Bruceine A_25514-31-2_DataSheet_MedChemExpress
BFH772_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:BFH772 is a potent oral VEGFR2 inhibitor, which is highly effective at targeting VEGFR2 kinase with an IC 50 value of 3 nM.IC50 & Target: IC50: 2.7±0.9 nM (hVEGFR2), 1.5±0.53 μM (mVEGFR2), 1.7±0.36 μM (hVEGFR1), 1.1±0.29 μM (hVEGFR3)[1]In Vitro: BFH772 is highly selective; apart from inhibiting VEGFR2 at 3 nM IC 50, it also targets B–RAF, RET, and TIE–2, albeit with atleast 40–fold lower potency. BFH772 is inactive (IC 50>10 μM; >2 μM for cKIT) against all other tyrosine specific– andserine/threonine–specific protein kinases tested. BFH772 inhibits VEGFR2 with IC 50 of 4.6±0.6 nM in CHO cells. BFH772 inhibits VEGFR2 with IC 50 of 3 nM in HUVEC cells. BFH772 inhibits the ligand induced autophosphorylation of RET, PDGFR, and KIT kinases,with IC 50 values ranging between 30 and 160 nM. BFH772 is selective (IC 50 values >0.5 μM) against the kinases of EGFR, ERBB2,INS–R, and IGF–1R and against the cytoplasmic BCR–ABL kinase. IC 50 of BFH772 (<0.01 nM, n=2) demonstrates that they abrogated VEGF induced proliferation at remarkably low nM concentrations [1].In Vivo: BFH772 at 3 mg/kg orally dosed once per day potently inhibits melanoma growth (by 54–90% for primary tumor and71–96% for metastasis growth) as depicted by treatment to control ratios. Dose–response curves of BFH772 at 0.3, 1, and 3 mg/kg demonstrate that even at the lowest concentrations, this naphthalene–1–carboxamide inhibits VEGF induced tissue weight and TIE–2 levels but only reaches statistical significance at 1 mg/kg and above [1].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[1]In vitro kinase assay is based on a filter binding assay, using the recombinant GST–fused kinase domainsexpressed in baculovirus and purified over glutathione–sepharose, γ–[33P]ATP as the phosphate donor, and poly(Glu:Tyr 4:1) peptide as the acceptor. Each GST–fused kinase is incubated under optimized buffer conditions [20 mM Tris–HCl buffer (pH 7.5), 1–3 mM MnCl 2, 3–10 mM MgCl 2, 3–8 μg/mL poly(Glu:Tyr 4:1), 0.25 mg/mL polyethylene glycol 20000, 8 μM ATP, 10 μM sodium vanadate, 1mM DTT] and 0.2 μCi γ–33P ATP in a total volume of 30 μL in the presence or absence of a test substance for 10 min at ambient temperature. The reaction is stopped by adding 10 mL of 250 mM EDTA. Using a 384–well filter system, half the volume istransferred onto an Immobilon–polyvinylidene difluoride membrane. The membrane is then washed extensively and dried, and scintillation counting is performed. IC 50s for compounds are calculated by linear regression analysis of the percentage inhibition [1].Cell Assay: BFH772 is dissolved in DMSO (10 mM) and stored, and then diluted with appropriate medium before use [1]. [1]DifferentBa/F3 cell lines rendered IL–3 independent by transduction with various constitutively active tyrosine kinases are grown in RPMI 1640 medium containing 10% fetal calf serum. For maintenance of parental Ba/F3 cells, the medium is additionally supplemented with 10 ng/mL interleukin–3 (IL–3). For proliferation assays, Ba/F3 cells are seeded on 96–well plates in triplicates at 10000 cells per well and incubated with various concentrations of compounds for 72 h followed by quantification of viable cells using a resazurin sodium salt dye reduction readout (commercially known as Alamar Blue assay). IC 50s are determined with the XLFit Excel Add–In using a four–parameter dose response model [1].Animal Administration: BFH772 is prepared in PEG200 100% (Mice)[1].Product Name:BFH772Cat. No.:HY-100419CAS No.:890128-81-1Molecular Formula:C 23H 16F 3N 3O 3Molecular Weight:439.39Target:VEGFR Pathway:Protein Tyrosine Kinase/RTK Solubility:DMSO: 7.75 mg/mLBFH772 is dissolved in N–methyl pyrrolidone/polyethylene glycol200 (30:70, v/v) (Rat)[1].[1]Mice[1]Female FVB mice weighing between 18 and 20 g are housed in groups of six. Porous chambers containing VEGF (2 μg/mL) in 0.5 mL of 0.8% w/v agar (containing heparin, 20 U/mL) are implanted subcutaneously in the flank of the mice (n=6 per group). VEGF induces the growth of vascularized tissue around the chamber. This response is dose–dependent and can be quantified by measuring the weight and TIE–2 levels of the tissue. Mice are treated either orally once daily with compounds or vehicle (PEG200 100%, 5 mL/kg) starting4–6 h before implantation of the chambers and continuing for 4 days. The animals are sacrificed for measurement of the vascularized tissues 24 h after the last dose. Tissue weight is taken and then a lysate prepared for TIE–2 ELISA analysis .Rat[1]Catheters are implanted into the femoral artery and vein of na?ve female rats strain OFA for BFH772, and BAW2881, or in the jugular vein and femoral artery in female Sprague–Dawley rats for compounds 4, 9, and 10. Animals are allowed to recover for 96 h and are housed in single cages with free access to food and water throughout the experiment. Female OFA rats received 2.5 mg/kg ofBAW2881 dissolved in ethanol/dimethylisosorbide/polyethylene glycol400/D5W (10/15/35/40 v/v) or 1 mg/kg of BFH772 dissolved in N–methyl pyrrolidone/polyethylene glycol200 (30:70, v/v) via injection into the femoral vein. D5W is glucose 5%/water (v/v). Oral administration: BAW2881 and BFH772 are formulated as a micronized suspension (dissolved/suspended in 0.5% carboxymethyl cellulose in distilled water) and administered by gavage to female OFA rats to deliver a dose of 25 mg/kg for BAW2881 or 3 mg/kg BFH772 (n=4 rats per group). For compounds 4, 9, and 10, female Sprague–Dawley rats at 8 weeks of age received an intraveno References:[1]. Bold G, et al. A Novel Potent Oral Series of VEGFR2 Inhibitors Abrogate Tumor Growth by Inhibiting Angiogenesis. J Med Chem. 2016 Jan 14;59(1):132–46.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
TALON Superflow Metal Affinity Resin说明书

Safety Data SheetRevision Date 2022-12-26Revision Number 91. IdentificationProduct identifier Product NameTALON Superflow Metal Affinity ResinOther means of identification Product Code635670 UN number or ID number UN3082SynonymsNo information availableRecommended use of the chemical and restrictions on use Identified uses No information available Restrictions on useNo information availableDetails of the supplier of the safety data sheetEmergency telephone number Emergency telephoneIn case of emergency, call PERS (Professional Emergency Resource Services) 1-800-633-8253 (US) or 801-629-0667 (international).2. Hazard(s) identificationProduct Classification Data Acute toxicity - Oral Category 2 CarcinogenicityCategory 1AHazards not otherwise classified (HNOC) Not applicable Label elements Supplier USA:Takara Bio USA, Inc. 2560 Orchard Parkway San Jose, CA 95131, USAPhone: 800.662.2566/888.251.6618 Web: DangerHazard statements Fatal if swallowed May cause cancerAppearance Pink slurry Physical state Paste / Gel Liquid Odor Alcohol Precautionary Statements - PreventionObtain special instructions before useDo not handle until all safety precautions have been read and understoodWear protective gloves/protective clothing/eye protection/face protectionWash face, hands and any exposed skin thoroughly after handlingDo not eat, drink or smoke when using this productPrecautionary Statements - ResponseIF exposed or concerned: Get medical advice/attentionSpecific treatment (see supplemental first aid instructions on this label)IF SWALLOWED: Immediately call a POISON CENTER or doctorRinse mouthPrecautionary Statements - StorageStore locked upPrecautionary Statements - DisposalDispose of contents/container to an approved waste disposal plantUnknown acute toxicity50.941 % of the mixture consists of ingredient(s) of unknown acute oral toxicityOther informationHarmful to aquatic life with long lasting effects. Harmful to aquatic life.3. Composition/information on ingredientsSubstanceNot applicable.MixtureChemical name CAS No Weight-% Trade secret Ethanol 64-17-5 10 - 20 *4. First-aid measuresDescription of first aid measuresGeneral advice Immediate medical attention is required. Show this safety data sheet to the doctor inattendance. IF exposed or concerned: Get medical advice/attention.Inhalation Remove to fresh air.Eye contact Rinse thoroughly with plenty of water for at least 15 minutes, lifting lower and upper eyelids.Consult a physician.Skin contact Wash skin with soap and water.Ingestion Get immediate medical advice/attention. Do NOT induce vomiting. Clean mouth with waterand drink afterwards plenty of water. Never give anything by mouth to an unconsciousperson.Most important symptoms and effects, both acute and delayedSymptoms No information available.Indication of any immediate medical attention and special treatment neededNote to physicians Treat symptomatically.5. Fire-fighting measuresSuitable Extinguishing Media Use extinguishing measures that are appropriate to local circumstances and thesurrounding environment.Large Fire CAUTION: Use of water spray when fighting fire may be inefficient.Unsuitable extinguishing media Do not scatter spilled material with high pressure water streams.Specific hazards arising from thechemicalNo information available.Explosion DataSensitivity to mechanical impact None.Sensitivity to static discharge None.Special protective equipment for fire-fighters Firefighters should wear self-contained breathing apparatus and full firefighting turnout gear. Use personal protection equipment.6. Accidental release measuresPersonal precautions, protective equipment and emergency proceduresPersonal precautions Ensure adequate ventilation.Other information Refer to protective measures listed in Sections 7 and 8.Methods and material for containment and cleaning upMethods for containment Prevent further leakage or spillage if safe to do so.Methods for cleaning up Pick up and transfer to properly labeled containers.7. Handling and storagePrecautions for safe handlingAdvice on safe handling Handle in accordance with good industrial hygiene and safety practice. Avoid contact withskin, eyes or clothing.Conditions for safe storage, including any incompatibilitiesStorage Conditions Store locked up. Keep containers tightly closed in a dry, cool and well-ventilated place.Keep out of the reach of children.8. Exposure controls/personal protectionControl parametersExposure LimitsChemical name ACGIH TLV OSHA PEL NIOSHEthanol 64-17-5 STEL: 1000 ppm TWA: 1000 ppmTWA: 1900 mg/m3(vacated) TWA: 1000 ppm(vacated) TWA: 1900 mg/m3IDLH: 3300 ppmTWA: 1000 ppmTWA: 1900 mg/m3Appropriate engineering controlsEngineering controls ShowersEyewash stationsVentilation systems.Individual protection measures, such as personal protective equipmentEye/face protection No special protective equipment required.Hand protection Wear suitable gloves.Skin and body protection Wear suitable protective clothing.Respiratory protection No protective equipment is needed under normal use conditions. If exposure limits areexceeded or irritation is experienced, ventilation and evacuation may be required. General hygiene considerations Do not eat, drink or smoke when using this product. Wash hands before breaks andimmediately after handling the product.9. Physical and chemical propertiesInformation on basic physical and chemical propertiesPhysical state Paste / Gel LiquidAppearance Pink slurryColor No information availableOdor AlcoholOdor Threshold No information availableProperty Values Remarks • MethodpH No data available None knownMelting point / freezing point No data available None knownBoiling point/boiling range (°C) No data available None knownFlash point No data available Open cupEvaporation Rate No data available None knownFlammability (solid, gas) No data available None knownFlammability Limit in Air None knownUpper flammability limit: No data availableLower flammability limit: No data availableVapor pressure No data available None knownVapor density No data available None knownRelative density No data available None knownWater solubility No data available None knownSolubility in other solvents No data available None knownPartition coefficient No data available None knownAutoignition temperature 363 °C / 685.4 °F None knownDecomposition temperature None knownKinematic viscosity No data available None knownDynamic Viscosity No data available None knownOther informationExplosive properties No information availableOxidizing properties No information availableSoftening point No information availableMolecular weight No information availableVOC content No information availableLiquid Density No information availableBulk Density No information available10. Stability and reactivityReactivity No information available.Chemical stability Stable under normal conditions.Possibility of hazardous reactions None under normal processing.Conditions to Avoid None known based on information supplied.Incompatible materials None known based on information supplied.Hazardous decomposition products None known based on information supplied.11. Toxicological informationInformation on likely routes of exposureProduct InformationInhalation Specific test data for the substance or mixture is not available.Eye contact Specific test data for the substance or mixture is not available.Skin contact Specific test data for the substance or mixture is not available.Ingestion Specific test data for the substance or mixture is not available. Fatal if swallowed. (based oncomponents).Symptoms related to the physical, chemical and toxicological characteristicsSymptoms No information available.Acute toxicityNumerical measures of toxicityThe following values are calculated based on chapter 3.1 of the GHS documentATEmix (oral) 9.81 mg/kgATEmix (inhalation-dust/mist) 573.50 mg/lUnknown acute toxicity50.941 % of the mixture consists of ingredient(s) of unknown acute oral toxicityComponent InformationChemical name Oral LD50 Dermal LD50 Inhalation LC50 Ethanol = 7060 mg/kg ( Rat ) - = 116.9 mg/L ( Rat ) 4 h64-17-5 = 133.8 mg/L ( Rat ) 4 hDelayed and immediate effects as well as chronic effects from short and long-term exposureSkin corrosion/irritation No information available.Serious eye damage/eye irritation No information available.Respiratory or skin sensitization No information available.Germ cell mutagenicity No information available.Carcinogenicity Contains a known or suspected carcinogen. Classification based on data available foringredients. May cause cancer.The table below indicates whether each agency has listed any ingredient as a carcinogen.Chemical name ACGIH IARC NTP OSHA Ethanol64-17-5A3 Group 1 Known XLegendACGIH (American Conference of Governmental Industrial Hygienists)A3 - Animal CarcinogenIARC (International Agency for Research on Cancer)Group 1 - Carcinogenic to HumansNTP (National Toxicology Program)Known - Known CarcinogenOSHA (Occupational Safety and Health Administration of the US Department of Labor)X - PresentReproductive toxicity No information available.STOT - single exposure No information available.STOT - repeated exposure No information available.Target organ effects Liver, Respiratory system, Eyes, Skin, Central nervous system, Blood, Reproductivesystem.Aspiration hazard No information available.Other adverse effects No information available.Interactive effects No information available.12. Ecological informationEcotoxicity Harmful to aquatic life with long lasting effects.Chemical name Algae/aquatic plants Fish Toxicity tomicroorganismsCrustaceaEthanol 64-17-5 - LC50: 12.0 - 16.0mL/L(96h, Oncorhynchus- LC50: 9268 - 14221mg/L(48h, Daphnia magna)mykiss)LC50: >100mg/L (96h, Pimephales promelas) LC50: 13400 - 15100mg/L (96h, Pimephalespromelas) EC50: =2mg/L (48h, Daphnia magna)Persistence and degradability No information available.Bioaccumulation There is no data for this product.Component InformationChemical name Partition coefficientEthanol64-17-5-0.35Other adverse effects No information available.13. Disposal considerationsWaste treatment methodsWaste from residues/unused products Dispose of in accordance with local regulations. Dispose of waste in accordance with environmental legislation.Contaminated packaging Do not reuse empty containers.California Hazardous Waste Status This product contains one or more substances that are listed with the State of California asa hazardous waste.14. Transport informationDOTUN number or ID number UN3082Proper shipping name Environmentally hazardous substance, liquid, n.o.s.Transport hazard class(es) 9Packing group IIISpecial Provisions 8, 146, 173, 335, 441, IB3, T4, TP1, TP29DOT Marine Pollutant NPDescription UN3082, Environmentally hazardous substance, liquid, n.o.s., 9, IIIEmergency Response GuideNumber171TDGUN number or ID number UN3082UN proper shipping name Environmentally hazardous substance, liquid, n.o.s.Transport hazard class(es) 9Packing group IIISpecial Provisions 16, 99Description UN3082, Environmentally hazardous substance, liquid, n.o.s. (Ethanol), 9, IIIMEXUN number or ID number UN3082UN proper shipping name Environmentally hazardous substance, liquid, n.o.s.Transport hazard class(es) 9Packing group IIITechnical Name EthanolDescription UN3082, Environmentally hazardous substance, liquid, n.o.s. (Ethanol), 9, IIISpecial Provisions 274, 331, 335ICAO (air)UN number or ID number UN3082UN proper shipping name Environmentally hazardous substance, liquid, n.o.s.Transport hazard class(es) 9Packing group IIIDescription UN3082, Environmentally hazardous substance, liquid, n.o.s. (Ethanol), 9, III Special Provisions A97, A158, A197, A215IATAUN number or ID number UN3082UN proper shipping name Environmentally hazardous substance, liquid, n.o.s.Transport hazard class(es) 9Packing group IIITechnical Name EthanolDescription UN3082, Environmentally hazardous substance, liquid, n.o.s. (Ethanol), 9, III Special Provisions A97, A158, A197ERG Code 9LUN number or ID number UN3082UN proper shipping name Environmentally hazardous substance, liquid, n.o.s.Transport hazard class(es) 9Packing group IIIEmS-No F-A, S-FSpecial Provisions 274, 335, 969Marine pollutant PDescription UN3082, Environmentally hazardous substance, liquid, n.o.s., 9, III, Marine Pollutant RIDUN number or ID number UN3082UN proper shipping name Environmentally hazardous substance, liquid, n.o.s.Transport hazard class(es) 9Packing group IIIClassification code M6Special Provisions 274, 335, 375, 601Description UN3082, Environmentally hazardous substance, liquid, n.o.s. (Ethanol), 9, IIIADRUN number or ID number UN3082UN proper shipping name Environmentally hazardous substance, liquid, n.o.s.Transport hazard class(es) 9Packing group IIIClassification code M6Tunnel restriction code (-)Special Provisions 274, 335, 601, 375Description UN3082, Environmentally hazardous substance, liquid, n.o.s. (Ethanol), 9, III, (-) ADNNotes Could not find a Marine Pollutant Name.UN number or ID number UN3082UN proper shipping name Environmentally hazardous substance, liquid, n.o.s.Transport hazard class(es) 9Packing group IIIClassification code M6Special Provisions 274, 335, 375, 601Description UN3082, Environmentally hazardous substance, liquid, n.o.s. (Ethanol, Cobalt), 9, III Equipment Requirements PP15. Regulatory informationInternational InventoriesTSCA -.