amber atom types
amber动力学常用参数说明

amber动力学常用参数说明个人日记2009-05-08 19:32:18 阅读130 评论1 字号:大中小订阅IMIN Flag to run minimization=0 No minimization (only do molecular dynamics;default)= 1 Perform minimization (and no molecular dynamics)=5 Read in a trajectory for analysis.NTX Option to read the initial coordinates, velocities and box size from the "inpcrd" file. The options 1-2 must be used when one is starting from minimized or model-built coordinates. If an MD restrt file is used as inpcrd, thenoptions 4-7 may be used.= 1 X is read formatted with no initial velocity information (default)= 2 X is read unformatted with no initial velocity information= 4 X and V are read unformatted.= 5 X and V are read formatted; box information will be read if ntb>0. The velocity information will only be used if irest=1.= 6 X, V and BOX(1..3) are read unformatted; in other respects, this is the same as option "5".=7 Same as option "5"; only included for backward compatibility with earlier versions of Amber. IREST Flag to restart the run.= 0 Noeffect (default)= 1 restart calculation. Requires velocities in coordinate input file, so you also may need to reset NTX if restarting MD.NTRX Format of the Cartesian coordinates for restraint from file "refc". Note: the program expects file "refc" to contain coordinates for all the atoms in the system. A subset for the actual restraints is selected by restraintmask in the controlnamelist.= 0 Unformatted (binary) form= 1 Formatted (ascii, default) formNTPR Every NTPR steps energy information will be printed in human-readable form to files "mdout" and "mdinfo". "mdinfo" is closed and reopened each time, so it always contains the most recent energy and temperature. Default 50.NTWR Every NTWR steps during dynamics, the "restrt" file will be written, ensuring that recovery from a crash will not be so painful. In any case, restrt is written ev ery NSTLIM steps for both dynamics and minimization calculations. If NTWR<0, a unique copy of the file, restrt_nstep, is written every abs(NTWR) steps. This option is useful if for example one wants to run free energy perturbations from multiple starting points or save a series of restrt files for minimization. Default 500.NTF Force evaluation. Note: If SHAKE is used (see NTC), it is not necessary to calculate forces for the constrained bonds.= 1 complete interaction is calculated (default)= 2 bond interactions involving H-atoms omitted (use with NTC=2)= 3 all the bond interactions are omitted (use with NTC=3)= 4 angle involving H-atoms and all bonds are omitted= 5 all bond and angle interactions are omitted= 6 dihedrals involving H-atoms and all bonds and all angle interactions are omitted= 7 all bond, angle and dihedral interactions are omitted= 8 all bond, angle, dihedral and non-bonded interactions are omittedNTB Periodic boundary. If NTB .EQ. 0 then a boundary is NOT applied regardless of any boundary condition information in the topology file. The value of NTB specifies whether constant volume or constant pressure dynamics will be used. Options for constant pressure are described in a separate section below.= 0 no periodicity is applied and PME is off= 1 constant volume (default)= 2 constant pressureIf NTB .NE. 0, there must be a periodic boundary in the topology file. Constant pressure is not used in minimization (IMIN=1, above). For a periodic system, constant pressure is the only way to equilibrate densityif the starting state is not correct. For example, the solvent packing scheme used in LEaP can result in a net void when solvent molecules are subtracted which can aggregate into "vacuum bubbles" in a constant volume run. Another potential problem are small gaps at the edges of the box. The upshot is that almost every system needs to be equilibrated at constant pressure (ntb=2, ntp>0) to get to a proper density. But be sure to equilibrate first (at constant volume) to something close to the final temperature, before turning on constant pressure.CUT This is used to specify the nonbonded cutoff, in Angstroms. For PME, the cutoff is used to limit direct space sum, and the default value of 8.0is usually a good value. When igb>0, the cutoff is used to truncate nonbonded pairs (on an atom-by-atom basis); here a larger value than the default is generally required. A separate parameter (RGBMAX) controls the maximum distance between atom pairs that will be considered in carrying out the pairwise summation involved in calculating the effective Born radii, see the generalized Born section below.IBELLY Flag for belly type dynamics.= 0 No belly run (default).= 1 Belly run. A subset of the atoms in the system will be allowed to move, and the coordinates of the rest will be frozen. The moving atoms are specified bellymask. This option is not available when igb>0. Note also that this option does not provide any significant speed advantage, and is maintained primarily for backwards compatibilitywith older version of Amber. Most applications should use the ntr variable instead to restrain parts of the system to stay close to some initial configuration.NTR Flag for restraining specified atoms in Cartesian space using a harmonic potential. The restrained atoms are determined by the restraintmask string. The force constant is given by restraint_wt. The coordinates are read in "restrt" format from the "refc" file (see NTRX, above). = 0 No position restraints (default) = 1 MD with restraint of specified atomsMAXCYC The maximum number of cycles of minimization. Default 1.NCYC If NTMIN is 1 then the method of minimization will be switched from steepest descent to conjugate gradient after NCYC cycles. Default 10.NSTLIM Number of MD-steps to be performed. Default 1.TEMP0Reference temperature at which the system is to be kept, if ntt > 0. Note that for temperatures above 300K, the step size should be reduced since increased distance traveled between evaluations can lead to SHAKE and other problems. Default 300.TEMPI Initial temperature. For the initial dynamics run, (NTX .lt. 3) the velocities are assignedfrom a Maxwellian distribution at TEMPI K. If TEMPI = 0.0, the velocities will be calculated from the forces instead. TEMPI has no effect if NTX .gt. 3. Default 0.0.NTT Switch for temperature scaling. Note that setting ntt=0 corresponds to the microcanonical (NVE) ensemble (which should approach the canonical one for large numbers of degrees of freedom). Some aspects of the "weak-coupling ensemble" (ntt=1) have been examined, and roughly interpolate between the microcanonical and canonical ensembles [63]. The ntt=2 and 3 options correspond to the canonical (constant T) ensemble. The ntt=4 option is included for historical reasons, but does not correspond to any of the traditionalensembles.= 0 Constant total energy classical dynamics (assuming that ntb<2, as should probably always be the case when ntt=0).= 1 Constant temperature, using the weak-coupling algorithm [64]. A single scaling factor is used for all atoms. Note that this algorithm just ensures that the total kinetic energy is appropriate for the desired temperature; it does nothing to ensure that the temperature is even over all parts of the molecule. Atomic collisions should serve to ensure an even temperature distribution, but this is not guaranteed, and can be a particular problem for generalized Born simulations, where there are no collisions with solvent. Other temperature coupling options (especially ntt=3) should probably be used for generalized Born simulations.= 2 Andersen temperature coupling scheme [65], in which imaginary "collisions" randomize the velocities to a distribution corresponding to temp0 every vrand steps. Note that in between these "massive collisions",the dynamics is Newtonian. Hence, time correlation functions (etc.) can be computed in these sections, and the results averaged over an initial canonical distribution. Note also that too high a collision rate (too small a value of vrand) will slow down the speed at which the molecules explore configuration space, whereas too low a rate means that the canonical distribution of energies will be sampled slowly. A discussion of this rate is given by Andersen [66].= 3 Use Langevin dynamics with the collision frequency γ given by gamma_ln, discussed below. Note that when γ has its default value of zero, this is the same as setting ntt = 0.GAMMA_LN The collision frequency γ , in ps-1, when ntt = 3. A simple Leapfrog integrator is used to propagate the dynamics, with the kinetic energy adjusted to be correct for the harmonic oscillator case [67,68]. Note that it is not necessary that γ approximate the physical collision frequency. In fact, it is often advantageous, in terms of sampling or stability of integration, to use much smaller values. Default is 0NTP Flag for constant pressure dynamics. This option should be set to 1 or 2 when Constant Pressure periodic boundary conditions are used (NTB = 2).= 0 Used with NTB not = 2 (default); no pressure scaling= 1 md with isotropic position scaling= 2 md with anisotropic (x-,y-,z-) pressure scaling: this should only be used with orthogonal boxes (i.e. with all angles set to 90). Anisotropic scaling is primarily intended for non-isotropic systems, such as membrane simulations, where the surface tensions are different in different directions; it is generally not appropriate for solutes dissolved in water.NTC Flag for SHAKE to perform bond length constraints [70]. (See also NTF in the Potential function section. In particular, typically NTF = NTC.) The SHAKE option should be used for most MD calculations. The size of the MDtimestep is determined by the fastest motions in the system. SHAKE removes the bond stretching freedom, which is the fastest motion, and consequently allows a larger timestep to be used. For water models, a special "three-point" algorithm is used [71]. Consequently, to employ TIP3P set NTF = NTC = 2. Since SHAKE is an algorithm based on dynamics, the minimizer is not aware of what SHAKE is doing; for this reason, minimizations generally should be carried out without SHAKE. One exception is short minimizations whose purpose is to remove bad contacts before dynamics can begin.= 1 SHAKE is not performed (default)= 2 bonds involving hydrogen are constrained= 3 all bonds are constrained (not available for parallel runs in sander)。
分子力学方法介绍

• 分子力学的势能函数表达方程很简单,其计算速度很快 约是半 经验量子化学计算方法速度的1000倍 ,能够用于生物大分子体 系的计算,
• 对于力场参数成熟的分子力学方法,已经可以达到很高的计算 精度,
2
• 目前,分子力学是模拟蛋白质、核酸等生物大分子结构和 性质以及配体和受体相互作用的常用方法,在生命科学领 域得到了广泛的应用,随着分子图形学的不断发展,分子 力学已经广泛应用于分子模型设计,当今优秀的分子设计 程序都将分子力学作为初始模型优化的主要方法,分子力 学和分子图形学已经充分地糅合在分子设计中,分子模型 的构建也是分子力学为主,分子力学方法是计算机辅助分 子设计中常用的方法,特别是在药物设计中,已经离不开 分子力学计算和模拟方法,
21
22
Functional Form
v()NVn(1cons()]
n0 2
: Torsional angle. n (multiplicity): Number of minima in a 360ºcycle. Vn: Correlates with the barrier height. (phase factor): Determines where the torsion passes through its minimum value.
Amber软件中动力学模拟的步骤

Molecular Dynamics simulation——从能量最小化到实际模拟 1 基本流程图1)概述前面我们已经得到了Amber 用来动力学模拟的prmtop 和inpcrd 文件,它们分别是参数文件和坐标文件。
我们先从一条命令说起来解释Amber 是如何做动力学模拟的: sander –O –i mdin –o mdout –p prmtop –c inpcrd –r rst –x mdcrd 动力学过程是一个连续地解牛顿运动方程的过程:上一个牛顿方程结束时,蛋白质中各原子的位置和速度保留给下一个牛顿方程,惟一改变的是原子的加速度,它会根据各种势能函数重新计算(势能随原子坐标改变:E=f (r,…))。
只不过每个牛顿方程的时间很短,短到fs (10-15s )级,Amber 软件提供的sander 主程序可以用来自动地做这样的数值计算。
它需要参数文件(prmtop )、坐标文件(inpcrd )、sander 程序配置文件(mdin )来启动运行,我们已经有了前两种文件,本节内容最主要的就是讲解如何配置我们需要的动力学模拟。
sander 程序运行过程中会输出临时文件(rst )保存坐标和速度,还有轨迹文件(mdcrd )。
2)动力学过程从基本流程图可以知道,一般的动力学过程也就可以分为三步:能量最小化(minimization)、体系平衡(equilibrium)、实际动力学模拟。
由于我们进行的初始结构来自晶体结构或同源建模,所以在分子内部存在着一定的结构张力,能量最小化就是真正的动力学之前释放这些张力,如果没有这个步骤,在动力学模拟开始之后,整个体系可能会因此变形、散架。
另外,由于动力学模拟的是真实的生物体环境,因此必须使研究对象升温升压到临界值,体系达到平衡,才能做实际的动力学模拟。
2 各流程输入文件要通过Amber软件做动力学模拟,需要明白如何去配置上述过程中的每一步。
一般来说就是指定一些键/值对。
分子模拟

