NCBI在线BLAST使用方法与结果详解
Blast分析报告

Blast分析报告引言Blast(Basic Local Alignment Search Tool)是一种常用的生物信息学工具,用于比对和比较生物序列。
本报告旨在分析和解释Blast结果,帮助读者理解序列的相似性和演化关系。
方法为了进行Blast分析,首先需要准备两个序列:查询序列和参考序列。
查询序列是我们要研究的序列,而参考序列是已知的序列。
Blast会将查询序列与参考序列进行比对,并计算序列之间的相似性。
在本次分析中,我们使用了NCBI(National Center for Biotechnology Information)提供的在线Blast工具。
具体的分析步骤如下:1.登录NCBI网站并进入Blast页面。
2.将查询序列输入到指定的文本框中。
3.选择参考序列数据库。
4.点击“运行Blast”按钮,等待分析结果。
结果经过Blast分析,我们获得了以下结果:1.序列相似性分析:Blast会将查询序列与参考序列进行比对,并计算序列之间的相似性。
结果以百分比的形式表示相似度。
较高的相似度表明序列之间有较高的共同点。
2.演化关系分析:Blast还可以帮助我们了解序列之间的演化关系。
通过比较序列中的保守区域和变异区域,我们可以推断序列的起源和演化路径。
讨论根据Blast分析结果,我们可以得出以下结论:1.查询序列与参考序列的相似性较高。
根据相似性百分比可以判断两个序列之间的关系,例如亲缘关系或功能相似性。
2.查询序列可能与参考序列在演化上存在一定的共同点。
通过比较序列中的保守区域和变异区域,我们可以推断序列的起源和演化路径。
3.查询序列与参考序列之间的差异可能与物种间的差异相关。
通过进一步的分析,可以探究这些差异对生物体功能的影响。
结论本次Blast分析报告旨在帮助读者理解序列的相似性和演化关系。
通过Blast工具,我们可以快速准确地比对和比较生物序列。
通过对结果的分析,我们可以推断序列的起源和演化路径,并进一步探究序列间的差异对生物体功能的影响。
NCBI在线BLAST使用方法与结果详解

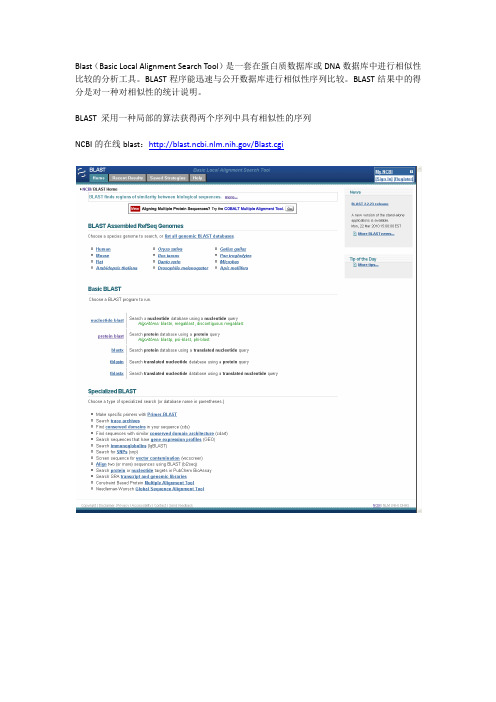
NCBI正在线BLAST使用要领与截止详解之阳早格格创做BLAST(Basic Local Alignment Search Tool)是一套正在蛋黑量数据库或者DNA数据库中举止相似性比较的分解工具.BLAST步调能赶快与公启数据库举止相似性序列比较.BLAST截止中的得分是对于一种对于相似性的统计证明.BLAST 采与一种局部的算法赢得二个序列中具备相似性的序列.Blast中时常使用的步调介绍:1、BLASTP是蛋黑序列到蛋黑库中的一种查询.库中存留的每条已知序列将逐一天共每条所查序列做一对于一的序列比对于.2、BLASTX是核酸序列到蛋黑库中的一种查询.先将核酸序列翻译成蛋黑序列(一条核酸序列会被翻译成大概的六条蛋黑),再对于每一条做一对于一的蛋黑序列比对于.3、BLASTN是核酸序列到核酸库中的一种查询.库中存留的每条已知序列皆将共所查序列做一对于一天核酸序列比对于.4、TBLASTN是蛋黑序列到核酸库中的一种查询.与BLASTX差异,它是将库中的核酸序列翻译成蛋黑序列,再共所查序列做蛋黑与蛋黑的比对于.5、TBLASTX是核酸序列到核酸库中的一种查询.此种查询将库中的核酸序列战所查的核酸序列皆翻译成蛋黑(每条核酸序列会爆收6条大概的蛋黑序列),那样屡屡比对于会爆收36种比对于阵列.底下是简直支配要领1,加进正在线BLAST界里,不妨采用blast特定的物种(如人,小鼠,火稻等),也不妨采用blast所有的核酸或者蛋黑序列.分歧的blast步调上头已经有了介绍.那里以时常使用的核酸库动做例子.2,粘揭fasta要领的序列.采用一个要比对于的数据库.闭于数据库的证明请瞅NCBI正在线blast数据库的简要证明.普遍的话参数默认.3,blast参数的树立.注意隐现的最大的截止数跟E值,E值是比较要害的.筛选的尺度.末尾会证明一下.4,注意一下您输进的序列少度.注意一下比对于的数据库的证明.5,blast截止的图形隐现.出啥佳道的.6,blast截止的形貌天区.注意分值与E值.分值越大越靠前了,E值越小也是那样.7,blast截止的仔细比对于截止.注意比对于到的序列少度.评介一个blast截止的尺度主要有三项,E值(Expect),普遍性(Identities),缺得或者拔出(Gaps).加上少度的话,便有四个尺度了.如图中隐现,比对于到的序列少度为1405,瞅Identities那一值,才匹配到1344bp,而输进的序列少度也是为1344bp(瞅上头的图),便证明比对于到的序列要少一面.由Qurey(起初1)战Sbjct(起初35)的起初位子可知,5'端是是多了一段的.偶尔也要注意3'端的.附:E值(Expect):表示随机匹配的大概性,E值越大,随机匹配的大概性也越大.E值交近整或者为整时,具原上便是实足匹配了.普遍性(Identities):或者相似性.匹配上的碱基数占总序列少的百分数.缺得或者拔出(Gaps):拔出或者缺得.用"—"去表示.。
在线blast的用法总结
Blast(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列NCBI的在线blast:/Blast.cgi本文详细出处参考:/475/举例一:核酸序列的比对1,进入在线blast界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
(补充介绍下:1、BLASTN【nucleotide blast】是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
2、BLASTP【protein blast】是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
3、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
)2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast 数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
BLAST使用方法