*Contact supplier for details. One or more substances in this product are either not listed on the US TSCA inventory, listed on the confidential US TSCA inventory or are otherwise exempted from inventory listing requirementsDSL/NDSL -.EINECS/ELINCS -.ENCS -.IECSC -.KECL -.PICCS -.AICS -.Legend:TSCA - United States Toxic Substances Control Act Section 8(b) InventoryDSL/NDSL - Canadian Domestic Substances List/Non-Domestic Substances ListEINECS/ELINCS - European Inventory of Existing Chemical Substances/European List of Notified Chemical SubstancesENCS - Japan Existing and New Chemical SubstancesIECSC - China Inventory of Existing Chemical SubstancesKECL - Korean Existing and Evaluated Chemical SubstancesPICCS - Philippines Inventory of Chemicals and Chemical SubstancesAICS - Australian Inventory of Chemical SubstancesUS Federal RegulationsSARA 313Section 313 of Title III of the Superfund Amendments and Reauthorization Act of 1986 (SARA). This product does not contain any chemicals which are subject to the reporting requirements of the Act and Title 40 of the Code of Federal Regulations, Part 372. SARA 311/312 Hazard CategoriesShould this product meet EPCRA 311/312 Tier reporting criteria at 40 CFR 370, refer to Section 2 of this SDS for appropriate classifications.CWA (Clean Water Act)This product does not contain any substances regulated as pollutants pursuant to the Clean Water Act (40 CFR 122.21 and 40 CFR 122.42).CERCLAThis material, as supplied, does not contain any substances regulated as hazardous substances under the Comprehensive Environmental Response Compensation and Liability Act (CERCLA) (40 CFR 302) or the Superfund Amendments and Reauthorization Act (SARA) (40 CFR 355). There may be specific reporting requirements at the local, regional, or state level pertaining to releases of this material.US State RegulationsCalifornia Proposition 65This product contains the following Proposition 65 chemicals:.Chemical name California Proposition 65Ethanol - 64-17-5 CarcinogenDevelopmentalCobalt - 7440-48-4 CarcinogenU.S. State Right-to-Know RegulationsChemical name New Jersey Massachusetts Pennsylvania EthanolX X X 64-17-5X X X Cobalt7440-48-4U.S. EPA Label InformationEPA Pesticide Registration Number Not applicable16. Other informationNFPA Health hazards 3 Flammability 1 Instability 0 Special hazards - HMIS Health hazards * 3 Flammability 1 Physical hazards 0 Personal protection X Chronic Hazard Star Legend * = Chronic Health HazardKey or legend to abbreviations and acronyms used in the safety data sheetLegend Section 8: EXPOSURE CONTROLS/PERSONAL PROTECTIONTWA Time weighted average STEL Short term exposure limitCeiling Maximum limit value * Skin designationKey literature references and sources for data used to compile the SDSAgency for Toxic Substances and Disease Registry (ATSDR)U.S. Environmental Protection Agency ChemView DatabaseEuropean Food Safety Authority (EFSA)EPA (Environmental Protection Agency)Acute Exposure Guideline Level(s) (AEGL(s))U.S. Environmental Protection Agency Federal Insecticide, Fungicide, and Rodenticide ActU.S. Environmental Protection Agency High Production Volume ChemicalsFood Research JournalHazardous Substance DatabaseInternational Uniform Chemical Information Database (IUCLID)Japan GHS ClassificationAustralia National Industrial Chemicals Notification and Assessment Scheme (NICNAS)NIOSH (National Institute for Occupational Safety and Health)National Library of Medicine's ChemID Plus (NLM CIP)National Library of Medicine’s PubMed database (NLM PUBMED)National Toxicology Program (NTP)New Zealand's Chemical Classification and Information Database (CCID)Organization for Economic Co-operation and Development Environment, Health, and Safety PublicationsOrganization for Economic Co-operation and Development High Production Volume Chemicals ProgramOrganization for Economic Co-operation and Development Screening Information Data SetWorld Health OrganizationRevision Date 2022-12-26Revision Note No information available.DisclaimerThe information provided in this Safety Data Sheet is correct to the best of our knowledge, information and belief at the date of its publication. The information given is designed only as a guidance for safe handling, use, processing, storage, transportation, disposal and release and is not to be considered a warranty or quality specification. The information relates only to the specific material designated and may not be valid for such material used in combination with any other materials or in any process, unless specified in the text.End of Safety Data Sheet。
Antibody structure, instability, and formulation

MINIREVIEWAntibody Structure,Instability,and FormulationWEI WANG,SATISH SINGH,DAVID L.ZENG,KEVIN KING,SANDEEP NEMAPfizer,Inc.,Global Biologics,700Chesterfield Parkway West,Chesterfield,Missouri63017Received14March2006;revised17May2006;accepted4June2006Published online in Wiley InterScience().DOI10.1002/jps.20727 ABSTRACT:The number of therapeutic monoclonal antibody in development hasincreased tremendously over the last several years and this trend continues.At presentthere are more than23approved antibodies on the US market and an estimated200ormore are in development.Although antibodies share certain structural similarities,development of commercially viable antibody pharmaceuticals has not been straightfor-ward because of their unique and somewhat unpredictable solution behavior.This articlereviews the structure and function of antibodies and the mechanisms of physical andchemical instabilities.Various aspects of formulation development have been examinedto identify the critical attributes for the stabilization of antibodies.ß2006Wiley-Liss,Inc.and the American Pharmacists Association J Pharm Sci96:1–26,2007Keywords:biotechnology;stabilization;protein formulation;protein aggregation;freeze drying/lyophilizationINTRODUCTIONProtein therapies are entering a new era with the influx of a significant number of antibody pharmaceuticals.Generally,protein drugs are effective at low concentrations with less side effects relative to small molecule drugs,even though,in rare cases,protein-induced antibody formation could be serious.1Therefore,this category of therapeutics is gaining tremendous momentum and widespread recognition both in small and large drugfirms.Among protein drug therapies,antibodies play a major role in control-ling many types of diseases such as cancer, infectious diseases,allergy,autoimmune dis-eases,and inflammation.Since the approval of thefirst monoclonal antibody(MAb)product -OKT-3in1986,more than23MAb drug products have entered the market(Tab.1).The estimated number of antibodies and antibody derivatives constitute20%of biopharmaceutical products currently in development(about200).2The global therapeutic antibody market was predicted to reach$16.7billion in2008.3There are several reasons for the increasing popularity of antibodies for commercial develop-ment.First,their action is specific,generally leading to fewer side effects.Second,antibodies may be conjugated to another therapeutic entity for efficient delivery of this entity to a target site, thus reducing potential side effects.For instance, Mylotarg is an approved chemotherapy agent composed of calicheamicin conjugated to huma-nized IgG4,which binds specifically to CD33for the treatment of CD33-positive acute myeloid leukemia.Another example is the conjugation of immunotoxic barnase with the light chain of the anti-human ferritin monoclonal antibody F11as potential targeting agents for cancer immuno-therapy.4Third,antibodies may be conjugated to radioisotopes for specific diagnostic purposes. Examples include CEA-Scan for detection of color-ectal cancer and ProstaScint for detection of prostate stly,technology advancement has made complete human MAb available,which are lessimmunogenic.JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY20071 Correspondence to:Wei Wang(Telephone:(636)-247-2111;Fax:(636)-247-5030;E-mail:wei.2.wang@pfi)Journal of Pharmaceutical Sciences,Vol.96,1–26(2007)Pharmacists AssociationT a b l e 1.C o m m e r c i a l M o n o c l o n a l A n t i b o d y P r o d u c t s#B r a n d n a m e M o l e c u l eM A bY e a r C o m p a n y R o u t e I n d i c a t i o n M A b C o n c B u f f e r E x c i p i e n t s S u r f a c t a n t p H1A v a s t i n B e v a c i z u m a bH u m a n i z e d I g G 1,149k D a2004G e n e t e c h a n d B i o O n c o l o g y I V i n f u s i o nM e t a s t a t i c c a r c i n o m a o f c o l o n o r r e c t u m ,b i n d s V E G F 100m g a n d 400m g /v i a l (25m g /m L )s o l u t i o n 5.8m g /m L m o n o b a s i c N a P h o s H 2O ;1.2m g /m L d i b a s i c N a P h o s a n h y d r o u s (4m L ,16m L fil l i n v i a l )60m g /m L a -T r e h a l o s e d i h y d r a t e (4m L ,16m L fil l i n v i a l )0.4m g /m L P S 20(4m L ,16m L fil l i n v i a l )6.22B e x x a rT o s i t u m o m a b a n d I -131T o s i t u m a b M u r i n e I g G 2l2003C o r i x a a n d G S KI V I n f u s i o nC D 20p o s i t i v e f o l l i c u l a r n o n H o d g k i n s l y m p h o m aK i t :14m g /m L M A b s o l u t i o n i n 35m g a n d 225m g v i a l s ;1.1m g /m L I 131-M A b s o l u t i o n10m M p h o s p h a t e (M A b v i a l )145m M N a C l ,10%w /v M a l t o s e ;I 131-M A b :5–6%P o v i d o n e ,1–2,9–15m g /m L M a l t o s e ,0.9m g /m L N a C l ,0.9–1.3m g /m L A s c o r b i c a c i d 7.23C a m p a t h A l e m t u z u m a bH u m a n i z e d ,I g G 1k ,150k D a2001I l e x O n c o l o g y ;M i l l e n i u m a n d B e r l e xI V i n f u s i o nB -c e l l c h r o n i c l y m p h o c y t i c l e u k e m i a ,CD 52-a n t i g e n 30m g /3m L s o l u t i o n3.5m g /3m L d i b a s i c N a P h o s ,0.6m g /3m L m o n o b a s i c K P h o s 24m g /3m L N a C l ,0.6m g /3m L K C l ,0.056m g /3m L N a 2E D T A 0.3m g /3m L P S 806.8–7.44C E A -S c a n (l y o )A c r i t u o m a b ;T c -99M u r i n e F a b ,50k D a1996I m m u n o m e d i c s I V i n j e c t i o n o r i n f u s i o nI m a g i n g a g e n t f o r c o l o r e c t a l c a n c e r1.25m g /v i a l L y o p h i l i z e d M A b .R e c o n s t i t u t e w 1m L S a l i n e w T c 99m 0.29m g /v i a l S t a n n o u s c h l o r i d e ,p o t a s s i u m s o d i u m t a r t r a t e t e t r a h y d r a t e ,N a A c e t a t e .3H 2O ,N a C l ,g l a c i a l a c e t i c a c i d ,H C l S u c r o s e5.75E r b i t u x C e t u x i m a bC h i m e r i c h u m a n /m o u s e I g G 1k ,152kD a 2004I m C l o n e a n d B M S I V i n f u s i o n T r e a t m e n t o fE GF R -e x p r e s s i n g c o l o r e c t a l c a r c i n o m a 100m g M A b i n 50m L ;2m g /m L s o l u t i o n1.88m g /m L D i b a s i c N a P h o s Á7H 2O ;0.42m g /m L M o n o b a s i c N a P h o s ÁH 2O8.48m g /m L N a C l 7.0–7.46H e r c e p t i n (l y o )T r a s t u z u m a bH u m a n i z e d I g G 1k1998G e n e t e c h I V i n f u s i o n M e t a s t a t i c b r e a s t c a n c e r w h o s e t u m o r o v e r e x p r e s s H E R 2p r o t e i n 440m g /v i a l ,21m g /m L a f t e r r e c o n s t i t u t i o n 9.9m g /20m L L -H i s t i d i n e H C l ,6.4m g /20m L L -H i s t i d i n e400m g /20m L a -T r e h a l o s e D i h y d r a t e 1.8m g /20m L P S 2067H u m i r a A d a l i m u m a bH u m a n I g G 1k ,148k D a2002C A T a n d A b b o t t S CR A p a t i e n t s n o t r e s p o n d i n g t o D M A R D s .B l o c k s T N F -a l p h a40m g /0.8m L s o l u t i o n (50m g /m L )0.69m g /0.8m L M o n o b a s i c N a P h o s Á2H 2O ;1.22m g /0.8m L D i b a s i c N a P h o s Á2H 2O ;0.24m g /0.8m L N a C i t r a t e ,1.04m g /0.8m L C i t r i c a c i d ÁH 2O 4.93m g /0.8m L N a C l ;9.6m g /0.8m L M a n n n i t o l 0.8m g /0.8m L P S 805.28L u c e n t i s R a n i b i z u m a bH u m a n i z e d I g G 1k f r a g m e n t2006G e n e n t e c h I n t r a v i t r e a l i n j e c t i o n A g e -r e l a t e d m a c u l a r d e g e n e r a t i o n (w e t )10m g /m L s o l u t i o n10m M H i s t i d i n e H C l10%a -T r e h a l o s e -D i h y d r a t e 0.01%P S 205.52WANG ET AL.JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY 2007DOI 10.1002/jps9M y l o t a r g (l y o )G e m t u z u m a b o z o g a m i c i nH u m a n i z e d I g G 4k c o n j u g a t e d w i t h c a l i c h e a m i c i n2000C e l l t e c h a n d W y e t h I V i n f u s i o nH u m a n i z e d A b l i n k e d t o c a l i c h e a m i c i n f o r t r e a t m e n t o f C D 33p o s i t i v e a c u t e m y e l o i d l e u k e m i a 5m g p r o t e i n -e q u i v a l e n t l y o p h i l i z e d p o w d e r /20-m L v i a l M o n o b a s i c a n d d i b a s i c N a P h o s p h a t e D e x t r a n 40,S u c r o s e ,N a C l 10O n c o S c i n tS a t u m o m a b p e n d e t i d eM u r i n e I g G 1k c o n j u g a t e d t o G Y K -D T P A1992C y t o g e n I V i n j e c t i o nI m a g i n g a g e n t f o r c o l o r e c t a l a n d o v a r i a n c a n c e r0.5m g c o n j u g a t e /m L s o l u t i o n (2m L p e r v i a l )P h o s p h a t e b u f f e r s a l i n e 6.011O r t h o c l o n e O K TM u r o m o m a b -C D 3M u r i n e ,I g G 2a ,170k D a1986O r t h o B i o t e c h I V i n j e c t i o nR e v e r s a l o f a c u t e k i d n e y t r a n s p l a n t r e j e c t i o n (a n t i C D 3-a n t i g e n )1m g /m L s o l u t i o n2.25m g /5m L m o n o b a s i c N a P h o s ,9.0m g /5m L d i b a s i c N a P h o s 43m g /5m L N a C l 1m g /m L P S 807Æ0.512P r o s t a S c i n tI n d i u m -111c a p r o m a b p e n d e t i d e M u r i n e I g G 1k -c o n j u g a t e d t o G Y K -D T P A1996C y t o g e n I V i n j e c t i o nI m a g i n g a g e n t f o r p r o s t a t e c a n c e r0.5m g c o n j u g a t e /m L s o l u t i o n (1m L p e r v i a l )P h o s p h a t e b u f f e r s a l i n e 5–713R a p t i v a (l y o )E f a l i z u m a bH u m a n i z e d I g G 1k2003X o m a a n d G e n e n t e c h S C C h r o n i c m o d e r a t e t o s e v e r e p l a q u e p s o r i a s i s ,b i n d s t o C D 11a s u b u n i t o f L F A -1150m g M A b /v i a l ;125m g /1.25m L (100m g /m L )a f t e r r e c o n s t i t u t i o n w i t h 1.3m L S W F I 6.8m g /v i a l L -H i s t i d i n e H C l ÁH 2O ;4.3m g /v i a l L -H i s t i d i n e123.2m g /v i a l S u c r o s e 3m g /v i a l P S 206.214R e m i c a d e (l y o )I n fli x i m a bC h i m e r i c h u m a n /m u r i n e M A b a g a i n s t T N F a l p h a (a p p .30%m u r i n e ,70%c o r r e s p o n d s t o h u m a n I g G 1h e a v y c h a i n a n d h u m a n k a p p a l i g h t c h a i n c o n s t a n t r e g i o n s )1998C e n t o c o r I V i n f u s i o nR A a n d C r o h n ’s d i s e a s e (a n t i T N F a l p h a )100m g /20-m L V i a l ,10m g /m L o n r e c o n s t i t u t i o n2.2m g /10m L M o n o b a s i c N a P h o s H 2O ,6.1m g /10m L D i b a s i c N a P h o s Á2H 2O 500m g /10m L S u c r o s e 0.5m g /10m L P S 807.215R e o P r o A b c i x i m a bF a b .C h i m e r i c h u m a n -m u r i n e ,48k D a 1994C e n t o c o r /L i l l y I V i n j e c t i o n a n d i n f u s i o n R e d u c t i o n o f a c u t e b l o o d c l o t r e l a t e d c o m p l i c a t i o n s 2m g /m L s o l u t i o n 0.01M N a P h o s p h a t e 0.15M N a C l 0.001%(0.01m g /m L )P S 807.216R i t u x a n R i t u x i m a bC h i m e r i c m o u s e /h u m a n I g G 1k w i t h m u r i n e l i g h t a n d h e a v y c h a i n v a r i a b l e r e g i o n (F a b d o m a i n ),145kD a1997I D E C a n d G e n e n t e c h I V i n f u s i o nN o n H o d g k i n ’s l y m p h o m a .