gsp-brochure-pdf
GSP 使用手册

序言北京万众国邦软件开发有限公司(原石家庄市万邦软件技术开发有限公司),是一家专门从事医药管理信息系统的研究与开发的高新技术企业,长期专注于医药管理系统的软件开发,并为客户提供从医药管理咨询、软件开发、技术服务的全方位解决方案。
经过多年的发展,在医药商业流通领域积累了丰富的行业经验,聚集了一批既精通计算机技术又具有丰富管理经验的专业人才,凭借多年的商业理论研究和丰富的行业实践经验,结合国内医药流通企业的实际需要,开发出了拥有自主知识产权的软件产品——"万众国邦医药管理软件"。
我们的业务范围已覆盖河北、河南、山东、山西、湖北、广东等地,并在各许多城市设有分支机构及代理网点,拥有完善的市场销售和技术服务网络,能为客户提供优质、快速、便捷的服务。
我们的战略目标:成为医药流通业领先软件供应商,企业使命:发展软件企业提高客户管理现代化,企业精神:敬业、团结、创新、服务。
我们的软件产品在国内已经拥有广泛的客户群,具有较高的市场占有率,受到新老客户的一致好评!为了更好的为医药行业服务,帮助广大客户顺利通过医药GSP认证,在我们研发、技术人员共同努力下,成功的开发出了《医药GSP管理系统》。
为了帮助客户准确、便捷地掌握系统功能、使用方法,解决客户在使用软件过程中一些疑难问题,我们编写了《医药GSP管理系统》操作说明书,如果通过阅读本说明书能使您在使用本产品时改善工作质量、提高工作效率,我们将深感荣幸!同时,对您在众多产品中选择我们的产品,我们对您表示由衷的感谢!另外,我公司又开发了一套专门针对连锁、分销、批发等企业的多功能性管理软件——连锁分销管理系统(B/S结构)。
“连锁分销管理系统”基于最新的多层架构体系,以连锁经营的管理思想为基础的一整套管理系统。
将现代化的经营理念融汇到具体经营管理的计算机软件之中,让您的管理即科学、快速、严谨而有序。
我们编印此书,是以《医药GSP管理系统》V8.0版为主编写的。
虎豹男装营运手册品牌、店铺运营培训资料

虎豹男装营运手册(初稿>A:品牌基本信息第一章公司企业文化与品牌简介一、公司简介二、1、品牌介绍2、品牌定位3、市场定位4、衣着场所5、产品结构6、价格定位第二章产品编码、面料知识一、公司商品编码规则<款号)按照我之前的标码经验编写<波司登瑞琦女装说明:商品基础资料建立方案是根据波司登瑞琦女装的产品特点及规范性和公司的业务需要,具体方案如下:A、商品编码示例图X X X X XXX产品流水号<第5-7位)款式大类代码<第4位)季节代码<第3位)年份代码<第2位)品牌代码<第1位)说明:定义名称注释明细序号位置名称商品编码编码总长度为7位取品牌+年份+季节+款式属性代码+ 产品流水号组成1 第1位品牌代码2 第2位年份代码3 第3位季节代码4 第4位款式大类代码5 第5-7位产品流水号B、商品属性编码商品属性为商品共有的信息,常有属性有品牌,品类,小类,年份,季节,系列,面料类别,风格,设计师等等,以下建议编码说明:b5E2RGbCAP <1)品牌名称注释明细序号代码名称品牌编码长度1位。
取字母编码1 H 虎豹服饰2 ……<2)年份属性名称注释明细序号代码名称年份编码长度为1位。
取字母(I,O,Z除外>编码1 Y 2009年2 A 2018年3 B 2018年4 C 2018年5 ………<3)季节属性名称注释明细序号代码名称季节编码长度为1位。
取数字编码1 0 不分季节2 2 春季3 6 夏季4 8 秋季5 9 冬季<4)大类属性名称注释明细序号代码名称款式大类编码长度为1位。
取数字编码1 0其它类(包/配饰/鞋袜>2 1 T恤类3 2 衬衫/上衣类4 3 裙装类5 4 裤子类<替换其他品类)6 5 背心/马夹类7 6 外套/风衣类8 7 …9 8 毛衫类10 9棉衣/羽绒衣类C、商品规格编码规则<1)颜色编码规则名称注释说明编码颜色编码颜色编码颜色颜色编码编码总长度为2位,由数字,可自行定简易规则编码如:00 均色23 天蓝90 杏01 黑24 浅兰02 白30 灰03 M白31 深灰10 红40 咖啡11 粉红41深咖啡12 桃红42浅咖啡13橡皮红50 黄20 蓝60 绿21 宝蓝61 草绿22 彩兰80 紫<2)尺码编码规则方案1:代码11 12 13 14 15 16 17 18衣服类均码XS S M L XL XXL 3XL代码21 22 23 24 25 26 27 28D、商品条形码XXXXXXXXXXXXX尺码代码:<第10-11位)颜色代码:<第8-9位)商品代码:<第1-7位)二、面料知识<以备单店负责人培训属下店员,提高店员产品知识。
PICS GMP Guide-part 2

PHARMACEUTICAL INSPECTION CO-OPERATION SCHEMEPE 009-11 (Part II)1 March 2014GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTSPART IIDeveloped by the International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use© PIC/S March 2014Reproduction prohibited for commercial purposes.Reproduction for internal use is authorised,provided that the source is acknowledged.Editor: PIC/S Secretariat14 rue du RoverayCH-1207 Genevae-mail:info@web site: TABLE OF CONTENTSPage 1.INTRODUCTION (1)1.1Objective (1)1.2Scope (1)2.QUALITY MANAGEMENT (4)2.1Principles (4)2.2Quality Risk Management (4)2.3Responsibilities of the Quality Unit(s) (5)2.4Responsibility for Production Activities (6)2.5Internal Audits (Self Inspection) (6)2.6Product Quality Review (6)3.PERSONNEL (7)3.1Personnel Qualifications (7)3.2Personnel Hygiene (7)3.3Consultants (8)4.BUILDINGS AND FACILITIES (8)4.1Design and Construction (8)4.2Utilities (9)4.3Water (9)4.4Containment (10)4.5Lighting (10)4.6 Sewage and Refuse (10)4.7Sanitation and Maintenance (10)5.PROCESS EQUIPMENT (11)5.1Design and Construction (11)5.2Equipment Maintenance and Cleaning (11)5.3Calibration (12)5.4Computerized Systems (13)6.DOCUMENTATION AND RECORDS (14)6.1Documentation System and Specifications (14)6.2Equipment Cleaning and Use Record (14)6.3Records of Raw Materials, Intermediates, API Labelling andPackaging Materials (15)6.4Master Production Instructions (Master Production and ControlRecords) (15)6.5Batch Production Records (Batch Production and ControlRecords) (16)6.6Laboratory Control Records (17)6.7Batch Production Record Review (17)7.MATERIALS MANAGEMENT (18)7.1General Controls (18)7.2Receipt and Quarantine (18)7.3Sampling and Testing of Incoming Production Materials (19)7.4Storage (20)7.5Re-evaluation (20)8.PRODUCTION AND IN-PROCESS CONTROLS (20)8.1Production Operations (20)8.2Time Limits (21)8.3In-process Sampling and Controls (21)8.4Blending Batches of Intermediates or APIs (22)8.5Contamination Control (23)9.PACKAGING AND IDENTIFICATION LABELLING OF APIS ANDINTERMEDIATES (23)9.1General (23)9.2Packaging Materials (23)9.3Label Issuance and Control (24)9.4Packaging and Labelling Operations (24)10.STORAGE AND DISTRIBUTION (25)10.1Warehousing Procedures (25)10.2Distribution Procedures (25)BORATORY CONTROLS (26)11.1 General Controls (26)11.2Testing of Intermediates and APIs (27)11.3Validation of Analytical Procedures - see Section 12. (27)11.4Certificates of Analysis (27)11.5Stability Monitoring of APIs (28)11.6Expiry and Retest Dating (28)11.7Reserve/Retention Samples (29)12.VALIDATION (29)12.1Validation Policy (29)12.2Validation Documentation (30)12.3Qualification (30)12.4Approaches to Process Validation (30)12.5Process Validation Program (31)12.6Periodic Review of Validated Systems (32)12.7Cleaning Validation (32)12.8Validation of Analytical Methods (33)13.CHANGE CONTROL (33)14.REJECTION AND RE-USE OF MATERIALS (34)14.1Rejection (34)14.2Reprocessing (34)14.3Reworking (35)14.4Recovery of Materials and Solvents (35)14.5Returns (35)PLAINTS AND RECALLS (36)16.CONTRACT MANUFACTURERS (INCLUDING LABORATORIES) (36)17.AGENTS, BROKERS, TRADERS, DISTRIBUTORS, REPACKERSAND RELABELLERS (37)17.1Applicability (37)17.2Traceability of Distributed APIs and Intermediates (37)17.3Quality Management (38)17.4Repackaging, Relabelling and Holding of APIs andIntermediates (38)17.5Stability (38)17.6Transfer of Information (38)17.7Handling of Complaints and Recalls (38)17.8Handling of Returns (39)18.SPECIFIC GUIDANCE FOR APIs MANUFACTURED BY CELLCULTURE/FERMENTATION (39)18.1General (39)18.2Cell Bank Maintenance and Record Keeping (40)18.3Cell Culture/Fermentation (41)18.4Harvesting, Isolation and Purification (42)18.5Viral Removal/Inactivation Steps (42)19.APIs FOR USE IN CLINICAL TRIALS (42)19.1General (42)19.2Quality (43)19.3Equipment and Facilities (43)19.4Control of Raw Materials (43)19.5Production (43)19.6Validation (44)19.7Changes (44)19.8Laboratory Controls (44)19.9Documentation (44)20.GLOSSARY (45)________________1. INTRODUCTION1.1 ObjectiveThis document (Guide) is intended to provide guidance regarding good manufacturing practice (GMP) for the manufacturing of active pharmaceutical ingredients (APIs) under an appropriate system for managing quality. It is also intended to help ensure that APIs meet the requirements for quality and purity that they purport or are represented to possess.In this Guide “manufacturing” include s all operations of receipt of materials, production, packaging, repackaging, labelling, relabelling, quality control, release, storage and distribution of APIs and the related controls. In this Guide the term “should” indicates recommendations that are expected to apply unless shown to be inapplicable, modified in any relevant annexes to the GMP Guide, or replaced by an alternative demonstrated to provide at least an equivalent level of quality assurance.The GMP Guide as a whole does not cover safety aspects for the personnel engaged in the manufacture, nor aspects of protection of the environment. These controls are inherent responsibilities of the manufacturer and are governed by national laws.This Guide is not intended to define registration requirements or modify pharmacopoeial requirements and does not affect the ability of the responsible competent authority to establish specific registration requirements regarding APIs within the context of marketing/manufacturing authorisations. All commitments in registration documents must be met.1.2 ScopeThis Guide applies to the manufacture of APIs for medicinal products for both human and veterinary use. It applies to the manufacture of sterile APIs only up to the point immediately prior to the APIs being rendered sterile. The sterilisation and aseptic processing of sterile APIs are not covered, but should be performed in accordance with the principles and guidelines of GMP as laid down in national legislations and interpreted in the GMP Guide including its Annex 1.In the case of ectoparasiticides for veterinary use, other standards than this Guide, that ensure that the material is of appropriate quality, may be used.This Guide excludes whole blood and plasma as the PIC/S GMP Guide for Blood Establishments lays down the detailed requirements for the collection and testing of blood. However, it does include APIs that are produced using blood or plasma as raw