BLAST使用方法一、BLAST的安装和准备工作2.获取待比对的序列文件,可以是FASTA格式的DNA或蛋白质序列。
二、BLAST的常用参数和选项1. Program:指定使用哪种BLAST程序(如BLASTn、BLASTp等)。
2. Database:指定使用哪个数据库进行比对。
3. Query:指定待比对的序列文件。
4. E-value:期望值。
一种描述比对结果误差率的指标,值越小表示结果越可信。
通常情况下,E-value小于0.01被认为是显著结果。
5. Word size:BLAST在比对时使用的核心词的长度。
长度越大表示查全率(sensitivity)越高,但速度会减慢。
6. Gap open:允许在比对过程中插入空位(如插入一个碱基)。
Gap open参数定义了开放一个空位的惩罚分数。
7. Gap extension:允许空位的延伸。
Gap extension参数定义了延伸一个空位的惩罚分数。
三、使用BLAST进行比对1.命令行方式:-打开命令行界面,并定位到BLAST软件的安装目录。
- 输入命令,指定BLAST程序、数据库、查询文件和其他参数。
例如:blastn -db nt -query query.fasta -out output.txt -evalue 0.01-运行命令,BLAST将开始进行比对并生成结果文件。
2.网页方式(以NCBIBLAST为例):- 打开NCBI网站的BLAST页面()。
-选择需要使用的BLAST程序(如BLASTn、BLASTp等)。
-上传待比对的序列文件,或者粘贴序列文本到输入框中。
-选择适当的数据库和其他参数。
-点击“BLAST”按钮,等待比对完成。
四、解读BLAST结果1. E-value:表示在随机比对中获得与查询序列相似度更高的结果的期望概率。
E-value越小表示比对结果越显著。
2. Bitscore:用于表示比对结果的质量。
Bitscore越高表示比对结果越可信。
NCBI在线BLAST使用方法与结果详解

NCBI在线BLAST使用方法与成果详解之袁州冬雪创作BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中停止相似性比较的分析工具.BLAST程序能迅速与公开数据库停止相似性序列比较.BLAST成果中的得分是对一种对相似性的统计说明. BLAST 采取一种部分的算法获得两个序列中具有相似性的序列.Blast中常常使用的程序先容:1、BLASTP是蛋白序列到蛋白库中的一种查询.库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对.2、BLASTX是核酸序列到蛋白库中的一种查询.先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成能够的六条蛋白),再对每条作一对一的蛋白序列比对.3、BLASTN是核酸序列到核酸库中的一种查询.库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对.4、TBLASTN是蛋白序列到核酸库中的一种查询.与BLASTX 相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对.5、TBLASTX是核酸序列到核酸库中的一种查询.此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会发生6条能够的蛋白序列),这样每次比对会发生36种比对阵列.下面是详细操纵方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列.分歧的blast程序上面已经有了先容.这里以常常使用的核酸库作为例子.2,粘贴fasta格式的序列.选择一个要比对的数据库.关于数据库的说明请看NCBI在线blast数据库的简要说明.一般的话参数默许.3,blast参数的设置.注意显示的最大的成果数跟E值,E 值是比较重要的.筛选的尺度.最后会说明一下.4,注意一下你输入的序列长度.注意一下比对的数据库的说明.5,blast成果的图形显示.没啥好说的.6,blast成果的描绘区域.注意分值与E值.分值越大越靠前了,E值越小也是这样.7,blast成果的详细比对成果.注意比对到的序列长度.评价一个blast成果的尺度主要有三项,E值(Expect),一致性(Identities),缺失或拔出(Gaps).加上长度的话,就有四个尺度了.如图中显示,比对到的序列长度为1405,看Identities这一值,才匹配到1344bp,而输入的序列长度也是为1344bp(看上面的图),就说明比对到的序列要长一点.由Qurey(起始1)和Sbjct(起始35)的起始位置可知,5'端是是多了一段的.有时也要注意3'端的.附:E值(Expect):暗示随机匹配的能够性,E值越大,随机匹配的能够性也越大.E值接近零或为零时,具本上就是完全匹配了.一致性(Identities):或相似性.匹配上的碱基数占总序列长的百分数.缺失或拔出(Gaps):拔出或缺失.用"—"来暗示.。
如何看懂NCBIBLAST输出结果

