
然而不排除存在偏差的可能性,因此我们不保证印刷品中所述内容与硬件和软件完全一致。
MPX2050GP;MPX2050DP;MPX2050D;MPX2050GSX;中文规格书,Datasheet资料

Pressure Range(1) Supply Voltage(2) Supply Current
Characteristics
Symbol
Min
Typ
Max
Unit
POP
0
—
50
kPa
VS
—
10
16
Vdc
Io
—
6.0
—
mAdc
Full Scale Span(3)
VFSS
38.5
40
41.5
mV
Offset(4) Sensitivity
MPX2050 Series
0 to 50 kPa (0 to 7.25 psi) 40 mV Full Scale Span (Typical)
Application Examples
• Pump/Motor Controllers • Robotics • Level Indicators • Medical Diagnostics • Pressure Switching • Non-Invasive Blood Presescale Semiconductor
MPX2050 Rev 9, 10/2008
+ 50 kPa On-Chip Temperature Compensated and Calibrated Silicon Pressure Sensors
The MPX2050 series devices are silicon piezoresistive pressure sensors providing a highly accurate and linear voltage output, directly proportional to the applied pressure. The sensor is a single, monolithic silicon diaphragm with the strain gauge and a thin-film resistor network integrated on-chip. The chip is laser trimmed for precise span and offset calibration and temperature compensation.
Guideline on quality of oral modified release products 2014.03.20

7 Westferry Circus ● Canary Wharf ● London E14 4HB ● United Kingdom20 March 2014 EMA/CHMP/QWP/428693/2013 Committee for Medicinal Products for Human Use (CHMP) Guideline on quality of oral modified release products Final Draft Agreed by QWPMay 2012 Adoption by CHMP for release for consultation19 July 2012 Start of public consultation 15 September 2012 End of consultation (deadline for comments)15 March 2013 Agreed by QWPFebruary 2014 Adoption by CHMPMarch 2014 Date for coming into effect6 months after publicationThis guideline together with the Guideline on Quality of Transdermal Patches replaces the Note for Guidance on Modified Release products: A: Oral dosage Forms B: Transdermal Dosage Forms. Part I (Quality).Keyword Oral dosage form, modified releaseGuideline on quality of oral modified release products Table of contents1. Introduction (3)1.1. Preamble (3)1.2. Scope (3)2. Prolonged release oral dosage forms (3)2.1. Development pharmaceutics (3)2.1.1. General remarks (3)2.1.2. Therapeutic objectives and principle of the release system (4)2.1.3. Development of dissolution methods (4)2.1.4. Discriminatory power of the dissolution test (6)2.1.5. Bioavailability study (6)2.1.6. Comparison of dissolution profiles (6)2.1.7. In vitro-in vivo comparison (7)2.2. Setting specifications (8)2.3. Control strategy (9)2.4. Variations to products (9)3. Delayed release dosage forms (10)3.1. General remarks (10)3.2. Development pharmaceutics (10)3.3. Setting specifications (11)3.4. Control strategy (11)3.5. Variations to products (11)ANNEX 1 (12)ANNEX 2 (15)1. Introduction1.1. PreamblePharmaceutical dosage forms may be developed in which the rate and/or place of release of active substance(s) has in some way been modified compared with conventional release formulations. Such modifications may have a number of objectives, such as maintaining therapeutic activity for an extended time, reducing toxic effects, protecting the active substance against degradation due to low pH, targeting the active substance to a predefined segment of the gastrointestinal tract for local treatment or targeting active substance release at specified time-points.This guideline covers the various parts of the application for marketing authorisation related to quality and should be read in conjunction with section II of this guideline relating to clinical aspects. Furthermore, it is clear that this Guideline cross-references to other quality guidelines and to official compendia.For clear definitions on the terminology used to describe different types of release models and other definitions, reference is made to Annex I.1.2. ScopeThis guideline concerns quality aspects, especially pharmaceutical development and in vitro testing, of dosage forms in which the release of active substance is modified. This guideline only covers prolonged release oral dosage forms and delayed release oral dosage forms with the principle of gastro-resistance. Pulsatile and accelerated release dosage forms are outside the scope of this guideline. Delayed release dosage forms with other principles, including those designed to release in a specific area of the gastrointestinal tract in response to a specific trigger (e.g. enzymes) or at specific time(s) after ingestion are not specifically addressed.Many principles discussed under section 2 with respect to prolonged release oral dosage forms will be relevant to other modified release dosage forms intended for oral administration or via other routes. 2. Prolonged release oral dosage forms2.1. Development pharmaceutics2.1.1. General remarksThe quality of a prolonged release dosage form is continuously improved during the development of a new drug product. The choice of the composition is made during the development based on small-scale batches and takes into account physicochemical properties of the active substance, stability and drug absorption characteristics throughout the gastrointestinal tract. As soon as the constituents are chosen, gradual scaling up of the manufacturing process will start. During this period it is reasonable to expect that adjustments will be necessary to reach full-scale production. These adjustments might be changes in composition, manufacturing processes, equipment or manufacturing site.In some cases these adjustments may have an effect on the properties of the drug product. It is therefore recommended that an in vitro dissolution test is developed which is able to detect changes which may have an effect on the efficacy or safety of the product.In other words, pharmaceutical development should establish the (qualitative or quantitative) link from pharmacokinetic parameters through in vivo drug release to in vitro dissolution rate.The formulation chosen in development should be evaluated under different dissolution conditions to determine its sensitivity/robustness to the expected physiological environment after administration. The discriminatory power of the test conditions chosen for routine control may be determined by comparison of the in vitro dissolution data and the bioavailability data of the different formulations. It is encouraged to establish an in vivo-in vitro correlation (IVIVC). With a level A IVIVC the dissolution test - after proper validation - can be used as a qualifying control method with in vivo relevance, while in the absence of a Level A IVIVC the dissolution test can be