AB
)2
+
k3AB
(ΔR
AB
)3
+
k
AB 4
(ΔR
AB
)4
+
...
{ 更多的参数 { 在若干情形下,极限性质是不对的 (如
3rd , 5th 展开情况…) { 优化时考虑要注意(长距离能量的截断 )
键伸缩能
z Morse势
Estr (ΔR AB ) = D[1 − eαΔR ]2
α = k / 2D
微观性质
uij
势能
r
En
=
n2h2 8ml 2
动能
EJ = hcBJ (J +1)
Eν
=
(v +
1 )hcv~ 2
分子特性
统计热力学
宏观性质
T,P U,H,A,G,S
μ,Cp,…
热力学性质
分子模拟
量子化学 实验数据
力场
分子力学 分子动力学 模特卡罗模拟
Force Field
力场
Typical I.R data
θ0
109.47 109.47 109.47 117.2 121.4 122.5
kθ (kcal mol-1 deg-1) 9.9×10-3 7.9×10-3 7.0×10-3 9.9×10-3 1.21×10-2 1.01×10-2
二面角扭转能
z A-B-C-D原子序中B-C键的角旋 转
z 与伸缩能和弯曲能间的差别
Force fields are empirical There is no “correct” form of a force field. Force fields are evaluated based solely on their performance.
英文版原子物理课件

1.1 Introduction
The origins of atomic physics :quantum mechanics Bohr model of the H This introductory chapter surveys some of the early ideas: Spectrum of atomic H and Bohr Theory Einstein's treatment of interaction of atom with light the Zeeman effect Rutherford scattering And so on
Shanxi University Atomic Physics
1.2 Spectrum of atomic hydrogen_3
Wavenumbers may seem rather old-fashioned but they are very useful in atomic physics
the characteristic spectrum for atoms is composed of discrete lines that are the ‘fingerprint' of the element.
In 1888, the Swedish professor J. Rydberg found that the spectral lines in hydrogen obey the following mathematical formula:
Shanxi University Atomic Physics
Lyman series: n’ = 2; 3; 4; … n = 1. Balmer (n = 2), Paschen series: (n = 3), Brackett (n = 4) and Pfund (n = 5)
matlab vmd程序

matlab vmd程序VMD (Visual Molecular Dynamics) 是一种用于分子动力学模拟和分子可视化的软件。
Matlab 并不是 VMD 的编程语言,但你可以使用 Matlab 来编写一些与 VMD 相关的程序。
下面是一个简单的例子,展示了如何使用 Matlab 来调用 VMD 进行分子可视化:```matlab% 创建一个 PDB 文件,用于输入到 VMDpdbFile = 'molecule.pdb';atomTypes = {'C', 'H', 'O', 'N'};atomCoords = [0, 0, 0; 1, 0, 0; 0, 1, 0; 0, 0, 1];atomNames = {'ATOM 1', 'ATOM 2', 'ATOM 3', 'ATOM 4'}; writepdb(pdbFile, atomTypes, atomCoords, atomNames);% 调用 VMD 进行分子可视化vmdCommand = 'vmd -pdb molecule.pdb';system(vmdCommand);```在上面的代码中,首先创建了一个名为 `molecule.pdb` 的 PDB 文件,其中包含了一些原子的类型、坐标和名称。
然后使用`vmd -pdb molecule.pdb` 命令调用 VMD 进行分子可视化。
你可以将上面的代码保存为 Matlab 脚本,并在 Matlab 中运行它。
请确保你已经在计算机上安装了 VMD 软件,并将 `vmd` 命令添加到系统的 PATH 环境变量中,以便能够在命令行中正常调用 VMD。
请注意,上面的例子只是一个简单的示例,你可以根据自己的需求和具体的分子模拟任务,编写更复杂的 Matlab 程序与VMD 进行交互。
gromacs 中文 索引文件

Gromacs的索引文件,即index文件Gromacs的索引文件,即index文件,由make_ndx程序生成,文件后缀为.ndx。
索引文件是gromacs最重要的概念文件,使用它可以在模拟过程中为所欲为。
举一个简单的例子,比如想详细了解HIV整合酶切割DNA的反应机理,使用量子力学模拟反应位点的反应过程,而分子其他部位使用一般分子动力学模拟。
于是我们就面临一个对模拟系统进行分割定义的问题,在gromacs中,就要用到索引文件。
基本的思路是这样的,在索引文件中,定义一个独立的组,这个组包括反应位点处所有原子。
在模拟的.mdp文件中,对这个组定义量子力学模拟,事情就是这么简单。
对蛋白进行量子力学模拟时,一般使用洋葱模型。
所谓洋葱模型,就是对反应位点使用量子机制,在反应位点一定的半径内,使用半量子力学机制,然后分子部分使用分子机制。
那么索引文件就定义一个使用量子力学的组,把需要引进量子机制的原子都放到这个组中;再定义一个半量子机制的组,同时放进需要半量子力学机制模拟的原子,再在.mdp文件中独自定义即可。
再举一个例子,比如说在进行SMD(Steered MolecularDynamics,这个我一直没有想到或者找到切恰的中文翻译方法,或许可以叫做牵引分子动力学??别扭!!)中,要对蛋白莫一个原子或者残基作用力,那么可以建立一个索引文件,在该文件中定义一个组,把要施力的残基或者原子放到该组中。
然后在.ppa文件中使用该组就行了。
如果我还没有说明白,那么看看gromacs的参考文件吧。
如果还是不明白,可以来找我,我免费培训。
^_^索引文件使用make_ndx命令产生,"make_ndx -h"可以看到全部的参数。
运行make_ndx 后,可以使用" r "命令选择残基," a "命令选择原子," name 命令多组进行改名。
可以使用" | "表示或运算," & "表示与运算。
Amber介绍

Basis SANDER Input
More SANDER
Analysis Programs
Ptraj
A program to process/analyse 3-D coordinates/trajectories outputted from the AMBER programs. Includes
Parameter information includes:
force constants necessary to describe the bond energy, angle energy, torsion energy, nonbonded interactions (van der Waals and electrostatics) other parameters for setting up the energy calculations (GB radii, FEP parameter sets)
Why AMBER?
One of the most widely used program for biomolecular studies, therefore an extensive user base Efficient parallel scaling implementation A wide variety of approaches suitable for different applications A completed suite of simulation tools
Preparatory Programs (LEaP)
LEaP
Includes a text-based interface – tleap and a graphical user interface – xleap
ASTM A376-02