如何看懂NCBIBLAST输出结果NCBI BLAST(Basic Local Alignment Search Tool)是一种用于比较生物序列之间的相似性的工具。
BLAST将一个查询序列与一个目标数据库中的序列进行比对,并输出比对结果。
下面将介绍如何看懂NCBI BLAST输出结果。
BLAST报告的不同部分提供了关于比对结果的详细信息。
以下是BLAST输出结果中的重要部分:1.查询信息:在输出结果的第一部分,会显示关于查询序列的信息,如查询序列的名称、长度以及描述。
这些信息可以帮助确认你是否正确提交了查询序列。
2.数据库信息:在查询信息的下方,输出结果会提供关于目标数据库的信息,包括数据库的名称、大小以及参与比对的序列数目。
这些信息可以帮助你了解比对参考的范围和样本数目。
3.参数信息:BLAST在进行比对时使用了一系列的参数,这些参数可以影响比对的灵敏度和特异性。
输出结果会显示用于比对的参数信息,包括比对算法、匹配得分、不匹配得分、开始扣分以及扩展扣分等。
这些参数提供了对比对结果的解释依据。
4.结果摘要:在参数信息的下方,会显示一个结果摘要表,提供了与查询序列最相似的多个数据库序列的信息。
这些信息包括数据库序列的名称、长度、比对得分以及比对的e值。
e值是一个表示比对结果的统计显著性的指标,越小表示比对结果越显著。
这些信息可以帮助你快速了解最相关的序列。
5.序列比对信息:在结果摘要之后,会显示每个比对的详细信息。
比对信息包括目标序列的名称和描述、比对长度、匹配得分、比对得分、e值以及比对图形。
比对图形以垂直线表示查询和目标序列之间的匹配,帮助你在比对中可视化相似区域。
6.比对统计信息:在序列比对信息之后,会显示比对的统计信息。
这些统计信息包括查询序列的覆盖率、比对序列的覆盖率以及总体比对得分。
这些信息对比对结果的解释和评估非常重要。
7.结果解释:在比对统计信息之后,会提供进一步解释和分析比对结果的信息。
NCBI在线BLAST使用方法与结果详解

NCBI在线BLAST使用方法与结果详解NCBI在线BLAST(Basic Local Alignment Search Tool)是一种广泛使用的生物信息学工具,用于比对和分析DNA、RNA或蛋白质序列。
它可以对已知和未知序列进行,找到与查询序列相似的序列,并提供有关相似性和功能的信息。
使用NCBI在线BLAST可以分为四个主要步骤:选择BLAST程序,输入查询序列,选择目标数据库,解析和分析结果。
第一步:选择BLAST程序NCBI提供了多种BLAST程序可供选择,包括BLASTN(DNA对DNA的比对)、BLASTP(蛋白质对蛋白质的比对)、BLASTX(DNA对蛋白质的比对)等。
根据实际需求选择相应的BLAST程序。
第二步:输入查询序列在查询序列的文本框中输入待比对的序列。
可以输入单个序列,也可以上传包含多个序列的文件。
如果输入的序列是DNA或RNA序列,需要选择相应的序列类型。
此外,还可以选择是否使用掩码序列或低复杂性筛选来优化比对结果。
第三步:选择目标数据库用户可以选择目标数据库来与查询序列相似的序列。
NCBI提供了多个常用的数据库,如nr(非冗余蛋白质数据库)、nt(核酸数据库)等。
此外,还可以选择特定的物种数据库来限制比对范围。
第四步:解析和分析结果在BLAST运行完成后,会生成一个结果页面,其中包含了比对结果的详细信息。
结果页面包括比对统计信息、序列比对图、E值、分数等。
通过分析这些信息,可以了解查询序列与目标数据库中的序列之间的相似性和可能的功能。
此外,NCBI在线BLAST还提供了一些高级选项,例如使用特定的算法或参数来进行比对、设置比对阈值、选择比对输出格式等。
这些选项可以根据实际需求进行调整。
总结起来,使用NCBI在线BLAST可以通过选择BLAST程序、输入查询序列、选择目标数据库以及解析和分析结果来比对和分析序列。
通过权衡算法和参数选择,在特定数据库中找到与查询序列相似的序列,从而获得有关其相似性和功能的信息。
NCBI在线版Blast使用(超详细奥)