materials. Finally, the Guide does not apply to bulk-packaged medicinal products. It applies to all other active starting materials subject to any derogations described in the annexes to the GMP Guide, in particular Annexes 2 to 7 where supplementary guidance for certain types of API may be found. The annexes will consequently undergo a review but in the meantime and only until this review is complete, manufacturers may choose to continue to use Part I of the basic requirements and the relevant annexes for products covered by those annexes, or may already apply Part II. Section 19 contains guidance that only applies to the manufacture of APIs used in the production of investigational medicinal products although it should be noted that its application in this case, although recommended, is not required in PIC/S countries.An “API Starting Material” is a raw material, intermediate, or an API that is used in the production of an API and that is incorporated as a significant structural fragment into the structure of the API. An API Starting Material can be an article of commerce, a material purchased from one or more suppliers under contract or commercial agreement, or produced in-house. API Starting Materials normally have defined chemical properties and structure.The manufacturer should designate and document the rationale for the point at which production of the API begins. For synthetic processes, this is known as the point at which "API Starting Materials" are entered into the process. For other processes (e.g. fermentation, extraction, purification, etc), this rationale should be established on a case-by-case basis. Table 1 gives guidance on the point at which the API Starting Material is normally introduced into the process.From this point on, appropriate GMP as defined in this Guide should be applied to these intermediate and/or API manufacturing steps. This would include the validation of critical process steps determined to impact the quality of the API. However, it should be noted that the fact that a manufacturer chooses to validate a process step does not necessarily define that step as critical.The guidance in this document would normally be applied to the steps shown in gray in Table 1. It does not imply that all steps shown should be completed. The stringency of GMP in API manufacturing should increase as the process proceeds from early API steps to final steps, purification, and packaging. Physical processing of APIs, such as granulation, coating or physical manipulation of particle size (e.g. milling, micronizing), should be conducted at least to the standards of this Guide.This GMP Guide does not apply to steps prior to the introduction of the defined "API Starting Material".Table 1: Application of this Guide to API Manufacturing2. QUALITY MANAGEMENT2.1 Principles2.10 Quality should be the responsibility of all persons involved in manufacturing. 2.11 Each manufacturer should establish, document, and implement an effectivesystem for managing quality that involves the active participation of management and appropriate manufacturing personnel.2.12 The system for managing quality should encompass the organisationalstructure, procedures, processes and resources, as well as activities necessary to ensure confidence that the API will meet its intended specifications for quality and purity. All quality related activities should be defined and documented.2.13 There should be a quality unit(s) that is independent of production and thatfulfils both quality assurance (QA) and quality control (QC) responsibilities. This can be in the form of separate QA and QC units or a single individual or group, depending upon the size and structure of the organization.2.14 The persons authorised to release intermediates and APIs should be specified.2.15 All quality related activities should be recorded at the time they are performed.2.16 Any deviation from established procedures should be documented andexplained. Critical deviations should be investigated, and the investigation and its conclusions should be documented.2.17 No materials should be released or used before the satisfactory completion ofevaluation by the quality unit(s) unless there are appropriate systems in place to allow for such use (e.g. release under quarantine as described in Section 10.20 or the use of raw materials or intermediates pending completion of evaluation).2.18 Procedures should exist for notifying responsible management in a timelymanner of regulatory inspections, serious GMP deficiencies, product defects and related actions (e.g. quality related complaints, recalls, regulatory actions, etc.).2.19 To achieve the quality objective reliably there must be a comprehensivelydesigned and correctly implemented quality system incorporating Good Manufacturing Practice, Quality Control and Quality Risk Management.2.2 Quality Risk Management2.20 Quality risk management is a systematic process for the assessment, control,communication and review of risks to the quality of the active substance. It can be applied both proactively and retrospectively.2.21 The quality risk management system should ensure that:-the evaluation of the risk to quality is based on scientific knowledge, experience with the process and ultimately links to the protection of thepatient through communication with the user of the active substance.-the level of effort, formality and documentation of the quality risk management process is commensurate with the level of risk.Examples of the processes and applications of quality risk management can be found, inter alia, in Annex 20.2.3 Responsibilities of the Quality Unit(s)2.30 The quality unit(s) should be involved in all quality-related matters.2.31 The quality unit(s) should review and approve all appropriate quality-relateddocuments.2.32 The main responsibilities of the independent quality unit(s) should not bedelegated. These responsibilities should be described in writing and should include but not necessarily be limited to:1. Releasing or rejecting all APIs. Releasing or rejecting intermediates foruse outside the control of the manufacturing company;2. Establishing a system to release or reject raw materials, intermediates,packaging and labelling materials;3. Reviewing completed batch production and laboratory control records ofcritical process steps before release of the API for distribution;4. Making sure that critical deviations are investigated and resolved;5. Approving all specifications and master production instructions;6. Approving all procedures impacting the quality of intermediates or APIs;7. Making sure that internal audits (self-inspections) are performed;8. Approving intermediate and API contract manufacturers;9. Approving changes that potentially impact intermediate or API quality;10. Reviewing and approving validation protocols and reports;11. Making sure that quality related complaints are investigated andresolved;12. Making sure that effective systems are used for maintaining andcalibrating critical equipment;13. Making sure that materials are appropriately tested and the results arereported;14. Making sure that there is stability data to support retest or expiry datesand storage conditions on APIs and/or intermediates where appropriate;andQuality management15. Performing product quality reviews (as defined in Section 2.6).2.4 Responsibility for Production ActivitiesThe responsibility for production activities should be described in writing, and should include but not necessarily be limited to:1. Preparing, reviewing, approving and distributing the instructions for theproduction of intermediates or APIs according to written procedures;2. Producing APIs and, when appropriate, intermediates according to pre-approved instructions;3. Reviewing all production batch records and ensuring that these arecompleted and signed;4. Making sure that all production deviations are reported and evaluatedand that critical deviations are investigated and the conclusions arerecorded;5. Making sure that production facilities are clean and when appropriatedisinfected;6. Making sure that the necessary calibrations are performed and recordskept;7. Making sure that the premises and equipment are maintained andrecords kept;8. Making sure that validation protocols and reports are reviewed andapproved;9. Evaluating proposed