used only as a quality control method.After completed scale-up it is reasonable to compare the laboratory/pilot scale batches with the full production scale batches in a bioavailability study if the scale-up factor exceeds 10 (compared to the laboratory/pilot scale biobatch) in order to verify that the dissolution test conditions chosen are appropriate for the release of clinical materials, scale-up and manufacture (see also 2.1.3., 2.1.4 and 2.1.5).2.1.2. Therapeutic objectives and principle of the release systemThe therapeutic objectives and rationale of the prolonged release product should be provided. Pharmacokinetic (e.g. AUC, C max, T max, t1/2) and physico-chemical characteristics of the active substance (e.g. solubility at different pH, partition coefficient, particle size, polymorphism) relevant to the development of the product should be given. Detailed information on the release controlling excipient(s) should be given. Reference is made to the guidelines on pharmaceutical development.The following characteristics of the prolonged release system should be described:•the manner in which prolonged release is intended to be achieved (membrane type, matrix, etc.); •the release mechanism and kinetics (diffusion, erosion, osmosis, etc. or a combination of these);•the system format e.g. single non-disintegrating unit, disintegrating tablet/capsule containing multiple-units of pellets, etc.It should be demonstrated that the prolonged release product maintains its drug release characteristics regardless of relevant variability in physiological conditions. Examples of such variability include gastric and intestinal transit time, food effect, pathological gastrointestinal fluid composition and concurrent alcoholic intake, if and where relevant.In general, prolonged release oral dosage forms should not have a score line because subdivision or other manipulation of modified release products may adversely affect the modified release properties of the dosage form, possibly leading to dose dumping. Any recommendation on subdivision of a modified release dosage form should be supported by scientific justification that the subdivision does not affect the modified release characteristics, including in vitro and/or in vivo data as appropriate.2.1.3. Development of dissolution methodsThe release rate should be tested in vitro by a dissolution test method. The development of a suitable dissolution test method should be based on the physicochemical in vitro and in vivo characteristics of the active substance and the drug product considering the mechanism of release.This in vitro dissolution test should be capable of:•discriminating between batches with respect to critical process parameters (CPP) which may have an impact on the desired bioavailability;•testing for batch to batch consistency of pivotal clinical, bioavailability and routine production batches;•determining stability of the relevant release characteristics of the product over the proposed shelf life and storage conditions.The prolonged release formulation should therefore be evaluated in vitro under various conditions (media, pH (normally pH range 1-7.5; in cases where it is considered necessary up to pH 8), apparatus, agitation, etc.). Testing conditions, including sampling time points and frequency providing the most suitable discrimination should be chosen.Suitable buffer capacity should be used to ensure that media pH is well controlled during the dissolution test. Otherwise it may be necessary to monitor the media pH throughout the test. If a surfactant is used in the dissolution medium, the amount needed should be justified. The choice of the surfactant should be discussed and its consistent batch to batch quality should be ensured.The inclusion of enzymes in the media is acceptable, and even encouraged, when justified (e.g., colonic delivery, gelatin capsules). If enzymes are added to the dissolution media, a rationale should be given for the type and concentration of enzymes added. Further, consistency of the batch to batch quality of the enzymes should be ensured including activity (IU/mg or IU/ml) or concentration (mg/ml) as appropriate. Note that the enzyme concentration of the SGF / SIF media prescribed in the Ph.Eur. are much higher than physiologically relevant values.Justified enzyme concentrations should be used when the enzymes constitute part of the dissolution control mechanism. The use of biorelevant media may improve the correlation to in vivo data and may detect a potential food effect.The volume of medium should preferably ensure sink conditions.For formulations having a zero order release kinetics (with or without lag time) a specification of the dissolution rate over time (per cent of label claim per hour) for a given interval should preferably be established (see also section 2.2). For this type of product, a graphical presentation of the dissolution rate versus time should be additionally presented in order to justify that the product can be regarded as a zero-order release formulation. For additional details with respect to the choice of apparatus, testing conditions, validation/qualification and acceptance criteria, reference is made to the Ph. Eur.Special attention should be paid to the importance of any variation in the active substance (e.g. particle size, polymorphism), release controlling excipient(s) (e.g. particle size, gelling properties) or manufacturing process with regard to its impact on the in vivo bioavailability.The assay method of the active substance in dissolution samples should be validated according to the relevant ICH guidelines "Validation of analytical procedures" and "Validation of analytical procedures: Methodology", with special attention to the stability of the active substance dissolved in the medium and effects from the excipients.Identical or, if not possible, comparable test conditions should be used for different strengths of the same product.Normally in development, individual dosage unit results, the mean value and a measure of variability (e.g. standard deviation or 95 % confidence interval) should be presented at each time point. Use of other statistical approaches must be justified. Dissolution profiles should be determined for all strengths and for any relevant changes in the composition and/or manufacturing process of the product during development.2.1.4. Discriminatory power of the dissolution testIt should be shown that the dissolution test under the chosen test conditions