Designation:A376/A376M–02a Used in USDOE-NE standards Standard Specification forSeamless Austenitic Steel Pipe for High-TemperatureCentral-Station Service1This standard is issued under thefixed designation A376/A376M;the number immediately following the designation indicates the yearof original adoption or,in the case of revision,the year of last revision.A number in parentheses indicates the year of last reapproval.A superscript epsilon(e)indicates an editorial change since the last revision or reapproval.1.Scope*1.1This specification2covers seamless austenitic steel pipe intended for high-temperature central-station service.Among the grades covered arefive H grades and two nitrogen grades (304N and316N)that are specifically intended for high-temperature service.1.2Optional supplementary requirements(S1through S10) are provided.These supplementary requirements specify addi-tional tests that will be made only when stated in the order, together with the number of such tests required.1.3Grades TP321and TP321H have lower strength require-ments for nominal wall thicknesses greater than3⁄8in.[9.5 mm].1.4The values stated in either inch-pound units or SI units are to be regarded separately as standard.Within the text,the SI units are shown in brackets.The values stated in each system are not exact equivalents;therefore,each system must be used independently of the bining values from the two systems may result in nonconformance with the specifi-cation.The inch-pound units shall apply unless the“M”designation of this specification is specified in the order.N OTE1—The dimensionless designator NPS(nominal pipe size)has been substituted in this standard for such traditional terms as“nominal diameter,”“size,”and“nominal size.”2.Referenced Documents2.1ASTM Standards:A262Practices for Detecting Susceptibility to Intergranu-lar Attack in Austenitic Stainless Steels3A941Terminology Relating to Steel,Stainless Steel,Re-lated Alloys,and Ferroalloys4A999/A999M Specification for General Requirements for Alloy and Stainless Steel Pipe4E112Test Methods for Determining Average Grain Size5 E213Practice for Ultrasonic Examination of Metal Pipe and Tubing6E381Method of Macroetch Testing Steel Bars,Billets, Blooms,and Forgings5E426Practice for Electromagnetic(Eddy-Current)Exami-nation of Seamless and Welded Tubular Products,Austen-itic Stainless Steel,and Similar Alloys62.2ASME Boiler and Pressure Vessel Code:Section IX Welding Qualifications72.3Other Standards:SNT-TC-1A Personnel Qualification and Certification in Nondestructive Testing83.Terminology3.1Definitions—For definitions of terms used in this speci-fication,refer to Terminology A941.4.Ordering Information4.1Orders for material to this specification should include the following,as required to describe the desired material adequately:4.1.1Quantity(feet,centimetres,or number of lengths), 4.1.2Name of material(seamless austenitic steel pipe), 4.1.3Grade(Table1),4.1.4Size(nominal size,or outside diameter and schedule number or average wall thickness),4.1.5Lengths(specific or random),(Permissible Variations in Length Section of Specification A999/A999M),4.1.6Endfinish(Ends Section of Specification A999/ A999M),4.1.7Optional requirements(Section9)(see Hydrostatic Test Requirements Section and the Permissible Variation in Weight for Seamless Pipe Section for weighing individual lengths,of Specification A999/A999M),(see10.6,repairing by welding;14.3,die stamping),1This specification is under the jurisdiction of ASTM Committee A01on Steel, Stainless Steel and Related Alloys and is the direct responsibility of Subcommittee A01.10on Stainless and Alloy Steel Tubular Products.Current edition approved Sept.10,2002.Published October2002.Originally published as A376–st previous edition A376/A376M–02.2For ASME Boiler and Pressure Vessel Code applications see related Specifi-cation SA-376in Section II of that Code.3Annual Book of ASTM Standards,V ol01.03.4Annual Book of ASTM Standards,V ol01.01.5Annual Book of ASTM Standards,V ol03.01.6Annual Book of ASTM Standards,V ol03.03.7Available from American Society of Mechanical Engineers(ASME Interna-tional),Three Park Ave.,New York,NY10016-5990.8Available from the American Society for Nondestructive Testing,P.O.Box 28518,1711Arlingate Ln.,Columbus,OH43228-0518.1*A Summary of Changes section appears at the end of this standard. Copyright©ASTM International,100Barr Harbor Drive,PO Box C700,West Conshohocken,PA19428-2959,United States.4.1.8Test report required (Certification Section of Specifi-cation A 999/A 999M),4.1.9Specification designation,and4.1.10Special requirements or any supplementary require-ments selected,or both.5.General Requirements5.1Material furnished to this specification shall conform to the applicable requirements of the current edition of Specifi-cation A 999/A 999M unless otherwise provided herein.6.Materials and Manufacture6.1Manufacture —At the manufacturer’s option,pipe may be either hot finished or cold finished,with a suitable finishing treatment,where necessary.6.2Heat Treatment :6.2.1All pipe shall be furnished in the heat-treated condi-tion unless the order specifically states that no final heat treatment shall be applied.When the order is furnished without final heat treatment,each pipe shall be stenciled “HT-O.”6.2.2As an alternate to final heat treatment in a continuous furnace or batch-type furnace,immediately following hot forming while the temperature of the pipes is not less than the specified minimum solution treatment temperature,pipes may be individually quenched in water or rapidly cooled by other means.6.2.3Grades TP304,TP304N,TP304LN,TP316,TP316N,TP316LN,TP321,TP347,TP348,16-8-2H,S 31725,and S 31726—Unless otherwise stated in the order,heat treatment shall consist of heating to a minimum temperature of 1900°F [1040°C]and quenching in water or rapidly cooling by other means. 6.2.3.1The purchaser may specify controlled structural or special service characteristics which shall be used as a guide for the most suitable heat treatment.If the final heat treatment is at a temperature under 1900°F [1040°C],each pipe shall be stenciled with the final heat treatment temperature in degrees Fahrenheit or Celsius after the suffix “HT.”6.2.4Grades TP304H,TP316H,TP321H,TP347H,and 16-8-2H —If cold working is involved in processing,the minimum solution-treating temperature for Grades TP321H and TP347H shall be 2000°F [1100°C],for Grades TP304H and TP316H,1900°F [1040°C],and for Grade 16-8-2H,1800°F [980°C].If the material is hot-rolled,the minimum solution-treating temperatures for Grades TP321H and TP347H shall be 1925°F [1050°C],for Grades TP304H and TP316H,1900°F [1040°C],and for Grade 16-8-2H,1800°F [980°C].6.2.5Grade S34565—Heat treatment shall consist of heat-ing to a temperature in the range of 2050°F [1120°C]minimum and 2140°F [1170°C]maximum,and quenching in water or rapidly cooling by other means.6.3A solution annealing temperature above 1950°F [1065°C]may impair the resistance to intergranular corrosion after subsequent exposure to sensitizing conditions in TP321,TP321H,TP347,TP347H,TP348,and TP348H.When speci-fied by the purchaser,a lower temperature stabilization or re-solution anneal shall be used subsequent to the initial high temperature solution anneal (see Supplementary Requirement S9).6.4The grain size of grades 304H,316H,321H,and 347H,as determined in accordance with Test Methods E 112,shall be No.7or coarser.TABLE 1Chemical RequirementsGradeUNS Desig-nationComposition,%CarbonMan-ganese,max Phos-phorus,maxSul-fur,max Sili-con,max NickelChromiumMolyb-denum Tita-nium Colum-bium Tan-talum Nitro-gen AOthersTP304S304000.08max 2.000.0450.0300.758.0–11.018.0–20.0..................TP304H S304090.04–0.10 2.000.0450.0300.758.0–11.018.0–20.0..................TP304N S304510.08max 2.000.0450.0300.758.0–11.018.0–20.0............0.10–0.16...TP304LN S304530.035max 2.000.0450.0300.758.0–11.018.0–20.0............0.10–0.16...TP316S316000.08max 2.000.0450.0300.7511.0–14.016.0–18.0 2.00–3.00...............TP316H S316090.04–0.10 2.000.0450.0300.7511.0–14.016.0–18.0 2.00–3.00...............TP316N S316510.08max 2.000.0450.0300.7511.0–14.016.0–18.0 2.00–3.00.........0.10–0.16...TP316LN S316530.035max 2.000.0450.0300.7511.0–14.016.0–18.0 2.00–3.00.........0.10–0.16...TP321S321000.08max 2.000.0450.0300.759.0–13.017.0–19.0...B ............TP321H S321090.04–0.10 2.000.0450.0300.759.0–13.017.0–19.0...C............TP347S347000.08max 2.000.0450.0300.759.0–13.017.0–19.0......D .........TP347H S347090.04–0.10 2.000.0450.0300.759.0–13.017.0–19.0......E .........TP348F S348000.08max 2.000.0450.0300.759.0–13.017.0–19.0......D0.10...Co 0.20max16-8-2H S168000.05–0.10 2.000.0450.0300.757.5–9.514.5–16.5 1.50–2.00..................S317250.030max 2.000.0450.0300.7513.5–17.518.0–20.0 4.0–5.0.........0.20max Cu 0.75max ...S317260.030max 2.000.0450.0300.7514.5–17.517.0–20.0 4.0–5.0.........0.10–0.20Cu 0.75max ...S345650.030max 5.0–7.00.0300.0101.016.0–18.023.0–25.04.0–5.0.........0.040–0.060Cb 0.10maxA The method of analysis for nitrogen shall be a matter of agreement between the purchaser and manufacturer.BThe titanium content shall be not less than five times the carbon content and not more than 0.70%.CThe titanium content shall be not less than four times the carbon content and not more than 0.70%.DThe columbium content shall be not less than ten times the carbon content and not more than 1.10%.EThe columbium content shall be not less than eight times the carbon content and not more than 1.10%.FThis grade is intended for special purposeapplications.7.Chemical Composition7.1The steel shall conform to the requirements as to chemical composition prescribed in Table 1.8.Product Analysis8.1At the request of the purchaser,an analysis of one billet from each heat or two pipes from each lot (Note 2)shall be made by the manufacturer.A lot of pipe shall consist of the following:NPS Designator Lengths of Pipe in Lot Under NPS 2400or fraction thereof NPS 2to NPS 5,incl 200or fraction thereof Over NPS 5100or fraction thereofN OTE 2—A lot shall consist of the number of lengths specified in 8.1of the same size and wall thickness from any one heat of steel.8.2The results of these analyses shall be reported to the purchaser or the purchaser’s representative,and shall conform to the requirements specified in Table 1.8.3If the analysis of one of the tests specified in Section 9does not conform to the requirements specified in Section 7,an analysis of each billet or pipe from the same heat or lot may be made,and all billets or pipe conforming to the requirements shall be accepted.9.Tensile Requirements9.1The material shall conform to the requirements as to tensile properties prescribed in Table 2.10.Workmanship,Finish,and Appearance10.1The pipe manufacturer shall explore a sufficient num-ber of visual surface imperfections to provide reasonable assurance that they have been properly evaluated with respect to depth.Exploration of all surface imperfections is not required but may be necessary to assure compliance with 10.2.10.2Surface imperfections that penetrate more than 121⁄2%of the nominal wall thickness or encroach on the minimum wall thickness shall be considered defects.Pipe with such defects shall be given one of the following dispositions:10.2.1The defect may be removed by grinding provided that the remaining wall thickness is within specified limits.10.2.2Repaired in accordance with the repair welding provisions of 10.6.10.2.3The section of pipe containing the defect may be cut off within the limits of requirements on length.10.2.4Rejected.10.3To provide a workmanlike finish and basis for evalu-ating conformance with 10.2,the pipe manufacturer shall remove by grinding the following:10.3.1Mechanical marks,abrasions (see Note 3),and pits,any of which imperfections are deeper than 1⁄16in.[1.6mm].N OTE 3—Marks and abrasions are defined as cable marks,dinges,guide marks,roll marks,ball scratches,scores,die marks,and so forth.10.3.2Visual imperfections commonly referred to as scabs,seams,laps,tears,or slivers found by exploration in accor-dance with 10.1to be deeper than 5%of the nominal wall thickness.10.4At the purchaser’s discretion,pipe shall be subject to rejection if surface imperfections acceptable under 10.2are not scattered,but appear over a large area in excess of what is considered a workmanlike finish.Disposition of such pipe shall be a matter of agreement between the manufacturer and the purchaser.10.5When imperfections or defects are removed by grind-ing,a smooth curved surface shall be maintained,and the wall thickness shall not be decreased below that permitted by this specification.The outside diameter at the point of grinding may be reduced by the amount so removed.10.5.1Wall thickness measurements shall be made with a mechanical caliper or with a properly calibrated nondestructive testing device of appropriate accuracy.In case of dispute,the measurement determined by use of the mechanical caliper shall govern.10.6Weld repair shall be permitted only subject to the approval of the purchaser and in accordance with Specification A 999/A 999M.10.7The finished pipe shall be reasonably straight.10.8The pipe shall be free of scale and contaminating iron particles.Pickling,blasting,or surface finishing is not manda-tory when pipe is bright annealed.The purchaser may request that a passivating treatment be applied.11.Hydrostatic or Nondestructive Electric Test11.1Each pipe shall be subjected to the Nondestructive Electric Test or the Hydrostatic Test.Unless specified by the purchaser,either test may be used at the option of the producer.11.2Hydrostatic Test —Each length of finished pipe shall be subjected to the hydrostatic test in accordance with Speci-fication A 999/A 999M,unless specifically exempted under the provisions of 11.3and 11.4.11.3For pipe sizes NPS 24and over,the purchaser,with the agreement of the manufacturer,may complete the hydrostatic test requirement with the system pressure test,which may beTABLE 2Tensile RequirementsGradeTensile A strength,min,ksi [MPa]Yield strength min,ksi[MPa]Elongation in 2in.or 50mm (or 4D)min,%LongitudinalTransverse TP304,TP304H,TP304LN,TP316,TP316H,TP316LN,TP347,TP347H,TP348,16-8-2H,S3172575[515]30[205]3525TP304N,TP316N,S3172680[550]35[240]3525S34565115[790]60[415]3530TP321,321H #3⁄8975[515]30[205]3525>3⁄89B70[480]25[170]3525A†For grade TP304,NPS8or larger,and in schedules 140and heavier,the required minimum tensile strength shall be 70ksi [480MPa].BPrior to the issuance of A 376/A 376M –88,the tensile and yield strength values were 75[520]and 30[210]respectively,for nominal wall greater than 3⁄8in.[9.5mm].†Editoriallycorrected.lower or higher than the specification test pressure,but in no case shall the test pressure be lower than the system design pressure.Each length of pipe furnished without the completed manufacturer’s hydrostatic test shall include with the manda-tory marking the letters“NH.”11.4Nondestructive Examination—Each pipe shall be ex-amined with a nondestructive test in accordance with Practice E213or Practice E426.Unless specifically called out by the purchaser,the selection of the nondestructive electric test will be at the option of the manufacturer.The range of pipe sizes that may be examined by each method shall be subject to the limitations in the scope of the respective practices.11.4.1The following information is for the benefit of the user of this specification:11.4.1.1The reference standards defined in11.10.1through 11.10.4are convenient standards for calibration of nondestruc-tive testing equipment.The dimensions of these standards should not be construed as the minimum size imperfection detectable by such equipment.11.4.1.2The ultrasonic testing(UT)can be performed to detect both longitudinally and circumferentially oriented de-fects.It should be recognized that different techniques should be employed to detect differently oriented imperfections.The examination may not detect short,deep,defects.11.4.1.3The eddy-current testing(ET)referenced in Prac-tice E426has the capability of detecting significant disconti-nuities,especially the short abrupt type.11.4.1.4A purchaser interested in ascertaining the nature (type,size,location,and orientation)of discontinuities that can be detected in the specific application of these examinations should discuss this with the manufacturer of the tubular product.11.5Time of Examination—Nondestructive testing for specification acceptance shall be performed after all mechani-cal processing,heat treatments,and straightening operations. This requirement does not preclude additional testing at earlier stages in the processing.11.6Surface Condition:11.6.1All surfaces shall be free of scale,dirt,grease,paint, or other foreign material that could interfere with interpretation of test results.The methods used for cleaning and preparing the surfaces for examination shall not be detrimental to the base metal or the surfacefinish.11.6.2Excessive surface roughness or deep scratches can produce signals that interfere with the test.11.7Extent of Examination:11.7.1The relative motion of the pipe and the transducer(s), coil(s),or sensor(s)shall be such that the entire pipe surface is scanned,except as in6.2.11.7.2The existence of end effects is recognized,and the extent of such effects shall be determined by the manufacturer, and,if requested,shall be reported to the purchaser.Other nondestructive tests may be applied to the end areas,subject to agreement between the purchaser and the manufacturer. 11.8Operator Qualifications—The test unit operator shall be certified in accordance with SNT-TC-1A,or an equivalent recognized and documented standard.11.9Test Conditions:11.9.1For eddy-current testing,the excitation coil fre-quency shall be chosen to ensure adequate penetration yet provide good signal-to-noise ratio.11.9.2The maximum eddy-current coil frequency used shall be as follows:On specified walls up to0.050in.—100KHz maxOn specified walls up to0.150in.—50KHz maxOn specified walls up to0.150in.—10KHz max11.9.3Ultrasonic—For examination by the ultrasonic method,the minimum nominal transducer frequency shall be 2.00MHz and the maximum nominal transducer size shall be 1.5in.11.9.3.1If the equipment contains a reject noticefilter setting,this shall remain off during calibration and testing unless linearity can be demonstrated at that setting.11.10Reference Standards:11.10.1Reference standards of convenient length shall be prepared from a length of pipe of the same grade,size(NPS,or outside diameter and schedule or wall thickness),surface finish,and heat treatment condition as the pipe to be examined.11.10.2For Ultrasonic Testing,the reference ID and OD notches shall be any one of the three common notch shapes shown in Practice E213,at the option of the manufacturer.The depth of each notch shall not exceed121⁄2%of the specified nominal wall thickness of the pipe or0.004in.,whichever is greater.The width of the notch shall not exceed twice the depth.Notches shall be placed on both the OD and ID surfaces.11.10.3For Eddy-Current Testing,the reference standard shall contain,at the option of the manufacturer,any one of the following discontinuities:11.10.3.1Drilled Hole—The reference standard shall con-tain three or more holes,equally spaced circumferentially around the pipe and longitudinally separated by a sufficient distance to allow distinct identification of the signal from each hole.The holes shall be drilled radially and completely through the pipe wall,with care being taken to avoid distortion of the pipe while drilling.One hole shall be drilled in the weld,if visible.Alternately,the producer of welded pipe may choose to drill one hole in the weld and run the calibration standard through the test coils three times with the weld turned at120°on each pass.The hole diameter shall vary with NPS as follows:NPS Designator Hole Diameter0.039in.(1mm)above1⁄2to11⁄40.055in.(1.4mm)above11⁄4to20.071in.(1.8mm)above2to50.087in.(2.2mm)above50.106in.(2.7mm)11.10.3.2Transverse Tangential Notch—Using a round tool orfile with a1⁄4-in.(6.4-mm)diameter,a notch shall befiled or milled tangential to the surface and transverse to the longitu-dinal axis of the pipe.Said notch shall have a depth not exceeding121⁄2%of the specified nominal wall thickness of the pipe or0.004in.(0.102mm),whichever is greater.11.10.3.3Longitudinal Notch—A notch0.031in.or less in width shall be machined in a radial plane parallel to the tube axis on the outside surface of the pipe,to have a depth not exceeding121⁄2%of the specified wall thickness of the pipeor0.004in.,whichever is greater.The length of the notch shall be compatible with the testing method.11.10.3.4More or smaller reference discontinuities,or both, may be used by agreement between the purchaser and the manufacturer.11.11Standardization Procedure:11.11.1The test apparatus shall be standardized at the beginning and end of each series of pipes of the same size (NPS or diameter and schedule or wall thickness),grade and heat treatment condition,and at intervals not exceeding4h. More frequent standardization may be performed at the manu-facturer’s option or may be required upon agreement between the purchaser and the manufacturer.11.11.2The test apparatus shall also be standardized after any change in test system settings;change of operator;equip-ment repair;or interruption due to power loss,process shut-down,or when a problem is suspected.11.11.3The reference standard shall be passed through the test apparatus at the same speed and test system settings as the pipe to be tested.11.11.4The signal-to-noise ratio for the reference standard shall be21⁄2to1or greater.Extraneous signals caused by identifiable causes such as dings,scratches,dents,straightener marks,and so forth,shall not be considered noise.The rejection amplitude shall be adjusted to be at least50%of full scale of the readout display.11.11.5If upon any standardization,the rejection amplitude has decreased by29%(3dB)of peak height from the last standardization,the pipe since the last calibration shall be rejected.The test system settings may be changed,or the transducer(s),coil(s)or sensor(s)adjusted,and the unit restan-dardized,but all pipe tested since the last acceptable standard-ization must be retested for acceptance.11.12Evaluation of Imperfections:11.12.1Pipes producing a signal equal to or greater than the lowest signal produced by the reference standard(s)shall be identified and separated from the acceptable pipes.The area producing the signal may be reexamined.11.12.2Such pipes shall be rejected if the test signal was produced by imperfections that cannot be identified or was produced by cracks or crack-like imperfections.These pipes may be repaired in accordance with Sections13and14.To be accepted,a repaired pipe must pass the same nondestructive test by which it was rejected,and it must meet the minimum wall thickness requirements of this specification.11.12.3If the test signals were produced by visual imper-fections such as:(1)Scratches,(2)Surface roughness,(3)Dings,(4)Straightener marks,(5)Cutting chips,(6)Steel die stamps,(7)Stop marks,or(8)Pipe reducer ripple.The pipe may be accepted based on visual examination provided the imperfection is less than0.004in.(0.1mm)or 121⁄2%of the specified wall thickness(whichever is greater).11.12.4Rejected pipe may be reconditioned and retested providing the wall thickness is not decreased to less than that required by this or the product specification.The outside diameter at the point of grinding may be reduced by the amount so removed.To be accepted,retested pipe shall meet the test requirement.11.12.5If the imperfection is explored to the extent that it can be identified as non-rejectable,the pipe may be accepted without further test providing the imperfection does not en-croach on the minimum wall thickness.12.Mechanical Tests Required12.1Transverse or Longitudinal Tension Test—The tension test shall be performed on1%of the pipe from each lot.N OTE4—The term“lot”applies to all pipe of the same nominal size and wall thickness(or schedule)which is produced from the same heat of steel and subjected to the samefinishing treatment in a continuous furnace or by directly obtaining the heat treated condition by quenching after hot forming.Whenfinal heat treatment is in a batch-type furnace,the lot shall include only that pipe which is heat treated in the same furnace charge.12.2Flattening Test—For pipe heat treated in a batch-type furnace,theflattening test shall be made on5%of the pipe from each heat-treated lot(see Note4).When heat treated by the continuous process or when treated condition is obtained directly by quenching after hot forming,this test shall be made on a sufficient number of pipe to constitute5%of the lot(Note 4)but in no case less than two pipes.13.Certification13.1In addition to the certification required by Specification A999/A999M,the certification for pipe furnished to this specification shall identify each length of pipe which is furnished without the manufacturer’s completed hydrostatic test,in accordance with11.3.14.Product Marking14.1In addition to the marking prescribed in Specification A999/A999M,the marking shall include the length,hydro-static test pressure,the ANSI schedule number,the heat number or manufacturer’s number by which the heat can be identified,the marking requirements of6.2,and,if applicable, NH when hydrotesting is not performed and ET when eddy-current testing is performed,or UT when ultrasonic testing is performed.14.2If the pipe conforms to any of the supplementary requirements specified in S1through S10,compliance shall be so indicated by adding the symbol“S”directly followed by the number of the applicable supplementary requirement to the marking prescribed in14.1.14.3No steel indentation stamping shall be done without the purchaser’s consent.15.Keywords15.1austenitic stainless steel;feedwater heater tubes;stain-less steel tube;steel tube;welded steeltubeSUPPLEMENTARY REQUIREMENTSFOR PIPE REQUIRING SPECIAL CONSIDERATION One or more of the following supplementary requirements shall apply only when specified in the purchase order.The purchaser may specify a different frequency of test or analysis than is provided in the supplementary requirement.Subject to agreement between the purchaser and manufacturer, retest and retreatment provisions of these supplementary requirements may also be modified.S1.Product AnalysisS1.1Product analysis shall be made on each length of pipe. Individual lengths failing to conform to the chemical compo-sition requirements shall be rejected.S2.Transverse Tension TestsS2.1A transverse tension test shall be made on a specimen from one end or both ends of each pipe NPS8and over in nominal diameter.If this supplementary requirement is speci-fied,the number of tests per pipe shall also be specified.If a specimen from any length fails to meet the required tensile properties(tensile,yield,and elongation),that length shall be rejected subject to retreatment in accordance with Specification A999/A999M and satisfactory retest.S3.Flattening TestS3.1Theflattening test of Specification A999/A999M shall be made on a specimen from one end or both ends of each pipe.Crop ends may be used.If this supplementary require-ment is specified,the number of tests per pipe shall also be specified.If a specimen from any length fails because of lack of ductility prior to satisfactory completion of thefirst step of theflattening test requirement that pipe shall be rejected subject to retreatment in accordance with Specification A999/ A999M and satisfactory retest.If a specimen from any length of pipe fails because of a lack of soundness that length shall be rejected,unless subsequent retesting indicates that the remain-ing length is sound.S4.Etching TestsS4.1The steel shall be homogeneous as shown by etching tests conducted in accordance with the appropriate portions of Method E381.Etching tests shall be made on a cross section from one end or both ends of each pipe and shall show sound and reasonably uniform material free from injurious lamina-tions,cracks,and similar objectionable defects.If this supple-mentary requirement is specified,the number of tests per pipe required shall also be specified.If a specimen from any length shows objectionable defects,the length shall be rejected, subject to removal of the defective end and subsequent retests indicating the remainder of the length to be sound and reasonably uniform material.S5.PhotomicrographsS5.1Photomicrographs at100diameters may be made from one end of each piece of pipe furnished in sizes6in.[152mm] and larger in the as-furnished condition.Such photomicro-graphs shall be suitably identified as to pipe size,wall thickness,piece number,and heat.Such photomicrographs are for information only,and shall show the actual metal structure of the pipe asfinished.S6.Ultrasonic TestS6.1Each piece of pipe may be ultrasonically tested to determine its soundness throughout the entire length of the pipe.Each piece shall be ultrasonically tested in a circumfer-ential direction in such a manner that the entire piece is scanned by the ultrasonic beam.The calibration standard shall be prepared from a section of pipe which has two notches,one in the inside surface and one in the outside surface.The notches shall be at least11⁄2-in.[38-mm]long and have a depth of3% of the wall thickness,or0.004in.[0.1mm],whichever is the greater.Any pipe showing an ultrasonic indication of greater amplitude than the amplitude of the indication from the calibration standard shall be subject to rejection.S7.Hot Ductility Test for Indicating WeldabilityS7.1A high-temperature ductility test may be made upon each heat of material supplied in heavy-wall pipe sections.An appropriate specimen shall be heated to an initial temperature, cooled100°F[50°C],then subjected to a tension test,and shall show a minimum reduction of area of60%.The initial temperature is that temperature50°F[30°C]below the tem-perature at which material exhibits zero ductility.Rejection of material shall not be based upon this test.S8.RetestsS8.1Upon the purchaser’s request,retests shall be made from sections of material removed from any part of the pipe. Failure to meet the requirements stated in this specification shall be cause for rejection.S9.Stabilization Heat TreatmentS9.1Subsequent to the solution anneal required in 6.4, Grades TP321,TP321H,TP347,TP347H,TP348,and TP348H shall be given a stabilization heat treatment at a temperature lower than that used for the initial solution annealing heat treatment.The temperature of stabilization heat treatment shall be at a temperature as agreed upon between the purchaser and vendor.S10.Intergranular Corrosion TestS10.1When specified,material shall pass intergranular corrosion tests conducted by the manufacturer in accordance with Practices A262,Practice E.N OTE S10.1—Practice E requires testing on the sensitized condition for low carbon or stabilized grades,and on the as-shipped condition for other grades.S10.2A stabilization heat treatment in accordance with Supplementary Requirement S9may be necessary and is permitted in order to meet this requirement for the grades containing titanium or columbium,particularly in their Hversions.。
分子力场的势函数形式

