设计确认SOP
设计确认、安装确认、运行确认、性能确认

设计确认(DQ)、安装确认(IQ)、运行确认(OQ)、性能确认(PQ)设备/仪器的选型(DQ):设计确认应当证明厂房、设施、设备的设计符合预定用途和要求;记录设施、设备系统按照GMP要求进行设计的书面证据,这些书面证据说明所设计的设施、设备系统适合于目的用途,并且所设计的设施、设备系统组元和单元依据现行的工程理论与实践原则与考量和用户要求。
1设计确认主要是对设备选型和订购设备的技术规格、技术参数和指标适用性的审查,由需求使用部门实施。
2确认主要的工艺参数,物料衡算数据选取所需设备,检验仪器要根据检验精密度要求选取所需仪器。
3 查找设备的说明书或参数介绍,考察设备是否适合生产工艺、检验精度、校准、维修保养、清洗方面的要求,是否符合GMP要求。
4 从技术和经济两项指标选择合适的供应商。
5 根据设备参数和工艺要求设计安装图纸,根据工艺检验精度要求确定检验仪器的精度要求。
6设计确认由设备需要部门直接的技术人员和工程人员合作完成。
7 确认设备的变更而导致的其它相关的变更,包括其它设备、厂房设施等的需要。
安装确认(IQ):安装确认应当证明厂房、设施、设备的建造和安装符合设计标准;安装确认应列出所有需要书面记录的可识别信息,包括:设备的名称、系统的描述、设备识别编号(硬件和软件)、地点、辅助设施的要求、连接和安装特点。
IQ应该核实设备与采购清单是否吻合,所有图纸、手册、备件、供货商信息和其它相关文件都要齐全。
1核对到厂生产设备或仪器、配件、配套设施、文件等是否符合选型要求,是否齐全。
2参照设备说明书和其他技术文件,核对该生产设备或仪器的各项参数是否符合设计要求、是否符合工艺流程和质量控制、检验精度要求。
检查设备是否符合GMP的要求。
3安装阶段,设备、管道、辅助设施和仪器均应按照设计图纸或厂家安装要求来进行安装和检查。
同时,安装确认应包括该设备或系统的组成部分,辅助管道和量器的检定。
4将供货单位的技术资料归档,收集制定有关操作文件、记录。
sop流程图制作

在分析流程图的过程中,我们发现了一些问题和瓶颈,包括流程不清晰、环节冗余、缺乏审核和审批 环节等。这些问题影响了流程的效率和执行效果,需要采取措施进行改进。
制定改进措施和方案
总结词
制定改进方案
详细描述
针对以上问题和瓶颈,我们制定了以下改进措施和方案
1. 优化流程图设计
重新梳理流程,简化环节,明确各个环节的职责和要求 。
收集和分析流程数据
收集流程相关数据
01
ห้องสมุดไป่ตู้
确定数据收集的范围和目标
在制作SOP流程图之前,需要明确收集哪些与流程相关的数据,如流
程步骤、时间、人力、资源等。
02
选择合适的数据收集工具
根据收集的数据类型和目标,选择合适的数据收集工具,如调查问卷
、工作日志、观察表等。
03
确保数据质量和准确性
在收集数据过程中,要确保数据的真实性和准确性,对数据进行校验
03
生产制造流程
包括原材料采购、生产计划、生产排 程、生产执行、质量检测等环节。
确定业务场景中的角色和职责
订单处理流程中的角色
订单处理员、审核员、仓库管理员、配送员等。
客户服务流程中的角色
客服代表、售后服务专员、投诉处理专员等。
生产制造流程中的角色
生产计划员、生产调度员、生产工人、质量检测员等。
03
实施改进措施和方案计划
建立项目小组
成立一个由流程图制作专家、业务骨干和项目管 理人员组成的项目小组,明确职责和任务。
制定改进计划
根据分析结果,制定详细的改进计划,包括改进 目标、实施步骤、时间安排和责任人等。
分析现状
对现有的SOP流程图进行全面分析,找出存在的 问题和改进点。
SOP标准化及制作技巧

SOP的标准格式
相关文件和参考资料
说明在阅读和操作此SOP前应了解和熟悉的文件和资料
设备示意图
用图片或照片的形式展示SOP所涉及的主要设备 编制并说明设备各部件的编号
15
SOP的标准格式
操作前准备
常用工具 列出本操作所涉及的操作及维修工具、防护 工具和清洁工具等; 相关记录 列出进行本操作所使用的记录; 安全措施 描述操作前实施的安全防护措施,包括佩戴 劳防用品,确认各项安全措施已经到位,了解操作过 程中发生安全事故的应急措施等; 准备工作 设备的点检、描述操作前需要就绪的工作, 包括作业内容和要点提示。
20
SOP编写要点
精确性 不但要正确,更加要精确 例: 某一个回流焊操作准备工作的SOP描述 正确:温度在控制在范围之内 精确:回流焊温度在范围之内,一级温度210℃、二级温度 230℃、三级270℃、四级220℃。温度公差为一级:。。要 点
准确数字来源 SIP、产品规格书
21
SOP编写要点
可操作性
7
SOP在公司文件中的位置
SOP属于品质管理体系文件中的三阶文件
手 册
程序 文件
规范/作业 指导书
报告表格
QM: Quality Manual QP: Quality Procedure SOP: Standard Operation Procedure WI: Working Instruction RF: Report/Form
SOP标准化及制作技巧
SOP现状情况
大家都懂得 文件统一性 受控管理都存在不顺畅与隐患
2
SOP的含义
SOP是哪几个英文单词的缩写?
Standard Operation Procedure(标准作业 指导书)
标准作业指导书(S.O.P.) 制作要领