is able to discriminate between batches with acceptable and non-acceptable release characteristics.Showing discriminatory power may be achieved in one of the following approaches in order of priority:•it is best practice to include batches which have failed to show acceptable pharmacokinetic parameters in vivo. Based on the dissolution results, meaningful specifications may be set to reject such batches due to their dissolution data. This may be supported quantitatively through a validated IVIVC, which has been developed under consideration of batches with unacceptable pharmacokinetic parameters; •in cases where there are no available batches showing non-acceptable in vivo behaviour, the dissolution data may be compared to the average results of the pharmacokinetic parameter (point estimates) of the in vivo studies. These data may be compared by checking the rank order of the results;•if neither of the first two approaches is feasible, the discriminatory power may be shown by deliberately varying an attribute of the active substance (e.g. particle size distribution), composition and/or manufacturing process parameters, in order to produce different in vitro dissolution behavior, without generating in vivo data for these batches. However such test procedures may lead to over-discrimination, i.e. even batches with acceptable in vivo performance may be rejected by the quality control method.2.1.5. Bioavailability studyA summary of the bioavailability studies should be given. The data should include information on pharmacokinetics (AUC0 → t(last), AUC0 →∞, C max, and where appropriate other relevant parameters (C min in steady state, partial AUC, C max/C min ratio, etc.); for generic products also the point estimates and 90% confidence intervals), manufacturing sites and dates, batch sizes and numbers, formulations and dissolution results of the batches used.Bioavailability studies should be performed with batches of 100,000 units or at least 10% of full production scale, whichever is greater, unless pivotal clinical studies have been performed with batches of this size. In this case bioavailability studies performed with batches of a smaller scale may be sufficient if these batches have been produced in a manner representative of the full scale manufacturing process. So, for example, if phase II trials (including PK/BA-studies) are conducted at a scale of 15 kg, the pivotal clinical trials (no BA data available) at a scale of 60 kg and full production scale is intended to be 600 kg, no additional BA-studies at a scale of 60 kg are required.2.1.6. Comparison of dissolution profilesOn several occasions dissolution profiles have to be compared for similarity, e.g. after scale-up or changes in composition and/or manufacturing process or in case of extrapolation of in vivo results to be applied for approval of different strengths. Similarity of dissolution profiles should be established with at least 12 individual values per time point. Consideration should be given to the sampling time points and frequency, taking into account the physicochemical in vitro and in vivo characteristics of the active substance and the mechanism of release of the drug product.In cases where an extrapolation of in vivo results is to be applied for approval of different strengths, if not all strengths of a test drug product are compared in vivo versus the reference, the dissolution of the other strengths of the test product will be compared to the strength of the test product used in the bioequivalence study.The profiles should be compared and their similarity may also need to be demonstrated by statistically justified methods using model-independent or model-dependent methods e.g. linear regression of the percentage dissolved at specified time points, statistical comparison of the parameters of the Weibull function or calculation of a similarity factor.2.1.7. In vitro-in vivo comparisonIn vitro dissolution testing is not only important as a necessary quality assurance for batch-to-batch consistency but also to indicate consistency within a batch (i.e. that individual dosage units will have the desired in vivo performance). By establishing a meaningful correlation between in vitro release characteristics and in vivo bioavailability parameters, the in vitro dissolution test can serve as a surrogate marker for in vivo behaviour and thereby confirm consistent therapeutic performance of batches from routine production. The variability of the data should be reported and discussed when establishing a correlation. In general the higher the variability in the data used to generate the in vitro-in vivo correlation (IVIVC), the less confidence can be placed in the model parameters’ estimates and the higher the uncertainty in the model-predictions for in vivo behaviour becomes.An established Level A IVIVC may reduce the number of in vivo studies during product development, be helpful in setting specifications and be used to facilitate certain regulatory decisions (e.g. scale-up and post-approval variations). Therefore, an attempt to develop such an IVIVC should be considered by the applicant. Furthermore, establishment of a Level A IVIVC gives confidence in the use of dissolution testing as a change control tool. Alternatively it may be acceptable to apply a mechanistic model for the in vitro in vivo comparison (e.g. using physiologically based pharmacokinetic models-(PBPK).Validation of a Level A IVIVC involves showing that it is sufficiently predictive. A Level A IVIVC is established based on for example a deconvolution technique, in which in vivo absorption or in vivo dissolution can be predicted from in vitro data (detailed in Annex 2). A validated Level A IVIVC allows the use of the associated in vitro dissolution test as a surrogate for an in vivo study, as the resulting in vivo concentration-time profile can be predicted using the in vitro dissolution data and the IVIVC equation. Implicit in this approach is that (1) such an IVIVC can only be reliably used for interpolation (explained below) and (2) a single IVIVC model must be applicable to all formulations used in development and validation of the model.Note that an IVIVC cannot serve as a basis for claiming bioequivalence between products from different MA applicants, based on in vitro data only.An IVIVC model should be used for interpolation within the range of data used in its development, rather than extrapolation outside of the range over which it is known to