changes in product, process or equipment; and10. Making sure that new and, when appropriate, modified facilities andequipment are qualified.2.5 Internal Audits (Self Inspection)2.50 In order to verify compliance with the principles of GMP for APIs, regularinternal audits should be performed in accordance with an approved schedule.2.51 Audit findings and corrective actions should be documented and brought to theattention of responsible management of the firm. Agreed corrective actions should be completed in a timely and effective manner.2.6 Product Quality Review2.60 Regular quality reviews of APIs should be conducted with the objective ofverifying the consistency of the process. Such reviews should normally be conducted and documented annually and should include at least:A review of critical in-process control and critical API test results;A review of all batches that failed to meet established specification(s);PersonnelA review of all critical deviations or non-conformances and relatedinvestigations;A review of any changes carried out to the processes or analyticalmethods;A review of results of the stability monitoring program;A review of all quality-related returns, complaints and recalls; andA review of adequacy of corrective actions.2.61The result of this review should be evaluated and an assessment made ofwhether corrective action or any revalidation should be undertaken. Reasonsfor such corrective action should be documented. Agreed corrective actionsshould be completed in a timely and effective manner.3. PERSONNEL3.1 Personnel Qualifications3.10 There should be an adequate number of personnel qualified by appropriateeducation, training and/or experience to perform and supervise the manufacture of intermediates and APIs.3.11 The responsibilities of all personnel engaged in the manufacture ofintermediates and APIs should be specified in writing.3.12 Training should be regularly conducted by qualified individuals and shouldcover, at a minimum, the particular operations that the employee performs and GMP as it relates to the employee's functions. Records of training should be maintained. Training should be periodically assessed.3.2 Personnel Hygiene3.20 Personnel should practice good sanitation and health habits.3.21 Personnel should wear clean clothing suitable for the manufacturing activitywith which they are involved and this clothing should be changed when appropriate. Additional protective apparel, such as head, face, hand, and arm coverings, should be worn when necessary, to protect intermediates and APIs from contamination.3.22 Personnel should avoid direct contact with intermediates or APIs.3.23 Smoking, eating, drinking, chewing and the storage of food should be restrictedto certain designated areas separate from the manufacturing areas.3.24 Personnel suffering from an infectious disease or having open lesions on theexposed surface of the body should not engage in activities that could result in compromising the quality of APIs. Any person shown at any time (either by medical examination or supervisory observation) to have an apparent illness or open lesions should be excluded from activities where the health condition could adversely affect the quality of the APIs until the condition is corrected orqualified medical personnel determine that the person's inclusion would not jeopardize the safety or quality of the APIs.3.3 Consultants3.30 Consultants advising on the manufacture and control of intermediates or APIsshould have sufficient education, training, and experience, or any combination thereof, to advise on the subject for which they are retained.3.31 Records should be maintained stating the name, address, qualifications, andtype of service provided by these consultants.4. BUILDINGS AND FACILITIES4.1 Design and Construction4.10 Buildings and facilities used in the manufacture of intermediates and APIsshould be located, designed, and constructed to facilitate cleaning, maintenance, and operations as appropriate to the type and stage of manufacture. Facilities should also be designed to minimize potential contamination. Where microbiological specifications have been established for the intermediate or API, facilities should also be designed to limit exposure to objectionable microbiological contaminants as appropriate.4.11 Buildings and facilities should have adequate space for the orderly placement ofequipment and materials to prevent mix-ups and contamination.4.12 Where the equipment itself (e.g., closed or contained systems) providesadequate protection of the material, such equipment can be located outdoors. 4.13 The flow of materials and personnel through the building or facilities should bedesigned to prevent mix-ups or contamination.4.14 There should be defined areas or other control systems for the followingactivities:Receipt, identification, sampling, and quarantine of incoming materials, pending release or rejection;Quarantine before release or rejection of intermediates and APIs;Sampling of intermediates and APIs;Holding rejected materials before further disposition (e.g., return, reprocessing or destruction);Storage of released materials;Production operations;Packaging and labelling operations; andLaboratory operations.4.15 Adequate, clean washing and toilet facilities should be provided for personnel.These washing facilities should be equipped with hot and cold water asappropriate, soap or detergent, air driers or single service towels. The washing and toilet facilities should be separate from, but easily accessible to, manufacturing areas. Adequate facilities for showering and/or changing clothes should be provided, when appropriate.4.16 Laboratory areas/operations should normally be separated from productionareas. Some laboratory areas, in particular those used for in-process controls, can be located in production areas, provided the operations of the production process do not adversely affect the accuracy of the laboratory measurements, and the laboratory and its operations do not adversely affect the production process or intermediate or API.4.2 Utilities4.20 All utilities that could impact on product quality (e.g. steam, gases, compressedair, and heating, ventilation and air conditioning) should be qualified and appropriately monitored and action should be taken when limits are exceeded.Drawings for these utility systems should be available.4.21 Adequate ventilation, air filtration and exhaust systems should be provided,where appropriate. These systems should be designed and constructed to minimise risks of contamination and cross-contamination and should include equipment for control of air pressure, microorganisms (if appropriate), dust, humidity, and temperature, as appropriate to the stage of manufacture.Particular attention should be given to areas where APIs are exposed to the environment.4.22 If air is recirculated to production areas, appropriate measures should be takento control risks of contamination and cross-contamination.4.23 Permanently installed pipework should be appropriately identified. This can beaccomplished by identifying individual lines, documentation, computer control systems, or alternative means. Pipework should be located to avoid risks of contamination of the intermediate or API.4.24 Drains should be of adequate size and should be provided with an air break ora suitable device to prevent back-siphonage, when appropriate.4.3 Water4.30 Water used in the manufacture of APIs should be demonstrated to be suitablefor its intended use.4.31 Unless otherwise justified, process water should, at a minimum, meet WorldHealth Organization (WHO) guidelines for drinking (potable) water quality.4.32 If drinking (potable) water is insufficient to assure API quality, and tighterchemical and/or microbiological water quality specifications are called for, appropriate specifications for physical/chemical attributes, total microbial counts, objectionable organisms and/or endotoxins should be established.4.33 Where water used in the process is treated by the manufacturer to achieve adefined quality, the treatment process should be validated and monitored with appropriate action limits.4.34 Where the manufacturer of a non-sterile API either intends or claims that it issuitable for use in further processing to produce a sterile drug (medicinal) product, water