SOP是最基本的也是最重要的职责,一份完整而且最新最标准的SOP不但可以规范生产流程而且影响整个公司的运作。很多资深的管理者这样概括一个公司:“一个公司有两本手册就可以了,一本是红本子(质量手册),一本是蓝本子(SOP),”可见SOP的延伸范围及重要性。
4. 站别: 标示此工作站位于工程段中第几站,以利排线。
5. 作业内容: 标示此工作站的工作项目一工作站的作业确认(优先作业)。
b. 作业顺序的排定。
c. 对与安规零件须标明。
6. 注意事项:标明每项工作项目的内容与要求。
(重点)
a. 关于注意事项的顺序编号须与作业内容对应。
(重点)
a. 关于版本编码的规定,用大写的英文字标示,如:”A”、”B”….。
b. 若26个字母用完,则改使用两个英文字,如:”AA”、”AB”….。
12. 判定: 注明作业的标准.
(重点)
a. 关于判定标示的顺序编号须与作业内容对应。
b. 判定的规范以客户的规定为优先;其次为设计的要求;再来为PH作业的规范。
2. 针对样品进行结构分析,将各工程段(SMT、AI、加工、插件、修补、组立)区分,并核对BOM分阶是否正确。
3. 确立零件及组件的加工方式与规格;安规零件的区分。
4. 制定生产的流程,及工作站的安排并编写流程图。
5. 确认工站的安排是否合理,是否有达到平衡工时,是否有重复确认及防止不良流出的功能。
如下是制作SOP(standard operation procedure/标准作业指导书)的一些数据,供参考.
标准作业指导书(S.O.P.) 制作要领
定义:
设计确认、安装确认、运行确认、性能确认

设计确认(DQ)、安装确认(IQ)、运行确认(OQ)、性能确认(PQ)设备/仪器的选型(DQ):设计确认应当证明厂房、设施、设备的设计符合预定用途和要求;记录设施、设备系统按照GMP要求进行设计的书面证据,这些书面证据说明所设计的设施、设备系统适合于目的用途,并且所设计的设施、设备系统组元和单元依据现行的工程理论与实践原则与考量和用户要求。
1设计确认主要是对设备选型和订购设备的技术规格、技术参数和指标适用性的审查,由需求使用部门实施。
2确认主要的工艺参数,物料衡算数据选取所需设备,检验仪器要根据检验精密度要求选取所需仪器。
3 查找设备的说明书或参数介绍,考察设备是否适合生产工艺、检验精度、校准、维修保养、清洗方面的要求,是否符合GMP要求。
4 从技术和经济两项指标选择合适的供应商。
5 根据设备参数和工艺要求设计安装图纸,根据工艺检验精度要求确定检验仪器的精度要求。
6设计确认由设备需要部门直接的技术人员和工程人员合作完成。
7 确认设备的变更而导致的其它相关的变更,包括其它设备、厂房设施等的需要。
安装确认(IQ):安装确认应当证明厂房、设施、设备的建造和安装符合设计标准;安装确认应列出所有需要书面记录的可识别信息,包括:设备的名称、系统的描述、设备识别编号(硬件和软件)、地点、辅助设施的要求、连接和安装特点。
IQ应该核实设备与采购清单是否吻合,所有图纸、手册、备件、供货商信息和其它相关文件都要齐全。
1核对到厂生产设备或仪器、配件、配套设施、文件等是否符合选型要求,是否齐全。
2参照设备说明书和其他技术文件,核对该生产设备或仪器的各项参数是否符合设计要求、是否符合工艺流程和质量控制、检验精度要求。
检查设备是否符合GMP的要求。
3安装阶段,设备、管道、辅助设施和仪器均应按照设计图纸或厂家安装要求来进行安装和检查。
同时,安装确认应包括该设备或系统的组成部分,辅助管道和量器的检定。
4将供货单位的技术资料归档,收集制定有关操作文件、记录。
SOP标准与制作技巧