NCBI在线版Blast使⽤(超详细奥)⾸先进⾏Blast类型的选择:blastp:将待查询的蛋⽩质序列及其互补序列⼀起对蛋⽩质序列数据库进⾏查询;blastn:将待查询的核酸序列及其互补序列⼀起对核酸序列数据库进⾏查询;blastx:先将待查询的核酸序列按六种可读框架(逐个向前三个碱基和逐个向后三个碱基读码)翻译成蛋⽩质序列,然后将翻译结果对蛋⽩质序列数据库进⾏查询;tblastn:先将核酸序列数据库中的核酸序列按六种可读框架翻译成蛋⽩质序列,然后将待查询的蛋⽩质序列及其互补序列对其翻译结果进⾏查询;tblastx:先将待查询的核酸序列和核酸序列数据库中的核酸序列按六种可读框架翻译成蛋⽩质序列,然后再将两种翻译结果从蛋⽩质⽔平进⾏查询。
基本步骤如下:1,进⼊在线blast界⾯,可以选择blast特定的物种(如下)。
不同的blast程序上⾯已经有了介绍。
这⾥以常⽤的Blast 中nucleotide blast作为例⼦。
Human ⼈类Mouse ⼩⿏Rat ⼤⿏Arabidopsis thaliana 拟南芥Oryza sativa ⽔稻Bos taurus ⽜Danio rerio 斑马鱼Drosophila melanogaster ⿊腹果蝇Gallus gallus 乌⾻鸡Pan troglodytes ⿊猩猩Microbes 微⽣物Apis mellifera 蜜蜂2,粘贴fasta格式的序列(可以是多条奥!!)或使⽤Accession number(s)、gi(s)(注意仅使⽤数字,不加上标志符gi)。
选择⼀个要⽐对的数据库,如果是⼈和⿏则进⾏相应的选择,否则选择Others中的nr/nt 。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
其他选项不是必选的,如Job Title就是这次⽐对的名字,随便起⼀个即可;Organism为物种,可以填⼊你想⽐对的物种(分类单元如green plant等)的名字(拉丁名字,输⼊⼏个字母后会出现索引的)。
(完整)NCBI在线BLAST使用方法与结果详解

(完整)NCBI在线BLAST使用方法与结果详解编辑整理:尊敬的读者朋友们:这里是精品文档编辑中心,本文档内容是由我和我的同事精心编辑整理后发布的,发布之前我们对文中内容进行仔细校对,但是难免会有疏漏的地方,但是任然希望((完整)NCBI在线BLAST使用方法与结果详解)的内容能够给您的工作和学习带来便利。
同时也真诚的希望收到您的建议和反馈,这将是我们进步的源泉,前进的动力。
本文可编辑可修改,如果觉得对您有帮助请收藏以便随时查阅,最后祝您生活愉快业绩进步,以下为(完整)NCBI在线BLAST使用方法与结果详解的全部内容。
NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
NCBI的在线BLAST:http://blast。
蛋白blast的操作方法

蛋白blast的操作方法
蛋白质BLAST是一种用于比对和识别已知蛋白质序列的工具。
下面是进行蛋白质BLAST的基本操作方法:
1. 打开NCBI(国家医学图书馆)的BLAST页面,网址为
2. 选择适合的蛋白质数据库。
在页面的“blastp”部分,可以选择不同的蛋白质数据库,如“nr”(NCBI非冗余蛋白质数据库),“pdb”(蛋白质数据银行)等。
3. 复制或输入要比对的蛋白质序列。
在页面的“Enter Query Sequence”部分,可以粘贴或输入要比对的蛋白质序列。
4. 设置其他参数。
可以根据需要设置其他参数,如过滤序列中的低复杂性区域,调整比对结果的显示方式等。
5. 点击“BLAST”按钮开始比对。
点击页面底部的“BLAST”按钮,系统将开始进行蛋白质比对。
6. 等待比对结果。
根据输入的蛋白质序列的长度和数据库的大小,比对过程可能需要一段时间。
可以在页面上监视比对进度。
7. 查看比对结果。
比对完成后,系统将显示比对结果。
可以查看比对的蛋白质序列,比对的统计信息和潜在的同源蛋白质。
8. 解释和分析比对结果。
根据比对结果,可以解释蛋白质的结构和功能,进一步分析和研究。
以上是蛋白质BLAST的基本操作方法,根据具体需求和研究目标,还可以进行更多的参数设置和高级分析。
NCBIBLAST比对结果详细分析