apply. This principle is particularly important for regulatory applications, such as justification of dissolution specification and biowaivers. This has important implications for the choice of formulations to be included in an IVIVC study.It is generally recommended to use formulations with widely varying in vitro dissolution profiles for IVIVC development and validation, since utilising formulations with only small differences in their in vitro dissolution profiles will limit the scope for widening of the specification range and the range for which a biowaiver can be justified. However, it is acknowledged that different release mechanisms or other biopharmaceutical factors may come into play at the formulation extremes, impacting on the relationship between in vitro and in vivo drug release and precluding generation of a single IVIVC equation which describes the behavior of all formulations within the range proposed for a biowaiver. Therefore, formulations should be chosen such that the same release mechanism is likely to control both the in vitro and in vivo release of drug. This will tend to limit the range of in vitro dissolution profiles used in practice for IVIVC development and validation.If an extreme formulation (i.e. one with the fastest or slowest in vitro dissolution of the formulations used in the IVIVC) is subsequently chosen for further development, it is advisable to extend the IVIVC validation range by generating in vivo data for another formulation (yet faster or slower, as the case may be) and using these data for external validation of the existing IVIVC or for redevelopment and validation of a new IVIVC.In other words, it is important that the intended target formulation is appropriately bracketed.2.2. Setting specificationsThe specification should be set using a discriminatory dissolution test.In general, a minimum of three points should be included in the specification on in vitro dissolution of an oral prolonged release product: an early time point to exclude dose dumping and/or to characterise a loading/initial dose (typically 20 to 30% dissolved), at least one point to ensure compliance with the shape of the dissolution profile (around 50% dissolved) and one to ensure that the majority of the active substance has been released (Q=80 %). If the maximum amount dissolved is less than 80%, the last time point should be the time when the plateau of the dissolution profile has been reached.For drug products showing a zero order release a specification of the dissolution rate/time for a given time interval may be more appropriate than the cumulative amount dissolved at a distinct time point. In cases where a zero order release kinetic is combined with a variable lag time, such a specification is mandatory. The method to determine the lag time is up to the applicant.The acceptable variation allowed around each time-point (upper and lower limits), can be determined in different ways:a. No IVIVC:The tolerance limits may be derived from the spread of in vitro dissolution data of batches with demonstrated acceptable in vivo performance (biobatch(es)), or by demonstrating bioequivalence between batches at the proposed upper and lower limit of the dissolution range (the so-called"side-batch" concept).Normally, the permitted range in release at any given time point should not exceed a total numerical difference of ±10% of the labelled content of active substance (i.e. a total variability of 20%: a requirement of 50±10% thus means an acceptable range from 40-60%), unless a wider range is supported by a bioequivalence study.b. Established Level A IVIVC:A validated Level A IVIVC allows in vitro dissolution data (in this case, proposed rather than observed data) to be used as a surrogate to an in vivo study of formulations at the proposed dissolution specification limits. Dissolution profiles are generated from the proposed limits using the established IVIVC that preferably includes an appropriate mathematical description of the in vitro dissolution behaviour (Weibull function, Hill, etc. as justified by the behaviour of formulations tested during product development) or, normally less usefully, based on release at different time points. The entire plasma concentration-time profile is calculated for the proposed upper and lower dissolution limits and the observed in vitro dissolution data for the to-be-marketed (reference) formulation utilising the validated IVIVC. The corresponding C max and the selected AUC parameter values are calculated for the proposed lower and upper limits and the reference formulation and the ratios calculated (upper to lower, upper to reference and lower to reference).The guiding principle of specification setting is that all batches within the lower and upper dissolution specification limits should be bioequivalent to one another. When bioequivalence is based on in vivo data,the acceptance range for the maximum difference in comparative data is 80-125%, based on confidence intervals around the mean C max and the selected AUC parameter. Although some methods of IVIVC analysis quantify biological variability (and allow prediction of confidence intervals), most methods predict mean concentration-time data only. Therefore, for BE predicted based on mean data (by use of dissolution data in lieu of in vivo data and supported by an IVIVC), the criteria for BE limits must necessarily be tighter i.e., the difference between the C max and the selected AUC parameter for the mean in vivo concentration-time data predicted for the upper and lower dissolution specification must be less than 20%. Limits based on a difference greater than 20% between the predicted C max and the selected AUC parameter for the upper and lower dissolution specifications must be justified.For drugs that are absorbed throughout the gastrointestinal tract, the AUC is often similar for formulations of widely varying dissolution rates and the specification is driven by C max, rather than AUC. In this case, the advantage of utilising an IVIVC for specification setting is that limits wider than ±10% in cumulative dissolution at particular time points may be possible, as not every time point has the same impact on C max. The sensitivity of C max to changes in dissolution depends on the pharmacokinetic properties of the drug (the shorter the half-life the greater the sensitivity to changes in dissolution) and the shape of the IVIVC relationship (i.e., whether in vitro or in vivo dissolution is