精品课件
六、SOP的标准格式
6.1 关键因素; 6.2 相关文件和参考资料; 6.3 设备示意图; 6.4 操作前准备; 6.5 操作步骤; 6.6 常见故障及简单排除方法; 6.7 紧急情况的处理; 6.8 流程图。
精品课件
6.1关键因素
➢ 环境因素 根据环境管理体系的要求排查出与本操作有关的因素; ➢ 安卫因素 根据安卫管理体系的要求排查出与本操作有关的因素; ➢ 卫生因素 根据HACCP管理体系的要求排查出与本操作有关的因素; ➢ 质量因素 根据技术文件的要求与本操作有关的质控点因素。
精品课件
6.5操作步骤
分为两栏 作业内容和要点提示; 作业内容应包括整个操作步骤的全部内容; 要点提示是对作业内容的强调和解释,是“最好知道”或 是“关键因素”的信息,但绝不是操作步骤。
6.6常见故障及简单排除方法
一个合格的操作员应了解操作设备的基本情况并熟悉一些 常见故障的简单操作方法; 分为三栏 a故障现象 b故障内容 c排除方法和使用工具; 简单明了,容易操作。
SOP标准与制作技巧、SOP的由来 三、SOP的重要作用 四、SOP在公司文件中的位置 五、SOP的制作 六、SOP的标准格式 七、SOP的编写要点 八、FT SOP编写注意事项 九、临时SOP管控 十、SOP增发
精品课件
一、SOP的含义
SOP是哪幾個英語單詞的縮寫?
1.SOP统一由制程管制部门制作(包括变更修改); 2.制程管制部门根据新机种试作完成之排站进行图片的拍摄 及作业内容来安排制作SOP,作为量产之作业依据; 3.SOP制作完成后需经制造、工程、品管签核确认OK后送至 文件管理室发行,使用单位根据生产需要申请使用; 4.相关单位签核重点
验证SOP的基本原则

验证SOP的基本原则目的:书面的验证SOP基本原则是为了帮助药物生产企业理解和保持验证规划符合GMP 要求。
责任:质保部部长负责起草和修订验证规程,相关部门负责人负责实施。
规程:1.SOP的目的和范围质量保证部门负责起草、升级和维护GMP,验证SOP要求逐步实施验证。
2.验证的定义⏹证明任何程序、生产工艺、设备、物料、活动或系统确实能导致预期结果,符合GMP基本原则的活动;⏹高保证提供某个特定工艺能符合预定标准、质量属性和特性的文件化证明;⏹获得和文件化证明一个方法能可靠的产生特定限度的预期结果;⏹证明生产和控制中的任何工艺、规程、活动、物料、系统、设备能获得设计和预定的结果。
3.验证的理由验证是商业中一种很好的规范,目的是新产品的第一次生产就能获得成功。
⏹政府规范GMP已在全球多个国家建立,GMP主要作为一个指导原则,并不提供如何去达到GMP要求,验证主计划和相关SOP实际上确定了责任,谁、什么时间、哪里以及有多少需要论证的。
⏹质量的保证由于有限的取样不能控制特定工艺的全部质量,验证提供了产品生产的质量一致性。
验证可减少日常生产中的解决纠纷,最终可降低顾客的设诉和产品召回。
⏹减少开支低水平的工艺通常会导致由于必要的重新检查、重新检测、重新加工和拒收带来的费用,验证可使用工艺最优化,从而使这些费用最小化。
4.验证的背景4.1历史自1970年以来,在医药产品的生产和质量控制中,验证变得日益重要,我国在1998年版GMP中已将验证单列一章,对制药企业进行了要求。
4.2法规要求药品管理法要求药品生产企业必须按照GMP组织生产,制药企业在遵守GMP的同时,还应该符合其他政策、指南、指导原则,这是一种好的管理和商业规范。
4.3上市要求由于服务对象(政府、医生、药剂师、病人等)对药品的安全性、有效性、效能以及消费的价值的更加关心,所以对制药行业的要求比原料变得要高。
制药行业产品质量必须符合健康和法规的要求,制药行业有义务验证工艺符合GMP要求。
设计审核确认单-SOP标准方法确认

设计审核确认单—标准方法确认
任务书编号
ISD/FOR-01300D5-02
项目名称
方案书编号
项目特性
申 请人
简述
申计任务书》、《设计方案/计划书》上的要求; l 是否满足国家、行业、企业的相关标准; l 文件格式及各项要素是否正确; l 依此方法作业能否达到测试标准上的各项要求; l 在满足测试标准要求的前题下,相关测试项目是否做到合理的删减, l 以简化检测步骤,提高检测效率,适应于大批量生产的需要;
l 测试方法应简明易懂,以适应不同层次的作业人员理解执行;
l QC、QA的测试项目及对应表单表格是否合理设计;
审 核 结果
审核意见及不 合 格 项 和改 善 要 求
开发部内部自审
品质部审核
生产部审核
上级主管审核
批注
合格,文件资料以正式文本发行存档。 不合格,按要求改善。 其它:
注:SIP SOP编制完后,由设计人员填写此表格,首先自审后,再由相关部门逐一审核,然后呈送上级主管处进行管理 审核。审核结果栏内用“√ × ? ”来表述“合格 不合格 欠缺”等结果。自审填于第一列 内, 其余类同。
临床试验机构工作制度、设计规范与SOP