NCBI BLAST比对结果详细分析
NCBI Blast结果-菜单与基本信息 1.下一步操作的菜单,你可以调整参数,重新比对、保存你的搜索条件以便下次比对、调整报告显示的参数,
以更符合你的要求、下载你比对的结果; 2.此次比对的标题,优先是你填写的,如果没有填写可能是你输入fasta序列头(大于号后面的),如果这个也没 有找到,NCBI 会自动生成一个; 3.你输入序列的信息,包括标识号、描述信息、类型、长度; 4.数据库的信息以及你选择的Blast程序; 5.查看其他报告,比如摘要、分类、距离树、结构、多重比对等。 Graphic Summary
Alignments 比对详细信息
1.比对上的序列信息;
2.比对的各种得分,这里不做一一说明,这里我最关注的是Identities,比对上(一致)的数字、一共有多少个,比 对上所占的比例。
3.具体的比对序列显示,一目了然,知道了哪些序列比对上了,哪些序列是不一样的,这里也要注意序列的位 置关系;
5.复选框,可以选择感兴趣的比对序列,在⑥处进行相应的操作;
BLAST地址: blastp&PAGE_TYPE=BlastSearch&SHOW_DEFAULTS=on&LINK_LOC=blasthome 比对用的例子:
>gi|16758036|ref|NP_445782.1| ribosomal protein L21 [Rattus norvegicus] MTNTKGKRRGTRYMFSRPFRKHGVVPLATYMRIYKKGDIVDIKGMGTVQKGMPHKCYHGKTGRVYNVTQH AVGIIVNKQVKGKILAKRINVRIEHIKHSKSRDSFLKRVKENDQKKKEAKEKGTWVQLNGQPAPPREAHF VRTNGKEPELLEPIPYEFMA 数据选择:nr 比对时间:2009年9月9日12:46:23 解读报告前需要掌握的概念
NCBI在线BLAST使用方法与结果详解之欧阳术创编

NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX 相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
NCBI的在线BLAST:/Blast.cgi下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
如何看懂NCBI-BLAST输出结果

如何看懂NCBI BLAST输出结果2010-11-13 10:38:11| 分类:生物信息分析| 标签:blast |字号大中小订阅本文转自:写在解读报告之前的,首先就使用Blast最终的目的是什么达成一致,Blast 是通过两两比对,找到数据库中与输入序列最相似的序列,或者说是最相似的序列片段。
那么我们看比对结果就是看Blast从数据库中找到哪些相似的序列,然后就是如何相似,这些相似又可以告诉我们哪些信息等。
当然Blast可以衍生出许多的用途,但都是建立在找到相似性序列(片段)的基础上的。
最新的BLAST结果报告解读,本文以BLASTP为例子,说明如何来解读BLAST 结果。
示例BLAST地址:比对用的例子:>gi|16758036|ref|NP_445782.1| ribosomal protein L21 [Rattus norvegicus] MTNTKGKRRGTRYMFSRPFRKHGVVPLATYMRIYKKGDIVDIKGMGTVQKG MPHKCYHGKTGRVYNVTQH AVGIIVNKQVKGKILAKRINVRIEHIKHSKSRDSFLKRVKENDQKKKEAKEKG TWVQLNGQPAPPREAHFVRTNGKEPELLEPIPYEFMA数据选择:nr比对时间:2009年9月9日12:46:23解读报告前需要掌握的概念:alignments 代表比对上的两个序列hits 表示两个序列比对上的片段Score 比对得分,如果序列匹配上得分,不一样,减分,分值越高,两个序列相似性越高E Value 值越小,越可信,相对的一个统计值。
Length 输入序列的长度Identities 一致性,就是两个序列有多少是一样的Query 代表输入序列Sbjct 代表数据库中的序列结果详细说明菜单与基本信息NCBI Blast结果-菜单与基本信息1.下一步操作的菜单,你可以调整参数,重新比对、保存你的搜索条件以便下次比对、调整报告显示的参数,以更符合你的要求、下载你比对的结果;2.此次比对的标题,优先是你填写的,如果没有填写可能是你输入fasta序列头(大于号后面的),如果这个也没有找到,NCBI会自动生成一个;3.你输入序列的信息,包括标识号、描述信息、类型、长度;4.数据库的信息以及你选择的Blast程序;5.查看其他报告,比如摘要、分类、距离树、结构、多重比对等。
在线blast的用法总结

在线b l a s t的用法总结-标准化文件发布号:(9556-EUATWK-MWUB-WUNN-INNUL-DDQTY-KIIBlast(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列NCBI的在线blast:/Blast.cgi本文详细出处参考:/475/举例一:核酸序列的比对1,进入在线blast界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
(补充介绍下:1、BLASTN【 nucleotide blast】是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
2、BLASTP【protein blast】是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
3、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
)2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
NCBI在线BLAST使用方法与结果详解

NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
没啥好说的。
6,blast结果的描述区域。
如何看懂NCBIBLAST输出结果