DREIDING, UFF以及TRIPOS等力场
ε
分子力学----分子力场的势函数形式
静电相互作用
静电相互作用 Electrostatic Contributions
点电荷法:通过经验规则或者量化计算确定每个原子上的 部分电荷(partial charge),两个原子之间的静电作用用 库仑公式来计算。 qq
分子力学----分子力场的势函数形式
二面角扭转能
二面角扭转能 Torsion Rotation
Vn ET [1 cos(n )] n 0 2
• Vn 为势垒高度(barrier height),定量描述了二面角旋转的难易程度; • N 为多重度(multiplicity),指键从0°到360°旋转过程中能量极小点的个数; • 为相因子(phase factor),指单键旋转通过能量极小值时二面角的数值。 • ω为扭转角度(torsion angle)
乙烷分子hcch二面角传统力场kollmangroup1984最初仅为蛋白质和核酸体系提供相应的原子类型和力场参数1990发展了适用于多糖模拟的力场参数homan1990harvardmacromolecularmechanicskarplusgroup1983适用于各种分子性质的计算和模拟对于从孤立的小分子到溶剂化的大生物体系的多种模拟体系都可以给出较好的结果但不适合于有机金属配合物cvff力场consistentvalenceforcefielddauberosguthorpegroup1988适用范围包括有机小分子和蛋白质体系口扩展后可用于某些无机体系的模拟如硅酸盐铝硅酸盐磷铝化合物主要用于预测分子的结构和结合自由能allingergroup1989包括mm2和mm3主要针对有机小分子函数形式比较复杂包含交叉项口也可用于生物大分子体系但是速度会比较慢eq第二代力场质之间的相互作用cff95除了适用于蛋白质和有机小分子体系还可用于有机高分子体系的模拟如聚碳酸酯及多糖pcff在cff91的基础上还适用于聚碳酸酯三聚氤胺甲醛树脂多糖核酸分子筛等其他无机和有机材料体系的模拟
lammps化学反应 -回复