5
• 格式
– 格式并无统一要求,但同一机构的所有SOP在编制和印刷形式上应尽 可能地保持一致,以利于查阅、检索和管理
– 封面页:文件的题目,文件号,版本号,起草或修订者,审核者, 批准者,执行日期,文件分发部门。
6
• 撰写要点
– 依据充分:SOP的内容应符合我国GCP、有关法规及药物临床研究技 术指导原则,符合国际通用的准则和指导原则
– 简明准确:内容条理清楚,简明准确,采取描述性语言,避免回顾 性或评论性的语言,避免容易引起歧义或含糊笼统的语言
– 可操作性强:起草时可参考有关文献、手册或仪器说明书的内容, 但也不可完全照搬,应当按照实际情况进行适当的修改。可操作性 强,所写内容应当经过适当培训就能够按照其内容进行操作
– 避免差错:SOP涉及的关键词、专业术语、计量单位和符号、有效数 字等应当按照国家有关标准或国际通用原则书写。避免使用已废弃 的或不规范的术语、计量单位、符号和汉字等。
7
• 实施
– SOP一经生效就具有内部法规性质,必须严格遵守 – SOP制订生效后要对有关人员进行培训,合格者才能上岗 – SOP放置地点要方便有关人员随时查阅参考 – 临床试验机构的所有人员都应当熟悉并遵循各自的SOP,对与SOP不
4
• 范围与内容
– 临床试验机构应当制定能够覆盖新药临床试验的所有全部工作的SOP, 使临床试验所有操作环节及管理环节都有相应的SOP
– 通用性SOP:临床试验方案及其附属文件设计的SOP,试验药物管理 的SOP,不良事件处理与报告的SOP,人员培训的SOP,数据管理的 SOP,实验室质量控制和仪器设备的SOP,临床试验机构质量保证系 统的SOP,文件资料管理的SOP,各类研究和管理人员工作职责的 SOP,等
药品GMP认证中SOP的制定规范及验证要求

制药企业的标准操作规程(SOP)或简称为标准操作程序,是制药企业文件系统的主要组成部分。
制定SOP的步骤制药企业一定要根据本企业药品生产的实际,围绕GMP的要求:1、制定编制SOP的大纲;2、确定具体的SOP题目;3、组织、动员、培训人员动手编写。
编写SOP的基本要求编写时一定要做到:1、全面、无漏项、不重复;2、科学、实用;3、文字精练、语言简明;4、可操作性强。
SOP属于标准类文件,每一个标准文件应有统一的格式文头。
文头内容包括:企业名称、文件名称、文件编号、制(修)定日期、审核日期、批准日期、执行日期、颁发部门、分发部门等。
文件分类6、高效过滤器检漏或性能检查的规则;7、水处理器安装的操作规程;8、设备维修操作规程和记录;9、无菌区控制的测试规程;10、湿粒混合机的维修和清洗规则;11、混合整粒器的维修和清洗规则;12、V-混合器的维修和清洗规则;13、包衣机的维修和清洗规则;14、洗瓶机和干燥机的维修和清洗规则;15、灭菌道的维修和清洗规程;16、标准自动联合贴签器的维修和清洗规则;17、标准传送器的维修规则;18、无菌区过滤系统的维修规则;19、粉碎机清洗和维修保养标准操作程序;20、计量仪器的管理程序;21、洁净区生产操作间清洁标准操作规程;22、非洁净区房间清洁标准操作规程;23、容器清洁标准操作规程;6、物料平衡管理操作规程;7、异常情况处理规程;8、不合格品管理与处理规程;9、中间控制操作规程;10、记录填写规范管理规程;11、清洁工具的清洁与管理规程;12、容器及设备的清洁与管理规程;13、清洁剂、消毒剂的配制与使用规程;14、地漏的清洁消毒规程;15、进风、回风装置清洁与管理规程;16、洁净区的清洁卫生与使用规程;17、清场管理规程;18、成品零头管理规程;19、工段间原辅料、半成品、成品的交接、储存与发放规程;20、废标签管理与销毁规程;21、生产区、仓库的废弃物处理规程;22、原辅料外包装的清洁、拆除操作规程;23、原辅料粉碎的操作规程;24、筛粉的操作规程;25、配料的操作规程;26、混合制粒的操作规程;27、沸腾干燥的操作规程;28、烘箱干燥的操作规程;29、整粒的操作规程;30、颗粒总混的操作规程;31、淀粉浆配制的操作规程;32、HPMC液配制的操作规程;33、压片的操作规程;34、胶囊充填的操作规程;35、包衣的操作规程;36、薄膜包衣的操作规程;37、糖浆配制的操作规程;38、明胶糖浆配制的操作规程;39、HPMC包衣液配制的操作规程;40、包装操作规程;41、包装工序清场的操作规程;等等。
SOP、SIP、OI判定标准