used in the final isolation and purification steps should be monitored and controlled for total microbial counts, objectionable organisms, and endotoxins.4.4 Containment4.40 Dedicated production areas, which can include facilities, air handling equipmentand/or process equipment, should be employed in the production of highly sensitizing materials, such as penicillins or cephalosporins.4.41 Dedicated production areas should also be considered when material of aninfectious nature or high pharmacological activity or toxicity is involved (e.g., certain steroids or cytotoxic anti-cancer agents) unless validated inactivation and/or cleaning procedures are established and maintained.4.42 Appropriate measures should be established and implemented to preventcross-contamination from personnel, materials, etc. moving from one dedicated area to another.4.43 Any production activities (including weighing, milling, or packaging) of highlytoxic non-pharmaceutical materials such as herbicides and pesticides should not be conducted using the buildings and/or equipment being used for the production of APIs. Handling and storage of these highly toxic non-pharmaceutical materials should be separate from APIs.4.5 Lighting4.50 Adequate lighting should be provided in all areas to facilitate cleaning,maintenance, and proper operations.4.6 Sewage and Refuse4.60Sewage, refuse, and other waste (e.g., solids, liquids, or gaseous by-productsfrom manufacturing) in and from buildings and the immediate surrounding area should be disposed of in a safe, timely, and sanitary manner. Containers and/or pipes for waste material should be clearly identified.4.7 Sanitation and Maintenance4.70 Buildings used in the manufacture of intermediates and APIs should be properlymaintained and repaired and kept in a clean condition.4.71 Written procedures should be established assigning responsibility for sanitationand describing the cleaning schedules, methods, equipment, and materials to be used in cleaning buildings and facilities.4.72 When necessary,written procedures should also be established for the use ofsuitable rodenticides, insecticides, fungicides, fumigating agents, and cleaning and sanitizing agents to prevent the contamination of equipment, raw materials, packaging/labelling materials, intermediates, and APIs.5. PROCESS EQUIPMENT5.1 Design and Construction5.10 Equipment used in the manufacture of intermediates and APIs should be ofappropriate design and adequate size, and suitably located for its intended use, cleaning, sanitization (where appropriate), and maintenance.5.11 Equipment should be constructed so that surfaces that contact raw materials,intermediates, or APIs do not alter the quality of the intermediates and APIs beyond the official or other established specifications.5.12 Production equipment should only be used within its qualified operating range. 5.13 Major equipment (e.g., reactors, storage containers) and permanently installedprocessing lines used during the production of an intermediate or API should be appropriately identified.5.14 Any substances associated with the operation of equipment, such as lubricants,heating fluids or coolants, should not contact intermediates or APIs so as to alter their quality beyond the official or other established specifications. Any deviations from this should be evaluated to ensure that there are no detrimental effects upon the fitness for purpose of the material. Wherever possible, food grade lubricants and oils should be used.5.15 Closed or contained equipment should be used whenever appropriate. Whereopen equipment is used, or equipment is opened, appropriate precautions should be taken to minimize the risk of contamination.5.16 A set of current drawings should be maintained for equipment and criticalinstallations (e.g., instrumentation and utility systems).5.2 Equipment Maintenance and Cleaning5.20 Schedules and procedures (including assignment of responsibility) should beestablished for the preventative maintenance of equipment.5.21 Written procedures should be established for cleaning of equipment and itssubsequent release for use in the manufacture of intermediates and APIs.Cleaning procedures should contain sufficient details to enable operators to clean each type of equipment in a reproducible and effective manner. These procedures should include:Assignment of responsibility for cleaning of equipment;Cleaning schedules, including, where appropriate, sanitizing schedules;。
GSP14第二部分 操作指南 第二章

myGS gSeries .121.
2
1. 2. 3. 4. 5. 6. 7. 8. 9. 10..50.myGS gSeries
11. 12. 13. 14. 15.
Join
XOR AND
Split AND
XOR
Assignment Strategy
Completion Strategy
Form Ref Biz Object Tool Custom Item
Notification
SubFlow
myGS gSeries .119.
Name Condition Type Condition
Redefinable Header Author Country key ISO 3166
Publication Status
UNDER_REVISION UNDER_TEST RELEASED
RELEASED
Version
External Packages Type Declarations Data Fields
myGS gSeries
.57.
IT
GSP Genersoft Platform GSP
Microsoft Vision GSP GSP
.50.
myGS gSeries
2.1
/
2. 1
myGS gSeries
.59.
2.1.
GSP GS
2.1.1.
2.2
.50.
myGS gSeries
2. 2
.50.
myGS gSeries
1.13.1
1.13.1
myGS gSeries .113.
新版GSP药品经营质量管理规范一ppt课件

• • • • • • •
2、劣药的定义: 药品成份的含量不符合国家药品标准 按劣药论处的六种情形: (一)未标明有效期或者更改有效期的; (二)不注明或者更改生产批号的; (三)超过有效期的; (四)直接接触药品的包装材料和容器未经批准 的; • (五)擅自添加着色剂、防腐剂、香料、矫味剂 及辅料的; • (六)其他不符合药品标准规定的。
反应,必须及时向当地省、自治区、直辖市人民政府药品监
• 试生产药品批准文号格式:国药试字+1位字母+8位数字 。 • 药品的批准文号中化学药品使用字母“H”, • 中药使用字母“Z”, • 生物制品使用字母“S”, • 体外化学诊断试剂使用字母“T”, • 药用辅料使用字母“F”, • 进口分包装药品使用字母“J”, • 通过国家药品监督管理局整顿的保健药品使用字母“B”。 • • • • • 国产保健食品批准文号格式为: 国食健字G+4位年代号+4位顺序号; 进口保健食品批准文号格式为: 国食健字J+4位年代号+4位顺序号, 其中“G”代表国家、国内生产。
• 消字号是卫生消毒用品生产单位在生产新品前, 经地方卫生部门审核批准后,取得的生产批准文 号。消字号产品都是用于杀灭或清除传播媒介上 的病原微生物,为提高公共卫生质量而批准的一 类产品。 • 器械为: 械字号 • 消毒制品为:消字号 • 化妆品为:妆字号
假药与劣药
• 1、假药的定义: • (一)药品所含成份与国家药品标准规定的成份 不符的; • (二)以非药品冒充药品或者以他种药品冒充此 种药品的。
药品包装标识、警示语
• 处方药: 请凭医师处方销售、购买和使用
• 非处方药:请仔细阅读药品使用说明书并 按说明使用或在药师指导下购买和使用• 甲类非处方药乙类非处药OTCOTC
brochure 的短语搭配

标题:brochure 的正确使用方法一、brochure 简介brochure 是一种常见的宣传材料,通常用于介绍公司、产品或活动。
它是一种页面较少、内容简洁的宣传资料,适合用于会议、展会、活动现场等场合发放。
二、brochure 的设计要点1. 清晰的标题:brochure 的标题要简洁明了,能够在短时间内吸引读者的注意力。
2. 简洁的内容:brochure 的内容应简洁明了,重点突出,不宜过于繁杂。
3. 吸引人的图片:精美的图片可以增加 brochure 的吸引力,但不宜过多,以免分散读者注意力。
4. 清晰的布局:brochure 的版面设计应清晰、整洁,方便读者阅读和获取信息。
三、brochure 的常用短语搭配1. 介绍公司/产品/活动:- "Wee to ourpany/ product/ event"- "Learn more about our services/ products"- "Discover the latest trends in our industry"2. 提供通信方式:- "Contact us for more information"- "Visit our website for details"- "Connect with us on social media"3. 引导行动:- "Call now for a free consultation"- "Join us at our uing event"- "Sign up for our newsletter"四、brochure 的常见错误用法1. 过于繁杂的内容:部分 brochure 可能会在介绍内容上过于详细,导致读者失去阅读的兴趣。
门店gsp操作流程及规范培训

门店gsp操作流程及规范培训Store GSP (General Store Procedure) operation process and standard training is essential for maintaining the efficiency and consistency of store operations. 门店GSP(一般店铺程序)操作流程和标准培训对于维持店铺运营的效率和一致性至关重要。
By following a clear and standardized process, it ensures that all staff members are on the same page when it comes to handling various tasks and responsibilities. 遵循明确和标准化的流程,可以确保所有员工在处理各种任务和责任时都能达成一致。
From opening and closing procedures to cash handling and customer service, having a well-defined GSP helps in maintaining quality and consistency in all aspects of store operations. 从开店到闭店程序再到现金处理和客户服务,拥有明确定义的GSP有助于在店铺运营的各个方面保持质量和一致性。
In addition to ensuring smooth operations, a standardized GSP also helps in mitigating risks and ensuring compliance with various regulations. 除了确保顺畅运营外,标准化的GSP还有助于减轻风险,并确保符合各种法规。
brochure

/earlyconnections/
Northwest Educational Technology Consortium Northwest Regional Educational Laboratory 101 SW Main, Suite 500 The contents of this publication were under grant #R302A000016 Portland, Oregon 97204 developedU.S. Department of Education. from the The contents of this publication do not necessarily reflect the policy of the 800-211-9435 Department or any other agency of the U.S. Government. This publication may / be reproduced without permission if the
then share their memories by writing or dictating captions, making drawings, or recording stories. The final projects can be viewed both on-screen and as a book. # Make an electronic slideshow of a class book. Each child composes a page with an illustration and a written or recorded sentence that supports the curriculum under study.