faster).2.3. Control strategyGeneral regulatory guidance on the establishment and justification of a control strategy for the drug product is given in other relevant guidelines. Particular attention should however be paid to the control of critical quality attributes that are required for the control of drug release.Pharmaceutical development should establish the link (qualitative or quantitative) from pharmacokinetic parameters through in vivo drug release to in vitro dissolution rate.In an enhanced pharmaceutical development environment, compliance with the dissolution requirement could be demonstrated by real time release testing (see Guideline on Real Time Release Testing). As the drug release rate may be susceptible to scale-up effects, it is particularly important that the drug release rate prediction algorithm is verified at the commercial scale.2.4. Variations to productsThe supporting data requirements for variations to the marketing authorisation will depend upon the significance of the change, whether or not a Level A IVIVC exists and whether or not the dissolution method/limits is to be changed. If bioavailability/bioequivalence data have not been submitted their absence should always be justified.When a Level A IVIVC has been established and the release specification is not changed, changes may be accepted on the basis of in vitro data, the therapeutic index of the active substance and predictive capability of the IVIVC. In this case, waiver of a bioequivalence study should be based on comparison of the predicted plasma concentration-time profiles and associated pharmacokinetic parameters for the formulations before and after changes, calculated utilising the in vitro data and the validated IVIVC.In general, bioavailability/bioequivalence data are needed for products with an established Level B or C correlation or no IVIVC, unless justification is provided for absence of such data.3. Delayed release dosage forms3.1. General remarksSeveral delayed release dosage forms have been identified by the Ph.Eur.: gastro-resistant capsules, tablets and granules. In this section, specific guidance is provided for gastro-resistant dosage forms. Products based on other principles can also often be classified as delayed release dosage forms, including those designed to release in a specific area of the gastrointestinal tract in response to a specific trigger (e.g. enzymes) or at a specific time after ingestion. Although the principles described herein for the pharmaceutical development, specifications and control strategy are also generally relevant for other delayed release dosage forms, specific guidance for those dosage forms would have to be developed based on the relevant formulation principle and mechanism of release.Note that in addition to the points addressed below, many of the principles discussed above under prolonged release oral dosage forms are also relevant to delayed release dosage forms.3.2. Development pharmaceuticsA summary of the bioavailability studies should be given. The data should include information on pharmacokinetics (AUC0 → t(last), AUC0 →∞, C max, and where appropriate other relevant parameters (e.g. partial AUC); for generic products also the point estimates and 90% confidence intervals), manufacturing sites and dates, batch sizes and numbers, formulations and dissolution results of the batches used.The rationale for the delayed release should be given, e.g. the protection of the gastric mucosa, the protection of the active substance against the influence of acidic gastric medium or intended release of the active substance in a predefined segment of the gastro-intestinal tract for local treatment, etc.The mechanism of release and choice of the excipient(s) responsible for the delayed release should be discussed e.g. targeting release at a given pH, susceptibility to enzymatic attack, erosion with time etc.Pharmaceutical development should establish the (qualitative or quantitative) link from pharmacokinetic parameters through in vivo drug release to in vitro dissolution rate.In principle two different types of formulations can be distinguished for delayed release products with respect to the behaviour in the stomach:•single unit non-disintegrating dosage forms;•disintegrating dosage forms containing multiple units of pellets.The development of single unit non-disintegrating gastro-resistant dosage forms is generally discouraged for gastro-resistant products since their residence time in the stomach is unpredictable and in general longer than disintegrating dosage forms which contain multiple units of pellets. Therefore, such single unit non-disintegrating dosage forms are liable to a higher risk of dose-dumping and/or erratic concentration profiles.If the SmPC requires the co-administration with food or does not exclude the co-administration with food, gastro-resistance should also be tested under conditions representative of fed state. For example, tests should be run at a higher pH (e.g. in the range 3-5) for both single unit non-disintegrating and disintegrating dosage forms with multiple units to determine resistance to release in the fed stomach. Most meals will temporarily buffer the pH in the stomach to 3 or above, so pH 2 would not be a sufficiently challenging test.。
v-Series 1Mpx相机说明书

Update to popular v-Series 1Mpx camera line Smaller, lighter-weight body Most popular signal connections on camera back panelOptional on-camera controlsFor the most current version visit www.vision Subject to change Rev Mar 2011PRELIMINARYPhantom v711Key Features:Custom-designed 1280x800 CMOS sensorExtreme Dynamic Range (EDR): two exposures per frameInternal Mechanical Shutter mechanism for hand-free/remote CSRs (not available on v211)Memory Segmentation: up to 63 segmentsNon-volatile, hot-swappable Phantom CineMag memory magazinesCineMag interface is standard. Not available on v211Range Data Input: embed tracker data into recorded cine file8GB, 16GB or 32GB of internal high-speed memory ISO (ISO 12232 SAT): 7000 mono, 2100 color Pixel Bit-Depth: 8-, 12-bit Gb Ethernetv611 and v711 models support a FAST option that provides frame rates of 1,000,000 fps or more as well as sub-microsecond exposure times (export controlled)Phantom v211, v311, v611, v711Key Benefits:WHEN IT’S TOO FAST TO SEE, AND TOO IMPORTANT NOT TO ®We’ve updated our popular 1 megapixel camera line. While the camera specifications stay the same, you’ll like what we’ve done with camera packaging and control.All