lammps化学反应-回复LAMMPS is a widely used software package for molecular dynamics simulations. It is designed to simulate the behavior of atoms and molecules at the atomic scale. One of the major applications of LAMMPS is in studying chemical reactions. In this article, we will explore the process of simulating chemical reactions using LAMMPS and understand the step-by-step approach involved.1. Preparing the System:The first step in simulating a chemical reaction with LAMMPS is to prepare the system. This involves setting up the initial configuration of atoms or molecules and defining the force field parameters. The force field is responsible for describing the interactions between atoms and determining their behavior during the simulation. Common force fields used in LAMMPS include the CHARMM, AMBER, and OPLS force fields.2. Defining the Reactions:Once the system is prepared, the next step is to define the chemical reactions to be simulated. This can be done by specifying the reactants, products, and the reaction mechanism. LAMMPS provides various options to specify the reactions, including usingbond-breaking and bond-making commands.3. Assigning Atomic Types:Before performing the simulation, each atom in the system needs to be assigned a type. This allows LAMMPS to correctly interpret the force field parameters for the atoms and their interactions. In some cases, the types may need to be modified to account for the changes in atomic connectivity during the reaction.4. Energy Minimization:After assigning the atomic types, the system is energy-minimized to remove any steric clashes or excessive energy. Energy minimization is an iterative process that adjusts the positions of the atoms to find a local minimum in the energy landscape. LAMMPS provides powerful energy minimization algorithms, such as the steepest descent and conjugate gradient methods.5. Equilibration:Once the system is energy-minimized, it needs to be equilibrated to bring it to a suitable starting state for the simulation. This involves allowing the system to evolve under constant temperature and pressure conditions until it reaches a dynamic equilibrium.LAMMPS provides various simulation settings to control the temperature and pressure during equilibration, including thermostats and barostats.6. Production Run:After equilibration, the system is ready for the production run, where the chemical reaction is simulated. The simulation is typically performed in an ensemble, such as the NVT or NPT ensemble, which allows the system to evolve while maintaining a constant number of particles, volume, and temperature.7. Analysis of Results:Once the simulation is completed, the results need to be analyzed to understand the behavior of the system during the chemical reaction. This can include analyzing the trajectories of the atoms, calculating reaction rates, monitoring the changes in bond lengths and angles, and studying the changes in the thermodynamic properties of the system.In conclusion, simulating chemical reactions using LAMMPS involves a systematic step-by-step approach, from preparing thesystem and defining the reactions to performing equilibration and the production run. Through this process, LAMMPS allows researchers to gain valuable insights into the behavior of atoms and molecules during chemical reactions, which can aid in the design and development of new materials and chemical processes.。
HyperChem基本操作.

HyperChem基本操作画原子1. 打开Element Table对话框。
这里有两种方法:在Build菜单中选择Default Element,或者双击Drawing工具。
Default Element对话框允许从周期表中选择缺省元素。
2. 如果单击Properties...按钮,将显示当前选择元素的物理属性。
也可以按下Shift键同时单击元素按钮,结果是一样的。
单击OK键,物理属性框消失。
3. 如果Allow Ions或者Explicit Hydrogens打开(用对勾选择),左键单击这些选项使其关闭。
4. 在缺省元素列表中选择Carbon,接着关闭元素对话框。
缺省元素将设置为碳。
当然也可以把打开的Default Element对话框移走,这样可以看到HyperChem工作区。
当画原子非常多的分子时,这是非常有效的。
5. 左键单击Drawing工具,把指针移到工作区。
6. 左键单击工作区左下角,将出现一个小圈,代表未成键的碳原子。
7. 在工作区不同位置画更多的原子。
画价键1. 把指针移到刚才画的第一个碳上。
2. 按下鼠标左键。
这是价键在第一个原子的位置。
3. 保持鼠标按钮按下的同时拖向工作区的顶端。
4. 放开鼠标按钮。
这是价键在第二个原子的位置。
一条线代表两个碳原子之间的价键。
5. 用仍旧停留在价键末端的指针, 用左键拖向工作区右下角。
6. 放开鼠标按钮。
这是第三个原子的位置。
7. 在空白工作区画六个价键,形成一个环。
现在你清楚了如何画原子和分子,并且学会了一些基本技巧。
选择原子在这个练习中,通过选择原子,你可以学到基本的选择技巧。
首先必须设置选择的级别[原子(atoms),基(residues),或分子(molecules)]。
这里设置为原子(atoms)。
1. 左键单击Select菜单。
2. 左键单击选择Atoms。
接下来,关闭Multiple Selections:1. 左键单击Select菜单。
Amber教程

研究案例——一种稳定蛋白质的全部原子结构预测和折叠模拟这段教程展示的是一个研究实例,像您演示如何重现下述文章中的研究工作:Simmerling, C., Strockbine, B., Roitberg, A.E., J. Am. Chem. Soc., 2002, 124, 11258-11259(/ja0273851)我们建议您在开始本教程前首先阅读上述文章,获得该蛋白的氨基酸序列及其他有用信息。
警告1: 本教程中的一些计算耗时很长,我使用了由16个1.3GHz cup的SGI Altix进行了27小时计算才完成整个工作,因此如果您没有足够的计算能力,我强烈建议您在重复本教程的过程中使用我为您提供的out文件,以使得您能够流畅地完成整个教程。
警告2: 如果您重复本教程,我们并不能保证您能够精确地重现我的计算结果,在计算过程中,不同结构的计算机会产生不同的近似误差,从而使得计算过程搜索的是相空间的不同部位,但是模拟的平均结果是大致相同的。
另外,尽管您完全重复了本教程也有可能无法获得论文中给出的结果,而且即便是我们自己也无法保证论文中的结果能够重现,这可能是因为我模拟的时间不够长,获取的仅仅是一个局部最小点,但是尽管如此,本教程的工作还是展示了蛋白折叠中一些有趣的行为。
背景这篇论文应用AMBER FF99力场和经典的全原子动力学对一个肽的折叠过程进行了模拟。
模拟的对象"trpcage"是一个由20个氨基酸构成的小肽,华盛顿大学的 Andersen已经对这个蛋白做过了结构优化,它是现在已知最小的能够显示两种不同折叠状态的蛋白,而且这个蛋白在室温下可以稳定存在。
该蛋白的小身量使得它成为模拟蛋白质折叠的绝嘉对象。
当最早的关于这个蛋白的折叠的计算结果出炉时,对这个蛋白结构的实验测定还没有完成,所以整个模拟过程是在没有实验数据作为指导的情况下完成的。
当蛋白的结构经由实验手段测定之后,人们惊喜地发现,计算机模拟的结果与实验测定的数值之间的RMSD值仅为1.4A。
amber实战

第一步:生成小分子模板蛋白质中各氨基酸残基的力参数是预先存在的,但是很多模拟过程会涉及配体分子,这些有机小分子有很高的多样性,他们的力参数和静电信息不可能预存在库文件中,需要根据需要自己计算生成模板。
amber中的antechamber 程序就是生成小分子模板的。
生成模板要进行量子化学计算,这一步可以由antechamber中附带的mopac完成,也可以由gaussian完成,这里介绍用gaussian计算的过程。
建议在计算前用sybyl软件将小分子预先优化,不要用gaussian优化,大基组从头计算进行几何优化花费的计算时间太长。
gaussian计算的输入文件可以用antechamber程序直接生成,生成后去掉其中关于几何优化的参数即可将小分子优化后的结构存储为mol2各式,上传到工作目录,用antechamber程序生成gaussian 输入文件,命令如下:antechamber -i 49.mol2 -fi mol2 -o 49.in -fo gzmat这样可以生成49.in文件,下载到windows环境,运行gaussian计算这个文件,如果发现计算时间过长或者内存不足计算中断,可以修改文件选择小一些的基组。
获得输出文件49.out之后将它上传到工作目录,再用antechamber生成模板,命令如下:antechamber -i 49.out -fi gout -o 49mod.mol2 -fo mol2 -c resp运行之后就会生成一个新的mol2文件,如果用看图软件打开这个文件会发现,原子的颜色很怪异,这是因为mol2的原子名称不是标准的原子名称,看图软件无法识别。
下面一步是检查参数,因为可能会有一些特殊的参数在gaff中不存在需要程序注入,命令如下:parmchk -i 49mod.mol2 -f mol2 -o 49mod这样那些特殊的参数就存在49mod这个文件中了第二步:处理蛋白质文件amber自带的leap程序是处理蛋白质文件的,他可以读入PDB格式的蛋白质文件,根据已有的力场模板为蛋白质赋予键参数和静电参数。
gromacs常见错误