医药行业专业英语词汇_非常有用_

FDA和EDQM术语:QC、QA、IPQC、JQE、DQA、SQE是什么?这些职位的全称都是什么?有什么区别?QC中文全称: 即英文QUALITY CONTROL的简称,中文意义是品质控制,质量检验。
其在ISO8402:1994的定义是“为达到品质要求所采取的作业技术和活动”。
有些推行ISO9000的组织会设置这样一个部门或岗位,负责ISO9000标准所要求的有关品质控制的职能,担任这类工作的人员就叫做QC人员,相当于一般企业中的产品检验员,包括进货检验员(IQC)、制程检验员(IPQC)、最终检验员(FQC)和出货检验员(OQC)。
QA中文全称:即英文QUALITY ASSURANCE 的简称, 中文意思是品质保证,质量保证。
其在ISO8402:1994中的定义是“为了提供足够的信任表明实体能够满足品质要求,而在品质管理体系中实施并根据需要进行证实的全部有计划和有系统的活动”。
有些推行ISO9000的组织会设置这样的部门或岗位,负责ISO9000标准所要求的有关品质保证的职能,担任这类工作的人员就叫做QA 人员。
IPQC:即英文In-process Quality Control 的简称, 中文意思是制程检验,担任这类工作的人员叫做制程检验员。
JQE:即英文Joint Qualit Engineer 的简称, 中文意思是品质工程师或客户端工程师,或客户端品质工程师,即供应商花钱雇用的为客户工作的品质工程师,是客户SQE的眼睛和耳朵。
iDQA:即英文Design Quality Assurance 的简称, 中文意思是设计品质保证,如DQA经理(设计品质认证经理)。
SQE:即英文Supplier Quality Engineer 的简称, 中文意思是供应商品质工程师。
此外,还有DQC:即英文Design Quality Control 的简称, 中文意思是设计品质控制。
CLINICAL TRIAL:临床试验 clinical trialANIMAL TRIAL:动物试验animal trialACCELERATED APPROV AL:加速批准 accelerated approvalSTANDARD DRUG:标准药物 standard drugINVESTIGATOR:研究人员;调研人员 investigatorPREPARING AND SUBMITTING:起草和申报 preparing and submittingSUBMISSION:申报;递交 submissionBENIFIT(S):受益 benifitRISK(S):受害riskDRUG PRODUCT:药物产品 drug productDR(drug substance)原料药 API(Active Pharmaceutical Ingrediet) 原料药又称:活性药物组分ESTABLISHED NAME:确定的名称 established nameGENERIC NAME:非专利名称 generic namePROPRIETARY NAME:专有名称; proprietary nameINN(INTERNATIONAL NONPROPRIETARY NAME):国际非专有名称international nonproprietaty name ADVERSE EFFECT:副作用 adverse effectADVERSE REACTION:不良反应adverse reactionPROTOCOL:方案 protocolARCHIV AL COPY:存档用副本 archival copy archival copyREVIEW COPY:审查用副本 rreview copyOFFICIAL COMPENDIUM:法定药典(主要指USP、 NF). Official compendiumUSP(THE UNITED STATES PHARMACOPEIA):美国药典 pharmacopeia英音:[,fɑ:məkə'pi:ə] pharmaceutical英音:[,fɑ:mə'sju:tikl]NF(NATIONAL FORMULARY):(美国)国家处方集 national formularyOFFICIAL=PHARMACOPEIAL= COMPENDIAL:药典的;法定的;官方的 compendial AGENCY:审理部门(指FDA) agencyIDENTITY:真伪;鉴别;特性identitySTRENGTH:规格;规格含量(每一剂量单位所含有效成分的量)strengthLABELED AMOUNT:标示量 labeled amountREGULATORY SPECIFICATION:质量管理规格标准(NDA提供) regulatory specification REGULATORY METHODOLOGY:质量管理方法 regulatory methodologyREGULATORY METHODS V ALIDATION:管理用分析方法的验证 validationmedical apparatus and instruments: 医疗器械pharmaceutical factory:药厂drugstore; chemist's shop; pharmacy:药店pharmacopeia: 药典prescription: 药方write out a prescription: 开药方drugstore; chemist's shop; pharmacy: 医药商店hospital pharmacy; disp(转载自第一范文网,请保留此标记。
GSP培训材料

一、GSP概述(一) GSP的由来1.我国GSP来源于日本。
第一部GSP於1984年发布,第二部GSP於1992年修改后发布,第三部GSP於2000年修改后发布,现行GSP是2010版GSP应属第四部。
(二) GSP的概念1.GSP的英文解释:Good Supply Practice好的供应规范2、GSP中文全称:《药品经营质量管理规范》。
3、2000版GSP的定义概括为:控制流通过程药品质量的规程。
4、质量标准是药品生产、检验、供应与使用的依据。
国家药品标准包括药典、部颁标准和地方标准上升后的国家药品标准(局颁标准)。
5、药品批准文号是指国家批准药品生产企业生产药品的文号,是药品生产合法性的标志。
《药品管理法》规定,生产药品“须经国务院药品监督管理部门批准,并发给药品批准文号”。
未取得批准文号而生产的药品按假药论处。
6、目前药品批准文号的统一格式为:国药准字H31020001国药准(试)字+1位字母+八位数字。
字母含义:H(化)Z(中成药)B(中药保健品)S(生物制品)T(体外生物诊断试剂)F(药用辅料)J (进口分装药品)数字含义:1-2位:10(卫生部);19、20(药监局);省级区域代码前两位。
3-4位:换发批准文号之年公元年号的后两位数字。
5-8位:顺序号二、企业实施GSP的核心内容(一)、购进1、进货质量管理程序:(1)、确定供货企业的法定资格及质量(首营企业)(2)、审核所购药品的合法性和质量(3)、验证销售人员合法资格(4)、对首营品种,填写“首次经营药品审批表”,并经质量管理机构(人)和企业主管领导审核批准。
(5)、签订有明确质量条款的购货合同(6)、按购货合同中质量条款执行2、购进药品的基本条件(1)、合法企业所生产或经营的药品(2)、具有法定的质量标准,即国家药品标准(3)、法定的批准文号和生产批号(4)、《进口药品注册证》、《进口药品通关单》或《进口药品检验报告书》复印件(5)、包装和标识符合有关规定和储运要求。
GMP文件常见缩写

GMP英语1.ABBREVIATED(NEW)DRUG:简化申请的新药2.AirLock 气闸3.ANDA(ABBREVIATED NEW DRUG APPLICATION):简化新药申请4.API(Active Pharmaceutical Ingrediet) 原料药又称:活性药物组分5.Authorized Person 授权人6.Batch Number/Lot-Number 批号7.Batch Numbering System 批次编码系统8.BATCH PRODUCTION RECORDS:生产批号记录9.BATCH PRODUCTION:批量生产;分批生产10.Batch Records 批记录11.Batch/Lot 批次12.Bulk Product 待包装品13.Calibration 校正14.CFR(CODE OF FEDERAL REGULATION):(美国)联邦法规15.Clean area 净区16.Consignmecnt(Delivery)托销药品17.DMF(DRUG MASTER FILE):药物主文件(持有者为谨慎起见而准备的保密资料,可以包括一个或多个人用药物在制备、加工、包装和贮存过程中所涉及的设备、生产过程或物品。
只有在DMF持有者或授权代表以授权书的形式授权给FDA,FDA在审查IND、NDA、ANDA时才能参考其内容)18.FDA(FOOD AND DRUG ADMINISTRATION):(美国)食品药品管理局19.HOLDER:DMF持有者20.IND(INVESTIGATIONAL NEW DRUG):临床研究申请(指申报阶段,相对于NDA而言);研究中的新药(指新药开发阶段,相对于新药而言,即临床前研究结束)RMED CONSENT:知情同意(患者对治疗或受试者对医疗试验了解后表示同意接受治疗或试验)22.NDA(NEW DRUG APPLICATION):新药申请23.OTC DRUG(OVER—THE—COUNTER DRUG):非处方药24.PANEL:专家小组25.PIC/S的全称为:Pharmaceutical Inspection Convention/Pharmaceutical InspectionCooperation Scheme, PIC/S(制药检查草案), 药品检查协会(PIC/S) ,也有人称PIC/S 为医药审查会议/合作计划(PIC/S)26.PIC的权威翻译:药品生产检查相互承认公约27.POST-OR PRE- MARKET SURVEILLANCE:销售前或销售后监督28.PRESCRIPTION DRUG:处方药29.TREATMENT IND:研究中的新药用于治疗GMP文件常见缩写ABPI Association of the British Pharmaceutical IndustryADR Adverse Drug ReactionAE Adverse EventAIM Active Ingredient ManufacturerANDA Abbreviated New Drug ApplicationANOVA Analysis of VarianceASM: Active Substance ManufacturerATC Anatomical Therapeutic ChemicalATX Animal Test Exemption CertificateBAN British Approved NameBIRA British Institute of Regulatory AffairsBNF British National FormularyBP British PharmacopoeiaC of A Certificate of AnalysisC of S Certificate of SuitabilityCENTRE FOR DRUG EVALUATION (CDE)Centre for Pharmaceutical Administration (CPA)CMS Concerned Member State 每个成员国COS Certificate of SuitabilityCPMP Committee for Proprietary Medicinal ProductsCRA Clinical Research AssociateCRF Case Report FormCRO Contract Research OrganisationCTA Clinical Trial ApplicationCTC Clinical Trial CertificateCTD Common Technical DocumentCTX