(a n t i C D 20-a n t i g e n )10m g /m L s o l u t i o n7.35m g /m L N a C i t r a t e Á2H 2O9m g /m L N a C l 0.7m g /m L P S 806.5(C o n t i n u e d )ANTIBODY FORMULATION3DOI 10.1002/jpsJOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY 200717S i m u l e c t (l y o )B a s i l i x i m a bC h i m a r i c I g G 1k ,144kD a1998N o v a r t i s I V i n j e c t i o n a n d i n f u s i o nP r e v e n t i o n o f a c u t e k i d n e y t r a n s p l a n t r e j e c t i o n ,I L -2r e c e p t o r a n t a g o n i s t10m g a n d 20m g /v i a l ,4m g /m L o n r e c o n s t i t u t i o n 3.61m g ,7.21m g M o n o b a s i c K P h o s ;0.50m g ,0.99m g N a 2H P O 40.8m g ,1.61m g N a C l ;10m g ,20m g S u c r o s e ;40m g ,80m g M a n n i t o l ;20m g 40m g G l y c i n e 18S y n a g i s (l y o )P a l i v i z u m a bH u m a n i z e d I g G 1k ,C D R o f m u r i n e M A b 1129,148k D a 1998M e d I m m u n e I M i n j e c t i o nP r e v e n t r e p l i c a t i o n o f t h e R e s p i r a t o r y s y n c y t i a l v i r u s (R S V )50m g a n d 100m g /v i a l ,100m g /m L o n r e c o n s t i t u t i o n47m M H i s t i d i n e ,3.0m M G l y c i n e 5.6%M a n n i t o l19T y s a b r i N a t a l i z u m a bH u m a i n z e d I g G 4k2004B i o g e n I D E C I V I n f u s i o nM S r e l a p s e 300m g /15m L s o l u t i o n 17.0m g M o n o b a s i c N a P h o s ÁH 2O ,7.24m g d i B a s i c N a P h o s Á7H 2O f o r 15m L 123m g /15m L N a C l3.0m g /15m L P S 806.120V e r l u m a N o f e t u m o m a b M u r i n e F a b 1996B o e h r i n g e r I n g e l h e i m a n d D u P o n t M e r c k I V i n j e c t i o n I m a g i n g a g e n t f o r l u n g c a n c e r10m g /m L s o l u t i o nP h o s p h a t e b u f f e r s a l i n e?21X o l a i r (l y o )O m a l i z u m a bH u m a n i z e d I g G 1k ,149k D aG e n e n t e c h w N o v a r t i s a n d T a n o xS CA s t h m a ,i n h i b i t s b i n d i n g o f I g E t o I g E r e c e p t o r F C e R I202.5m g /v i a l ,D e l i v e r 150m g /1.2m L o n r e c o n s t i t u t i o n w i t h 1.4m L S W F I 2.8m g L H i s t i d i n e H C l ÁH 2O ;1.8m g L H i s t i d i n e145.5m g S u c r o s e 0.5m g P S 2022Z e n a p a x D a c l i z u m a bH u m a n i z e d I g G 1,144k D a1997R o c h e I V i n f u s i o nP r o p h y l a x i s o f a c u t e o r g a n r e j e c t i o n i n p a t i e n t s r e c e i v i n g r e n a l t r a n s p l a n t s .I n h i b i t s I L -2b i n d i n g t o t h e T a c s u b u n i t o f I L -2r e c e p t o r c o m p l e x 25m g /5m L M A b S o l u t i o n3.6m g /m L M o n o b a s i c N a P h o s ÁH 2O ;11m g /m L D i b a s i c N a P h o s Á7H 2O4.6m g /m L N a C l 0.2m g /m L P S 806.923Z e v a l i nI b r i t u m o m a b -T i u x e t a nM u r i n e I g G 1k -t h i o u r e a c o v a l e n t l i n k a g e t o T i u x e t a nI D E C I V i n f u s i o nC D 20a n t i g e n .(K i t w i t h Y t t e r i u m -90i n d u c e s c e l l u l a r d a m a g e b y b e t a e m i s s i o n )3.2m g /2m L s o l u t i o n 09%N a C l 7.1T a b l e 1.(C o n t i n u e d )#B r a n d n a m e M o l e c u l eM A bY e a r C o m p a n y R o u t e I n d i c a t i o n M A b C o n c B u f f e r E x c i p i e n t s S u r f a c t a n t p H4WANG ET AL.JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY 2007DOI 10.1002/jpsDevelopment of commercially viable antibody pharmaceuticals has,however,not been straight-forward.This is because the behavior of antibodies seems to vary,even though they have similar structures.In attempting to address some of the challenges in developing antibody therapeutics, Harris et al.5reviewed the commercial-scale formulation and characterization of therapeutic recombinant antibodies.In a different review, antibody production and purification have been discussed.2Nevertheless,the overall instability and stabilization of antibody drug candidates have not been carefully examined in the litera-ture.This article,not meant to be exhaustive, intends to review the structure and functions of antibodies,discuss their instabilities,and sum-marize the methods for stabilizing/formulating antibodies.ANTIBODY STRUCTUREAntibodies(immunoglobulins)are roughly Y-shaped molecules or combination of such molecules(Fig.1). Their structures are divided into two regions—the variable(V)region(top of the Y)defining antigen-binding properties and the constant(C)region (stem of the Y),interacting with effector cells and molecules.Immunoglobulins can be divided into five different classesÀIgA,IgD,IgE,IgM,and IgG based on their C regions,respectively desig-nated as a,d,e,m,and g(five main heavy-chain classes).6Most IgGs are monomers,but IgA and IgM are respectively,dimmers and pentamers linked by J chains.IgGs are the most abundant,widely used for therapeutic purposes,and their structures will be discussed as antibody examples in detail.Primary StructureThe structure of IgGs have been thoroughly reviewed.6The features of the primary structure of antibodies include heavy and light chains, glycosylation,disulfide bond,and heterogeneity. Heavy and Light ChainsIgGs contain two identical heavy(H,50kDa)and two identical light(L,25kDa)chains(Fig.1). Therefore,the total molecular weight is approxi-mately150kDa.There are several disulfide bonds linking the two heavy chains,linking the heavy and light chains,and residing inside the chains (also see next section).IgGs are further divided into several subclasses—IgG1,IgG2,IgG3,and IgG4(in order of relative abundance in human plasma),with different heavy chains,named g1, g2,g3,and g4,respectively.The structural differences among these subtypes are the number and location of interchain disulfide bonds and the length of the hinge region.The light chains consist of two types—lambda(l)and kappa(k). In mice,the average of k to l ratio is20:1,whereas it is2:1in humans.6The variable(V)regions of both chains cover approximately thefirst 110amino acids,forming the antigen-binding (Fab)regions,whereas the remaining sequences are constant(C)regions,forming Fc(fragment crystallizable)regions for effector recognition and binding.6The N-terminal sequences of both the heavy and light chains vary greatly between different antibodies.It was suggested that the conserved sequences in human IgG1antibodies Figure1.Linear(upper panel)and steric(lower panel)structures of immunoglobulins(IgG).ANTIBODY FORMULATION5DOI10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY2007are approximately95%and the remaining5% is variable and creates their antigen-binding specificity.5The V regions are further divided into three hypervariable sequences(HV1,HV2,and HV3)on both H and L chains.In the light chains,these are roughly from residues28to35,from49to59,and from92to103,respectively.6Other regions are the framework regions(FR1,FR2,FR3,and FR4).The HV regions are also called the complementarity determining regions(CDR1,CDR2,and CDR3). While the framework regions form the b-sheets, the HV sequences form three loops at the outer edge of the b barrel(also see Section2.2).Disulfide BondsMost IgGs have four interchain disulfide bonds—two connecting the two H chains at the hinge region and the other two connecting the two L chains to the H chains.6Exceptions do exist.Two disulfide bonds were found in IgG1and IgG4 linking the two heavy chain in the hinge region but four in IgG2.7In IgG1MAb,HC is linked to the LC between thefifth Cys(C217)of HC and C213on the LC.In IgG2and IgG4MAbs,it is the third Cys of HC(C123)linking to the LC.7A disulfide bond between HC C128and LC C214 was found for mouse catalytic monoclonal anti-bodies(IgG2a).8IgGs have four intrachain disulfide bonds, residing in each domain of the H and L chains, stabilizing these domains.The intrachain disul-fide bonds in V H and V L are required in functional antigen binding.9Native IgG MAbs should not have any free sulfhydryl groups.7However, detailed examination of the free sulfhydryl groups in recombinant MAbs(one IgG1,two IgG2,and one IgG4)suggests presence of a small portion of free sulfhydryl group(approximately0.02mol per mole of IgG2or IgG4MAb and0.03for IgG1.7In rare cases,a free cysteine is found.A nondisulfide-bonded Cys at residue105was found on the heavy chain of a mouse monoclonal antibody,OKT3 (IgG2a).10OligosaccharidesThere is one oligosaccharide chain in IgGs.6This N-linked biantennary sugar chain resides mostly on the conserved Asn297,which is buried between the C H2domains.5,11For example,the oligosaccharide resides on Asn-297of the C H2 domain of chimeric IgG1and IgG3molecules12but on Asn299in a monoclonal antibody,OKT3 (IgG2a).10The oligosaccharide,often microheter-ogeneous,is typically fucosylated in antibodies produced in CHO or myeloma cell lines5and may differ in other cell lines.2,11There are many factors that dictate the nature of the glycan microheterogenity on IgGs.These include cell line,the bioreactor conditions and the nature of the downstream processing.An additional oligo-saccharide can be found in rare cases.A human IgG produced by a human-human-mouse hetero-hybridoma contains an additional oligosaccharide on Asn75in the variable region of its heavy chain.13In addition,O-linked carbohydrates could also exist in this antibody.Proper glycosylation is critical for correct functioning of antibodies.11It was demonstrated that removal of the oligosaccharide in IgGs(IgG1 and IgG3)made them ineffective in binding to C1q, in binding to the human Fc g RI and activating C; and generally more sensitive to most proteases than their corresponding wild-type IgGs(one exception).12This is because the binding site on IgG for C1q,thefirst component of the complement cascade,is localized in the C H2domains.11 Furthermore,the glycosylation can affect the antibody conformation.12Oligosaccharides in other regions can also play a critical role.Removal of an oligosaccharide in a Fv region of the CBGA1antibody resulted in a decreased antigen-binding activity in several ELISA systems.13In addition,this oligosaccharide might play critical role in reducing the antigenicity of the protein.14The sugar composition of the oligosaccharide is also critical in antibody functions.It has been shown that a low fucose(Fuc)content in the complex-type oligosaccharide in a humanized chimeric IgG1is responsible for a50-fold higher antibody-dependent cellular cytotoxicity(ADCC) compared with a high Fuc counterpart.15 HeterogeneityPurified antibodies are heterogeneous in struc-ture.This is true for all monoclonal antibodies (MAbs)due to differences in glycosylation pat-terns,instability during production,and terminal processing.5For example,five charged isoforms were found in recombinant humanized monoclo-nal antibody HER2as found by capillary iso-electric focusing(cIEF)and sodium dodecyl sulfate–capillary gel electrophoresis(SDS–CGE).16Six separate bands were focused under6WANG ET AL.JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY2007DOI10.1002/jpsIEF for two mouse monoclonal antibodies IgG2a (k)and IgG1(k).17A mature monoclonal antibody, OKT3(IgG2a),contain cyclized N-terminus (pyroglutamic acid,À17D)in both H and L chains, processed C-terminus(no Lys,À128D)of the H chains,and a small amount of deamidated form.10 Similar observation was also reported for a huma-nized IgG1(k).18In rare cases,gene cross-over may lead to formation of abnormal heavy chains.For example,a purified monoclonal anti-IgE antibody contains a small amount of a variant H chain, which had16fewer amino acid residues than the normal H chain(position is between Arg108of the L chain and Ala124of the H chain).19 Secondary and Higher-Order StructureThe basic secondary and higher-order structural features of IgGs have been reviewed.6Only a small portion of the three-dimensional structures of IgGs has been solved.20The antibody’s secon-day structure is formed as the polypeptide chains form anti-parallel b-sheets.The major type of secondary structure in IgGs is these b-sheets and its content is roughly70%as measured by FTIR.21The light chain consists of two and the heavy chain contains four domains,each about 110amino acid long.6,20All these domains have similar folded structures—b barrel,also called immunoglobulin fold,which is stabilized by a disulfide bond and hydrophobic interaction(pri-mary).These individual domains($12kDa in size)interact with one another(V H and V L;C H1 and C L;and between two C H3domains except the carbohydrate-containing C H2domain)and fold into three equal-sized spherical shape linked by a flexible hinge region.These three spheres form a Y shape(mostly)and/or a T shape.22The less globular shape of IgGs is maintained both by disulfide bonds and by strong noncovalent interactions between the two heavy chains and between each of the heavy-chain/light-chain pairs.23Through noncovalent interactions,a less stable domain becomes more stable,and thus,the whole molecule can be stabilized.24A detailed study indicates that the interaction between two CH3domains are dominated by six contact residues,five of these residues(T366,L368, F405,Y407,and K409)forming a patch at the center of the interface.25These noncovalent interactions are spatially oriented such that variable domain exchange(switching V H and V L; inside-out IgG;ioIgG)induces noncovalent multimerization.26The six hypervariable regions in CDR(L1,L2, L3,H1,H2,and H3)form loops of a few predictable main-chain conformations(or canonical forms), except H3loop,which has too many variations in conformation to be predicted accurately.27,28 There is a slight difference in the loop composition and shape between the two types of light chains.20 However,no functional difference was found in antibodies having l or k chain.6Basic Functions of AntibodiesThe basic functions of antibodies have been reviewed.6There are two functional areas in IgGs—the V and C regions.The V regions of the two heavy and light chains offer two identical antigen-binding sites.The binding of the two sites (bivalent)can be independent of each other and does not seem to depend on the C region.29The exact antigen-binding sites are the CDR regions with participation of the frame work regions.30 Binding of antigens seems through the induced-fit mechanism.31,32The induced-fit mechanism allows multispecificity and polyreactivity.It has been suggested that about5–10residues usually contribute significantly to the binding energy.32 The C regions of antibodies have three main effector functions(1)being recognized by receptors on immune effector cells,initiating antibody-dependent cell cytotoxicities(ADCC),(2)binding to complement,helping to recruit activated pha-gocytes,and(3)being transported to a variety of places,such as tears and milk.6In addition,C domains also modulate in vivo stability.23,29,33The function of Fc is affected by the structure of Fab. Variable domain exchange(switching V H and V L; inside-out IgG;ioIgG)affected Fc-associated func-tions such as serum half-life and binding to protein G and Fc g RI.26The hinge region providesflexibility in bivalent antigen binding and activation of Fc effector functions.26Two chimeric IgG3antibodies lacking a genetic hinge but with Cys residues in CH2 regions was found to be deficient in their inter-molecular assembly,and both IgG3D HþCys and IgG3D Hþ2Cys lost greatly their ability to bind Fc g RI and failed to bind C1q and activate the complement cascade.34Alternative Forms of AntibodiesIn addition to species-specific antibodies,other antibody forms are generated to meet various needs.In the early development of antibody therapies,antibodies were made from murineANTIBODY FORMULATION7DOI10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY2007sources.However,these antibodies easily elicit formation of human anti-mouse antibody (HAMA).Therefore,humanized chimeric antibo-dies were generated.Chimeric monoclonal anti-bodies(60–70%human)are made of mouse variable regions and human constant regions.2 Such antibodies can still induce formation of human anti-chimeric antibody(HACA).Highly humanized antibodies,CDR-grafted antibodies, are made by replacing only the human CDR with mouse CDR regions(90–95%human).2These antibodies are almost the same in immunogeni-city potential as completely human antibodies, which may illicit formation of human anti-human antibody(HAHA).Other alternative forms of antibodies have also been generated and these different forms have been reviewed.35Treatment with papain would cleave the N-terminal side of the disulfide bonds and generate two identical Fab fragments and one Fc fragment.Fab0s are50kDa(V HþC H1)/ (V LþC L)heterodimers linked by a single disul-fide bond.Treatment with pepsin cleaves the C-terminal side of the disulfide bonds and pro-duces a F(ab)02fragment.The remaining H chains were cut into several small fragments.6Cleavage by papain occurs at the C-terminal side of His-H22836or His-H227.37Reduction of F(ab0)2will produce two Fab0.23Fv fragments are noncovalent heterodimers of V H and V L.Stabilization of the fragment by a hydrophilicflexible peptide linker generates single-chain Fv(scFvs).2Fragments without constant domains can also be made into domain antibodies (dAbs).These scFvs are25–30kDa variable domain (V HþV L)dimers joined by polypeptide linkers of at least12residues.Shorter linkers(5–10residues)do not allow pairing of the variable domains but allow association with another scFv form a bivalent dimer (diabody)(about60kDa,or trimer:triabody about 90kDa).38Two diabodies can be further linked together to generate bispecific tandem diabody (tandab).39Disulfide-free scFv molecules are rela-tively stable and useful for intracellular applica-tions of antibodies—‘‘intrabodies.’’38The smallest of the antibody fragments is the minimal recognition unit(MRU)that can be derived from the peptide sequences of a single CDR.2ANTIBODY INSTABILITYAntibodies,like other proteins,are prone to a variety of physical and chemical degradation path-ways,although antibodies,on the average,seem to be more stable than other proteins.Antibody instabilities can be observed in liquid,frozen,and lyophilized states.The glycosylation state of an antibody can significantly affect its degradation rate.40In many cases,multiple degradation path-ways can occur at the same time and the degrada-tion mechanism may change depending on the stress conditions.41These degradation pathways are divided into two major categories—physical and chemical instabilities.This section will explore the possible degradation pathways of antibodies and their influencing factors.Physical InstabilityAntibodies can show physical instability via two major pathways—denaturation and aggregation. DenaturationAntibodies can denature under a variety of conditions.These conditions include temperature change,shear,and various processing steps. Compared with other proteins,antibodies seem to be more resistant to thermal stress.They may not melt completely until temperature is raised above708C,21,42,43while most other mesophilic proteins seem to melt below708C.44Shear may cause antibody denaturation.For example,the antigen-binding activity of a recombinant scFv antibody fragment was reduced with afirst-order rate constant of0.83/h in a buffer solution at a shear of approximately20,000/s.45Lyophilization can denature a protein to var-ious extents.An anti-idiotypic antibody(MMA 383)in a formulation containing mannitol,sac-charose,NaCl,and phosphate was found to loose its in vivo immunogenic properties(only10–20% of normal response rate)upon lyophilization.46 Since the protein showed no evidence of degrada-tion after lyophilization,no change in secondary structure by CD(29%b-sheet,14%a-helix,and 57%‘‘other’’),the loss of activity was attributed to the conformational change.Indeed,tryptophan fluorescence properties were different between the lyophilized and unlyophilized antibodies.46 AggregationAntibody aggregation is a more common manifes-tation of physical instability.The concentration-dependent antibody aggregation was considered the greatest challenge to developing protein formulations at higher concentrations.47This is8WANG ET AL.JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY2007DOI10.1002/jps。