如何看懂NCBIBLAST输出结果NCBI BLAST(Basic Local Alignment Search Tool)是一种用于比较两个生物序列的计算工具,常用于基因组学和蛋白质研究中。
NCBI BLAST输出结果提供了在数据库中找到的最相关的序列匹配结果,以及其他有关这些匹配结果的统计信息。
为了更好地理解NCBI BLAST输出结果,需要了解输出结果的基本结构、匹配结果的意义以及其他可能对研究有帮助的信息。
NCBIBLAST的输出结果通常分为四个主要部分:1.查询标题和信息;2.对于每个匹配,给出的序列标识、匹配得分和e值;3.每个匹配的序列比对详情,包括片段的位置和相似性比例;4.其他比对相关的统计信息。
3.序列比对详情:这部分提供了每个匹配的序列比对详情,包括匹配片段的起始位置、终止位置以及相似性比例。
使用这些信息,你可以直观地了解到均匀性、差异性和缺失的区域。
比对结果通常以比对序列为中心,显示查询序列和匹配序列之间的相似结果,有助于确定匹配的准确性和可靠性。
4. 其他统计信息:NCBI BLAST的输出结果还通常提供一些其他统计信息,这些信息可能包括数据库中的匹配数目、查询和匹配序列的长度、比对片段的数量以及在给定匹配中存在的Gap数量等。
这些信息可以帮助你更好地了解匹配结果的分布和偏差,进一步评估匹配结果的可靠性。
要正确解读NCBIBLAST的输出结果,可以参考以下几点:2.关注匹配得分和e值:匹配得分高或e值低的结果表示匹配结果越显著,但这并不意味着其他结果就不重要。
可以根据实际需要筛选和排序结果。
3. 注意比对细节:仔细查看比对细节,了解匹配序列和查询序列之间的具体差异和相似性。
比对中的Gap、匹配片段的位置等信息可以帮助理解匹配的准确性和局限性。
4.与现有知识结合:将NCBIBLAST的结果与已知的生物信息学数据库和文献中的相关结果进行比较和分析,有助于验证和解释BLAST的结果。
如何看懂NCBI-BLAST输出结果
如何看懂NCBI BLAST输出结果2010-11-13 10:38:11| 分类:生物信息分析| 标签:blast |字号大中小订阅本文转自:写在解读报告之前的,首先就使用Blast最终的目的是什么达成一致,Blast 是通过两两比对,找到数据库中与输入序列最相似的序列,或者说是最相似的序列片段。
那么我们看比对结果就是看Blast从数据库中找到哪些相似的序列,然后就是如何相似,这些相似又可以告诉我们哪些信息等。
当然Blast可以衍生出许多的用途,但都是建立在找到相似性序列(片段)的基础上的。
最新的BLAST结果报告解读,本文以BLASTP为例子,说明如何来解读BLAST 结果。
示例BLAST地址:比对用的例子:>gi|16758036|ref|NP_445782.1| ribosomal protein L21 [Rattus norvegicus] MTNTKGKRRGTRYMFSRPFRKHGVVPLATYMRIYKKGDIVDIKGMGTVQKG MPHKCYHGKTGRVYNVTQH AVGIIVNKQVKGKILAKRINVRIEHIKHSKSRDSFLKRVKENDQKKKEAKEKG TWVQLNGQPAPPREAHFVRTNGKEPELLEPIPYEFMA数据选择:nr比对时间:2009年9月9日12:46:23解读报告前需要掌握的概念:alignments 代表比对上的两个序列hits 表示两个序列比对上的片段Score 比对得分,如果序列匹配上得分,不一样,减分,分值越高,两个序列相似性越高E Value 值越小,越可信,相对的一个统计值。
Length 输入序列的长度Identities 一致性,就是两个序列有多少是一样的Query 代表输入序列Sbjct 代表数据库中的序列结果详细说明菜单与基本信息NCBI Blast结果-菜单与基本信息1.下一步操作的菜单,你可以调整参数,重新比对、保存你的搜索条件以便下次比对、调整报告显示的参数,以更符合你的要求、下载你比对的结果;2.此次比对的标题,优先是你填写的,如果没有填写可能是你输入fasta序列头(大于号后面的),如果这个也没有找到,NCBI会自动生成一个;3.你输入序列的信息,包括标识号、描述信息、类型、长度;4.数据库的信息以及你选择的Blast程序;5.查看其他报告,比如摘要、分类、距离树、结构、多重比对等。
NCBI在线BLAST使用方法与结果详解

NCBI在线BLAST使用方法与结果详解NCBI(National Center for Biotechnology Information)是一个包含大量基因组学、生物信息学等相关数据和工具的数据库。
其中,BLAST (Basic Local Alignment Search Tool)是一种常用的序列比对工具,可用于在数据库中搜索相似序列。
一、BLAST简介BLAST是一种基于序列比对的方法,可用于确定一给定序列与数据库中序列的相似性。
其工作原理是将查询序列与数据库中的序列进行比对,并生成一个比对得分来衡量它们之间的相似程度。
通过BLAST的结果,可以获得序列的匹配位置、长度、相似性等信息,从而帮助研究人员进行更深入的生物学研究。
二、使用方法1. 打开NCBI网站首先,打开浏览器,输入NCBI的网址(https:///),进入NCBI的官方网站。
2. 进入BLAST页面在NCBI的主页上,找到“BLAST”或“BLAST and Alignments”选项,并点击进入BLAST页面。
3. 输入查询序列在BLAST页面上,找到“Enter Query Sequence”或“Enter accession number, gi, or FASTA sequence”等文本框,将需要查询的序列输入其中。
可以直接复制粘贴序列,或选择上传文件的方式输入。
4. 选择数据库在BLAST页面上,找到“Choose Search Set”或“Database”等选项,选择需要比对的数据库。
NCBI提供了多个数据库,如“nr”(非冗余蛋白数据库)、“nt”(非冗余核酸数据库)等,根据研究需要选择合适的数据库。
5. 设置参数根据需要,可以通过“Algorithm parameters”等选项来设置比对参数,如设置匹配的阈值、比对的方式等。
6. 运行BLAST设置完成后,点击“BLAST”或“Run BLAST”等按钮运行BLAST。
如何使用NCBI中的Blast