models feature a widescreen 1280 x 800 CMOS sensor – 25% wider than most competitive models – allowing you to keep moving targets in-frame longer and see more of the event you are recording. The wide sensor also allows you to get true 1280 x 720 HD images from a 1Mpx camera.With a pixel size of 20 microns and improved quantum efficiency, these cameras have the sensitivity you need for even the most challenging lighting conditions.Minimum exposure times of 1-2 microseconds (depending upon model) eliminate blur and allow you to see the smallest of details.With throughput specifications ranging from 2 gigapixels-per-sec (Gpx/s) to7Gpx/s, there is a model to meet your frame-rate requirements. At 2Gpx/s, the v211 can take over 2000 frames-per-second (fps) at full resolution. A 7Gpx/s camera (the v711) can take over 7000 fps at full resolution (7530 fps, actually!) Top speeds at reduced resolution range from 300,000 fps to 1,400,000 fps depending on camera model.All cameras support both 8- and 12-bit pixel depth. Smaller bit-depth gives you more recording time and smaller files. Greater bit-depth gives you more gray levels and finer detail. With the greater latitude of 12 bits, you can pull more detail out of the image, an essential requirement for most motion analysis applications.Phantom’s high-accuracy timing system means improved frame rate, frame synchronization and exposure accuracy. And a frame-synchronization (F-SYNC) signal is now available on a dedicated BNC connector on the camera connector panel for easier cabling and increased signal integrity.Of course, all camera models offer the Extreme Dynamic Range feature – pioneered by Vision Research. This gives you the ability to get two different exposures within a single frame so areas that would otherwise be overexposed contain image detail. And, with Auto Exposure, the camera adjusts to changing lighting conditions automatically.There is an internal mechanical shutter (not available on the v211) that can cut off all light to the sensor when doing a session-specific black reference (CSR). You can now do remote CSRs through software control without the need to manually cover the lens! With the optional Canon EOS lens mount installed you get remote control over lens aperture and focus, too. This enables complete remote control in environments where you cannot easily access the camera.All models come with 8GB, 16GB or 32GB internal high-speed memory. Segmenting memory allows you to divide this into up to 63 segments so you can take multiple shots back-to-back without the need to download data from the camera.Or, record directly to our Phantom CineMag non-volatile, hot-swappable memory magazines. They mount on the CineMag interface of compatible cameras. Continuously record full resolution cines into a CineMag at up to780 fps. That’s just over 2 minutes into the 128GB CineMag, 4.25 minutes into the 256GB, or 8.5 minutes into the 512GB version. Or, record at even higher speeds into camera RAM, then manually or automatically move your recording to the CineMag. With CineMag storage you get maximum data protection and an ideal storage medium forsecure environments.Throughput:v211 - 2Gpx/s v311 - 3Gpx/s v611 - 6Gpx/s v711 - 7Gpx/sPhantom v311Phantom v611when it’s too fast to see, and too important not to.PRELIMINARY100 Dey RoadWayne, NJ 07470 USA +1.973.696.4500**************************www.visionFocusedSince 1950, Vision Research has been shooting, designing, and manufacturing high-speed cameras. Our single focus is to invent, build, and support the most advanced cameras possible.AMETEK Vision Research’s digital high-speed cameras are subject to the export licensing jurisdiction of the Export Administration Regulations. As a result, the export, transfer, or re-export of these cameras to a country embargoed by the United States is strictly prohibited. Likewise, it is prohibited under the Export Administration Regulations to export, transfer, or re-export AMETEK Vision Research’s digital high-speed cameras to certain buyers and/or end users.Customers are also advised that some models of AMETEK Vision Research’s digital high-speed cameras may require a license from the U.S. Department of Commerce to be: (1) exported from the United States; (2) transferred to a foreign person in the United States; or (3) re-exported to a third country. Interested parties should contact the U.S. Department of Commerce to determine if an export or a re-export license is required for their specific transaction.Additional Features:Size (without lens, CineMag or handle): 11.5 x 5.5 x 5.0 inches (L x W x H); 29.2 x 14 x 12.7 cmWeight (without lens or CineMag): 11.75 lb; 5.33 kg Temperature and Humidity: 0°C - 40°C @ 8% to 80% RH Shock: 30G, half sine wave, 11 ms, 10 times all axes (without CineMag or lens)Vibration: 25G, 5-500 Hz, all axes without CineMagPhantom v211, v311, v611, v711Move the CineMag to a CineStation connected to a PC and view, edit, and save your recordings using the Phantom Camera Control software included with the camera. Keep the recordings in their original raw cine format, or convert them to TIFF, QuickTime, AVI, or other popular formats. Move files from the CineStation to a disk or video recorder via 10Gb Ethernet; 4:4:4 HD-SDI, or Component Video outputs.When using the camera on a tracking mount, elevation and azimuth data can be transferred to the camera and associated with image frames through our unique Range Data interface.View your recordings immediately in a variety of formats either through the HD-SDI ports on the camera, or through the component video port. There are two HD-SDI ports on the camera which can be configured in a variety of ways including 4:4:4 dual-link and simultaneous play/record (on some models).The cameras can be controlled with the feature-rich PCC software, the Phantom RCU, or the new (optional) on-camera controls.Phantom v311。
美国MOTOROLA MPX系列硅压力传感器说明书

美国MOTOROLA压力传感器美国MOTOROLA公司的MPX系列硅压力传感器,主要以气压测量为主,适合用于医疗器械,气体压力控制等领域,输出数字信号。
其测量方式可分为:表压(GP)、绝压(A、AP)、差压(D、DP)型。
在宽温度范围工作时需外加补偿网络和信号调整电路。
具体型号分类而定名称:MPX2010DP 名称:MPX5700DP MPX5700GP 名称:MPX2100AP名称:MPX5500DP 名称:MPX5100AP 名称:MPX5050DP名称:MPX5010DP 名称:MPX4115AP 名称:MPX2200A 名称:MPX2200AP 名称:MPXH6115A6U 名称:MPX4250DP名称:MPX4115A 名称:MPX2202DP 名称:MPX2102AP名称:MPX2053GP 名称:MPXY8300A6U 压力传感器 名称:触力型压力传感器 FSG15N1A 名称:硅压力传感器 MPXH6115A 名称:MPX5700DP 硅压力传感器 名称:MPX53GP 硅压力传感器 名称:压力传感器FPM07 名称:轮胎压力传感器TP015 名称:轮胎压力传感器NPP301名称:Freescale 压力传感器 MPX2010DP商斯达实业传感器与智能控制分公司专门从事各种进口传感器的营销工作,代理多家欧美知名公司的产品。