稳定性英文版

HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationFLUOXETINE HClC17H18F3NO•HClM.W. = 345.79CAS — 59333-67-4STABILITY INDICATINGA S S A Y V A L I D A T I O NMethod is suitable for:ýIn-process controlþProduct ReleaseþStability indicating analysis (Suitability - US/EU Product) CAUTIONFLUOXETINE HYDROCHLORIDE IS A HAZARDOUS CHEMICAL AND SHOULD BE HANDLED ONLY UNDER CONDITIONS SUITABLE FOR HAZARDOUS WORK.IT IS HIGHLY PRESSURE SENSITIVE AND ADEQUATE PRECAUTIONS SHOULD BE TAKEN TO AVOID ANY MECHANICAL FORCE (SUCH AS GRINDING, CRUSHING, ETC.) ON THE POWDER.ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationTABLE OF CONTENTS INTRODUCTION........................................................................................................................ PRECISION............................................................................................................................... System Repeatability ................................................................................................................ Method Repeatability................................................................................................................. Intermediate Precision .............................................................................................................. LINEARITY................................................................................................................................ RANGE...................................................................................................................................... ACCURACY............................................................................................................................... Accuracy of Standard Injections................................................................................................ Accuracy of the Drug Product.................................................................................................... VALIDATION OF FLUOXETINE HCl AT LOW CONCENTRATION........................................... Linearity at Low Concentrations................................................................................................. Accuracy of Fluoxetine HCl at Low Concentration..................................................................... System Repeatability................................................................................................................. Quantitation Limit....................................................................................................................... Detection Limit........................................................................................................................... VALIDATION FOR META-FLUOXETINE HCl (POSSIBLE IMPURITIES).................................. Meta-Fluoxetine HCl linearity at 0.05% - 1.0%........................................................................... Detection Limit for Fluoxetine HCl.............................................................................................. Quantitation Limit for Meta Fluoxetine HCl................................................................................ Accuracy for Meta-Fluoxetine HCl ............................................................................................ Method Repeatability for Meta-Fluoxetine HCl........................................................................... Intermediate Precision for Meta-Fluoxetine HCl......................................................................... SPECIFICITY - STABILITY INDICATING EVALUATION OF THE METHOD............................. FORCED DEGRADATION OF FINISHED PRODUCT AND STANDARD..................................1. Unstressed analysis...............................................................................................................2. Acid Hydrolysis stressed analysis..........................................................................................3. Base hydrolysis stressed analysis.........................................................................................4. Oxidation stressed analysis...................................................................................................5. Sunlight stressed analysis.....................................................................................................6. Heat of solution stressed analysis.........................................................................................7. Heat of powder stressed analysis.......................................................................................... System Suitability stressed analysis.......................................................................................... Placebo...................................................................................................................................... STABILITY OF STANDARD AND SAMPLE SOLUTIONS......................................................... Standard Solution...................................................................................................................... Sample Solutions....................................................................................................................... ROBUSTNESS.......................................................................................................................... Extraction................................................................................................................................... Factorial Design......................................................................................................................... CONCLUSION...........................................................................................................................ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationBACKGROUNDTherapeutically, Fluoxetine hydrochloride is a classified as a selective serotonin-reuptake inhibitor. Effectively used for the treatment of various depressions. Fluoxetine hydrochloride has been shown to have comparable efficacy to tricyclic antidepressants but with fewer anticholinergic side effects. The patent expiry becomes effective in 2001 (US). INTRODUCTIONFluoxetine capsules were prepared in two dosage strengths: 10mg and 20mg dosage strengths with the same capsule weight. The formulas are essentially similar and geometrically equivalent with the same ingredients and proportions. Minor changes in non-active proportions account for the change in active ingredient amounts from the 10 and 20 mg strength.The following validation, for the method SI-IAG-206-02 , includes assay and determination of Meta-Fluoxetine by HPLC, is based on the analytical method validation SI-IAG-209-06. Currently the method is the in-house method performed for Stability Studies. The Validation was performed on the 20mg dosage samples, IAG-21-001 and IAG-21-002.In the forced degradation studies, the two placebo samples were also used. PRECISIONSYSTEM REPEATABILITYFive replicate injections of the standard solution at the concentration of 0.4242mg/mL as described in method SI-IAG-206-02 were made and the relative standard deviation (RSD) of the peak areas was calculated.SAMPLE PEAK AREA#15390#25406#35405#45405#55406Average5402.7SD 6.1% RSD0.1ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::PRECISION - Method RepeatabilityThe full HPLC method as described in SI-IAG-206-02 was carried-out on the finished product IAG-21-001 for the 20mg dosage form. The method repeated six times and the relative standard deviation (RSD) was calculated.SAMPLENumber%ASSAYof labeled amountI 96.9II 97.8III 98.2IV 97.4V 97.7VI 98.5(%) Average97.7SD 0.6(%) RSD0.6PRECISION - Intermediate PrecisionThe full method as described in SI-IAG-206-02 was carried-out on the finished product IAG-21-001 for the 20mg dosage form. The method was repeated six times by a second analyst on a different day using a different HPLC instrument. The average assay and the relative standard deviation (RSD) were calculated.SAMPLENumber% ASSAYof labeled amountI 98.3II 96.3III 94.6IV 96.3V 97.8VI 93.3Average (%)96.1SD 2.0RSD (%)2.1The difference between the average results of method repeatability and the intermediate precision is 1.7%.HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationLINEARITYStandard solutions were prepared at 50% to 200% of the nominal concentration required by the assay procedure. Linear regression analysis demonstrated acceptability of the method for quantitative analysis over the concentration range required. Y-Intercept was found to be insignificant.RANGEDifferent concentrations of the sample (IAG-21-001) for the 20mg dosage form were prepared, covering between 50% - 200% of the nominal weight of the sample.Conc. (%)Conc. (mg/mL)Peak Area% Assayof labeled amount500.20116235096.7700.27935334099.21000.39734463296.61500.64480757797.52000.79448939497.9(%) Average97.6SD 1.0(%) RSD 1.0ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::RANGE (cont.)The results demonstrate linearity as well over the specified range.Correlation coefficient (RSQ)0.99981 Slope11808.3Y -Interceptresponse at 100%* 100 (%) 0.3%ACCURACYACCURACY OF STANDARD INJECTIONSFive (5) replicate injections of the working standard solution at concentration of 0.4242mg/mL, as described in method SI-IAG-206-02 were made.INJECTIONNO.PEAK AREA%ACCURACYI 539299.7II 540599.9III 540499.9IV 5406100.0V 5407100.0Average 5402.899.9%SD 6.10.1RSD, (%)0.10.1The percent deviation from the true value wasdetermined from the linear regression lineHPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::ACCURACY OF THE DRUG PRODUCTAdmixtures of non-actives (placebo, batch IAG-21-001 ) with Fluoxetine HCl were prepared at the same proportion as in a capsule (70%-180% of the nominal concentration).Three preparations were made for each concentration and the recovery was calculated.Conc.(%)Placebo Wt.(mg)Fluoxetine HCl Wt.(mg)Peak Area%Accuracy Average (%)70%7079.477.843465102.27079.687.873427100.77079.618.013465100.0101.0100%10079.6211.25476397.910080.8011.42491799.610079.6011.42485498.398.6130%13079.7214.90640599.413080.3114.75632899.213081.3314.766402100.399.618079.9920.10863699.318079.3820.45879499.418080.0820.32874899.599.4Placebo, Batch Lot IAG-21-001HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::VALIDATION OF FLUOXETINE HClAT LOW CONCENTRATIONLINEARITY AT LOW CONCENTRATIONSStandard solution of Fluoxetine were prepared at approximately 0.02%-1.0% of the working concentration required by the method SI-IAG-206-02. Linear regression analysis demonstrated acceptability of the method for quantitative analysis over this range.ACCURACY OF FLUOXETINE HCl AT LOW CONCENTRATIONThe peak areas of the standard solution at the working concentration were measured and the percent deviation from the true value, as determined from the linear regression was calculated.SAMPLECONC.µg/100mLAREA FOUND%ACCURACYI 470.56258499.7II 470.56359098.1III 470.561585101.3IV 470.561940100.7V 470.56252599.8VI 470.56271599.5(%) AverageSlope = 132.7395299.9SD Y-Intercept = -65.872371.1(%) RSD1.1HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationSystem RepeatabilitySix replicate injections of standard solution at 0.02% and 0.05% of working concentration as described in method SI-IAG-206-02 were made and the relative standard deviation was calculated.SAMPLE FLUOXETINE HCl AREA0.02%0.05%I10173623II11503731III10103475IV10623390V10393315VI10953235Average10623462RSD, (%) 5.0 5.4Quantitation Limit - QLThe quantitation limit ( QL) was established by determining the minimum level at which the analyte was quantified. The quantitation limit for Fluoxetine HCl is 0.02% of the working standard concentration with resulting RSD (for six injections) of 5.0%. Detection Limit - DLThe detection limit (DL) was established by determining the minimum level at which the analyte was reliably detected. The detection limit of Fluoxetine HCl is about 0.01% of the working standard concentration.ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::VALIDATION FOR META-FLUOXETINE HCl(EVALUATING POSSIBLE IMPURITIES)Meta-Fluoxetine HCl linearity at 0.05% - 1.0%Relative Response Factor (F)Relative response factor for Meta-Fluoxetine HCl was determined as slope of Fluoxetine HCl divided by the slope of Meta-Fluoxetine HCl from the linearity graphs (analysed at the same time).F =132.7395274.859534= 1.8Detection Limit (DL) for Fluoxetine HClThe detection limit (DL) was established by determining the minimum level at which the analyte was reliably detected.Detection limit for Meta Fluoxetine HCl is about 0.02%.Quantitation Limit (QL) for Meta-Fluoxetine HClThe QL is determined by the analysis of samples with known concentration of Meta-Fluoxetine HCl and by establishing the minimum level at which the Meta-Fluoxetine HCl can be quantified with acceptable accuracy and precision.Six individual preparations of standard and placebo spiked with Meta-Fluoxetine HCl solution to give solution with 0.05% of Meta Fluoxetine HCl, were injected into the HPLC and the recovery was calculated.HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::META-FLUOXETINE HCl[RECOVERY IN SPIKED SAMPLES].Approx.Conc.(%)Known Conc.(µg/100ml)Area in SpikedSampleFound Conc.(µg/100mL)Recovery (%)0.0521.783326125.735118.10.0521.783326825.821118.50.0521.783292021.55799.00.0521.783324125.490117.00.0521.783287220.96996.30.0521.783328526.030119.5(%) AVERAGE111.4SD The recovery result of 6 samples is between 80%-120%.10.7(%) RSDQL for Meta Fluoxetine HCl is 0.05%.9.6Accuracy for Meta Fluoxetine HClDetermination of Accuracy for Meta-Fluoxetine HCl impurity was assessed using triplicate samples (of the drug product) spiked with known quantities of Meta Fluoxetine HCl impurity at three concentrations levels (namely 80%, 100% and 120% of the specified limit - 0.05%).The results are within specifications:For 0.4% and 0.5% recovery of 85% -115%For 0.6% recovery of 90%-110%HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::META-FLUOXETINE HCl[RECOVERY IN SPIKED SAMPLES]Approx.Conc.(%)Known Conc.(µg/100mL)Area in spikedSample Found Conc.