The vast 巨大的majority of error messages generated by GROMACS are descriptive 解释的, informing the user where the exact error lies. Some errors that arise are noted below, along with more details on what the issue is and how to solve it.1. 1. General1. 1.1. Cannot allocate memory2. 2. pdb2gmx1. 2.1. Residue 'XXX' not found in residue topology database2. 2.2. Long bonds and/or missing atoms3. 2.3. Chain identifier 'X' was used in two non-sequential blocks4. 2.4. WARNING: atom X is missing in residue XXX Y in the pdb file5. 2.5. Atom X in residue YYY not found in rtp entry3. 3. grompp1. 3.1. Found a second defaults directive file2. 3.2. Invalid order for directive defaults3. 3.3. System has non-zero total charge4. 3.4. Incorrect number of parameters5. 3.5. Number of coordinates in coordinate file does not match topology6. 3.6. Fatal error: No such moleculetype XXX7. 3.7. T-Coupling group XXX has fewer than 10% of the atoms8. 3.8. The cut-off length is longer than half the shortest box vector or longer than thesmallest box diagonal element. Increase the box size or decrease rlist9. 3.9. Unknown left-hand XXXX in parameter file10.3.10. Atom index (1) in bonds out of bounds4. 4. mdrun1. 4.1. Stepsize too small, or no change in energy. Converged to machine precision, butnot to the requested precision2. 4.2. LINCS/SETTLE/SHAKE warnings3. 4.3. 1-4 interaction not within cut-off4. 4.4. Simulation running but no output5. 4.5. Can not do Conjugate Gradients with constraints6. 4.6. Pressure scaling more than 1%7. 4.7. Range Checking error8. 4.8. X particles communicated to PME node Y are more than a cell length out of thedomain decomposition cell of their charge group9. 4.9. There is no domain decomposition for n nodes that is compatible with the givenbox and a minimum cell size of x nmGeneralCannot allocate memoryThe executed script has attempted to assign memory to be used in the calculation, but is unable to due to insufficient memory.Possible solutions are:•install more memory in the computer.•use a computer with more memory.•reduce the scope of the number of atoms selected for analysis.•reduce the length of trajectory file being processed.•in some cases confusion between Ångström and nm may lead to users wanting to generate a water box that is 103 times larger than what they think it is (e.g.genbox).The user should bear in mind that the cost in time and/or memory for various activities will scale with the number of atoms/groups/residues N or the simulation length T as order N, N log N, or N2 (or maybe worse!) and the same for T, depending on the type of activity. If it takes a long time, have a think about what you are doing, and the underlying algorithm (see the manual, man page, or use the -h flag for the utility), and see if there's something sensible you can do that has better scaling properties. pdb2gmxResidue 'XXX' not found in residue topology残基不存在databaseThis means that the force field {you have selected while running pdb2gmx} does not have an entry in the residue database for XXX. The residue database entry is necessary both for stand-alone molecules (e.g. formaldehyde甲醛) or a peptide (standard or non-standard). This entry defines the atom types, connectivity, bonded and non-bonded interaction types for the residue and is necessary to use pdb2gmx to build a .top file. A residue database entry may be missing simply because the database does not contain the residue at all, or because the name is different.For new users, this error appears because they are running pdb2gmx blindly盲目的on a PDB file they have without consideration of the contents of the file. A force fieldis not something that is magical, it can only deal with molecules or residues (building blocks) that are provided in the residue database or included otherwise.If you want to use pdb2gmx to automatically generate your topology, you have to ensure that the appropriate .rtp entry is present within the desired force field and has the same name as the building block you are trying to use. If you call your molecule "HIS," then pdb2gmx will not magically build a random molecule; it will try to build histidine, based on the [ HIS ] entry in the .rtp file, so it will look for the exact atomic entries for histidine, no more no less.If you want a topology for an arbitrary molecule, you cannot use pdb2gmx (unless you build the .rtp entry yourself). You will have to build it by hand, or use another program (such as x2top or one of the scripts contributed by users) to build the .top file.If there is not an entry for this residue in the database, then the options for obtaining the force field parameters are:•see if there is a different name being used for the residue in the residue database and rename as appropriate,•parameterize the residue / molecule yourself,•find a topology file for the molecule, convert it to an .itp file and include it in your .top file,•use another force field which has parameters available for this,•search the primary literature for publications for parameters for the residue that are consistent with the force field that is being used.Long bonds and/or missing atomsThere are probably atoms missing earlier in the .pdb file which makes pdb2gmx go crazy. Check the screen output of pdb2gmx, as it will tell you which one is missing. Then add the atoms in your pdb file, energy minimization will put them in the right place, or fix the side chain with e.g. the WhatIF program.Chain identifier 'X' was used in two non-sequential非次序的blocksThis means that within the coordinate file fed to pdb2gmx, the X chain has been split 断裂, possibly by the incorrect insertion of one molecule within another. The solution is simple: move the inserted molecule to a location within the file so that it is not splitting another molecule.This message may also mean that the same chain identifier has been used for two separate chains. In that case, rename the second chain to a unique identifier.WARNING: atom X is missing in residue XXX Y in the pdb fileRelated to the long bonds/missing atoms error above, this error is usually quite obvious in its meaning. That is, pdb2gmx expects certain atoms within the given residue, based on the entries in the force field .rtp file. There are several cases to which this error applies:•Missing hydrogen atoms; the error message may be suggesting that an entry in the .hdb file is missing. More likely, the nomenclature 名称of yourhydrogen atoms simply does not match what is expected by the .rtp entry. In this case, use -ignh to allow pdb2gmx to add the correct hydrogens for you, or re-name the problematic问题的atoms.• A terminal residue (usually the N-terminus) is missing H atoms; this usually suggests that the proper -ter option has not been supplied or chosen properly.In the case of the AMBER force fields, nomenclature is typically the problem.N-terminal and C-terminal residues must be prefixed by N and C,respectively. For example, an N-terminal alanine 丙氨酸should not belisted in the .pdb file as ALA, but rather NALA, as specified in the ffamberinstructions.•Atoms are simply missing in the structure file provided to pdb2gmx; look for REMARK 465 and REMARK 470 entries in the .pdb file. These atoms willhave to be modeled in using external software. There is no GROMACS tool to re-construct incomplete models.Contrary to what the error message says, the use of the option -missing is almost always inappropriate. The -missing option should only be used to generate specialized topologies for amino acid-like molecules to take advantage of .rtp entries. If you find yourself using -missing in order to generate a topology for a protein or nucleic acid, don't; the topology produced is likely physically unrealistic.Atom X in residue YYY not found in rtp entryIf you are attempting to assemble a topology using pdb2gmx, the atom names are expected to match those found in the .rtp file that define the building block(s) in your structure. If you get this error, simply re-name the atoms in your coordinate file appropriately.gromppFound a second defaults directive fileThis is caused by the [defaults] directive appearing more than once in the topology or force field files for the system - it can only appear once. A typical cause of this is a second defaults being set in an included topology file, .itp, that has been sourced from somewhere else. For specifications on how the topology files work, see GROMACS Manual, Section 5.6.[ defaults ]; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ1 1 no 1.0 1.0One solution is to simply comment out (or delete) the lines of code out in the file where it is included for the second time i.e.,;[ defaults ]; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ;1 1 no 1.0 1.0A better approach to finding a solution is to re-think what you are doing. The [defaults] directive should only be appearing at the top of your .top file where you choose the force field. If you are trying to mix two force fields, then you are asking for trouble. If a molecule .itp file tries to choose a force field, then whoever produced it is asking for trouble.Invalid无效的order for directive defaultsThis is a result of defaults being set in the topology or force field files in the inappropriate location; the [defaults] section can only appear once and must be the first directive in the topology. The [defaults] directive is typically present in the force field file (ffgmx.itp, ffoplsaa.itp, etc), and is added to the topology when you #include this file in the system topology.The "invalid order" error can also pertain to 适合any of the other topology directives. In these cases, the error is usually a result of simply inappropriately re-organizing the topology (hence "out of order" or "out of sequence," as the error implies). A common problem is placing position restraint files for multiple molecules out of order. Recall that a position restraint file can only belong to the [ moleculetype ] block that contains it. For example:WRONG:#include "topol_A.itp"#include "topol_B.itp"#include "ligand.itp"#ifdef POSRES#include "posre_A.itp"#include "posre_B.itp"#include "ligand_posre.itp"#endifRIGHT:#include "topol_A.itp"#ifdef POSRES#include "posre_A.itp"#endif#include "topol_B.itp"#ifdef POSRES#include "posre_B.itp"#endif#include "ligand.itp"#ifdef POSRES#include "ligand_posre.itp"#endifSystem has non-zero total chargeNotifies you that counter-ions反离子may be required for the system to neutralize the charge or there may be problems with the topology.If the charge is a non-integer, then this indicates that there is a problem with the topology. If pdb2gmx has been used, then look at the right hand comment column of the atom listing, which lists the cumulative 累计charge. This should be an integer after every residue (and/or charge group where applicable). This will assist in finding the residue where things start departing from integer values. Also check the capping groups that have been used.If the charge is already close to an integer, then the difference is caused by rounding errors and not a major problem.Note for PME users: It is possible to use a uniform neutralizing background charge in PME to compensate for a system with a net background charge. There is probably nothing wrong with this in principle, because the uniform charge will not perturb 不安the dynamics. Nevertheless, it is standard practice to actually add counter-ions to make the system net neutral.Incorrect number of parametersLook at the topology file for the system. You've not given enough parameters for one of the bonded definitions.Sometimes this also occurs if you've mangled损坏the Include File Mechanism or the topology file format (see: GROMACS Manual Chapter 5) when you edited the file.Number of coordinates in coordinate file does not match topologyThis is pointing out that, based on the information provided in the topology file, .top, the total number of atoms or particles within the system does not match exactly with what is provided within the coordinate file, often a .gro or a .pdb.The most common reason for this is simply that the user has failed to update the topology file after solvating or adding additional molecules to the system, or made a typographical error in the number of one of the molecules within the system. Ensure that the end of the topology file being used contains something like the following, that matches exactly with what is within the coordinate file being used, in terms of both numbers and order of the molecules:[ molecules ]; Compound #molProtein 1SOL 10189NA+ 10In a case when grompp can't find any any atoms in the topology file at all (number of coordinates in coordinate file (conf.gro, 648) does not match topology (topol.top, 0)) and that error is preceded by warnings like:calling /lib/cpp...sh: /lib/cpp: No such file or directorycpp exit code: 32512Tried to execute: '/lib/cpp -I/usr/local/gromacs-...The '/lib/cpp' command is defined in the .mdp filethen your system's C preprocessor, cpp, is not being found or run correctly. One reason might also be that the cpp variable is not properly set in the .mdp file. As of GROMACS version 4.0, grompp contains its own preprocessor, so this error should not occur.This error can also occur when the .mdp file has been edited under Windows, and your cpp is intolerant of the mismatch between Windows and Unix end-of-line characters. If it is possible that you have done this, try running your .mdp file through the standard Linux dos2unix utility.Fatal error: No such moleculetype XXXEach type of molecule in your [ molecules ] section of your .top file must have a corresponding [ moleculetype ] section defined previously, either in the .top file or an included.itp file. See GROMACS Manual section 5.6.1 for the syntax句法description. Your .top file doesn't have such a definition for the indicated molecule. Check the contents of the relevant files, and pay attention to the status of #ifdef and / or #include statements.T-Coupling group XXX has fewer than 10% of the atomsIt is possible to specify separate thermostats恒温器(temperature coupling groups) for each and every molecule type within a simulation. This is a particularly bad practice employed by many new users to Molecular Dynamics Simulations. Doing so is a bad idea, as you can introduce errors and artifacts that are hard to predict. In most cases it is best to have all molecules within a single group, using system. If separate coupling groups are required, then ensure that they are of "sufficient size" and combine molecule types that appear together within the simulation. For example, for a protein in water with counter-ions, one would likely want to use Protein and Non-Protein. The cut-off length is longer than half the shortest box vector or longer than the smallest box diagonal element. Increase the box size or decrease rlistThis error is generated in the cases as noted within the message. The dimensions of the box are such that an atom will interact with itself (when using periodic boundary conditions), thus violating the minimum image convention. Such an event is totally unrealistic and will introduce some serious artefacts. The solution is again what is noted within the message, either increase the size of the simulation box so that it is at an absolute minimum twice the cut-off length in all three dimensions (take care here if are using pressure coupling, as the box dimensions will change over time and if they decrease even slightly, you will still be violating the minimum image convention) or decrease the cut-off length (depending on the force field utilised, this may not be an option).Unknown left-hand XXXX in parameter filegrompp has found an unknown term in the .mdp file fed to it. You should check the spelling of XXXX and look for typographical排字上的errors. Be aware that quite a few run parameters changed between GROMACS 3.x and GROMACS 4.x and the output from grompp will sometimes offer helpful commentary about these situations.Atom index (1) in bonds out of boundsThis kind of error looks likeFatal error:[ file spc.itp, line 32 ]Atom index (1) in bonds out of bounds (1-0).This probably means that you have inserted topologysection "settles" in a part belonging to a differentmolecule than you intended to. in that case move the"settles" section to the right molecule.This error is fairly self-explanatory. You should look at your .top file and check that all of the [molecules] sections contain all of the data pertaining to that molecule, and no other data. That is, you cannot #include another molecule type (.itp file) before the previous [moleculetype] has ended. Consult the examples in chapter 5 of the manual for information on the required ordering of the different [sections]. Pay attention to the contents of any files you have included with #include directives.This error can also arise if you are using a water model that is not enabled for use with your chosen force field by default. For example, if you are attempting to use the SPC water model with an AMBER force field, you will see this error. The reason is that, in spc.itp, there is no #ifdef statement defining atom types for any of the AMBER force fields. You can either add this section yourself, or use a different water model. mdrunStepsize too small, or no change in energy. Converged to machine precision, but not to the requested precisionThis is not an error as such. It is simply informing you that during the energy minimization process it reached the limit possible to minimize the structure with your current parameters. It does not mean that the system has not been minimized fully, butin some situations that may be the case. If the system has a significant amount of water present, then a E pot of the order of -105 to -106 is typically a reasonable value for almost all cases, e.g. starting a Molecular Dynamics simulation from the resulting structure. Only for special purposes, such as normal mode analysis type of calculations, it may be required to minimize further.Further minimization may be achieved by using a different energy minimization method or by making use of double precision-enabled GROMACS.LINCS/SETTLE/SHAKE warningsSometimes, when running dynamics, mdrun may suddenly stop (perhaps after writing several pdb files) after a series of warnings about the constraint algorithms (e.g. LINCS, SETTLE or SHAKE) are written to the log file. These algorithms often used to constrain bond lengths and/or angles. When a system is blowing up (i.e. exploding due to diverging forces), the constraints are usually the first thing to fail. This doesn't necessarily mean you need to troubleshoot the constraint algorithm. Usually it is a sign of something more fundamentally wrong (physically unrealistic) with your system. Perhaps you didn't minimize well enough, have a bad starting structure/steric clashes, are using too large a timestep, or are doing particle insertion in free energy calculations without using soft core. This can also be caused by a single water molecule that is isolated from the other water molecules, somewhere within the system.1-4 interaction not within cut-offSome of your atoms have moved so two atoms separated by three bonds are separated by more than the cut-off distance. This is BAD. Most importantly, do not increase your cut-off! This error actually indicates that the atoms have very large velocities, which usually means that (part of) your molecule(s) is (are) blowing up. If you are using LINCS for constraints, you probably also already got a number of LINCS warnings. When using SHAKE this will give rise to a SHAKE error, which halts暂停your simulation before the "1-4 not within cutoff" error can appear.There can be a number of reasons for the large velocities in your system. If it happens at the beginning of the simulation, your system might be not equilibrated well enough (e.g. it contains some bad contacts). Try a(nother) round of energy minimization to fix this. Otherwise you might have a very high temperature, and/or a timestep that is toolarge. Experiment with these parameters until the error stops occurring. If this doesn't help, check the validity of the parameters in your topology!Simulation running but no outputNot an error as such, but mdrun appears to be chewing up CPU time but nothing is being written to the output files. There are a number of reasons why this may occur: •Your simulation might simply be (very) slow, and since output is buffered减轻, it can take quite some time for output to appear in the respective files. Ifyou are trying to fix some problems and you want to get output as fast aspossible, you can set the environment variable LOG_BUFS to 0 by usingsetenv LOG_BUFS 0, this disables output buffering. Use unsetenvLOG_BUFS to turn buffering on again.•Something might be going wrong in your simulation, causing e.g.not-a-numbers (NAN) to be generated (these are the result of e.g. division by zero). Subsequent calculations with NAN's will generate floating pointexceptions which slow everything down by orders of magnitude. On a SGIsystem this will usually result in a large percentage of CPU time beingdevoted to 'system' (check it with osview, or for a multi-processor machinewith top and osview).•You might have all nst* parameters (see your .mdp file) set to 0, this will suppress阻止most output.•Your disk might be full. Eventually this will lead to mdrun crashing, but since output is buffered, it might take a while for mdrun to realize it can't write.•You are running an executable可执行的compiled with MPI support (e.g.LAM) and did not start the LAM daemon无交互后台程序(lamboot). SeeLAM documentation.Can not do Conjugate结合Gradients with constraintsThis means you can't do energy minimization with the conjugate gradient algorithm 共轭斜度算法if your topology has constraints defined - see here.Pressure scaling more than 1%This error tends to be generated when the simulation box begins to oscillate 摆动(due to large pressures and / or small coupling constants), the system starts to resonate 共振and then crashes. This can mean that the system isn't equilibrated sufficiently before using pressure coupling. Therefore, better / more equilibration may fix the issue.It is recommended to observe the system trajectory prior and during the crash. This may indicate if a particular part of the system / structure is the problem.In some cases, if the system has been equilibrated sufficiently, this error can mean that the pressure coupling constant, tau_p, is too small (particularly when using the Berendsen weak coupling method). Increasing that value will slow down the response to pressure changes and may stop the resonance from occuring.This error can also appear when using a timestep that is too large, e.g. 5 fs, in the absence of constraints and / or virtual sites.Range Checking errorThis usually means your simulation is blowing up. Probably you need to do better energy minimization and/or equilibration and/or topology design.X particles communicated to PME node Y are more than a cell length out of the domain decomposition cell of their charge groupThis is another way that mdrun tells you your system is blowing up. In GROMACS version 4.0, domain decomposition was introduced to divide the system into regions containing nearby atoms (for more details, see the manual or the GROMACS 4 paper). If you have particles that are flying across the system, you will get this fatal error. The message indicates that some piece of your system is tearing apart (hence out of the "cell of their charge group"). Refer to the Blowing Up page for advice on how to fix this issue.There is no domain decomposition for n nodes that is compatible with the given box and a minimum cell size of x nmThis means you tried to run a parallel calculation并行计算, and when mdrun tried to partition your simulation cell into chunks 相当大的数量for each processor, it couldn't. The minimum cell size is controlled by the size of the largest charge group or bonded interaction and the largest of rvdw, rlist and rcoulomb, some other effects of bond constraints, and a safety margin. Thus it is not possible to run a small simulation with large numbers of processors. So, if grompp warned you about a large charge group, pay attention and reconsider its size. mdrun prints a breakdown of how it computed this minimum size in the .log file, so you can perhaps find a cause there. If you didn't think you were running a parallel calculation, be aware that from 4.5, GROMACS uses thread-based parallelism by default. To prevent this, you can either give mdrun the "-nt 1" command line option, or build GROMACS so that it will not use threads线. Otherwise, you might be using an MPI-enabled GROMACS and not be aware of the fact.。
lammps_data文件