Clinical Trials ExemptionDDD Defined Daily DoseDGC Daily Global ComparisonDIA Drug Information AssociationDMF Drug Master FileDrug Registration Branch (DR, Product Evaluation & Registration Division, CPA EDQM (European Directorate for the Quality of Medicines) 欧洲联盟药品质量指导委员会EEA 欧洲经济地区EGMA European Generics Medicine AssociationELA Established Licence ApplicationEMEA European Medicines Evaluation AgencyEMEA (European Agency for the Evaluation of Medicinal Products)欧洲联盟药品评价机构EP European PharmacopoeiaEPAR European Public Assessment ReportsESRA European Society of Regulatory Affairs European Pharmacopoeia Commission 欧洲药典委员会FDAFDA Food and Drug Administrationfinal evaluation report (FER)free sale certificates (FSCs)GCP Good Clinical PracticeGCP药品临床研究管理规范GLP Good Laboratory PracticeGLP 药品临床前安全性研究质量管理规范GMP Good Manufacturing PracticeGMP 药品生产质量管理规范GSP药品销售管理规范Health Sciences Authority (HSA)HSA’s Medicines Advisory Committee (MAC)IB Investigators BrochureICH International Conference for Harmonisation IDMC Independent Data-Monitoring CommitteeIEC Independent Ethics CommitteeIND Investigational New DrugINN International Non-proprietary Name International Conference on Harmonisation (ICH) IPC In Process ControlIRB Institutional Review BoardLICENCE HOLDERMA Marketing AuthorisationMAA Marketing Authorisation ApplicationMAA上市申请MAH Marketing Authorisation HolderMAH 销售许可持有者MCA Medicines Control AgencyMHW Ministry of Health and Welfare (Japan)MR Mutual RecognitionMRA 美国与欧盟的互认协议MRAs (Mutual Recognition Agreements) 互相認證同意MRFG Mutual Recognition Facilitation Group MRP Mutual Recognition ProcedureNAS New Active SubstanceNCE New Chemical EntityNDA New Drug Applicationnew chemical entities (NCEs)new drug applications (NDAs)NSAID Non Steroidal Anti Inflammatory DrugNTA Notice To ApplicantsOOS Out of SpecificationOTC Over The CounterPAGB Proprietary Association of Great BritainPh Eur European PharmacopoeiaPIL Patient Information LeafletPL Product LicencePOM Prescription Only MedicinePRODUCT OWNERPSU Periodic Safety UpdatesQA Quality AssuranceQC Quality ControlRAJ Regulatory Affairs JournalRMS Reference Member StateRMS相互认可另一成员国RSD Relative Standard DeviationRx Prescription OnlySAE Serious Adverse EventSMF Site Master FileSOP Standard Operating ProcedureSOP (STANDARD OPERATION PROCEDURE)标准运作程序SPC/SmPC Summary of Product Characteristicssummary of product characteristics(SPC)Therapeutic Goods Administration (TGA)USP US PharmacopoeiaVMF Veterinary Master FileVPC Veterinary Products CommitteeA.A.A Addition and Amendments 增补和修订AC Air Conditioner 空调器ADR Adverse Drug Reaction 药物不良反应AFDO Association of Food and Drug Officials 食品与药品官员协会(美国)ACC Accept 接受AQL Acceptable Quality Level 合格质量标准ADNA Abbreviated New Drug Application 简化的新药申请BOM Bill of Material 物料清单BPC Bulk pharmaceutical Chemiclls 原料药CBER Center for Biologics Evaluation Research 生物制品评价与研究中心CFU Colony Forming Unet 菌落形成单位DMF Drug Master File 药品管理档案CDER Cemter for Drug Evaluation amd Research 药物评价与研究中心CI Corporate Identity (Image) 企业识别(形象)CIP Cleaning in Place 在线清洗CSI Consumer Safety Insepctor 消费者安全调查员CLP Cleaning Line Procedure 在线清洗程序DAL Defect Action Level 缺陷作用水平DEA Drug Enforcement Adminestration 管制药品管理DS Documentation Systim 文件系统FDA Food and Drug Administration 食品与药品管理局(美国)GATT General Agreemernt on Tariffs and Trade 关贸总协会GMP Good Manufacturing Practice Gvp 药品生质量管理规范GCP Good Clinical Practice 药品临床实验管理规范GLP Good Laboratory Practice 实验室管理规范GSP Good Supply Practice 药品商业质量规范GRP Gook RaTAIL Practice 药品零业质量管理规范GAP Good Agriculture Practice 药材生产管理规范GVP Gook Validation Prctice 验证管理规范GUP Gook Use Practice 药品重用规范HVAC Heating Ventilation Air Conditioning 空调净化系统ISO Intematonal Organization for Standardization 车际标准化组织MOU Memorandum of Understanding 谅解备忘录PF Porduction File 生产记录用表格OTC Over the Counter (Drug) 非处方药品PLA Product License Application 产品许可申请QA Quality Assurance 质量保证QC Quality Control 质量控制QMP Quality Management Procedure 质量管理程序SDA State Drug Administration 国家药品监督管理局SMP Standard Managmert Procedure 标准管理程序SOP Standard Operating Procedure 标准操作程序TQC Tatal Quality Control 全面质量管理USA Uneted States Pharmacopeia 美国药典。
GSP系统使用手册

GSP管理信息系统V3.0使用手册(客户端)镇江未来软件有限公司二零零七年九月未经未来软件有限公司事先书面许可,本手册的任何部分不得以任何形式进行增删,改编,节选,翻译,翻印,或仿制。
本手册全部内容未来软件有限公司可能随时加以更改,此类更改将不另外通知。
本手册著作权归属于镇江未来软件有限公司版权所有翻制必究2007年9月第一次印刷目录第一章 (6)产品介绍 (6)安装手册 (7)使用手册 (20)第二章 (26)2.1 系统初始化 (26)2.2 权限维护 (26)2.4 其他分类维护 (27)2.5 药品信息设置 (28)第三章 (32)3.1 采购到货 (32)3.2 质量检验 (33)3.3 采购入库 (33)3.4 采购退货 (33)3.5 采购到货查询 (34)3.6 采购退货查询 (34)第四章 (35)4.1 零售出库 (35)4.2 零售退货 (36)4.3零售退货便捷窗口 (37)4.4 调价单 (37)4.5 营业员零售日报 (37)4.6 零售日报表 (38)4.7 利润表 (38)4.8 利润明细表 (38)4.9 零售退货查询 (39)第五章 (40)5.1 期初库存 (40)5.2 盘点表 (40)5.3 入库单 (41)5.4 出库单 (41)第六章 (43)6.1 入库查询 (43)6.2 入库统计 (43)6.3 出库查询 (44)6.4 出库统计 (44)6.5 库存查询 (44)6.6 药品有效期预警 (45)6.7 药品上下限预警 (45)6.8 拆零销售记录查询 (45)第七章 (47)7.1 GSP日常办公 (47)7.1.1 首营药品审批表 (47)7.1.2 首营企业审批表 (49)7.1.3 合格供货方档案表 (50)7.1.4 药品质量信息登记及分析记录 (50)7.1.5 企业员工健康检查汇总表 (51)7.1.6 质量管理制度执行情况检查考核记录 (52)7.1.7 培训计划一览表 (53)7.1.8培训实施记录表 (55)7.1.9 员工个人培训教育档案 (55)7.2 质量检验管理 (56)7.2.1中药饮片装斗复核记录 (56)7.2.2 药品拆零销售记录 (56)7.2.3 处方药调配销售记录 (57)7.2.4 购进药品退回记录 (57)7.2.5 药品购进质量验收记录 (57)7.2.6 药品拒收报告单 (58)7.2.7 药品不良反应报告表 (60)7.2.8 药品购进记录单 (60)7.3 库存养护管理 (61)7.3.1药品陈列环境和存储条件检查记录 (61)7.3.2 近效期药品催销表 (62)7.3.3 重点养护品种确定表 (62)7.3.4 陈列药品质量检验记录 (63)7.3.5 中药饮片在库养护记录 (64)7.3.6 陈列.存储.重点药品质量和包装检查养护记录 (65)7.3.7 药品质量查询.处理通知单 (66)7.3.8 药品解除停售通知单 (67)7.3.9药品停售通知单 (68)7.4 不合格药品管理 (69)7.4.1 不合格药品销毁记录 (69)7.4.2 不合格药品台帐 (70)7.4.3 不合格药品报损审批表 (71)7.4.4 不合格药品报告表 (72)7.5 设施设备管理 (73)7.5.1 温湿度记录 (73)7.5.2 仪器设备定期检查记录 (74)第一章产品介绍GSP是英文Good Supplying Practice缩写,直译为“良好的药品供应规范”,在我国称为“药品经营质量管理规范”。