如何使用NCBI中的BlastNCBI(National Center for Biotechnology Information)是一个提供生物信息学数据库和工具的综合性资源平台。
其中,BLAST(Basic Local Alignment Search Tool)是一种经典的序列比对工具,用于比对和分析DNA、RNA和蛋白质序列的相似性。
使用NCBI中的BLAST可以有多种方式,包括在线使用和本地使用。
下面将对这两种使用方式进行详细介绍。
一、在线使用NCBIBLASTNCBI提供了一个在线的BLAST界面,用户可以直接在浏览器中使用。
具体步骤如下:1. 打开NCBI网站,点击"Blast"选项卡,然后选择需要比对的序列类型,例如,DNA、蛋白质或者其他。
2. 复制并粘贴待比对的序列到"Enter Query Sequence"文本框中。
或者,您也可以选择上传一个FASTA格式的文件。
3.选择适当的数据库。
NCBI提供了多个数据库供选择,根据您的研究目的选择合适的数据库。
4.配置其他参数。
您可以选择不同的比对算法、设置匹配参数、设定范围等。
5.点击"BLAST"按钮开始比对。
该过程可能需要一些时间,取决于比对数据的大小和服务器的负载情况。
6.一旦比对完成,系统将生成一个结果页面,显示比对结果。
您可以查看比对的统计信息、序列相似性分析、注释信息等。
7.针对一些结果,您可以选择进一步分析和操作,例如,设计引物、进行序列比对、构建进化树等。
二、本地使用NCBIBLAST3.准备待比对的序列,并保存到FASTA格式的文件中。
4.打开终端或命令提示符,并导航到BLAST软件的安装目录。
5. 运行BLAST命令。
根据您的比对需求,运行适当的BLAST命令,例如,“blastn”用于DNA比对,”blastp”用于蛋白质比对。
6.设置适当的输入参数,包括查询序列文件、目标数据库、比对算法等。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
没啥好说的。
6,blast结果的描述区域。
注意分值与E值。
分值越大越靠前了,E值越小也是这样。
7,blast结果的详细比对结果。
注意比对到的序列长度。
评价一个blast结果的标准主要有三项,E值(Expect),一致性(Identities),缺失或插入(Gaps)。
加上长度的话,就有四个标准了。
如图中显示,比对到的序列长度为1405,看Identities这一值,才匹配到1344bp,而输入的序列长度也是为1344bp(看上面的图),就说明比对到的序列要长一点。
由Qurey(起始1)和Sbjct(起始35)的起始位置可知,5'端是是多了一段的。
有时也要注意3'端的。
附:E值(Expect):表示随机匹配的可能性,E值越大,随机匹配的可能性也越大。
E值接近零或为零时,具本上就是完全匹配了。
一致性(Identities):或相似性。
匹配上的碱基数占总序列长的百分数。
缺失或插入(Gaps):插入或缺失。
用"—"来表示。
1、请你自我介绍一下自己好吗?回答提示:一般人回答这个问题过于平常,只说姓名、年龄、爱好、工作经验,这些在简历上都有。
其实,企业最希望知道的是求职者能否胜任工作,包括:最强的技能、最深入研究的知识领域、个性中最积极的部分、做过的最成功的事,主要的成就等,这些都可以和学习无关,也可以和学习有关,但要突出积极的个性和做事的能力,说得合情合理企业才会相信。
企业很重视一个人的礼貌,求职者要尊重考官,在回答每个问题之后都说一句“谢谢”,企业喜欢有礼貌的求职者。
2、你觉得你个性上最大的优点是什么?回答提示:沉着冷静、条理清楚、立场坚定、顽强向上、乐于助人和关心他人、适应能力和幽默感、乐观和友爱。
我在北大青鸟经过一到两年的培训及项目实战,加上实习工作,使我适合这份工作。
3、说说你最大的缺点?回答提示:这个问题企业问的概率很大,通常不希望听到直接回答的缺点是什么等,如果求职者说自己小心眼、爱忌妒人、非常懒、脾气大、工作效率低,企业肯定不会录用你。
绝对不要自作聪明地回答“我最大的缺点是过于追求完美”,有的人以为这样回答会显得自己比较出色,但事实上,他已经岌岌可危了。
企业喜欢求职者从自己的优点说起,中间加一些小缺点,最后再把问题转回到优点上,突出优点的部分,企业喜欢聪明的求职者。
4、你对薪资的要求?回答提示:如果你对薪酬的要求太低,那显然贬低自己的能力;如果你对薪酬的要求太高,那又会显得你分量过重,公司受用不起。