6、格式定义
根据3中三阶规范化标准文件的制作要求,特定义文件如下: 6.1 SOP 主要针对部门:工程部 文件构架(正文): ×× ×× ×× ××作业标准规范 1、目的 规范工程部××参数设定,确保××的规范性,保证产品质 量。 2、适用范围 适用于×× ××工序。 3、权责 工程部严格按照工艺要求,进行工艺参数设定,若有调 整依据经批准的【工程变更变更通知书】执行; 4、作业程序 相关工艺参数设定,可用表格显示。(如温度、压强、 时间等)
××
按顺序检查
按过程检查
6、格式定义
6.3 SIP主要针对部门:品管检验 文件构架(正文):
×× ×× ×× ××检验标准
1、目的 为××检验提供依据,确保××符合要求。 2、适用范围 适用于本公司××检验作业。 3、权责 品管部按本标准要求进行××检验,最终达成工艺 要求和品质保证。 4、检验标准(用图表表示如下:)
4.2区别
4.2.1、文件性质区别 SOP:生产之前制程工程师写出的标准技 术文件 ,是ISO控制文件; OI: 管理性文件,是现场用来管理生产 用的文件; SIP: Standard Inspection Process 标准检 验过程。
4.2区别
4.2.2、适用对象区别 SOP:用来给专业技术人员,I. E. 工程師与 初学者参考用的; (主要指工程部 参 数设定) OI:是给已经训练过、考核通过的人看的, 每天工作时看的,并且每天看还有用的; (主要指车间生产制造程序) SIP:是针对经过培训之检验人员检验判定 标准的规范。(主要指品管部门)
4.2区别
3.2.3、用途区别 SOP:只告诉你要做什么 ,但不会告诉你具 体怎么做告诉你怎么做; OI: 告诉你怎么做+注意事项,将不稳当的 地方写在注意事项里,将容易犯错的 细节体现出来,起到提醒的作用,防 止人犯错; SIP:只告诉你检验的标准是什么,但不会告 诉你要做什么、怎么做。
确认
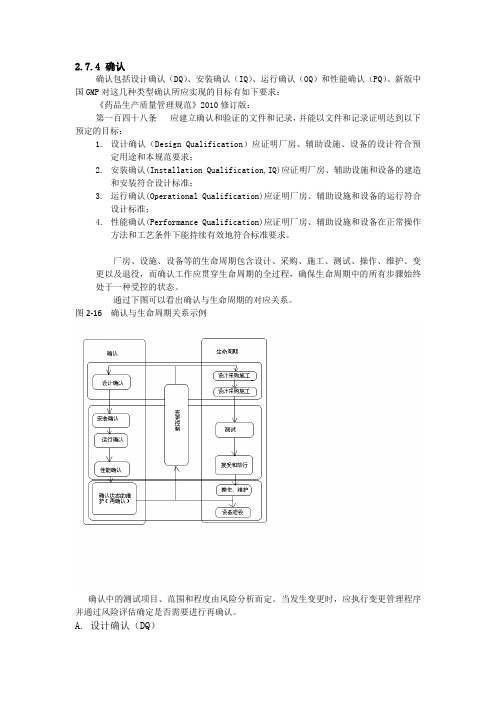
2.7.4 确认确认包括设计确认(DQ)、安装确认(IQ)、运行确认(OQ)和性能确认(PQ)。
新版中国GMP对这几种类型确认所应实现的目标有如下要求:《药品生产质量管理规范》2010修订版:第一百四十八条应建立确认和验证的文件和记录,并能以文件和记录证明达到以下预定的目标:1.设计确认(Design Qualification)应证明厂房、辅助设施、设备的设计符合预定用途和本规范要求;2.安装确认(Installation Qualification,IQ)应证明厂房、辅助设施和设备的建造和安装符合设计标准;3.运行确认(Operational Qualification)应证明厂房、辅助设施和设备的运行符合设计标准;4.性能确认(Performance Qualification)应证明厂房、辅助设施和设备在正常操作方法和工艺条件下能持续有效地符合标准要求。
厂房、设施、设备等的生命周期包含设计、采购、施工、测试、操作、维护、变更以及退役,而确认工作应贯穿生命周期的全过程,确保生命周期中的所有步骤始终处于一种受控的状态。
通过下图可以看出确认与生命周期的对应关系。
图2-16 确认与生命周期关系示例确认中的测试项目、范围和程度由风险分析而定。
当发生变更时,应执行变更管理程序并通过风险评估确定是否需要进行再确认。
A.设计确认(DQ)新的厂房、设施、设备确认的第一步为设计确认(DQ).设计确认是有文件记录的对厂房、设施、设备等的设计所进行的审核活动,目的是确保设计符合用户所提出的各方面需求,经过批准的设计确认是后续确认活动(如安装确认,运行确认,性能确认)的基础。
通常,设计确认中包括以下的项目:用户需求说明文件(User Requirement Specification,URS)用户需求说明文件时从用户角度对厂房、设施、设备等所提出的要求。
需求的程度和细节应与风险、复杂程度相匹配,其中可以针对待设计的厂房、设施、设备等考虑以下内容:◆法规方面的要求(GMP要求、环保要求等)◆安装方面的要求和限制(尺寸、材质、动力类型、洁净级别等)◆功能方面的要求◆文件方面的要求(供应商应提供的文件及格式要求,如图纸、维护计划、使用说明、备件清单等)下表为建议的用户需求说明文件模板,具体内容可根据实际需要进行增减。
药品GMP认证中SOP的制定规范及验证要求