lammps_data⽂件⼀、notes:1、不在data⽂件⾥写“#”(注释),否则,容易出错;2、前两⾏不⽤写东西(建议);3、相互作⽤系数可以不⽤写在data⾥边(如pair_coeff等),可有可⽆,but for pair_coeff,最好还是在data⽂件中设定,因为它⽐在in⽂件中设定更加⽅便、简洁。
⼆、主体框架(必须要有):1.数⽬:原⼦、键、⾓、⼆⾯⾓、⾮⼆⾯⾓的的总数⽬2.类型:原⼦、键、⾓、⼆⾯⾓、⾮⼆⾯⾓3.box的⼤⼩:x、y、z4.masses(质量)5.Atoms6.Bonds7.Angles8....三、example(转载:/s/blog_b48a7ac30102w4pr.html)Here is a sample file with annotations in parenthesis and lengthy sections replaced by dots (...). Note that the blank lines are important in this example.LAMMPS Description (1st line of file)100 atoms (this must be the 3rd line, 1st 2 lines are ignored)95 bonds (# of bonds to be simulated)50 angles (include these lines even if number = 0)30 dihedrals20 impropers5 atom types (# of nonbond atom types)10 bond types (# of bond types = sets of bond coefficients)18 angle types20 dihedral types (do not include a bond,angle,dihedral,improper type2 improper types line if number of bonds,angles,etc is 0)-0.5 0.5 xlo xhi (for periodic systems this is box size,-0.5 0.5 ylo yhi for non-periodic it is min/max extent of atoms)-0.5 0.5 zlo zhi (do not include this line for 2-d simulations)Masses1 mass...N mass (N = # of atom types)Bond Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of bond types) Angle Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of angle types) Dihedral Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of dihedral types) Improper Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of improper types) BondBond Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of angle types) BondAngle Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of angle types) MiddleBondTorsion Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of dihedral types)AngleTorsion Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of dihedral types) AngleAngleTorsion Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of dihedral types) BondBond13 Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of dihedral types) AngleAngle Coeffs1 coeff1 coeff2 ......N coeff1 coeff2 ... (N = # of improper types) Atoms1 molecule-tag atom-type q x y z nx ny nz (nx,ny,nz are optional - ... see "true flag" input command)...N molecule-tag atom-type q x y z nx ny nz (N = # of atoms) Velocities1 vx vy vz......N vx vy vz (N = # of atoms)Bonds1 bond-type atom-1 atom-21 angle-type atom-1 atom-2 atom-3 (atom-2 is the center atom in angle)...N angle-type atom-1 atom-2 atom-3 (N = # of angles)Dihedrals1 dihedral-type atom-1 atom-2 atom-3 atom-4 (atoms 2-3 form central bond)...N dihedral-type atom-1 atom-2 atom-3 atom-4 (N = # of dihedrals)Impropers1 improper-type atom-1 atom-2 atom-3 atom-4 (atom-2 is central atom)...N improper-type atom-1 atom-2 atom-3 atom-4 (N = # of impropers)注意:data⽂件不仅能够表⽰坐标,键,键⾓等,⽽且可以输⼊初始速度,键系数等信息。
Surflex-Dock
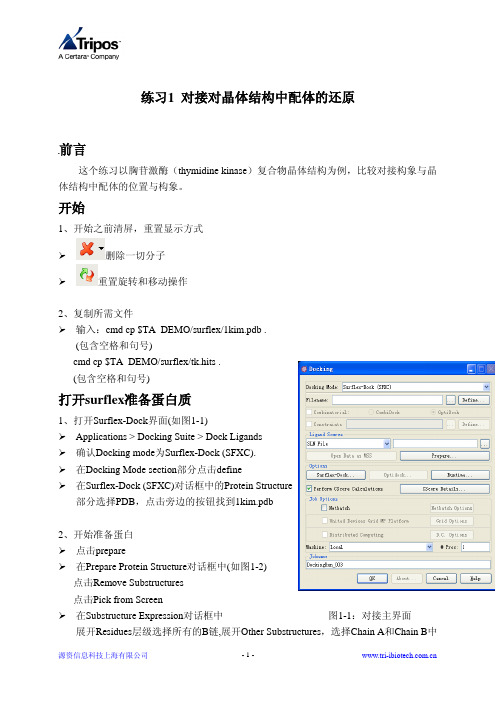
练习1 对接对晶体结构中配体的还原前言这个练习以胸苷激酶(thymidine kinase)复合物晶体结构为例,比较对接构象与晶体结构中配体的位置与构象。
开始1、开始之前清屏,重置显示方式删除一切分子重置旋转和移动操作2、复制所需文件输入:cmd cp $TA_DEMO/surflex/1kim.pdb .(包含空格和句号)cmd cp $TA_DEMO/surflex/tk.hits .(包含空格和句号)打开surflex准备蛋白质1、打开Surflex-Dock界面(如图1-1)Applications > Docking Suite > Dock Ligands确认Docking mode为Surflex-Dock (SFXC).在Docking Mode section部分点击define在Surflex-Dock (SFXC)对话框中的Protein Structure部分选择PDB,点击旁边的按钮找到1kim.pdb2、开始准备蛋白点击prepare在Prepare Protein Structure对话框中(如图1-2)点击Remove Substructures点击Pick from Screen在Substructure Expression对话框中图1-1:对接主界面展开Residues层级选择所有的B链,展开Other Substructures,选择Chain A和Chain B中的SO4_3,选择Waters,点击OK回到Select Substructures对话框,点击OK删除所选项目。
3、从复合物中提取配体点击Extract Ligand Substructures.在Select Substructures对话框选择A/THM1,点击OK4、分析蛋白结构和对接准备(又回到Prepare Protein Structure对话框),点击Analyze Selected Structure点击与Repair Backbone, Repair Sidechain, and Termini Treatment相关的show按钮找到相关残基的位置。
Amber使用笔记B3