(µg/100mL)Recovery (%)[0.4%]0.4174.2614283182.66104.820.4174.2614606187.11107.370.4174.2614351183.59105.36[0.5%]0.5217.8317344224.85103.220.5217.8316713216.1599.230.5217.8317341224.81103.20[0.6%]0.6261.3918367238.9591.420.6261.3920606269.81103.220.6261.3920237264.73101.28RECOVERY DATA DETERMINED IN SPIKED SAMPLESHPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::REPEATABILITYMethod Repeatability - Meta Fluoxetine HClThe full method (as described in SI-IAG-206-02) was carried out on the finished drug product representing lot number IAG-21-001-(1). The HPLC method repeated serially, six times and the relative standard deviation (RSD) was calculated.IAG-21-001 20mg CAPSULES - FLUOXETINESample% Meta Fluoxetine % Meta-Fluoxetine 1 in Spiked Solution10.0260.09520.0270.08630.0320.07740.0300.07450.0240.09060.0280.063AVERAGE (%)0.0280.081SD 0.0030.012RSD, (%)10.314.51NOTE :All results are less than QL (0.05%) therefore spiked samples with 0.05% Meta Fluoxetine HCl were injected.HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::Intermediate Precision - Meta-Fluoxetine HClThe full method as described in SI-IAG-206-02 was applied on the finished product IAG-21-001-(1) .It was repeated six times, with a different analyst on a different day using a different HPLC instrument.The difference between the average results obtained by the method repeatability and the intermediate precision was less than 30.0%, (11.4% for Meta-Fluoxetine HCl as is and 28.5% for spiked solution).IAG-21-001 20mg - CAPSULES FLUOXETINESample N o:Percentage Meta-fluoxetine% Meta-fluoxetine 1 in spiked solution10.0260.06920.0270.05730.0120.06140.0210.05850.0360.05560.0270.079(%) AVERAGE0.0250.063SD 0.0080.009(%) RSD31.514.51NOTE:All results obtained were well below the QL (0.05%) thus spiked samples slightly greater than 0.05% Meta-Fluoxetine HCl were injected. The RSD at the QL of the spiked solution was 14.5%HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationSPECIFICITY - STABILITY INDICATING EVALUATIONDemonstration of the Stability Indicating parameters of the HPLC assay method [SI-IAG-206-02] for Fluoxetine 10 & 20mg capsules, a suitable photo-diode array detector was incorporated utilizing a commercial chromatography software managing system2, and applied to analyze a range of stressed samples of the finished drug product.GLOSSARY of PEAK PURITY RESULT NOTATION (as reported2):Purity Angle-is a measure of spectral non-homogeneity across a peak, i.e. the weighed average of all spectral contrast angles calculated by comparing all spectra in the integrated peak against the peak apex spectrum.Purity Threshold-is the sum of noise angle3 and solvent angle4. It is the limit of detection of shape differences between two spectra.Match Angle-is a comparison of the spectrum at the peak apex against a library spectrum.Match Threshold-is the sum of the match noise angle3 and match solvent angle4.3Noise Angle-is a measure of spectral non-homogeneity caused by system noise.4Solvent Angle-is a measure of spectral non-homogeneity caused by solvent composition.OVERVIEWT he assay of the main peak in each stressed solution is calculated according to the assay method SI-IAG-206-02, against the Standard Solution, injected on the same day.I f the Purity Angle is smaller than the Purity Threshold and the Match Angle is smaller than the Match Threshold, no significant differences between spectra can be detected. As a result no spectroscopic evidence for co-elution is evident and the peak is considered to be pure.T he stressed condition study indicated that the Fluoxetine peak is free from any appreciable degradation interference under the stressed conditions tested. Observed degradation products peaks were well separated from the main peak.1® PDA-996 Waters™ ; 2[Millennium 2010]ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationFORCED DEGRADATION OF FINISHED PRODUCT & STANDARD 1.UNSTRESSED SAMPLE1.1.Sample IAG-21-001 (2) (20mg/capsule) was prepared as stated in SI-IAG-206-02 and injected into the HPLC system. The calculated assay is 98.5%.SAMPLE - UNSTRESSEDFluoxetine:Purity Angle:0.075Match Angle:0.407Purity Threshold:0.142Match Threshold:0.4251.2.Standard solution was prepared as stated in method SI-IAG-206-02 and injected into the HPLC system. The calculated assay is 100.0%.Fluoxetine:Purity Angle:0.078Match Angle:0.379Purity Threshold:0.146Match Threshold:0.4272.ACID HYDROLYSIS2.1.Sample solution of IAG-21-001 (2) (20mg/capsule) was prepared as in method SI-IAG-206-02 : An amount equivalent to 20mg Fluoxetine was weighed into a 50mL volumetric flask. 20mL Diluent was added and the solution sonicated for 10 minutes. 1mL of conc. HCl was added to this solution The solution was allowed to stand for 18 hours, then adjusted to about pH = 5.5 with NaOH 10N, made up to volume with Diluent and injected into the HPLC system after filtration.Fluoxetine peak intensity did NOT decrease. Assay result obtained - 98.8%.SAMPLE- ACID HYDROLYSISFluoxetine peak:Purity Angle:0.055Match Angle:0.143Purity Threshold:0.096Match Threshold:0.3712.2.Standard solution was prepared as in method SI-IAG-206-02 : about 22mg Fluoxetine HCl were weighed into a 50mL volumetric flask. 20mL Diluent were added. 2mL of conc. HCl were added to this solution. The solution was allowed to stand for 18 hours, then adjusted to about pH = 5.5 with NaOH 10N, made up to volume with Diluent and injected into the HPLC system.Fluoxetine peak intensity did NOT decrease. Assay result obtained - 97.2%.ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationSTANDARD - ACID HYDROLYSISFluoxetine peak:Purity Angle:0.060Match Angle:0.060Purity Threshold:0.099Match Threshold:0.3713.BASE HYDROLYSIS3.1.Sample solution of IAG-21-001 (2) (20mg/capsule) was prepared as per method SI-IAG-206-02 : An amount equivalent to 20mg Fluoxetine was weight into a 50mL volumetric flask. 20mL Diluent was added and the solution sonicated for 10 minutes. 1mL of 5N NaOH was added to this solution. The solution was allowed to stand for 18 hours, then adjusted to about pH = 5.5 with 5N HCl, made up to volume with Diluent and injected into the HPLC system.Fluoxetine peak intensity did NOT decrease. Assay result obtained - 99.3%.SAMPLE - BASE HYDROLYSISFluoxetine peak:Purity Angle:0.063Match Angle:0.065Purity Threshold:0.099Match Threshold:0.3623.2.Standard stock solution was prepared as per method SI-IAG-206-02 : About 22mg Fluoxetine HCl was weighed into a 50mL volumetric flask. 20mL Diluent was added. 2mL of 5N NaOH was added to this solution. The solution was allowed to stand for 18 hours, then adjusted to about pH=5.5 with 5N HCl, made up to volume with Diluent and injected into the HPLC system.Fluoxetine peak intensity did NOT decrease - 99.5%.STANDARD - BASE HYDROLYSISFluoxetine peak:Purity Angle:0.081Match Angle:0.096Purity Threshold:0.103Match Threshold:0.3634.OXIDATION4.1.Sample solution of IAG-21-001 (2) (20mg/capsule) was prepared as per method SI-IAG-206-02. An equivalent to 20mg Fluoxetine was weighed into a 50mL volumetric flask. 20mL Diluent added and the solution sonicated for 10 minutes.1.0mL of 30% H2O2 was added to the solution and allowed to stand for 5 hours, then made up to volume with Diluent, filtered and injected into HPLC system.Fluoxetine peak intensity decreased to 95.2%.ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationSAMPLE - OXIDATIONFluoxetine peak:Purity Angle:0.090Match Angle:0.400Purity Threshold:0.154Match Threshold:0.4294.2.Standard solution was prepared as in method SI-IAG-206-02 : about 22mg Fluoxetine HCl were weighed into a 50mL volumetric flask and 25mL Diluent were added. 2mL of 30% H2O2 were added to this solution which was standing for 5 hours, made up to volume with Diluent and injected into the HPLC system.Fluoxetine peak intensity decreased to 95.8%.STANDARD - OXIDATIONFluoxetine peak:Purity Angle:0.083Match Angle:0.416Purity Threshold:0.153Match Threshold:0.4295.SUNLIGHT5.1.Sample solution of IAG-21-001 (2) (20mg/capsule) was prepared as in method SI-IAG-206-02 . The solution was exposed to 500w/hr. cell sunlight for 1hour. The BST was set to 35°C and the ACT was 45°C. The vials were placed in a horizontal position (4mm vials, National + Septum were used). A Dark control solution was tested. A 2%w/v quinine solution was used as the reference absorbance solution.Fluoxetine peak decreased to 91.2% and the dark control solution showed assay of 97.0%. The difference in the absorbance in the quinine solution is 0.4227AU.Additional peak was observed at RRT of 1.5 (2.7%).The total percent of Fluoxetine peak with the degradation peak is about 93.9%.SAMPLE - SUNLIGHTFluoxetine peak:Purity Angle:0.093Match Angle:0.583Purity Threshold:0.148Match Threshold:0.825 ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationSUNLIGHT (Cont.)5.2.Working standard solution was prepared as in method SI-IAG-206-02 . The solution was exposed to 500w/hr. cell sunlight for 1.5 hour. The BST was set to 35°C and the ACT was 42°C. The vials were placed in a horizontal position (4mm vials, National + Septum were used). A Dark control solution was tested. A 2%w/v quinine solution was used as the reference absorbance solution.Fluoxetine peak was decreased to 95.2% and the dark control solution showed assay of 99.5%.The difference in the absorbance in the quinine solution is 0.4227AU.Additional peak were observed at RRT of 1.5 (2.3).The total percent of Fluoxetine peak with the degradation peak is about 97.5%. STANDARD - SUNLIGHTFluoxetine peak:Purity Angle:0.067Match Angle:0.389Purity Threshold:0.134Match Threshold:0.8196.HEAT OF SOLUTION6.1.Sample solution of IAG-21-001-(2) (20 mg/capsule) was prepared as in method SI-IAG-206-02 . Equivalent to 20mg Fluoxetine was weighed into a 50mL volumetric flask. 20mL Diluent was added and the solution was sonicated for 10 minutes and made up to volume with Diluent. 4mL solution was transferred into a suitable crucible, heated at 105°C in an oven for 2 hours. The sample was cooled to ambient temperature, filtered and injected into the HPLC system.Fluoxetine peak was decreased to 93.3%.SAMPLE - HEAT OF SOLUTION [105o C]Fluoxetine peak:Purity Angle:0.062Match Angle:0.460Purity Threshold:0.131Match Threshold:0.8186.2.Standard Working Solution (WS) was prepared under method SI-IAG-206-02 . 4mL of the working solution was transferred into a suitable crucible, placed in an oven at 105°C for 2 hours, cooled to ambient temperature and injected into the HPLC system.Fluoxetine peak intensity did not decrease - 100.5%.ED. N0: 04Effective Date:APPROVED::。
FDA批准的精准医疗诊断体外器械一览表List of Cleared or Approved Companion Diagnostic Devices
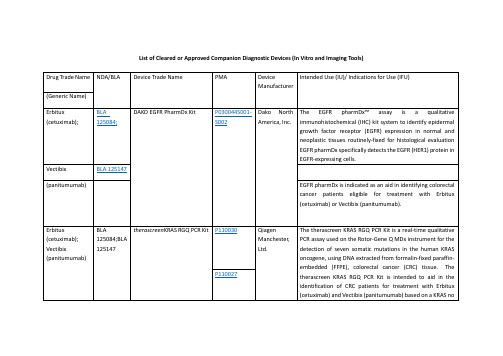
Drug Trade Name
NDA/BLA
Device Trade Name
PMA
Device Manufacturer
Intended Use (IU)/ Indications for Use (IFU)
(imatinibmesylate)
NDA 021588
The c-KitpharmDxis indicated as an aid in the differential diagnosis of gastrointestinal stromal tumors (GIST). After diagnosis of GIST, results from c-KitpharmDxmay be used as an aid in identifying those patients eligible for treatment withGleevec/Glivec(imatinibmesylate).
(deferasirox)
Gilotrif
NDA 201292
therascreenEGFR RGQ PCR Kit
P120022
QiagenManchester, Ltd.
ThetherascreenEGFR RGQ PCR Kit is a real-time PCR test for the qualitative detection of exon 19 deletions and exon 21 (L858R) substitution mutations of the epidermal growth factor receptor (EGFR) gene in DNA derived from formalin-fixed paraffin-embedded (FFPE) non-small cell lung cancer (NSCLC) tumor tissue. The test is intended to be used to select patients with NSCLC for whom GILOTRIF (afatinib), an EGFR tyrosine kinase inhibitor (TKI), is indicated. Safety and efficacy of GILOTRIF (afatinib) have not been established in patients whose tumors have L861Q, G719X, S768I, exon 20 insertions, and T790M mutations, which are also detected by thetherascreenEGFR RGQ PCR Kit.
7. Endotoxin LALTests

Charles River Endosafe
2
Woo Jung BSC Inc.
August 25, 2003
LAL Discoveries by Bang and Levin
Described role of endotoxin in coagulation of Limulus blood
Prepared Endotoxin - responsive lysate from Amoebocytes
Endotoxicity
ENDOTOXIN CAUSES HUMAN TISSUE TO RELEASE INFLAMMATORY MEDIATORS INFLAMMATION INDUCES A VARIETY OF TISSUE DAMAGE SHOCK and MULTIPLE ORGAN DYSFUNCTION MAY OCCUR
Endotoxins and Pyrogens
Pyrogens are fever-inducing agents in humans and animals
include endotoxin, gram + cell debris, fungi
Endotoxins are components from the outer membrane of gram-negative bacteria
Clotting Enzyme
Liquid Coagulogen
M++ pH=7.2
Clotted Coagulin Gel
Summary of Gel Clot Test
Endpoint sought by 180 inversion of sample tube
机械通气临床应用指南(中华重症医学分会2024)

机械通气临床应用指南中华医学会重症医学分会(2024年)引言重症医学是探讨危重病发生发展的规律,对危重病进行预防和治疗的临床学科。
器官功能支持是重症医学临床实践的重要内容之一。
机械通气从仅作为肺脏通气功能的支持治疗起先,经过多年来医学理论的发展及呼吸机技术的进步,已经成为涉及气体交换、呼吸做功、肺损伤、胸腔内器官压力及容积环境、循环功能等,可产生多方面影响的重要干预措施,并主要通过提高氧输送、肺脏爱护、改善内环境等途径成为治疗多器官功能不全综合征的重要治疗手段。
机械通气不仅可以依据是否建立人工气道分为“有创”或“无创”,因为呼吸机具有的不同呼吸模式而使通气有众多的选择,不同的疾病对机械通气提出了具有特异性的要求,医学理论的发展及循证医学数据的增加使对呼吸机的临床应用更加趋于有明确的针对性和规范性。
在这种条件下,不难看出,对危重病人的机械通气制定规范有明确的必要性。
同时,多年临床工作的积累和多中心临床探讨证据为机械通气指南的制定供应了越来越充分的条件。
中华医学会重症医学分会以循证医学的证据为基础,采纳国际通用的方法,经过广泛征求看法和建议,反复仔细探讨,达成关于机械通气临床应用方面的共识,以期对危重病人的机械通气的临床应用进行规范。
重症医学分会今后还将依据医学证据的发展及新的共识对机械通气临床应用指南进行更新。
指南中的举荐看法依据2024年ISF提出的Delphi分级标准(表1)。
指南涉及的文献依据探讨方法和结果分成5个层次,举荐看法的举荐级别依据Delphi分级分为A E级,其中A 级为最高。
表1 Delphi分级标准举荐级别A 至少有2项I级探讨结果支持B 仅有1项I级探讨结果支持C 仅有II级探讨结果支持D 至少有1项III级探讨结果支持E 仅有IV级或V探讨结果支持探讨课题分级I 大样本,随机探讨,结果清楚,假阳性或假阴性的错误很低II 小样本,随机探讨,结果不确定,假阳性和/或假阴性的错误较高III 非随机,同期比照探讨IV 非随机,历史比照和专家看法V 病例报道,非比照探讨和专家看法危重症患者人工气道的选择人工气道是为了保证气道通畅而在生理气道与其他气源之间建立的连接,分为上人工气道和下人工气道,是呼吸系统危重症患者常见的抢救措施之一。
MEDICA EasyRA

REF 10213-4 4 x 24mL总蛋白 (TP)楔形瓶,每瓶含试剂可用量 24mL。
预期用途EasyRA® TP 试剂目的是用于通过 MEDICA EasyRA 临床化学分析仪进行人血清或血浆(采用肝素锂作为抗凝血剂)中总蛋白的定量测定。
仅用于体外诊断用途。
仅供专业人员使用。
摘要和说明1, 2, 3在人血清中,血清蛋白主要包括白蛋白(占总蛋白的50-60%),其余部分包括α1-、α2-、β- 和γ-球蛋白。
总蛋白的浓度对维持血液和组织之间水的正常平衡与交换是很重要的。
血清蛋白浓度较低,可能是由于吸收不良、合成受损引起的,或是由于出血或过度分解代谢导致蛋白损失而引起的,如肾病和肝病中观察到的情况,这种情况可能导致低蛋白血症。
在高免疫球蛋白血症(如多发性骨髓瘤和感染)或脱水情况下,可能发生高蛋白血症。
方法的原理本分析法采用双缩脲反应测定血清蛋白,其中碱性溶液中的二价铜离子与化合物(含 2 个或多个、与碳原子结合的酰胺基或肽基)反应,形成一种有色络合物4。
碱性 pH血清蛋白质类 + Cu++———— 紫色络合物紫色络合物以分光光度法在 550nm 处测定,以 700nm 作为空白波长。
在较宽的线性范围内,该络合物的密度与蛋白质浓度成正比5。
试剂五水合硫酸铜0.3g/dL氢氧化钠0.8g/dL碘化钾、酒石酸钾钠和叠氮化钠作为防腐剂。
注意事项1.当处理任何实验室试剂时,应遵守良好实验室安全规范(CLSI, GP17-A2)。
2.氢氧化钠具有腐蚀性,能引起烧伤。
不要用嘴吸取试剂。
如果吞服,应立即就医处理。
避免眼部和皮肤接触。
如发生眼部接触,立即用大量水清洗眼睛,并就医处理。
如果发生皮肤接触,用水清洗皮肤至少 15 分钟。
3.本试剂含叠氮化钠 <0.1%;叠氮化钠可与铅和铜水管反应,形成高爆炸性金属叠氮化物。
请参考化学品安全说明书中的危险、危害和安全信息。
4.就任何诊断试验方法而言,其结果应根据所有其它试验结果和患者的临床状况加以解释。
安捷伦产品目录

15
Real-Time PCR
16
Mx3000P QPCR System
17
Brilliant III Ultra-Fast SYBR Green QPCR and QRT-PCR Reagents
18
Brilliant III Ultra-Fast QPCR and QRT-PCR Reagents
Agilent / STRATAGENE
Agilent website: /genomics
Welgene | Agilent Stratagene
威健股份有限公司 | Stratagene 總代理
Table of Content
Table of Contents
/ XL1-Red Competent Cells SoloPack Gold Supercompetent Cells
/ TK Competent Cells Specialty Cells
/ Classic Cells / Fine Chemicals For Competent Cells
適用於 UNG 去汙染或 bisulphite
sequencing
適用於 TA Cloning
最高敏感性
取代傳統 Taq 的好選擇
-
2
威健股份有限公司 | Stratagene 總代理
PCR Enzyme & Instrument
Agilent SureCycler 8800
市場上領先的 cycling 速度和 sample 體積 10 ~ 100 μL 簡易快速可以選擇 96 well 和 384 well 操作盤 優秀的溫控設備讓各個 well 都能保持溫度的穩定 七吋的高解析度觸控螢幕讓操作上更為簡便 可以透過網路遠端操控儀器及監控儀器 Agilent 專業的技術支援可以幫助您應對各種 PCR 的問題
CHIR-99021_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:CHIR–99021 is a GSK–3α/β inhibitor with IC 50 of 10 nM/6.7 nM; > 500–fold selectivity for GSK–3 versus its closest homologs CDC2 and ERK2, as well as other protein kinases.IC50 & Target: IC50: 10 nM/6.7 nM (GSK–3α/β)[1]In Vitro: CHIR 99021inhibits human GSK–3β with K i values of 9.8 nM [1]. CHIR 99021 is a small organic molecule that inhibits GSK3α and GSK3β by competing for their ATP–binding sites.In vitro kinase assays reveal that CHIR 99021 specifically inhibits GSK3β (IC 50=~5 nM) and GSK3α (IC 50=~10 nM), with little effect on other kinases [2]. In the presence of CHIR–99021 the viability of the ES–D3 cells is reduced by 24.7% at 2.5 μM, 56.3% at 5 μM, 61.9% at 7.5 μM and 69.2% at 10 μM CHIR–99021 with an IC 50 of 4.9μM [3].In Vivo: In ZDF rats, a single oral dose of CHIR 99021 (16 mg/kg or 48 mg/kg) rapidly lowers plasma glucose, with a maximal reduction of nearly 150 mg/dl 3–4 h after administration [1]. CHIR99021 (2 mg/kg) given once, 4 h before irradiation, significantly improves survival after 14.5 Gy abdominal irradiation (ABI). CHIR99021 treatment significantly blocks crypt apoptosis andaccumulation of p–H2AX + cells, and improves crypt regeneration and villus height. CHIR99021 treatment increases Lgr5+ cellsurvival by blocking apoptosis, and effectively prevents the reduction of Olfm4, Lgr5 and CD44 as early as 4 h [4].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[2]Kinases are purified from SF9 cells through use of their His or Glu tag. Glu–tagged proteins are purified, and His–tagged proteins are purified. Kinase assays are performed in 96–well plates with appropriate peptide substrates in a 300–μL reaction buffer (variations on 50 mM Tris–HCl, pH 7.5, 10 mM MgCl 2, 1 mM EGTA, 1 mMdithiothreitol, 25 mMβ–glycerophosphate, 1mM NaF, and 0.01% bovine serum albumin). Peptides has K m values from 1 to 100 μM. CHIR 99021 or CHIR GSKIA is added in 3.5μL of Me 2SO, followed by ATP to a final concentration of 1 μM. After incubation, triplicate 100–μL aliquots are transferred to Combiplate 8 plates containing 100 μL/well of 50 μM ATP and 20 mM EDTA. After 1 hour, the wells are rinsed five times with phosphate–buffered saline, filled with 200 μL of scintillation fluid, sealed, and counted in a scintillation counter 30 min later. All of the steps are at room temperature. The percentage of inhibition is calculated as 100×(inhibitor–no enzyme control)/(Me 2SO control–no enzyme control)[2].Cell Assay: CHIR 99021 is dissolved in DMSO and stored, and then diluted with appropriate media before use [3].[3]The viability of the mouse ES cells is determined after exposure to different concentrations of GSK3 inhibitors for three days using the MTT assay.The decrease of MTT activity is a reliable metabolism–based test for quantifying cell viability; this decrease correlates with the loss of cell viability. 2,000 cells are seeded overnight on gelatine–coated 96–well plates in LIF–containing ES cell medium. On the next day the medium is changed to medium devoid of LIF and with reduced serum and supplemented with 0.1–1 μM BIO, or 1–10 μM SB–216763, CHIR–99021 or CHIR–98014. Basal medium without GSK3 inhibitors or DMSO is used as control. All tested conditions are analyzed in triplicates [3].Product Name:CHIR–99021Cat. No.:HY-10182CAS No.:252917-06-9Molecular Formula:C 22H 18Cl 2N 8Molecular Weight:465.34Target:GSK–3; GSK–3; Autophagy Pathway:Stem Cell/Wnt; PI3K/Akt/mTOR; Autophagy Solubility:DMSO: ≥ 5.1 mg/mLAnimal Administration: CHIR 99021 is formulated as solutions in 20 mM citrate–buffered 15% Captisol or as fine suspensions in0.5% carboxymethylcellulose (Rat)[1].CHIR 99021 is prepared in DMSO and diluted (Mice)[4].[1][4]Rat[1]Primary hepatocytes from male Sprague Dawley rats that weighed <140 g are prepared and used 1–3 h after isolation. Aliquotsof 1×106cells in 1 mL of DMEM/F12 medium plus 0.2% BSA and CHIR 99021(orally at 16 or 48 mg/kg) or controls are incubated in 12–well plates on a low–speed shaker for 30 min at 37°C in a CO2–enriched atmosphere, collected by centrifugation and lysed by freeze/thaw in buffer A plus 0.01% NP40; the GS assay is again performed.Mice[4]Mice 6–10 weeks old are used. The PUMA+/+ and PUMA–/– littermates on C57BL/6 background (F10) and Lgr5–EGFP(Lgr5–EGFP–IRES–creERT2) mice are subjected to whole body irradiation (TBI), or abdominal irradiation (ABI). Mice are injected intraperitoneally (i.p.) with 2 mg/kg of CHIR99021 4 h before radiation or 1 mg/kg of SB415286 28 h and 4 h before radiation. Mice are sacrificed to collect small intestines for histology analysis and western blotting. All mice are injected i.p. with 100 mg/kg of BrdU before sacrifice.References:[1]. Ring DB, et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. 2003 Mar;52(3):588–95.[2]. Bennett CN, et al. Regulation of Wnt signaling during adipogenesis. J Biol Chem. 2002 Aug 23;277(34):30998–1004.[3]. Naujok O, et al. Cytotoxicity and activation of the Wnt/beta–catenin pathway in mouse embryonic stem cells treated with four GSK3 inhibitors.BMC Res Notes. 2014 Apr 29;7:273.[4]. Wang X, et al. Pharmacologically blocking p53–dependent apoptosis protects intestinal stem cells and mice from radiation. Sci Rep. 2015 Apr 10;5:8566.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