涉及压力、温度、湿度、电流、液位、磁阻、霍尔、流量、称重、光纤、倾角、扭矩、气体、光电、位移、触力、红外、速度、加速度等多种产品。
广泛应用于航空航天、医疗器械(如血压计)、工业控制、冶金化工、汽车制造、教育科研等领域。
商斯达实业代理的品牌产品主要有:压 力:Kulite、ACSI、Honeywell、Entran、Gems、Dwyer、SSI、Smi、Senstronics、Intersema、Motorola、 NAIS、E+H、Fujikura、Dytran、APM称重测力:Transcell、HBM、Interface、Thamesside、Philips、Entran 温 湿 度:Honeywell、Dwyer流 量:Gems、Dwyer、Honeywell、Folwline、WorldMagnetics 液 位:Honeywell、Siccom、Gems、Dwyer、Kulite、SSI 加 速 度:Entran、Silicondesigns、Dytran 压力开关:ACSI、Gems、Dwyer、台湾矽微航空器材:TexTech 隔音材料、Honeywell 薄膜加热片、DigirayX 射线探伤仪 仪 表:Honeywell、Transcell、东辉、上润、AD、东崎商斯达实业 除代理上述产品外,还有几条传感器生产线,一条压力传感器组装线,可为用户提供各种用途的、特殊要求的配套产品。
教你玩转MP3视频(MTV、AMV、MPV、SMV格式的概念与区别

教你玩转MP3视频(MTV、AMV、MPV、SMV格式的概念与区别由于各种原因,目前视频MP3播放器的存储容量大多在512MB 以下,加上屏幕小、电池续航时间有限以及支持的视频解码技术存在版权等因素的限制,所以视频MP3对目前主流的视频格式如RMVB、WMV和***I等视频格式大多不支持,厂商一般都为自己的视频MP3播放器开发了体积小、适于小屏幕MP3播放的特殊视频格式。
现在视频MP3播放器专用的视频格式还没有统一的标准,在视频MP3播放器的发展过程中,一些公司研发出了多种可用于视频MP3播放器播放的视频文件格式。
就目前而言,一款视频MP3播放器大多只支持一两种视频格式,再加上各种视频格式文件所能达到的文件大小、画质各不相同,所以有必要先来了解一下这些视频格式的基本知识。
这对于购买适合自已的视频MP3播放器有很大帮助。
下面就来看看目前不同视频MP3播放器支持的主流视频格式。
1.MTV格式:这是视频MP3较早支持播放的视频格式,也是目前最常见的视频格式。
但其缺点也很明显,文件体积大,一分钟的MTV 格式文件约占12MB的容量,而且画面质量也欠佳。
MTV视频格式正逐渐被表现更好的AMV视频格式所取代。
2.AMV格式:相对于MTV格式来说,AMV视频格式比MTV视频格式有着更好的压缩比例以及画面质量。
通过AMV转换工具转换出来的影音文件一分钟的容量约为1.8MB。
也就是说,256MB的MP3播放器可存放130分钟的AMV格式的电影,这样用户就可将一部电影从头看到尾,真正让视频MP3播放器成为“电影播放机”,所以AMV视频格式正被广泛应用。
3.MPV格式:MPV是由柯达、HP、LG、奥林巴斯、飞利浦、三星和索尼公司等多家消费电子厂商为消费类电子产品和数字成像技术(音频、图像和视频)制定的一个标准。
MPV格式文件是由音频流和运动图像构成的多媒体文件,音频流的格式仍然选用了当今流行的 MP3 音频编码格式。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
MPX12Rev 11, 07/2009Freescale Semiconductor © Freescale Semiconductor, Inc., 2007-2009. All rights reserved.10 kPa Uncompensated Silicon Pressure SensorsThe MPX12 series silicon piezoresistive pressure sensors provide a very accurate and linear voltage output, directly proportional to the applied pressure. This standard, low cost, uncompensated sensor permits manufacturers to design and add their own external temperaturecompensating and signal conditioning networks. Compensation techniques are simplified because of the predictability of Freescale's single element strain gauge design.Features•Low Cost•Patented Silicon Shear Stress Strain Gauge Design •Ratiometric to Supply Voltage•Easy to Use Chip Carrier Package Options •Gauge Options•Durable Epoxy PackageORDERING INFORMATIONDevice NamePackage Options Case No.# of PortsPressure Type Device Marking None SingleDualGaugeDifferentialAbsoluteUnibody Package (MPX12 Series)MPX12D Tray 344••MPX12D MPX12DP Tray 344C ••MPX12DP MPX12GP Tray 344B ••MPX12GPSmall Outline Package (MPXV12 Series)MPXV12DP Tray 1351••MPXV12DP MPXV12GP Tray 1369••MPXV12GP MPXV12GW6U Rail 1735••MPXV12GW MPXV12GW7U Rail 1560••MPXV12GW MPAK Package (MPXM12 Series)MPXM12GS Rail 1320A ••MPXM12GS MPXM12GST1Tape & Reel1320A••MPXM12GSMPX120 to 10 kPa (0 to 1.45 psi)55 mV Full Scale Span(Typical)SeriesApplication Examples•Air Movement Control•Environmental Control Systems •Level Indicators •Leak Detection•Medical Instrumentation •Industrial Controls•Pneumatic Control Systems •RoboticsMPX12SensorsSMALL OUTLINE PACKAGESMPX12GPCASE 344B-01MPX12D CASE 344-15MPX12DP CASE 344C-01UNIBODY PACKAGESMPXV12DP CASE 1351-01MPXV12GP CASE 1369-01MPXV12GW7U CASE 1560-02MPXV12GW6U CASE 1735-02MPAK PACKAGEMPXM12GS/GST1CASE 1320A-02MPX12SensorsOperating CharacteristicsTable 1. Operating Characteristics (V S = 3.0 Vdc, T A = 25°C unless otherwise noted, P1 > P2)CharacteristicSymbol Min Typ Max Unit Differential Pressure Range (1)1. 1.0 kPa (kiloPascal) equals 0.145 psi.P OP 0—10kPa Supply Voltage (2)2.Device is ratiometric within this specified excitation range. Operating the device above the specified excitation range may induce additional error due to device self-heating. V S — 3.0 6.0Vdc Supply Current I o — 6.0—mAdc Full Scale Span (3)3.Full Scale Span (V FSS ) is defined as the algebraic difference between the output voltage at full rated pressure and the output voltage at the minimum related pressure.V FSS 455570mV Offset (4)4.Offset (V OFF ) is defined as the output voltage at the minimum rated pressure.V off 02035mV Sensitivity ΔV/ΔP — 5.5—mV/kPa Linearity—–0.5— 5.0%V FSS Pressure Hysteresis (6) (0 to 10 kPa)——±0.1—%V FSS Temperature Hysteresis (–40°C to +125°C)——±0.5—%V FSS Temperature Coefficient of Full Scale Span TCV FSS –0.22—–0.16%V FSS /°C Temperature Coefficient of Offset TCV off —±15—μV/°C Temperature Coefficient of Resistance TCR 0.21—0.27%Z in /°C Input Impedance Z in 400—550ΩOutput ImpedanceZ out 750—1250ΩResponse Time (5) (10% to 90%)5.Response Time is defined as the time form the incremental change in the output to go from 10% to 90% of its final value when subjected to a specified step change in pressure. t R — 1.0—ms Warm-Up Time (6)6.Warm-up Time is defined as the time required for the product to meet the specified output voltage after the pressure is stabilized.——20—ms Offset Stability (7)7.Offset stability is the product’s output deviation when subjected to 1000 hours of Pulsed Pressure, Temperature Cycling with Bias Test.——±0.5—%V FSSMPX12SensorsMaximum RatingsFigure 1 shows a block diagram of the internal circuitry integrated on a pressure sensor chip.Figure 1. Uncompensated Pressure Sensor SchematicVoltage Output versus Applied Differential PressureThe output voltage of the differential or gauge sensor increases with increasing pressure applied to the pressure side (P1) relative to the vacuum side (P2). Similarly, outputvoltage increases as increasing vacuum is applied to the vacuum side (P2) relative to the pressure side (P1).Table 2. Maximum Ratings (1)1.Exposure beyond the specified limits may cause permanent damage or degradation to the device.RatingSymbol Value Unit Maximum Pressure (P1 > P2)P MAX 75kPa Burst Pressure (P1 > P2)P BURST 100kPa Storage Temperature T STG –40 to +125°C Operating TemperatureT A–40 to +125°C1234GND+V OUT–V OUT+V SSensing ElementMPX12SensorsTemperature CompensationFigure 2 shows the typical output characteristics of the MPX12 series over temperature.Because this strain gauge is an integral part of the silicon diaphragm, there are no temperature effects due todifferences in the thermal expansion of the strain gauge and the diaphragm, as are often encountered in bonded strain gauge pressure sensors. However, the properties of the strain gauge itself are temperature dependent, requiring that the device be temperature compensated if it is to be used over an extensive temperature range.Temperature compensation and offset calibration can be achieved