最终灭菌小容量注射剂主要的SOP
14、装箱工序标准操作规程; 15、批号管理规程; 16、人员净化标准操作规程; 17、物料净化标准操作规程; 18、生产前准备标准操作规程; 19、清场标准操作规程; 20、洁净室清洁卫生标准操作规程; 21、洗涤液、消毒剂使用标准操作规程;
整理ppt
最终灭菌小容量注射剂主要的SOP
整理ppt
最终灭菌小容量注射剂主要的SOP
1、配料的操作规程; 2、容器清洗及处理标准操作规程; 3、滤器清洗及处理标准操作规程; 4、滤膜安装处理及气泡点测定标准
操作规程; 5、输药管道清洗及处理标准操作规
程; 6、药液过滤标准操作规程;
整理ppt
最终灭菌小容量注射剂主要的SOP
7、安瓿洗涤、干燥标准操作规程; 8、灌封工序标准操作规程; 9、灭菌工序标准操作规程; 10、灯检工序标准操作规程; 11、印包工序标准操作规程; 12、印字机标准操作规程; 13、包装机标准操作规程;
整理ppt
五、物料处理的SOP
6、半成品、成品取样标准操作程序; 7、原材料取样标准操作程序; 8、原材料出入库管理程序; 9、原材料验收、化验规程; 10、原材料发放、记帐规则; 11、包装材料验收、检验、入库程序;
整理ppt
五、物料处理的SOP
12、包装材料发放、记帐规则; 13、标签验收、入库、记帐程序; 14、标签发放、记帐规则; 15、成品验收、入库、记帐规则; 16、成品销售规则;
验证文件应包括验证方案验证报告评价和建议批准人证方案验证报告评价和建议批准人验证工作的要求验证工作的要求在我国在我国药品生产质量管理规范药品生产质量管理规范19981998年修订的年修订的第二十一第二十一对生产激对生产激素类抗肿瘤类化学药品的设备和空气素类抗肿瘤类化学药品的设备和空气净化系统净化系统三十五三十五对用于生产和检对用于生产和检验的仪器仪表量具衡器等验的仪器仪表量具衡器等三十三十对生产设备对生产设备七十一条七十一条对工艺对工艺用水中对验证也提出了不同的要求
药品GMP认证中SOP的制定规范及验证要求

与民同富 与家同兴 与国同强
编写SOP的基本要求
编写时一ห้องสมุดไป่ตู้要做到: 1、全面、无漏项、不重复; 2、科学、实用; 3、文字精练、语言简明; 4、可操作性强。
与民同富 与家同兴 与国同强
口服固体制剂生产主要的SOP
27、沸腾干燥的操作规程; 28、烘箱干燥的操作规程; 29、整粒的操作规程; 30、颗粒总混的操作规程; 31、淀粉浆配制的操作规程; 32、HPMC液配制的操作规程; 33、压片的操作规程;
与民同富 与家同兴 与国同强
口服固体制剂生产主要的SOP
2、验证工作是质量保证的一种手段, 质量保证靠“验证”实现对GMP的承 诺。
与民同富 与家同兴 与国同强
验证工作的要求
三、我国GMP对验证的要求
我国GMP(2010年修订)对验证单独列为 一章(第七章):
第一百三十八条:企业的厂房、设施、设备和 检验仪器应当经过确认,应当采用经过验证的生 产工艺、操作规程和检验方法进行生产、操作和 检验,并保持持续的验证状态。
一、通用技术方面的SOP
9、容器的使用规程; 10、生产区墙壁和地面的保养维护管理规程; 11、职工体检规划; 12、计量管理制度和实施办法; 13、用户意见处理规程; 14、退货处理规程(紧急退货处理程); 15、原料和包装材料供应厂家选择与质量审计规 程;
与民同富 与家同兴 与国同强
一、通用技术方面的SOP
程; 等等。
与民同富 与家同兴 与国同强
四、质量控制的SOP
1、质量控制通用规则; 2、标准溶液配制的通用规则; 3、质量控制无菌区的操作规程; 4、实验室安全规则; 5、实验动物的护理、使用和处理规程; 6、原料、中间体和成品的处理程序; 7、对批量生产中某批失败的调查和分析;
URS、FAT、SAT、DQ、IQ、OQ、PQ、SOP什么意思