Amuvatinib_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Amuvatinib (MP–470) is a potent and multi–targeted inhibitor of c–Kit, PDGFRα and Flt3 with IC50 of 10 nM, 40 nM and 81 nM,respectively.IC50 Value: 10 nM(c–KitD816H); 40 nM(PDGFRαV561D); 81 nM(Flt3D835Y) [1]Target: c–Kit; PDGFRα; FLT3in vitro: The hydrochloride salt of MP–470 also inhibits several mutants of c–Kit, including c–KitD816V, c–KitD816H, c–KitV560G, and c–KitV654A, as well as a Flt3 mutant (Flt3D835Y) and two PDGFRα mutants (PDGFRαV561D and PDGFRαD842V), with IC50 of 10 nM to8.4 μM. MP–470 potently inhibits the proliferation of OVCAR–3, A549, NCI–H647, DMS–153, and DMS–114 cells, with IC50 of 0.9μM–7.86 μM [1]. MP–470 also inhibits c–Kit and PDGFRα, with IC50 values of 31 μM and 27 μM, respectively. MP–470 demonstrates potent cytotoxicity against MiaPaCa–2, PANC–1, and GIST882 cells, with IC50 of 1.6 μM to 3.0 μM. MP–470 also binds to and inhibits several c–Kit mutants, including c–KitK642E, c–KitD816V, and c–KitK642E/D816V [2]. In MDA–MB–231 cells, MP–470 (1 μM) inhibits tyrosine phosphorylation of AXL [3]. In LNCaP and PC–3, but not DU145 cells, MP–470 exhibits cytotoxicity with IC50 of 4 μM and 8μM, respectively, and induces apoptosis at 10 μM. In LNCaP cells, MP–470 (10 μM) elicits G1 arrest and decreases phosphorylation of Akt and ERK1/2 [4].in vivo: In mice xenograft models of HT–29, A549, and SB–CL2 cells, MP–470 (10 mg/kg–75 mg/kg via i.p. or 50 mg/kg–200 mg/kg via p.o.) inhibits tumor growth [1]. In mice bearing LNCaP xenograft, MP–470 (20 mg/kg) combined with Erlotinib significantly induces tumor growth inhibition (TGI) [4].PROTOCOL (Extracted from published papers and Only for reference)Cell assay [4]For routine analysis of apoptosis, treated cells are examined for apoptotic morphology using a fluorescence staining technique. Briefly,cells are exposed to DMSO or differing doses of MP470, Erlotinib, or IM (1 to 10 μM) for 24 h and are harvested by trypsinization.After staining with a mixed dye solution containing 100 mg/ml each acridine orange and ethidium bromide the morphology of the cells is observed by fluorescence microscopy, and the number of apoptotic cells is quantified. In all cases a minimum of 200 cells are counted for each sample. Using Annexin V staining to detect apoptosis, treated cells are harvested by trypsinization and rinsed with cold PBS once. After centrifugation for 5 min, cells are resuspended in 500 μl of 1× Annexin V binding buffer and then added 1 μl of Annexin V–FITC and 1 μl of Propidium Iodide. After incubation for 5 min at room temperature in the dark, the samples were analyzed by flow cytometry.Animal administration [4]Forty eight 6–7 week–old SCID male mice are injected with 2× 107 LNCaP cells subcutaneously into the right hind flank. One month after inoculation, when tumors reach a volume of ~100 mm3, animals are divided randomly (pair–matched) into four test groups eachProduct Name:Amuvatinib Cat. No.:HY-10206CAS No.:850879-09-3Molecular Formula:C 23H 21N 5O 3S Molecular Weight:447.51Target:c–Kit; FLT3; PDGFR Pathway:Protein Tyrosine Kinase/RTK; Protein Tyrosine Kinase/RTK; Protein Tyrosine Kinase/RTK Solubility:10 mM in DMSOwith 12 mice: control group (DMSO), Erlotinib (80 mg/kg) group, MP470 (50 mg/kg) group and Erlotinib plus MP470 group. TKIs is administered IP daily from days 1 to 24. The control group is injected with 5% DMSO. A second study is also conducted with MP470 at 10 mg/kg and 20 mg/kg with 80 mg/kg Erlotinib to assess for biological efficacy (pharmacodynamics) and efficacy with 12 mice per group with the control arm of 5% DMSO. The length (L) and width (W) of the subcutaneous tumors are measured by calipers and the tumor volume (TV) was calculated as: TV = (L × W2)/2. Mice are sacrificed at the end of treatment (2–3/group), end of study or if they reached 2000 mm3 at any time during the study. Excised tumors are either fixed in paraffin or snap frozen for immunohistochemical analysis.References:[1]. Bearss DJ, et al. US Patent, US/2008/0226747.[2]. Hurley LH, et al. World Patent, WO/2005/037825.[3]. Mahadevan D, et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene, 2007, 26(27), 3909–3919.[4]. Qi W, et al. MP470, a novel receptor tyrosine kinase inhibitor, in combination with Erlotinib inhibits the HER family/PI3K/Akt pathway and tumor growth in prostate cancer. BMC Cancer, 2009, 9, 142.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
MedBio_193829-96-8_Cortistatin 14资料说明
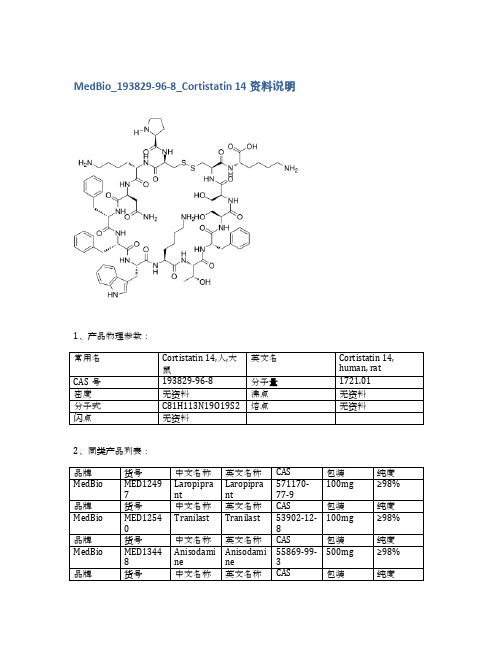
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED13351
Losartan Carboxylic Acid
Losartan Carboxylic Acid
124750-92-1
中文名称
英文名称
CAS
包装
纯度
MedBio
MED13366
3-chloro-5-hydroxy BA
CAS
包装
纯度
MedBio
MED12497
Laropiprant
Laropiprant
571170-77-9
100mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED12540
Tranilast
Tranilast
53902-12-8
100mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
5mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED12697
CRF (human, rat)
CRF (human, rat)
86784-80-7
10mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED12969
2-PMDQ
2-PMDQ
139047-55-5
50mg
纯度
MedBio
MED13448
Bioanalytical Method ValidationGuidance for Indust

Guidance for Industry Bioanalytical Method ValidationU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Veterinary Medicine (CVM)May 2001BPGuidance for Industry Bioanalytical Method ValidationAdditional copies are available from:Drug Information Branch (HFD-210)Center for Drug Evaluation and Research (CDER)5600 Fishers Lane, Rockville, MD 20857 (Tel) 301-827-4573Internet at /cder/guidance/index.htmorCommunications Staff (HFV-12)Center for Veterinary Medicine (CVM)7500 Standish Place, Rockville, MD 20855 (Tel) 301–594-1755Internet at /cvmU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Veterinary Medicine (CVM)May 2001BPTable of ContentsI.INTRODUCTION (1)II.BACKGROUND (1)A.F ULL V ALIDATION (2)B.P ARTIAL V ALIDATION (2)C.C ROSS-V ALIDATION (3)III.REFERENCE STANDARD (4)IV.METHOD DEVELOPMENT: CHEMICAL ASSAY (4)A.S ELECTIVITY (4)B.A CCURACY, P RECISION, AND R ECOVERY (5)C.C ALIBRATION/S TANDARD C URVE (5)D.S TABILITY (6)E.P RINCIPLES OF B IOANALYTICAL M ETHOD V ALIDATION AND E STABLISHMENT (8)F.S PECIFIC R ECOMMENDATIONS FOR M ETHOD V ALIDATION (10)V.METHOD DEVELOPMENT: MICROBIOLOGICAL AND LIGAND-BINDING ASSAYS (11)A.S ELECTIVITY I SSUES (11)B.Q UANTIFICATION I SSUES (12)VI.APPLICATION OF VALIDATED METHOD TO ROUTINE DRUG ANALYSIS (13)A CCEPTANCE C RITERIA FOR THE R UN (15)VII.DOCUMENTATION (16)A.S UMMARY I NFORMATION (16)B.D OCUMENTATION FOR M ETHOD E STABLISHMENT (17)C.A PPLICATION TO R OUTINE D RUG A NALYSIS (17)D.O THER I NFORMATION (19)GLOSSARY (20)GUIDANCE FOR INDUSTRY1Bioanalytical Method ValidationI.INTRODUCTIONThis guidance provides assistance to sponsors of investigational new drug applications (INDs), new drug applications (NDAs), abbreviated new drug applications (ANDAs), and supplements in developing bioanalytical method validation information used in human clinical pharmacology, bioavailability (BA), and bioequivalence (BE) studies requiring pharmacokinetic (PK) evaluation. This guidance also applies to bioanalytical methods used for non-human pharmacology/toxicology studies and preclinical studies. For studies related to the veterinary drug approval process, this guidance applies only to blood and urine BA, BE, and PK studies.The information in this guidance generally applies to bioanalytical procedures such as gas chromatography (GC), high-pressure liquid chromatography (LC), combined GC and LC mass spectrometric (MS) procedures such as LC-MS, LC-MS-MS, GC-MS, and GC-MS-MS performed for the quantitative determination of drugs and/or metabolites in biological matricessuch as blood, serum, plasma, or urine. This guidance also applies to other bioanalytical methods, such as immunological and microbiological procedures, and to other biological matrices, such as tissue and skin samples.This guidance provides general recommendations for bioanalytical method validation. The recommendations can be adjusted or modified depending on the specific type of analytical method used. II.BACKGROUND1 This guidance has been prepared by the Biopharmaceutics Coordinating Committee in the Center for Drug Evaluation and Research (CDER) in cooperation with the Center for Veterinary Medicine (CVM) at the Food and Drug Administration.This guidance has been developed based on the deliberations of two workshops: (1) Analytical Methods Validation: Bioavailability, Bioequivalence, and Pharmacokinetic Studies (held on December 3B5, 19902 ) and (2) Bioanalytical Methods Validation C A Revisit With a Decade of Progress (held on January 12B14, 20003).Selective and sensitive analytical methods for the quantitative evaluation of drugs and their metabolites (analytes) are critical for the successful conduct of preclinical and/or biopharmaceutics and clinical pharmacology studies. Bioanalytical method validation includes all of the procedures that demonstrate that a particular method used for quantitative measurement of analytes in a given biological matrix, such as blood, plasma, serum, or urine, is reliable and reproducible for the intended use. The fundamental parameters for this validation include (1) accuracy, (2) precision, (3) selectivity, (4) sensitivity, (5) reproducibility, and (6) stability. Validation involves documenting, through the use of specific laboratory investigations, that the performance characteristics of the method are suitable and reliable for the intended analytical applications. The acceptability of analytical data corresponds directly to the criteria used to validate the method.Published methods of analysis are often modified to suit the requirements of the laboratory performing the assay. These modifications should be validated to ensure suitable performance of the analytical method. When changes are made to a previously validated method, the analyst should exercise judgment as to how much additional validation is needed. During the course of a typical drug development program, a defined bioanalytical method undergoes many modifications. The evolutionary changes to support specific studies and different levels of validation demonstrate the validity of an assay’s performance. Different types and levels of validation are defined and characterized as follows:A.Full Validation•Full validation is important when developing and implementing a bioanalytical method for the first time.•Full validation is important for a new drug entity.• A full validation of the revised assay is important if metabolites are added to an existing assay for quantification.B.Partial ValidationPartial validations are modifications of already validated bioanalytical methods. Partial validation can range from as little as one intra-assay accuracy and precision determination to a nearly full2 Workshop Report: Shah, V.P. et al., Pharmaceutical Research: 1992; 9:588-592.3 Workshop Report: Shah, V.P. et al., Pharmaceutical Research: 2000; 17:in press.validation. Typical bioanalytical method changes that fall into this category include, but are not limited to:•Bioanalytical method transfers between laboratories or analysts•Change in analytical methodology (e.g., change in detection systems)•Change in anticoagulant in harvesting biological fluid•Change in matrix within species (e.g., human plasma to human urine)•Change in sample processing procedures•Change in species within matrix (e.g., rat plasma to mouse plasma)•Change in relevant concentration range•Changes in instruments and/or software platforms•Limited sample volume (e.g., pediatric study)•Rare matrices•Selectivity demonstration of an analyte in the presence of concomitant medications•Selectivity demonstration of an analyte in the presence of specific metabolitesC.Cross-ValidationCross-validation is a comparison of validation parameters when two or more bioanalytical methods are used to generate data within the same study or across different studies. An example of cross-validation would be a situation where an original validated bioanalytical method serves as thereference and the revised bioanalytical method is the comparator. The comparisons should be done both ways.When sample analyses within a single study are conducted at more than one site or more than one laboratory, cross-validation with spiked matrix standards and subject samples should be conducted at each site or laboratory to establish interlaboratory reliability. Cross-validation should also be considered when data generated using different analytical techniques (e.g., LC-MS-MS vs.ELISA4) in different studies are included in a regulatory submission.All modifications should be assessed to determine the recommended degree of validation. The analytical laboratory conducting pharmacology/toxicology and other preclinical studies for regulatory submissions should adhere to FDA=s Good Laboratory Practices (GLPs)5 (21 CFR part 58) and to sound principles of quality assurance throughout the testing process. The bioanalytical method for human BA, BE, PK, and drug interaction studies must meet the criteria in 21 CFR 320.29. The analytical laboratory should have a written set of standard operating procedures (SOPs) to ensure a complete system of quality control and assurance. The SOPs should cover all aspects of analysis from the time the sample is collected and reaches the laboratory until the results of the analysis are reported. The SOPs also should include record keeping, security and chain of sample custody4 Enzyme linked immune sorbent assay5 For the Center for Veterinary Medicine, all bioequivalence studies are subject to Good Laboratory Practices.(accountability systems that ensure integrity of test articles), sample preparation, and analytical tools such as methods, reagents, equipment, instrumentation, and procedures for quality control and verification of results.The process by which a specific bioanalytical method is developed, validated, and used in routine sample analysis can be divided into (1) reference standard preparation, (2) bioanalytical method development and establishment of assay procedure, and (3) application of validated bioanalytical method to routine drug analysis and acceptance criteria for the analytical run and/or batch. These three processes are described in the following sections of this guidance.III.REFERENCE STANDARDAnalysis of drugs and their metabolites in a biological matrix is carried out using samples spiked with calibration (reference) standards and using quality control (QC) samples. The purity of the reference standard used to prepare spiked samples can affect study data. For this reason, an authenticated analytical reference standard of known identity and purity should be used to prepare solutions of known concentrations. If possible, the reference standard should be identical to the analyte. When this is not possible, an established chemical form (free base or acid, salt or ester) of known purity can be used. Three types of reference standards are usually used: (1) certified reference standards (e.g., USP compendial standards); (2) commercially supplied reference standards obtained from a reputable commercial source; and/or (3) other materials of documented purity custom-synthesized by an analytical laboratory or other noncommercial establishment. The source and lot number, expiration date, certificates of analyses when available, and/or internally or externally generated evidence of identity and purity should be furnished for each reference standard.IV.METHOD DEVELOPMENT: CHEMICAL ASSAYThe method development and establishment phase defines the chemical assay. The fundamental parameters for a bioanalytical method validation are accuracy, precision, selectivity, sensitivity, reproducibility, and stability. Measurements for each analyte in the biological matrix should be validated. In addition, the stability of the analyte in spiked samples should be determined. Typical method development and establishment for a bioanalytical method include determination of (1) selectivity, (2) accuracy, precision, recovery, (3) calibration curve, and (4) stability of analyte in spiked samples.A.SelectivitySelectivity is the ability of an analytical method to differentiate and quantify the analyte in thepresence of other components in the sample. For selectivity, analyses of blank samples of theappropriate biological matrix (plasma, urine, or other matrix) should be obtained from at leastsix sources. Each blank sample should be tested for interference, and selectivity should be ensured at the lower limit of quantification (LLOQ).Potential interfering substances in a biological matrix include endogenous matrix components, metabolites, decomposition products, and in the actual study, concomitant medication and other exogenous xenobiotics. If the method is intended to quantify more than one analyte, each