2023-冷链药品的GSP管理培训课件

目录
(一)
山东疫苗事件
(二)
冷链基础知识
(三)
冷链管理质量体系
(四)
冷链流程及环节控制
〔一〕从山东疫苗事件说起
• 2021年3月,山东警方披露庞红卫母女非法经营25种二类 疫苗,未经严格冷链存储运输销往全国24个省市,涉案金 额达5.7亿元。
• 截至2021年11月,批准逮捕涉嫌非法经营等犯罪嫌疑人 324人、起诉68人、第一批问责公职人员357人,立案侦查 涉及职务犯罪的有100人。 除此,该事件引发药品流通监 管体制的深刻变革。
质量体系各要素
1.组织机构
• 质量管理机构职责: • 〔一〕催促相关部门和岗位人员执行药品管理的法律法规及本标准;
〔二〕组织制订质量管理体系文件,并指导、监督文件的执行; 〔三〕负责对供货单位和购货单位的合法性、购进药品的合法性以及供货单位销售人员、 购货单位采购人员的合法资格进行审核,并根据审核内容的变化进行动态管理; 〔四〕负责质量信息的收集和管理,并建立药品质量档案; 〔五〕负责药品的验收,指导并监督药品采购、储存、养护、销售、退货、运输等环节 的质量管理工作; 〔六〕负责不合格药品确实认,对不合格药品的处理过程实施监督; 〔七〕负责药品质量投诉和质量事故的调查、处理及报告;
• 冷藏箱与泡沫箱相比,具有重复利用性。
保温箱
有源制冷
外接显示
自带显示
蓄冷剂 • 被动制冷剂无毒、无腐蚀、无害、无污染 • 1、冰袋
• 2、冰盒/冰排
隔温装置
第一百零四条 企业应当根据药品的温度控制要求,在运输过程中采取 必要的保温或者冷藏、冷冻措施。
运输过程中,药品不得直接接触冰袋、冰排等蓄冷剂,防止对药品 质量造成影响。
GSP操作说明书

步骤 3:基本资料填完成后点击右边功能框的保存按钮。如下图。
3、初始化库存余额
步骤 1:打开软件操作界面——单击基本信息——单击初始化库存 余额。如下图:
4
对全部高中资料试卷电气设备,在安装过程中以及安装结束后进行高中资料试卷调整试验;通电检查所有设备高中资料电试力卷保相护互装作置用调与试相技互术关,系电,力根通保据过护生管高产线中工敷资艺设料高技试中术卷资,配料不置试仅技卷可术要以是求解指,决机对吊组电顶在气层进设配行备置继进不电行规保空范护载高与中带资负料荷试下卷高问总中题体资,配料而置试且时卷可,调保需控障要试各在验类最;管大对路限设习度备题内进到来行位确调。保整在机使管组其路高在敷中正设资常过料工程试况中卷下,安与要全过加,度强并工看且作护尽下关可都于能可管地以路缩正高小常中故工资障作料高;试中对卷资于连料继接试电管卷保口破护处坏进理范行高围整中,核资或对料者定试对值卷某,弯些审扁异核度常与固高校定中对盒资图位料纸置试,.卷保编工护写况层复进防杂行腐设自跨备动接与处地装理线置,弯高尤曲中其半资要径料避标试免高卷错等调误,试高要方中求案资技,料术编试交写5、卷底重电保。要气护管设设装线备备置敷4高、调动设中电试作技资气高,术料课中并3中试、件资且包卷管中料拒含试路调试绝线验敷试卷动槽方设技作、案技术,管以术来架及避等系免多统不项启必方动要式方高,案中为;资解对料决整试高套卷中启突语动然文过停电程机气中。课高因件中此中资,管料电壁试力薄卷高、电中接气资口设料不备试严进卷等行保问调护题试装,工置合作调理并试利且技用进术管行,线过要敷关求设运电技行力术高保。中护线资装缆料置敷试做设卷到原技准则术确:指灵在导活分。。线对对盒于于处调差,试动当过保不程护同中装电高置压中高回资中路料资交试料叉卷试时技卷,术调应问试采题技用,术金作是属为指隔调发板试电进人机行员一隔,变开需压处要器理在组;事在同前发一掌生线握内槽图部内 纸故,资障强料时电、,回设需路备要须制进同造行时厂外切家部断出电习具源题高高电中中源资资,料料线试试缆卷卷敷试切设验除完报从毕告而,与采要相用进关高行技中检术资查资料和料试检,卷测并主处且要理了保。解护现装场置设。备高中资料试卷布置情况与有关高中资料试卷电气系统接线等情况,然后根据规范与规程规定,制定设备调试高中资料试卷方案。
商品说明书 - Guru99 Excel Formula手册
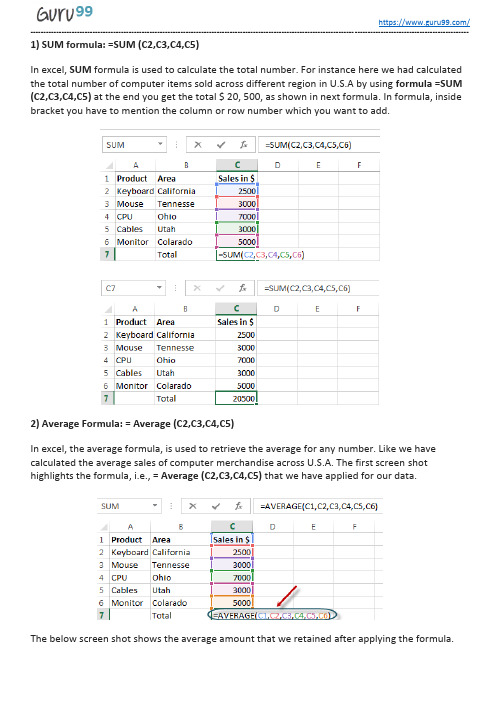
1) SUM formula: =SUM (C2,C3,C4,C5)In excel, SUM formula is used to calculate the total number. For instance here we had calculated the total number of computer items sold across different region in U.S.A by using formula =SUM (C2,C3,C4,C5) at the end you get the total $ 20, 500, as shown in next formula. In formula, inside bracket you have to mention the column or row number which you want to add.2) Average Formula: = Average (C2,C3,C4,C5)In excel, the average formula, is used to retrieve the average for any number. Like we have calculated the average sales of computer merchandise across U.S.A. The first screen shot highlights the formula, i.e., = Average (C2,C3,C4,C5) that we have applied for our data.The below screen shot shows the average amount that we retained after applying the formula.3) SumIF formula = SUMIF (A2:A7,“Items wanted”, D2:D7)The SumIF gives the total number of any items for selected ranges. For instance here we want to calculate only the total sales amount for software items, to do that we will apply the formulaas =SUMIF (A2:A7, “software”, D2:D7).Here A2 and A7 defines the range for software and same way we can find sales amount for hardware. (A2:A7, “hardware”, D2:D7).Below screen-shot show the total sale amount of hard-ware and soft-ware in the table.4) COUNTIF Formula: COUNTIF(D2:D7, “Function”)COUNTIF function offers wide application; you can apply the formula according. Here we have taken a simple example of COUNTIF function, where our motive is to find the total number of cells whose value is greater than $3000. In order to know that we will apply theformula =COUNTIF(D2:D7,”3000”).Below screen shot shows the total number of cells that has value greater than 3000.5) Concatenate Function: =CONCATENATE(C4,Text, D4, Text,…)Concatenate function is used in excel to connect different segment or text to display as a single sentence. For example, here we want to display text as “NewYork has the highest sale of 12000 dollars”, for that we will use the formula =CONCATENATE(C4,”has the highest sale of”,D4,dollar”).When you execute the formula and display the text as show in below screen-shot6) Int Formula: int (this number)Int formula is used to remove integer from the number like we have demonstrated over here in below example.7) MAX Formula: =Max(D2:D7)This excel formula will retain the cells that have the highest value in the column, for example, here we want to know the highest value for computer items, and it retains the value $12000. Likewise, you can execute same formula to get a minimum value, in the formula you have to replace Max with Min.Below, screen shot shows the highest value in the column.8) Factorial Formula= FACT(number)Factorial formula will return the factorial of the number. To know the factorial number for 3, we use this formula. You can use this formula to know the probability for any number, here we will have factor 3=3x2x1.9) VLookup Formula = Vlookup(value, range, and get me value in the column, is my list sorted) VLookup formula is used when you know anyone detail of any object or person and, you retain other formation based on that detail. For