URS、FAT、SAT、DQ、IQ、OQ、PQ、SOP什么意思确认(qualification)和验证(validation)是制药企业基本的质量活动,并且已经成为法规要求。
确认与验证的范围和定义有所不同:确认主要针对设备、人员和供应商;而验证则是将经过确认的人员、设备、物料、软件、程序等整合在一起,证明整个工艺或方法能够达到既定目的;因此,确认是验证的步骤之一,通常用“验证”来统称确认和验证活动。
验证在中国推广已经有超过十年的时间,大大提高了制药企业的质量保证水平。
目前国外的验证又有新的发展趋势,越来越科学合理。
笔者结合对多个国内制药企业审计中发现的问题,以压片机的确认为例,讨论确认的组织、连接、检查项目以及一些难点的解决方案。
首先,需要明确法规对验证的要求,对验证过程中涉及的文件做一个分类:哪一些是法规强制药设备URS、IQ、OQ 和PQ 的组织和连接第 2 页共 9 页制要求的 GMP 文件?所谓的GMP 文件一是在法规中明确指出的文件,二是为达到法规符合性而产生的文件。
比如,21CFR211.56(d)中规定的“sanitization procedures”(清洁卫生程序)和21CFR211.101(c)(2)中规定的“batch production record”(批生产记录)[1];“qualification”(确认)和“validation”(验证)的文件和记录也属于GMP 文件。
GMP 文件的编制和内容应符合法规要求,遵循一定程序通过,并且有记录证明其得到有效执行。
[9]根据良好工程规范(GEP)和良好管理实践等产生的其它一些文件,并非达到法规符合性所必须的。
这部分文件包括用户规格要求(URS)和试车文件(commissioning)等。
各个国家之间的法规也有区别,例如,验证主计划(VMP)和设计确认(DQ)在欧盟属于法规要求的文件[2],在美国则属于良好工程规范的文件。
图1 显示确认和验证过程中涉及的文件之间的关系。
验证与确认的区别

验证(Verification)与确认(Validation)的区别验证:我们正确地构造了产品吗?(注重过程-由QA负责)确认:我们构造了正确的产品吗?(注重结果-由QC负责)说法一:(2)“验证(Verification)”的涵义通过提供客观证据对规定要求已得到满足的认定。
(2)“确认(Validation)”的涵义通过提供客观证据对特定的预期用途或应用要求已得到满足的认定。
(3)“验证”和“确认”之区别“验证”和“确认”都是认定。
但是,“验证”表明的是满足规定要求,而“确认”表明的是满足预期用途或应用要求,说简单点,“确认”就是检查最终产品是否达到顾客使用要求。
(4)“设计和开发”中“设计验证”和“设计确认”之区别在于:设计验证的目的是检查设计输出是否满足设计输入的规定要求。
设计确认的目的是检查设计形成的最终产品是否达到顾客的使用要求。
说法二:1.“确认”是要证明所提供的(或将要提供的)产品适合其预计的用途,而“验证”则是要查明工作产品是否恰当地反映了规定的要求。
换句话说,验证要保证“做得正确”,而确认则要保证“做的东西正确”。
2.验证注重“过程”,确认注重“结果”3.(Verification) ---Are we producing the product right?(Validation) ---Are we producing the right product?说法三:1.什么是验证?验证就是要用数据证明我们是不是在正确的制造产品。
注意这里强调的是过程的正确性2.什么是确认?确认就是要用数据证明我们是不是制造了正确的产品。
注意这里强调的是结果的正确性。
3.验证和确认是一个广泛的概念,感兴趣的读者可以参考IEEE Std 1012-1998 。
验证:验证检查某样东西是否符合之前已定好的标准,如:文档评审,要检查的东西是文档,检查标准就是文档的评审标准,又如:测试软件,要检查的东西就是软件,检查的标准就是软件的规格说明,包括功能说明,性能要求等。
制药企业设备验证