analyte should be tested to ensure that there is no interference.B.Accuracy, Precision, and RecoveryThe accuracy of an analytical method describes the closeness of mean test results obtained by the method to the true value (concentration) of the analyte. Accuracy is determined by replicate analysis of samples containing known amounts of the analyte. Accuracy should be measured using a minimum of five determinations per concentration. A minimum of three concentrations in the range of expected concentrations is recommended. The mean value should be within 15% of the actual value except at LLOQ, where it should not deviate by more than 20%. The deviation of the mean from the true value serves as the measure of accuracy.The precision of an analytical method describes the closeness of individual measures of an analyte when the procedure is applied repeatedly to multiple aliquots of a single homogeneous volume of biological matrix. Precision should be measured using a minimum of five determinations per concentration. A minimum of three concentrations in the range of expected concentrations is recommended. The precision determined at each concentration level should not exceed 15% of the coefficient of variation (CV) except for the LLOQ, where it should not exceed 20% of the CV. Precision is further subdivided into within-run, intra-batch precision or repeatability, which assesses precision during a single analytical run, and between-run, inter-batch precision or repeatability, which measures precision with time, and may involve different analysts, equipment, reagents, and laboratories.The recovery of an analyte in an assay is the detector response obtained from an amount of the analyte added to and extracted from the biological matrix, compared to the detector response obtained for the true concentration of the pure authentic standard. Recovery pertains to the extraction efficiency of an analytical method within the limits of variability. Recovery of the analyte need not be 100%, but the extent of recovery of an analyte and of the internal standard should be consistent, precise, and reproducible. Recovery experiments should be performed by comparing the analytical results for extracted samples at three concentrations (low, medium, and high) with unextracted standards that represent 100% recovery.C.Calibration/Standard CurveA calibration (standard) curve is the relationship between instrument response and known concentrations of the analyte. A calibration curve should be generated for each analyte in thesample. A sufficient number of standards should be used to adequately define the relationship between concentration and response. A calibration curve should be prepared in the same biological matrix as the samples in the intended study by spiking the matrix with known concentrations of the analyte. The number of standards used in constructing a calibration curve will be a function of the anticipated range of analytical values and the nature of theanalyte/response relationship. Concentrations of standards should be chosen on the basis of the concentration range expected in a particular study. A calibration curve should consist of a blank sample (matrix sample processed without internal standard), a zero sample (matrix sample processed with internal standard), and six to eight non-zero samples covering the expected range, including LLOQ.1.Lower Limit of Quantification (LLOQ)The lowest standard on the calibration curve should be accepted as the limit ofquantification if the following conditions are met:C The analyte response at the LLOQ should be at least 5 times the responsecompared to blank response.C Analyte peak (response) should be identifiable, discrete, and reproducible witha precision of 20% and accuracy of 80-120%.2.Calibration Curve/Standard Curve/Concentration-ResponseThe simplest model that adequately describes the concentration-response relationshipshould be used. Selection of weighting and use of a complex regression equation should be justified. The following conditions should be met in developing a calibration curve:C#20% deviation of the LLOQ from nominal concentrationC#15% deviation of standards other than LLOQ from nominal concentrationAt least four out of six non-zero standards should meet the above criteria, including the LLOQ and the calibration standard at the highest concentration. Excluding thestandards should not change the model used.D.StabilityDrug stability in a biological fluid is a function of the storage conditions, the chemical properties of the drug, the matrix, and the container system. The stability of an analyte in a particular matrix and container system is relevant only to that matrix and container system and should not be extrapolated to other matrices and container systems. Stability procedures should evaluate the stability of the analytes during sample collection and handling, after long-term (frozen at theintended storage temperature) and short-term (bench top, room temperature) storage, and after going through freeze and thaw cycles and the analytical process. Conditions used in stability experiments should reflect situations likely to be encountered during actual sample handling and analysis. The procedure should also include an evaluation of analyte stability in stock solution.All stability determinations should use a set of samples prepared from a freshly made stock solution of the analyte in the appropriate analyte-free, interference-free biological matrix. Stock solutions of the analyte for stability evaluation should be prepared in an appropriate solvent at known concentrations.1.Freeze and Thaw StabilityAnalyte stability should be determined after three freeze and thaw cycles. At least three aliquots at each of the low and high concentrations should be stored at the intendedstorage temperature for 24 hours and thawed unassisted at room temperature. Whencompletely thawed, the samples should be refrozen for 12 to 24 hours under the sameconditions. The freeze–thaw cycle should be repeated two more times, then analyzedon the third cycle. If an analyte is unstable at the intended storage temperature, thestability sample should be frozen at -700C during the three freeze and thaw cycles.2.Short-Term Temperature StabilityThree aliquots of each of the low and high concentrations should be thawed at roomtemperature and kept at this temperature from 4 to 24 hours (based on the expectedduration that samples will be maintained at room temperature in the intended study) and analyzed.3.Long-Term StabilityThe storage time in a long-term stability evaluation should exceed the time between the date of first sample collection and the date of last sample analysis. Long-term stabilityshould be determined by storing at least three aliquots of each of the low and highconcentrations under the same conditions as the study samples. The volume of samples should be sufficient for analysis on three separate occasions. The concentrations of allthe stability samples should be compared to the mean of back-calculated values for the standards at the appropriate concentrations from the first day of long-term stabilitytesting.4.Stock Solution StabilityThe stability of stock solutions of drug and the internal standard should be evaluated at room temperature for at least 6 hours. If the stock solutions are refrigerated or frozenfor the relevant period, the stability should be documented. After completion of thedesired storage time, the stability should be tested by comparing the instrumentresponse with that of freshly prepared solutions.5.Post-Preparative StabilityThe stability of processed samples, including the resident time in the autosampler, should be determined. The stability of the drug and the internal standard should be assessedover the anticipated run time for the batch size in validation samples by determiningconcentrations on the basis of original calibration standards.Although the traditional approach of comparing analytical results for stored samples with those for freshly prepared samples has been referred to in this guidance, other statistical approaches based on confidence limits for evaluation of an analyte=s stability in abiological matrix can be used. SOPs should clearly describe the statistical method andrules used. Additional validation may include investigation of samples from dosedsubjects.E.Principles of Bioanalytical Method Validation and Establishment•The fundamental parameters to ensure the acceptability of the performance of a bioanalytical method validation are accuracy, precision, selectivity, sensitivity,reproducibility, and stability.• A specific, detailed description of the bioanalytical method should be written. This can be in the form of a protocol, study plan, report, and/or SOP.•Each step in the method should be investigated to determine the extent to which environmental, matrix, material, or procedural variables can affect the estimation of analyte in the matrix from the time of collection of the material up to and including the time ofanalysis.•It may be important to consider the variability of the matrix due to the physiological nature of the sample. In the case of LC-MS-MS-based procedures, appropriate steps should be taken to ensure the lack of matrix effects throughout the application of the method,especially if the nature of the matrix changes from the matrix used during method validation.• A bioanalytical method should be validated for the intended use or application. All experiments used to make claims or draw conclusions about the validity of the methodshould be presented in a report (method validation report).•Whenever possible, the same biological matrix as the matrix in the intended samples should be used for validation purposes. (For tissues of limited availability, such as bone marrow, physiologically appropriate proxy matrices can be substituted.)•The stability of the analyte (drug and/or metabolite) in the matrix during the collection process and the sample storage period should be assessed, preferably prior to sampleanalysis.•For compounds with potentially labile metabolites, the stability of analyte in matrix from dosed subjects (or species) should be confirmed.•The accuracy, precision, reproducibility, response function, and selectivity of the method for endogenous substances, metabolites, and known degradation products should beestablished for the biological matrix. For selectivity, there should be evidence that thesubstance being quantified is the intended analyte.•The concentration range over which the analyte will be determined should be defined in the bioanalytical method, based on evaluation of actual standard samples over the range,including their statistical variation. This defines the standard curve.• A sufficient number of standards should be used to adequately define the relationship between concentration and response. The relationship between response and concentration should be demonstrated to be continuous and reproducible. The number of standards used should be a function of the dynamic range and nature of the concentration-responserelationship. In many cases, six to eight concentrations (excluding blank values) can define the standard curve. More standard concentrations may be recommended for nonlinear than for linear relationships.•The ability to dilute samples originally above the upper limit of the standard curve should be demonstrated by accuracy and precision parameters in the validation.•In consideration of high throughput analyses, including but not limited to multiplexing, multicolumn, and parallel systems, sufficient QC samples should be used to ensure control of the assay. The number of QC samples to ensure proper control of the assay should be determined based on the run size. The placement of QC samples should be judiciously considered in the run.•For a bioanalytical method to be considered valid, specific acceptance criteria should be set in advance and achieved for accuracy and precision for the validation of QC samples over the range of the standards.F.Specific Recommendations for Method Validation•The matrix-based standard curve should consist of a minimum of six standard points, excluding blanks, using single or replicate samples. The standard curve should cover the entire range of expected concentrations.•Standard curve fitting is determined by applying the simplest model that adequately describes the concentration-response relationship using appropriate weighting and statistical tests for goodness of fit.•LLOQ is the lowest concentration of the standard curve that can be measured with acceptable accuracy and precision. The LLOQ should be established using at least five samples independent of standards and determining the coefficient of variation and/orappropriate confidence interval. The LLOQ should serve as the lowest concentration on the standard curve and should not be confused with the limit of detection and/or the low QC sample. The highest standard will define the upper limit of quantification (ULOQ) of an analytical method.•For validation of the bioanalytical method, accuracy and precision should be determined using a minimum of five determinations per concentration level (excluding blank samples).The mean value should be within ±15% of the theoretical value, except at LLOQ, where it should not deviate by more than ±20%. The precision around the mean value should not exceed 15% of the CV, except for LLOQ, where it should not exceed 20% of the CV.Other methods of assessing accuracy and precision that meet these limits may be equally acceptable.•The accuracy and precision with which known concentrations of analyte in biological matrix can be determined should be demonstrated. This can be accomplished by analysis ofreplicate sets of analyte samples of known concentrations C QC samples C from anequivalent biological matrix. At a minimum, three concentrations representing the entire range of the standard curve should be studied: one within 3x the lower limit of quantification (LLOQ) (low QC sample), one near the center (middle QC), and one near the upperboundary of the standard curve (high QC).•Reported method validation data and the determination of accuracy and precision should include all outliers; however, calculations of accuracy and precision excluding values that are statistically determined as outliers can also be reported.•The stability of the analyte in biological matrix at intended storage temperatures should be established. The influence of freeze-thaw cycles (a minimum of three cycles at twoconcentrations in triplicate) should be studied.•The stability of the analyte in matrix at ambient temperature should be evaluated over a time period equal to the typical sample preparation, sample handling, and analytical run times.•Reinjection reproducibility should be evaluated to determine if an analytical run could be reanalyzed in the case of instrument failure.•The specificity of the assay methodology should be established using a minimum of six independent sources of the same matrix. For hyphenated mass spectrometry-basedmethods, however, testing six independent matrices for interference may not be important.In the case of LC-MS and LC-MS-MS-based procedures, matrix effects should beinvestigated to ensure that precision, selectivity, and sensitivity will not be compromised.Method selectivity should be evaluated during method development and throughout methodvalidation and can continue throughout application of the method to actual study samples.•Acceptance/rejection criteria for spiked, matrix-based calibration standards and validation QC samples should be based on the nominal (theoretical) concentration of analytes.Specific criteria can be set up in advance and achieved for accuracy and precision over therange of the standards, if so desired.V.METHOD DEVELOPMENT: MICROBIOLOGICAL AND LIGAND-BINDING ASSAYSMany of the bioanalytical validation parameters and principles discussed above are also applicable to microbiological and ligand-binding assays. However, these assays possess some unique characteristics that should be considered during method validation.A.Selectivity IssuesAs with chromatographic methods, microbiological and ligand-binding assays should be shown to be selective for the analyte. The following recommendations for dealing with two selectivity issues should be considered:1.Interference From Substances Physiochemically Similar to the Analyte•Cross-reactivity of metabolites, concomitant medications, or endogenouscompounds should be evaluated individually and in combination with the analyteof interest.•When possible, the immunoassay should be compared with a validated reference method (such as LC-MS) using incurred samples and predetermined criteria foragreement of accuracy of immunoassay and reference method.。
Bruceine A 25514-31-2 GlpBio