Amber使用笔记B3- Case Study: Folding TRP Cage (Advanced analysis and clustering)---Written by PanLu Introduction:1)"Trpcage" ,含有20个氨基酸残基,是可以显示两态折叠性质且在室温下稳定的最小的一个氨基酸;Section 1:创建构型1)建一个线性的多肽链时,Leap工具不会自动识别这个链的两端,因此,我们要明确这个链的N端(在氨基酸前加一个N)和C端(在氨基酸前加一个C);使用命令:肽链名= sequence {氨基酸按顺序排列}2)文献上使用的leaprc.ff99力场在高版本的Amber11中没有,我使用了leaprc.ff99SB这个力场;3)分别保存了一个lib文件(saveoff命令,AmberTools13 Reference Manual P125,save UNITs and PARMSETs using the Object File Format, 这个文件是用来干什么的?)和一个pdb文件(savepdb命令,便于VMD可视化)。
Section 2:创建prmtop和inpcrd文件1)文献中提到使用了FF99力场,但Amber6版本并没有FF99力场,作者实际使用了修改了phi/psi二面角参数的FF99力场;由于这种的修正可能会导致glycine参数化的问题,在Amber9版本中这种修正被删除,而FF99力场也被FF99SB力场替代;2)在生成prmtop文件和inpcrd文件之前,先使用了loadoff命令调用了lib文件,然后使用loadamberparams命令上载了一个替代的二面角参数文件,之后才使用saveamberparm命令保存prmtop和inpcrd文件;3)在我们的工作中,直接使用了FF99SB力场,不考虑这种二面角的修正;Section 3:优化Section 4:heating1) 既然已经在前面做了结构优化,为什么不直接做动力学,而要先做heating ? 2) “MD simulations of 100 ns were performed at 300 K, but all were kineticallytrapped on this time scale, showing strong dependence on initial conditions and failing to converge to similar conformational ensembles. We therefore increased the temperature to 325 K.” 因此,在这个工作中,没有在300K 温度下做MD ,而是将温度升至325K;3) 由于我们的初始结构是人工构建而不是实验的晶体结构,在开始heating 时相比实验结构会不太稳定,所以,在heating 过程中,该工作time step 设置为0.5fs ,而在平衡之后的MD 过程,time step 为2fs ;4) Heating 过程分7步,可以发现从300K~325K 过程,花了较长的时间和步数,为什么?5)6)Tutorial中提供的csh脚本在我们的服务器上并不适合,作业并不能依次提交,因此,我们还是手动的一个作业一个作业地提交;7)使用了一个节点4个cpu并行的计算,大约10个小时可完成一个作业,并行化的使用为:Export DO_PARALLEL=’mpirun –np 4’DO_PARALLEL $AMBERHOME/exe/sander ……Section 5:Production MD1)在做完之前一系列的准备工作之后,可以开始在325K下的long production(?)MD模拟。
gromacs中有机溶剂力场选择

gromacs中有机溶剂力场选择在GROMACS(GROningen MAchine for Chemical Simulations)中,选择合适的有机溶剂力场通常涉及到在模拟系统中使用正确的力场参数。
GROMACS支持多种力场,而力场的选择通常取决于模拟系统中的分子种类。
以下是一些GROMACS中常用的力场以及适用的情境:1.GROMOS Force Field:•适用于生物大分子如蛋白质、核酸等的模拟。
2.OPLS-AA (Optimized Potential for Liquid Simulations AllAtom):•适用于溶液中的有机小分子和生物大分子。
3.CHARMM (Chemistry at HARvard Molecular Mechanics):•适用于生物分子、溶液中的小分子以及膜蛋白等。
4.AMBER (Assisted Model Building with Energy Refinement):•适用于生物分子、蛋白质、核酸等,也有适用于溶液中小分子的版本。
5.GROMACS提供的其他力场:•GROMACS还包含一些其他的力场,如Lennard-Jones力场、Buckingham力场等,可以用于一些特殊情况或自定义模拟系统。
在选择力场时,你需要考虑模拟系统的性质、分子的种类、溶剂等因素。
力场的参数通常在相关文献中有详细描述,你可以查阅相应的力场手册或原始文献以获取参数信息。
此外,GROMACS的官方网站和文档也提供了一些关于力场选择和使用的指导。
在创建模拟系统时,你需要使用GROMACS工具如pdb2gmx来为你的分子生成拓扑文件,并确保使用正确的力场选项。
在模拟运行时,使用mdrun命令,确保正确选择了力场参数文件。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Table 25 . Atom types--AMBER (Page 1 of 4)
general class atom
type1
description
hydrogen types
H amide or imino hydrogen
HC explicit hydrogen attached to carbon
HO hydrogen on hydroxyl oxygen
HS hydrogen attached to sulfur
HW hydrogen in water
H2 amino hydrogen in NH2
H3 hydrogen of lysine or arginine (positively charged) all-atom carbon types2
C sp2carbonyl carbon and aromatic carbon with hydroxyl substituent in tyrosine
CA sp2aromatic carbon in 6-membered ring with 1 substituent
CB sp2aromatic carbon at junction between 5- and 6-membered rings
CC sp2aromatic carbon in 5-membered ring with 1 substituent and next to a nitrogen
CK sp2 aromatic carbon in 5-membered ring between 2 nitrogens and bonded to 1 hydrogen (in purine)
CM sp2 same as CJ but one substituent
CN sp2aromatic junction carbon in between 5- and 6-membered rings
CQ sp2carbon in 6-membered ring of purine between 2 NC nitrogens and bonded to 1 hydrogen
CR sp2 aromatic carbon in 5-membered ring between 2 nitrogens and bonded to 1 H (in his)
CT sp3 carbon with 4 explicit substituents
CV sp2 aromatic carbon in 5-membered ring bonded to 1 N and bonded to an explicit hydrogen
CW sp2 aromatic carbon in 5-membered ring bonded to 1 N-H and bonded to an explicit hydrogen
C* sp2aromatic carbon in 5-membered ring with 1 substituent
united carbon types3
CD sp2aromatic carbon in 6-membered ring with 1 hydrogen
CE sp2 aromatic carbon in 5-membered ring between 2 nitrogens with 1 hydrogen (in purines)
CF sp2 aromatic carbon in 5-membered ring next to a nitrogen without a hydrogen
CG sp2aromatic carbon in 5-membered ring next to an N-H CH sp3 carbon with 1 hydrogen
CI sp2 carbon in 6-membered ring of purines between 2 NC nitrogens
CJ sp2 carbon in pyrimidine at positions 5 or 6 (more pure double bond than aromatic with 1 hydrogen)
CP sp2 aromatic carbon in 5-membered ring between 2 nitrogens with one hydrogen (in his)
C2 sp3 carbon with 2 hydrogens
C3 sp3 carbon with 3 hydrogens nitrogen types
N sp2 nitrogen in amide group
NA sp2nitrogen in 5-membered ring with hydrogen attached
NB sp2 nitrogen in 5-membered ring with lone pairs NC sp2 nitrogen in 6-membered ring with lone pairs
NT sp3 nitrogen with 3 substituents
N2 sp2 nitrogen in base NH2 group or arginine NH2 N3 sp3 nitrogen with 4 substituents
N* sp2nitrogen in purine or pyrimidine with alkyl group attached
oxygen types
O carbonyl oxygen
OH alcohol oxygen
OS ether or ester oxygen
OW water oxygen
O2 carboxyl or phosphate nonbonded oxygen sulfur types
S sulfur in disulfide linkage or methionine
SH sulfur in cystine
phosphorus
P phosphorus in phosphate group
ion types
CU copper ion (Cu+2)
CØ calcium ion (Ca+2)
I iodine ion (I-)
IM chlorine ion (Cl-)
MG magnesium ion (Mg+2)
QC cesium ion (Cs+)
QK potassium ion (K+)
QL lithium ion (Li+)
QN sodium ion (Na+)
QR rubidium ion (Rb+)
other
LP lone pair
1From Weiner et al. (1984) and Weiner et al. (1986).2 Non-hydrogen-containing carbons.
3 United-atom carbons with implicit inclusion of hydrogens.。