example here we have an example of the keyboard, where you know the retail price of the keyboard but you don’t know how much total sale it made in California by selling keyboard. To know that you will use =Vlookup(20,D2:D7,2,False). This formula will give you the total sale amount based on the retail price. While applying this formula you have ensure that whatever you are placing as ref, must be unique, for example you are looking for any particular employee with its ID number it should not be allotted to others otherwise it will show an error.When formula is executed, the total sale amount shown is $250010) IF function formula: IF (E2>2000, correct/Incorrect)Here we have used IF function; this function is used when you want to refer whether the following condition met is correct or incorrect. Here we have used “good” as any sales made greater than 2000 should be remarked as good. Likewise, you can set this as “bad”, “correct” or “incorrect”.Below table shows when we applied our formula it highlighted cell as “good”.Which other formulas were asked to you in an interview? Let us know in comments below - Guru99 Provides FREE ONLINE TUTORIAL on Various courses likeJava MIS MongoDB BigData Cassandra Web Services SQLite JSP Informatica Accounting SAP Training Python Excel ASP Net HBaseProjectTest Management Business Analyst Ethical Hacking PMP ManagementLive Project SoapUI Photoshop Manual Testing Mobile Testing Data Warehouse R Tutorial Tableau DevOps AWSSoftware Jenkins Agile Testing RPA JUnitEngineering Selenium CCNA AngularJS NodeJS PLSQL。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
MORE INFORMATION
conta cting GSP
For more information please visit the program website or contact us at the email address or telephone number listed below. Program website: http:/ / gsp.c.u-tokyo.ac.jp/ Email: office@gsp.c.u-tokyo.ac.jp Telephone: +81-3-5465-8220
STUDYING
courses with GSP
The working language of GSP is English. Students in GSP belong to one of four departments in the Graduate School of Arts and Sciences? Language and Information Sciences, Interdisciplinary Cultural Studies, Area Studies, or Advanced Social and International Studies? and take courses with GSP in order to satisfy their course requirements. In addition, students are strongly encouraged to participate in GSP-sponsored research workshops and to pursue intensive courses and internship opportunities so as to further learning and scholarly exchange. Students in GSP also have access to courses offered in English or Japanese at other graduate schools in the university.
The Ph.D. program is a three-year course designed to develop researchers capable of making a significant contribution to scholarship in fields in the humanities, social sciences, and beyond. Students in the course are expected to develop and pursue an original research topic that touches on the cultural, social, political, ecological, or ethical aspects of human existence in global society. In the process, they will develop their own research expertise and experience. The program is aimed, in particular, at those who intend to seek careers as researchers or advanced professionals in fields related to the study of global society.
GRADUATE PROGRAM ON GL OBAL SOCI ETY
Gr aduate School of Ar ts and Sciences at the Univer sity of Tokyo
APPLYING
Байду номын сангаасa pplica tion schedule
1. Applications are accepted in January. 2. The screening process takes place in February. 3. All applicants are notified of the results of their applications by the end of March. 4. The academic year begins in September. Full details on admission eligibility and application requirements can be found in the Application Guidelines available on the GSP program website.
G ra d u a teS c h o o lo fA rtsa n dS c ien c es T h eU n ivers ityo fT o kyo 3-8-1 K o m a b a ,M eg u ro -ku ,T o kyo153-8902
PROGRAM OF STUDY
a bout GSP
The M.A. program is a two-year course intended for students with an interest in the cultural, social, political, ecological, and ethical aspects of human existence in global society. It aims to provide an in-depth understanding and broad knowledge of the changing situations in Japan and the Asian region, and global society in general. Students in the program will also cultivate the skills needed to shape agendas in our globalized world. The program is especially designed for those seeking an M.A. degree before or after entering the business world, government service, the non-governmental sector, or the media in fields related to global society.
M.A. PROGRAM
overview
PH.D. PROGRAM
overview
The Graduate Program on Global Society (GSP) focuses on the cultural, social, political, ecological, and ethical dimensions of human existence in the age of globalization. The program offers a curriculum based on critical thinking, practical action, and creative spirit that extends beyond the disciplinary borders of the humanities and social sciences so as to better prepare the next generation to meet the challenges and to take advantage of the possibilities of our changing global society. It aims to cultivate in its students the qualities of leadership necessary to manage the global issues we confront today and to adapt to those that we will face in the future.