Peptides, Inhibitors, AgonistsProduct Data SheetProduct Name: Bruceine ACat. No.:GC17948Chemical Name: (1R,2S,3S,3aS,3a1R,4R,6aR,7aR,11aS,11bR)-methyl 1,2,9-trihydroxy-8,11a-dimethyl-4-((3-methylbutanoyl)oxy)-5,10-dioxo-2,3,3a,4,5,6a,7,7a,10,11,11a,11b-dodecahydro-1H-3,3a1-CHEMICAL PROPERTIESCas No.: 25514-31-2Molecular Formula: C26H34O11Molecular Weight: 522.54Storage: PowderSolubility: Soluble in DMSOChemical Structure:BackgroundIC50: 7.7 nM against the parasitesCanine babesiosis is a tick-borne disease caused by the intraerythrocytic apicomplexan parasites, Babesia gibsoni and B. canis. Clinical signs of B. gibsoni infection are anemia, fever, thrombocytopenia, splenomegaly, lymphadenopathy, and lethargy. Bruceine A, a natural quassinoid compound extracted from the Brucea javanica (L.) Merr. dried fruits, was reported to have antibabesial activity.In vitro: Bruceine A inhibited the in-vitro Babesia gibsoni growth in canine erythrocytes at lower concentration compared with the antibabesial drug diminazene aceturate and killed the parasites within 24 hr at a concentration of 25 nM [1].In vivo: Oral administration of bruceine A at a dosage of 6.4 mg/kg/day for 5 days resulted in no clinical findings in a dog. An untreated dog developed typical acute babesiosis symptoms including high fever, severe anemia, and complete loss of appetite and movement. However, the two bruceine A-treated dogs maintained their healthy conditions throughout the experimental period of 4 weeks [1].Clinical trials: Currenlty there is no clinical data available.Research Update1. Effect of Hydrofluoric Acid Concentration and Etching Time on Bond Strength to Lithium Disilicate Glass Ceramic. Oper Dent. 2017 Nov/Dec;42(6):606-615. doi: 10.2341/16-215-L. Epub 2017 Jul 14. PMID:28708007AbstractThe aim of this study was to evaluate the influence of different concentrations of hydrofluoric acid (HF) associated with varied etching times on the microshear bo nd strength (μSBS) of a resin cement to a lithium disilicate glass ceramic. Two hundred seventy-five ceramic blocks (IPS e.max Press [EMX], Ivoclar Vivadent), measuring 8 mm × 3 mm thickness, were randomly distributed into fiv e groups according to the HF concentrations (n=50): 1%, 2.5%, 5%, 7.5%, and 10%.2. Does acid etching morphologically and chemically affect lithium disilicate glass ceramic surfaces? J Appl Biomater Funct Mater. 2017 Jan 26;15(1):e93-e100. doi: 10.5301/jabfm.5000303. PMID:27647389AbstractBACKGROUND: This study evaluated the surface morphology, chemical composition and adhesiveness of lithium disilicate glass ceramic after acid etching with hydrofluoric acid or phosphoric acid.METHODS: Lithium disilicate glass ceramic specimens polished by 600-grit silicon carbide paper were subjected to one or a combination of these surface treatments: airborne particle abrasion with 50-μm alumina (AA), etching with 5% hydrofluoric acid (HF) or 36% phosphoric acid (Phos), and application of silane coup ling agent (Si).3. Fatigue failure load of feldspathic ceramic crowns after hydrofluoric acid etching at different concentrations. J Prosthet Dent. 2018 Feb;119(2):278-285. doi: 10.1016/j.prosdent.2017.03.021. Epub 2017 May 26. PMID:28552291AbstractSTATEMENT OF PROBLEM: Hydrofluoric acid etching modifies the cementation surface of ceramic restorations, which is the same surface where failure is initiated. Information regarding the influence of hydrofluoric acid etching on the cyclic loads to failure of ceramic crowns is lacking.PURPOSE: The purpose of this in vitro study was to evaluate the influence of different hydrofluoric acid concentrations on the fatigue failure loads of feldspathic ceramic crowns.。
(完整版)美国药典USP31(921)翻译版(上)

(921 ) WATER DETERMINATION 水分测定Many Pharmacopeial articles either are hydrates or contain water in adsorbed form. As a result, the determination of the water content is important in demonstrating compliance with thePharmacopeial standards. Generally one of the methods given below is called for in the individual monograph, depending upon the nature of the article. In rare cases, a choice is allowed between two methods. When the article contains water of hydration, the Method I (Titrimetric), the Method II (Azeotropic), or the Method III (Gravimetric) is employed, as directed in the individual monograph, and the requirement is given under the heading Water.很多药典物品要么是水合物,要么含有处丁吸附状态的水.因此,测定水分含量对丁证实与药典标准的符合性是很重要的.通常,在具体的各论中会根据该物品的性质,要求使用下面假设干方法中的一个. 偶尔,会允许在2个方法中任选一个.当该物品含有水合状态的水,根据具体各论中的规定,使用方法I (滴定测量法)、方法II (包沸测量法)、或方法III (重量分析法),这个要求在标题水分项下给出.The heading Loss on drying (see Loss on Drying 731 ) is used in those cases where the loss sustained on heating may be not entirely water.在加热时的持续失重可能不全是水分的情况下,使用标题枯燥失重(见十燥失重<731>).METHOD I (TITRIMETRIC) 方法I (滴定测量法)Determine the water by Method Ia, unless otherwise specified in the individual monograph. 除非具体各论中另有规定,使用方法Ia来测定水分.Method Ia (Direct Titration) 方法Ia (直接滴定)Principle — The titrimetric determination of water is based upon the quantitative reaction of water with an anhydrous solution of sulfur dioxide and iodine in the presence of a buffer that reacts with hydrogen ions.原理:水分的滴定法检测是基丁水与二氧化硫的无水溶液以及存在丁缓冲液中与氢离子反响的碘之间的定量反响.In the original titrimetric solution, known as Karl Fischer Reagent, the sulfur dioxide and iodine are dissolved in pyridine and methanol. The test specimen may be titrated with the Reagent directly, or the analysis may be carried out by a residual titration procedure. The stoichiometry of the reactionis not exact, and the reproducibility of a determination depends upon such factors as the relativeconcentrations of the Reagent ingredients, the nature of the inert solvent used to dissolve the test specimen, and the technique used in the particular determination. Therefore, an empirically standardized technique is used in order to achieve the desired accuracy. Precision in the method is governed largely by the extent to which atmospheric moisture is excluded from the system. The titration of water is usually carried out with the use of anhydrous methanol as the solvent for the test specimen; however, other suitable solvents may be used for special or unusual test specimens. 在最初的滴定测量溶液〔即卡尔•费休试剂〕中,二氧化硫和碘溶解于嚅噬和甲醇中.该供试样品可以用该试剂直接滴定,或者可以使用残留滴定程序来进行该分析. 此反响的化学计算法不够准确,并且检测的重现性取决于某些因素,例如该试剂成分的相对浓度、用于溶解供试样品的惰性溶剂的性质、用于具体测定的方法等.因此,需要应用根据经验得到的标准化方法,以便实现预期的准确性.该方法中的精密度很大程度上取决于将大气湿度从该系统中排除的程度. 进行水分滴定通常使用无水甲醇作为供试样品的溶剂;但是,可以将其他适当的溶剂用于特殊或不常见的供试样品.Apparatus — Any apparatus may be used that provides for adequate exclusion of atmospheric moisture and determination of the endpoint. In the case of a colorless solution that is titrated directly, the endpoint may be observed visually as a change in color from canary yellow to amber. The reverse is observed in the case of a test specimen that is titrated residually. More commonly, however, the endpoint is determined electrometrically with an apparatus employing a simple electrical circuit that serves to impress about 200 mV of applied potential between a pair of platinum electrodes immersed in the solution to be titrated. At the endpoint of the titration a slight excess of the reagent increases the flow of current to between 50 and 150 microamperes for 30 seconds to 30 minutes, depending upon the solution being titrated. The time is shortest for substances that dissolve in the reagent. With some automatic titrators, the abrupt change in current or potential at the endpoint serves to close a solenoid-operated valve that controls the buret delivering the titrant. Commercially available apparatus generally comprises a closed system consisting of one or two automatic burets and a tightly covered titration vessel fitted with the necessary electrodes and a magnetic stirrer. The air in the system is kept dry with a suitable desiccant, and the titration vessel may be purged by means of a stream of dry nitrogen or current of dry air.仪器:任何能够充分排除大气湿度,并能测定终点的仪器.在直接向无色溶液滴定的情况下,可以通过从淡黄色到琥珀色的颜色改变来观察此终点. 在向供试样品作残留滴定的情况下, 会观察到与此相反的情况.但是,更常见的情况是,使用仪器,利用其中的简单电路在浸没在待滴定溶液中的一对白金电极上加上200mV的应用电压,从而以电势滴定来测定终点.在滴定终点,稍微过量的该试剂会使电流提升到50和150微安培,并维持30秒到30分钟,具体时间取决于被滴定的溶液. 溶解于该试剂中的物质所用时间是最短的. 在一些自动滴定仪上,在该终点出现的电流或电压的忽然变化会使由螺线管操纵的阀门关闭,该阀门限制者输送滴定剂的滴定管. 市场上销售的仪器通常包含一个封闭系统,其中由一个或两个自动滴定管、一个配备了必须的电极和磁力搅拌器的严密覆盖的滴定容器组成.通过适当的十燥器使系统内空气保持十燥, 并且该滴定容器可以通过十燥氮气流或十燥空气流来进行净化.Reagent — Prepare the Karl Fischer Reagent as follows. Add 125 g of iodine to a solution containing 670 mL of methanol and 170 mL of pyridine, and cool. Place 100 mL of pyridine in a 250-mL graduated cylinder, and, keeping the pyridine cold in an ice bath, pass in dry sulfur dioxide until the volume reaches 200 mL. Slowly add this solution, with shaking, to the cooled iodine mixture. Shake to dissolve the iodine, transfer the solution to the apparatus, and allow the solution to stand overnight before standardizing. One mL of this solution when freshly prepared is equivalent to approximately 5 mg of water, but it deteriorates gradually; therefore, standardize it within 1 hour before use, or daily if in continuous use. Protect from light while in use. Store any bulk stock of the reagent in a suitably sealed, glass-stoppered container, fully protected from light, and under refrigeration.试剂:按下面方法配制卡尔•费休试剂.参加125克碘至含有670mL甲醇和170mL嚅噬的溶液中, 放凉.将100mL嚅噬置于一个250mL量筒中,将该嚅噬置于冰浴中以保持冰冷,送入十燥二氧化硫直到体积到达200mL.伴随摇动,缓慢将此溶液参加到放凉后的碘混合物中.摇动以使碘溶解, 转移此溶液至该仪器,并在标准化之前将该溶液静置过夜.在刚刚配制之后, 1mL此溶液相当于约5mg水,但是会逐渐变差;因此,在使用前1个小时,或在连续使用时每日,对其进行标准化.使用中需避光.将该试剂的散装存货保存于适当密闭的玻璃塞容器中,完全避光,并冷藏.A commercially available, stabilized solution of Karl Fischer type reagent may be used. Commercially available reagents containing solvents or bases other than pyridine or alcohols other than methanol may be used also. These may be single solutions or reagents formed in situ by combining the components of the reagents present in two discrete solutions. The diluted Reagent called for in some monographs should be diluted as directed by the manufacturer. Either methanol or other suitable solvent, such as ethylene glycol monomethyl ether, may be used as the diluent.可以使用市场上销售的卡尔•费休类型试剂的稳定溶液.也可以使用市场上销售的试剂,其中含有除了嚅噬之外的溶剂或盐基,或除了甲醇之外的醇类.这些可以是通过合并存在于两个独立溶液中的试剂组成局部,在现场形成的单一的溶液或试剂.如果某些各论中要求使用稀释后的试剂,那么应当根据生产商的规定稀释.可以使用甲醇或其他适当溶剂,例如乙二醇一甲酰,作为稀释剂.Test Preparation — Unless otherwise specified in the individual monograph, use an accurately weighed ormeasured amount of the specimen under test estimated to contain 2 to 250 mg of water. The amount of water depends on the water equivalency factor of the Reagent and on the method of endpoint determination. In most cases, the minimum amount of specimen, in mg, can be estimated using the formula:供试配制品:除非在具体各论中另有规定,使用数量经过精确称定或称量的供试样品,其中应含水2至250mg.水的数量取决丁试剂的水当量因子和终点测定的方法.在大多数情况下,可以使用此公式估计以毫克计的供试样品的最小量:FCV/KFin which F is the water equivalency factor of the Reagent, in mg per mL; C is the used volume, in percent, of the capacity of the buret; V is the buret volume, in mL; and KF is the limit or reasonable expected water content in the sample, in percent. C is between 30% and 100% for manual titration, and between 10% and 100% for the instrumental method endpoint determination. 其中,F是试剂的水当量因子,单位为毫克每毫升;C是滴定管容量中所使用的体积〔为白分比〕;V是滴定管体积,以毫升计;KF是样品中限度或合理预期的水含量,为白分比.对丁手动滴定, C 值在30%至100%之间,而对丁仪器方法终点测定,其在10%至100%之间.Where the specimen under test is an aerosol with propellant, store it in a freezer for not less than 2 hours, open the container, and test 10.0 mL of the well-mixed specimen. In titrating the specimen,□determine the endpoint at a temperature of 10 or higher.如果供试样品是带有推进剂的气雾剂〔烟雾剂、气溶胶〕,将其存放丁冷冻室中不少丁2小时,翻开容器,并检验10.0mL混合均匀的样品.在滴定该样品过程中,在10口或更高的温度下确定反响终点.Where the specimen under test is capsules, use a portion of the mixed contents of not fewer than 4 capsules. 如果该供试样品为胶囊,使用不少丁4个胶囊的混合内容物的一局部.Where the specimen under test is tablets, use powder from not fewer than 4 tablets ground to a fine powder in an atmosphere of temperature and relative humidity known not to influence the results.如果该供试品为片剂,在不会影响检验结果的温度和相对湿度环境中, 将不少丁4片磨碎至细粉末.Where the monograph specifies that the specimen under test is hygroscopic, use a dry syringe to inject an appropriate volume of methanol, or other suitable solvent, accurately measured, into a tared container, and shake to dissolve the specimen. Using the same syringe, remove the solution from the container and transferit to a titration vessel prepared as directed for Procedure. Repeat the procedure with a second portion of methanol, or other suitable solvent, accurately measured, add this washing to the titration vessel, and immediately titrate. Determine the water content, in mg, of a portion of solvent of the same total volume as that used to dissolve the specimen and to wash the container and syringe, as directed for Standardization of Water Solution for Residual Titrations, and subtract this value from the water content, in mg, obtained in the titration of the specimen underotest. Dry the container and its closure at 100 for 3 hours, allow to cool in a desiccator, and weigh. Determine the weight of specimen tested from the difference in weight from the initial weight of the container.如果该各论中显示此供试样品易吸湿, 使用一个枯燥注射器,注射经过精确称量的适当体积的甲醇或其他适当溶剂,至一个已称过皮重的容器,并摇动以使该样品溶解.使用同一个注射器,从该容器中吸出此溶液并转移至根据步骤项下规定准备的一个滴定容器.使用精确称量的第二局部甲醇或其他适当溶剂,重复该步骤,将此洗液参加至滴定容器,并马上滴定.取与用丁溶解样品以及洗涤容器和注射器的溶剂同样体积的一局部溶剂, 根据用丁残留滴定的水溶液的标准化项下的规定,测定溶剂中的水分含量〔以mg为单位〕,并从得自供试样品滴定的水分含量〔以mg为单位〕中减去此数值.在100“温度条件下将这些容器及其盖子枯燥3小时,在枯燥器中静置至凉,并称重.根据与该容器初始重量的差距,来确定试验所用的样品重量.Standardization of the Reagent ——Place enough methanol or other suitable solvent in the titration vessel to cover the electrodes, and add sufficient Reagent to give the characteristic endpoint color, or 100 50 microamperes of direct current at about 200 mV of applied potential.试剂的标准化:将足够的甲醇或其他适当溶剂置丁滴定容器,以覆盖电极,并参加充足的试剂,以产生典型终点颜色,或者在约200mV应用电压下产生100 土50微安培直流电.For determination of trace amounts of water 〔less than 1%〕, it is preferable to use waterReagent with a equivalency factor of not more than 2.0. Sodium tartrate may be used as a convenient waterreference substance. Quickly add 75 to 125 mg of sodium tartrate 〔C 4H4Na2Q e 2H2O〕, accurately weighed by difference, and titrate to the endpoint. The water equivalence factor F, in mg of water per mL of reagent, is given by the formula:为了检测痕量水份〔少丁1%〕最好使用水当量因子不超过 2.0的试剂.可以使用洒石酸钠作为便捷的水标准物质.快速参加精密称定的75至125mg洒石酸钠〔C4H4Na2Q e 2H2O〕,并滴定至终点. 在下面的公式中给出了水平■衡因子F的计算方法〔单位为以每毫升试剂中毫克水〕:2〔18.02/230.08〕〔 W/V〕,in which 18.02 and 230.08 are the molecular weights of water and sodium tartrate dihydrate, respectively; W is the weight, in mg, of sodium tartrate dihydrate; and V is the volume, in mL, of the Reagent consumed in the second titration.其中,18.02和230.08是水和洒石酸钠二水合物的分子量;W是洒石酸钠二水合物的重量〔单位mg〕; V是第二次滴定中消耗的试剂体积〔单位mL〕.For the precise determination of significant amounts of water 〔1% or more〕, use Purified Water as the reference substance. Quickly add between 25 and 250 mg of water, accurately weighed by difference, from a weighing pipet or from a precalibrated syringe or micropipet, the amount taken being governed by the reagent strength and the buret size, as referred to under VolumetricApparatus 31 . Titrate to the endpoint. Calculate the water equivalence factor, F, in mg of water per mL of reagent, by the formula:为了精确测定显著水分含量〔1%或更多〕,使用纯洁水作为标准物质.从称重移液器或者经过预校准的注射器或微量移液器中,快速参加经过精密称定的25至50mg水,参加数量需取决丁该试剂的水平和滴定管的大小,参见称量器具<31>.滴定至终点.使用下面公式,计算水分平衡因子 F 〔单位为每毫升试剂中毫克水〕.W/V,in which W is the weight, in mg, of the water; and V is the volume, in mL, of the reagent required. 其中W是水的重量〔单位mg〕 ; V是需要的试剂体积〔单位mL〕.Procedure — Unless otherwise specified, transfer 35 to 40 mL of methanol or other suitable solvent to the titration vessel, and titrate with the Reagent to the electrometric or visual endpoint to consume any moisture that may be present. 〔Disregard the volume consumed, since it does not enter into the calculations.〕Quickly add the Test Preparation, mix, and again titrate with theReagent to the electrometric or visual endpoint. Calculate the water content of the specimen, in mg, taken by the formula:步骤:除非另有规定,转移35至40mL甲醇或其他适当溶剂至滴定容器,并使用该试剂进行滴定至测电法或视觉观察的终点,以消耗掉可能存在的任何水分. 〔不要理会消耗的体积,由于其不会带入计算.〕快速参加供试配制液,混匀,并再次使用试剂滴定至测电法或视觉观察的终点.使用下面的公式,计算样品中的水分含量:SF,in which S is the volume, in mL, of the Reagent consumed in the second titration; and F is the water equivalence factor of the Reagent.其中,S是在第二次滴定中消耗掉的试剂体积〔单位mL〕 ; F是该试剂的水平衡因子.。
牛布病(Brucellosis)说明书定性
牛布病(Brucellosis)酶联免疫分析(ELISA)试剂盒使用说明书本试剂仅供研究使用目的:本试剂盒用于检测牛血清,血浆及相关液体样本中布病(Brucellosis)水平,感染人的血液学诊断。
实验原理:本试剂盒采用双抗体夹心酶联免疫法(ELISA)测定标本中牛布病(Brucellosis)。
用纯化的牛布病(Brucellosis)抗体包被微孔板,制成固相抗体,可与样品中布病(Brucellosis)相结合,经洗涤除去未结合的抗体和其他成分后再与HRP标记的布病(Brucellosis)抗体结合,形成抗体-抗原-酶标抗体复合物,经过彻底洗涤后加底物TMB显色。
TMB在HRP酶的催化下转化成蓝色,并在酸的作用下转化成最终的黄色。
用酶标仪在450nm波长下测定吸光度(OD 值),与CUTOFF值相比较,从而判定标本中牛布病(Brucellosis)的存在与否。
样本处理及要求:1. 血清:室温血液自然凝固10-20分钟,离心20分钟左右(2000-3000转/分)。
仔细收集上清,保存过程中如出现沉淀,应再次离心。
2. 血浆:应根据标本的要求选择EDTA或柠檬酸钠作为抗凝剂,混合10-20分钟后,离心20分钟左右(2000-3000转/分)。
仔细收集上清,保存过程中如有沉淀形成,应该再次离心。
3. 尿液:用无菌管收集,离心20分钟左右(2000-3000转/分)。
仔细收集上清,保存过程中如有沉淀形成,应再次离心。
胸腹水、脑脊液参照实行。
4. 细胞培养上清:检测分泌性的成份时,用无菌管收集。
离心20分钟左右(2000-3000转/分)。
仔细收集上清。
检测细胞内的成份时,用PBS(PH7.2-7.4)稀释细胞悬液,细胞浓度达到100万/ml左右。
通过反复冻融,以使细胞破坏并放出细胞内成份。
离心20分钟左右(2000-3000转/分)。
仔细收集上清。
保存过程中如有沉淀形成,应再次离心。
5. 组织标本:切割标本后,称取重量。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
References: [1]. Nakao R, et al. Evaluation of efficacy of bruceine A, a natural quassinoid compound extracted from a medicinal plant, Brucea javanica, for canine babesiosis. J Vet Med Sci. 2009 Jan;71(1):3341.
Product Data Sheet
Product Name: CAS No.: Cat. No.: MWt: Formula: Purity :
Bruceine A 25514-31-2 HY-N0841 522.54 C26H34O11 >98%
Solubility:
DMSO
Mechanisms: Pathways:Anti-infection; Target:Antiparasitic Biological Activity: Bruceine A(NSC310616; Dihydrobrusatol) is a natural quassinoid compound extracted from the dried fruits of Brucea javanica (L.); are potential candidates for the treatment of canine babesiosis. IC50 value: Target: Bruceine A inhibited the in vitro growth of Babesia gibsoni in canine erythrocytes at lower concentration compared with the standard antibabesial drug diminazene aceturate and killed the parasites within 24 hr at a concentration of 25 nM. Oral administration of bruceine A at a dosage of 6.4 mg/kg/day for 5 days resulted in no clinical findings in a dog with normal ranges of hematological and biochemical values in the blood. Three dogs g were infected with B. g gibsoni and two of them were treated with bruceine A at a dosage of 6.4 mg/kg/day for 6 days from day 5 post-infection....
Caution: Not fully tested. For research purposes only Medchemexpress LLp x e m e h c d e m . w w w : b e AW Sm Uo ,c 0 4. 5s 8s 0e r p Jx Ne ,m n e oh t c e cd ne i rm P @ ,o y f an Wi : l ni oa sm n iE k l i W 8 1