rather simply with additional resistive components, or by designing your system using the MPX2010D series sensor.Several approaches to external temperaturecompensation over both –40 to +125°C and 0 to +80°C ranges are presented in Applications Note AN840.LINEARITYLinearity refers to how well a transducer's output follows the equation: V OUT = V OFF + sensitivity x P over the operating pressure range (Figure 3). There are two basic methods for calculating nonlinearity: (1) end point straight line fit or (2) a least squares best line fit. While a least squares fit gives the “best case” linearity error (lower numerical value), the calculations required are burdensome.Conversely, an end point fit will give the “worst case” error (often more desirable in error budget calculations) and the calculations are more straightforward for the user.Freescale’s specified pressure sensor linearities are based on the end point straight line method measured at the midrange pressure.Figure 2. Output vs. Pressure DifferentialFigure 3. Linearity Specification ComparisonPressure DifferentialO u t p u t (m V d c )8070605040302010000.32.00.64.00.96.01.28.0101.5PSI kPaSpan Range (Typ)Offset (Typ)V S = 3 V DC P1 > P2-40°C+25°C+125°CO u t p u t (m V d c )70605040302010080MPX12SensorsFigure 4. Cross-Sectional Diagram (not to scale)Figure 4 illustrates the differential/gauge die. A gel isolates the die surface and wire bonds from the environment, while allowing the pressure signal to be transmitted to the silicon diaphragm.Operating characteristics, internal reliability andqualification tests are based on use of dry clean air as thepressure media. Media other than dry clean air may have adverse effects on sensor performance and long termreliability. Contact the factory for information regarding media compatibility in your application.PRESSURE (P1)/VACUUM (P2) SIDE IDENTIFICATION TABLEFreescale designates the two sides of the pressure sensor as the Pressure (P1) side and the Vacuum (P2) side. The Pressure (P1) side is the side containing gel which isolates the die from the environment. Freescale’s MPx12 series is designed to operate with positive differential pressure applied, P1 > P2.The Pressure (P1) side may be identified by using the following tableGel Die CoatWire BondDieP1Stainless Steel CapThermoplasticCaseDie BondDifferential SensingElementP2Lead FramePart Number Case TypePressure (P1) SideIdentifier MPX12D 344Stainless Steel Cap MPX12DP 344C Side with Part Marking MPX12GP 344B Side with Port Attached MPXV12DP 1351Side with Part MarkingMPXV12GP 1369Side with Port MPXV12GW6U 1735 Side with Port MPXV12GW7U 1560 Side with Port MPXM12GS/GST11320ASide with Port AttachedISSUE BUNIBODY PACKAGEMPX12 SensorsMPX12SensorsCASE 1351-01ISSUE ASMALL OUTLINE PACKAGEMPX12 SensorsISSUE ASMALL OUTLINE PACKAGEMPX12SensorsISSUE OSMALL OUTLINE PACKAGEMPX12 SensorsISSUE BSMALL OUTLINE PACKAGEMPX12SensorsISSUE BSMALL OUTLINE PACKAGEMPX12 SensorsISSUE BSMALL OUTLINE PACKAGEMPX12SensorsISSUE DSMALL OUTLINE PACKAGEMPX12 SensorsISSUE DSMALL OUTLINE PACKAGEMPX12SensorsISSUE DSMALL OUTLINE PACKAGEMPX12 SensorsISSUE AMPAK PACKAGEMPX12SensorsCASE 1320A-02ISSUE AMPAK PACKAGEMPX12 SensorsMPX12Rev. 11How to Reach Us:Home Page:Web Support:/support USA/Europe or Locations Not Listed:Freescale Semiconductor, Inc.Technical Information Center, EL5162100 East Elliot Road Tempe, Arizona 852841-800-521-6274 or +/supportEurope, Middle East, and Africa:Freescale Halbleiter Deutschland GmbH Technical Information Center Schatzbogen 781829 Muenchen, Germany +44 1296 380 456 (English)+46 8 52200080 (English)+49 89 92103 559 (German)+33 1 69 35 48 48 (French)/supportJapan:Freescale Semiconductor Japan Ltd.Headquarters ARCO Tower 15F1-8-1, Shimo-Meguro, Meguro-ku,Tokyo 153-0064Japan0120 191014 or +81 3 5437 9125support.japan@ Asia/Pacific:Freescale Semiconductor China Ltd.Exchange Building 23F No. 118 Jianguo Road Chaoyang District Beijing 100022 China+86 010 5879 8000@For Literature Requests Only:Freescale Semiconductor Literature Distribution Center 1-800-441-2447 or +1-303-675-2140Fax: +1-303-675-2150LDCForFreescaleSemiconductor@Information in this document is provided solely to enable system and software implementers to use Freescale Semiconductor products. There are no express or implied copyright licenses granted hereunder to design or fabricate any integrated circuits or integrated circuits based on the information in this document.Freescale Semiconductor reserves the right to make changes without further notice to any products herein. Freescale Semiconductor makes no warranty, representation or guarantee regarding the suitability of its products for any particular purpose, nor does Freescale Semiconductor assume any liability arising out of the application or use of any product or circuit, and specifically disclaims any and all liability, including without limitation consequential or incidental damages. “Typical” parameters that may beprovided in Freescale Semiconductor data sheets and/or specifications can and do vary in different applications and actual performance may vary over time. All operating parameters, including “Typicals”, must be validated for each customer application by customer’s technical experts. Freescale Semiconductor does not convey any license under its patent rights nor the rights of others. Freescale Semiconductor products are not designed, intended, or authorized for use as components in systems intended for surgical implant into the body, or other applications intended to support or sustain life, or for any other application in which the failure of the Freescale Semiconductor product could create a situation where personal injury or death may occur. Should Buyer purchase or use Freescale Semiconductor products for any such unintended orunauthorized application, Buyer shall indemnify and hold Freescale Semiconductor and its officers, employees, subsidiaries, affiliates, and distributors harmless against all claims, costs, damages, and expenses, and reasonable attorney fees arising out of, directly or indirectly, any claim of personal injury or death associated with such unintended or unauthorized use, even if such claim alleges that Freescale Semiconductor was negligent regarding the design or manufacture of the part. Freescale™ and the Freescale logo are trademarks of Freescale Semiconductor, Inc.All other product or service names are the property of their respective owners.© Freescale Semiconductor, Inc. 2009. All rights reserved.。