c、安装确认内容: (1)检查及登记设备生产的厂商名称、设备名称、型号、生产厂商编 号及生产日期、公司内部设备登记号。 (2)安装地点及安装状况。 (3)设备规格标准是否符合设计要求。 (4)计量、仪表准确性和精确度。 (5)设备相应的公用工程和建筑设施的配套。 (6)部件及备件的配套与清单。 (7)制订或确认清洗规程及记录格式。 (8)制订或确认校正、维护、保养、运行的SOP草案及记录表格式草案。
c、性能确认内容: (1)空白料或代用品试生产。 (2)产品实物试生产。 (3)进一步考察运行确认中参数的稳定性。 (4)产品质量检验。 (5)提供试生产产品与该设备有关的SOP资料。 (6)为证明可靠性和重现性,至少需进行连续三批的试生产。
5、数据填写及附件要求
a、设备确认过程中定量检测项目在报告中必须采用定量数 据填写,并附相应原始记录或复印件。 b、所有附件必须编号并在报告中有明确的索引。
c、运行确认内容: (1)按SOP草案对设备的单机或系统进行空载试车。 (2)考察设备运行参数的波动性。 (3)对仪表确认前各进行一次校验,以确定其可靠性。 (4)试验/测试应在一种或一组运行条件之下进行,包括设备运行的上 下限,必要时采用“最差条件”进行确认。 (5)设备运行的稳定性。 (6)SOP草案的适用性。运行确认完成后,应当建立必要的操作、清洁、 校准和预防性维护保养的操作规程,并对相关人员培训。
a、安装确认文件: (1)设备规格标准及使用说明书 (2)设备安装图及质量验收标准 (3)设备各部件及备件的清单 (4)设备安装相应公用工程和建筑设施 (5)安装、操作、清洁的SOP及相关记录格式 b、新增项目由工程设备部组织,设备动力部及外来安装单位相关人员参
与;旧设备改造或更换由设备使用部门组织、设备动力部、工程设备部
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Design Qualification Doc. No.: QAS1. Purpose目的This SOP provides a procedure for the design qualification in XYZ. This proceduredefines the approach,working structure of design qualification and content ofdesign qualification report.本SOP旨在为XYZ工厂的设计确认工作提供一个标准规程。
明确设计去人的方法、工作结构以及文件内容。
2. Scope范围This procedure applies to all new projects, facilities, equipment and operations which have potential GMP and regulation impact in ANBISON. This includes but not limited to:本规程适用于ANBISON工厂所有对GMP法规符合性有潜在影响的设备、操作等的新的投资项目。
包括但不仅限于以下:·Process equipment/facilities/utilities工艺过程的设备/设施/公共辅助系统·Testing equipment/facilities/utilities检验用设备/设施/公共辅助系统·Computerized systems计算机系统Legacy computerized systems should also follow this design qualification procedure.现有计算机系统,也必须遵循设计审核程序。
The cactivities for computerized system design qualification should also follow SOPVAL022<Computerized System Validation>relevant sections except this SOP.Design Qualification Doc. No.: QAS 关于计算机系统的设计阶段的相关工作,除本SOP的规定外,还应参照VAL022《计算机系统验证》的相关章节执行。
3. Responsibility责任Project leader: Project leader ensures all the opinions of Production, Engineering, EHS, Quality Assurance and Validation are covered during design review process.Ensure all the related documents are available for design review.项目负责人:项目负责人负责保证在设计审核过程中,相应的生产、工程维护、安全环保、质量和验证人员的意见、建议均已被包括涵盖。
保证所有相关文件的及时完成,以供审核。
Validation person:Give training to all the personnel who are possible involved in the design review qualification. Ensure design review qualification is implementedaccording to this procedure. Review and approve DQ report.验证人员负责:对所有可能直接介入DQ 过程的人员进行本规程的培训。
确保DQ 按照本规程进行。
对DQ 报告进行审核及批准。
4. Definitions定义:URS:User Requirement Specification 用户需求标准FS:Function Specification 功能标准DS:Design Specification 设计标准DQ:Design Qualification 设计确认VMP:Validation Master Plan 验证主计划RTM:Requirement Traceability Matrix 需求追踪矩阵ERES:Electronic Record & Electronic Signature 电子记录和电子签名5.Safety Notification安全注意事项N/A 不适用于本规程。
Design Qualification Doc. No.: QAS6. Procedure程序:6.1 Basic principles of Design Qualification设计确认(DQ)的基本原则:Design development is the process that ensures user requirements andGMP/regulatory expectations has been incorporated in the design. Design documents in this process include Functional Specification (FS) and Design Specification (DS).These design documents are basic evidences for design qualifications.在设计中将用户需求与GMP法规要求相结合的过程叫做设计开发。
此阶段产生的设计文件包括功能标准(FS)及设计标准(DS)。
这些设计文件将成为进行设计确认最基本证据。
DQ is a process to complete and document design reviews to demonstrate that all user requirements, regulatory requirements and quality aspects have been fullyconsidered in the design of facility, plant, equipment or services. Consideration and recommendations of related departments should be considered at the early design stage.DQ 就是完成并记录设计审核的过程,以证明在厂房、设施、设备或公共辅助系统的设计中已经对所有的用户需求、法规要求和质量关注进行了全面地考虑。
各相关部门的意见、建议应在提出设计要求之初即被纳入。
The people who are responsible for design qualification must have technicalknowledge, typical engineering system experience and familiar with systemoperating interaction and know the approach of design qualification.DQ 应由具备相应技术知识和工程系统典型问题经验,了解系统的操作联系和掌握设计确认方法的人员进行。
6.2 DQ process (reference appendix 1)DQ流程(参见附录1)6.2.1 When a project is established, such as equipment purchasing, introducingor modifying, project owner will be assigned by department manager.当新的建造、购买、引进或改造项目产生时,应由使用部门的经理为其指定项目负责人。
6.2.2 Project owner should raise change control application for the new project.项目负责人为新的项目填写变更控制申请。
Design Qualification Doc. No.: QAS6.2.3 Project owner should prepare User Requirements Specification (URS) after discussion with the experts from Engineering, Process and Quality department.项目负责人与工程,工艺及质量方面的专家讨论后起草用户需求标准(URS)。
6.2.4 Potential supplier develops the system according to the URS of XYZ. Provides Functional Specification based on URS, briefly describe thesystem function. Provides the detailed Design Specification (DS).潜在供应商根据XYZ 提供的URS 进行设计开发。
根据URS 提供功能标准(FS),进行简单的功能描述。
提供详细的设计标准(DS)。
6.2.5 Project owner should check the design documents provided by supplieragainst URS and DQ review principle in relevant VMP to verify whetherrequirements can be met or not.项目负责人根据URS、供应商提供的设计文件以及已经在VMP 中预先规定的DQ 审核的原理对设计是否符合于其需求进行审核确认。
6.2.6 Project owner complete the DQ report according to review output.项目负责人根据审核结果完成DQ 报告。
6.2.7 The system should be redesigned to modify the defect if there are any major design defects according to conclusion in DQ report. Design qualification will be done after modification until URS requirements can be satisfied.根据DQ 报告的结论确定是否有重要的设计缺陷。