一些雇主通常都事先对求聘的职位定下开支预算,因而他们第一次提出的价钱往往是他们所能给予的最高价钱,他们问你只不过想证实一下这笔钱是否足以引起你对该工作的兴趣。
回答样本一:我对工资没有硬性要求,我相信贵公司在处理我的问题上会友善合理。
我注重的是找对工作机会,所以只要条件公平,我则不会计较太多。
回答样本二:我受过系统的软件编程的训练,不需要进行大量的培训,而且我本人也对编程特别感兴趣。
因此,我希望公司能根据我的情况和市场标准的水平,给我合理的薪水。
回答样本三:如果你必须自己说出具体数目,请不要说一个宽泛的范围,那样你将只能得到最低限度的数字。
最好给出一个具体的数字,这样表明你已经对当今的人才市场作了调查,知道像自己这样学历的雇员有什么样的价值。
5、你对加班的看法?回答提示:实际上好多公司问这个问题,并不证明一定要加班,只是想测试你是否愿意为公司奉献。
回答样本:如果工作需要我会义不容辞加班,我现在单身,没有任何家庭负担,可以全身心的投入工作。
但同时我也会提高工作效率,减少不必要的加班。
6、如果通过这次面试我们录用了你,但工作一段时间却发现你根本不适合这个职位,你怎么办?回答提示:一段时间发现工作不适合我,有两种情况:①如果你确实热爱这个职业,那你就要不断学习,虚心向领导和同事学习业务知识和处事经验,了解这个职业的精神内涵和职业要求,力争减少差距;②你觉得这个职业可有可无,那还是趁早换个职业,去发现适合你的,你热爱的职业,那样你的发展前途也会大点,对单位和个人都有好处。
7、谈谈你对跳槽的看法?回答提示:①正常的“跳槽”能促进人才合理流动,应该支持。
②频繁的跳槽对单位和个人双方都不利,应该反对。
8、工作中难以和同事、上司相处,你该怎么办?回答提示:①我会服从领导的指挥,配合同事的工作。
②我会从自身找原因,仔细分析是不是自己工作做得不好让领导不满意,同事看不惯。
还要看看是不是为人处世方面做得不好,如果是这样的话我会努力改正。
③如果我找不到原因,我会找机会跟他们沟通,请他们指出我的不足,有问题就及时改正。
④作为优秀的员工,应该时刻以大局为重,即使在一段时间内,领导和同事对我不理解,我也会做好本职工作,虚心向他们学习,我相信,他们会看见我在努力,总有一天会对我微笑的。
9、你对于我们公司了解多少?回答提示:在去公司面试前上网查一下该公司主营业务。
如回答:贵公司有意改变策略,加强与国外大厂的OEM合作,自有品牌的部分则透过海外经销商。
10、最能概括你自己的三个词是什么?回答提示:我经常用的三个词是:适应能力强,有责任心和做事有始终,结合具体例子向主考官解释,11、你的业余爱好是什么?回答提示:找一些富于团体合作精神的,这里有一个真实的故事:有人被否决掉,因为他的爱好是深海潜水。
主考官说:因为这是一项单人活动,我不敢肯定他能否适应团体工作。
12、作为被面试者给我打一下分?回答提示:试着列出四个优点和一个非常非常非常小的缺点(可以抱怨一下设施,没有明确责任人的缺点是不会有人介意的)。
13、你为什么要离开原来的公司?回答提示:①回答这个问题时一定要小心,就算在前一个工作受到再大的委屈,对公司有多少的怨言,都千万不要表现出来,尤其要避免对公司本身主管的批评,避免面试官的负面情绪及印象。
建议此时最好的回答方式是将问题归咎在自己身上,例如觉得工作没有学习发展的空间,自己想在面试工作的相关产业中多加学习,或是前一份工作与自己的生涯规划不合等等,回答的答案最好是积极正面的。
②我希望能获得一份更好的工作,如果机会来临,我会抓住。
我觉得目前的工作,已经达到顶峰,即沒有升迁机会。
14、你欣赏哪种性格的人?回答提示:诚实、不死板而且容易相处的人、有“实际行动”的人。
15、你通常如何对待别人的批评?回答提示:①沈默是金,不必说什么,否则情况更糟,不过我会接受建设性的批评。
②我会等大家冷靜下来再讨论。
16、怎样对待自己的失败?回答提示:我们大家生来都不是十全十美的,我相信我有第二个机会改正我的错误。
17、你为什么愿意到我们公司来工作?回答提示:对于这个问题,你要格外小心,如果你已经对该单位作了研究,你可以回答一些详细的原因,像“公司本身的高技术开发环境很吸引我。
”、“我同公司出生在同样的时代,我希望能够进入一家与我共同成长的公司。
”、“你们公司一直都稳定发展,在近几年来在市场上很有竞争力。
”、“我认为贵公司能够给我提供一个与众不同的发展道路。
”这都显示出你已经做了一些调查,也说明你对自己的未来有了较为具体的远景规划。
18、对这项工作,你有哪些可预见的困难?回答提示:①不宜直接说出具体的困难,否则可能令对方怀疑应聘者不行。
②可以尝试迂回战术,说出应聘者对困难所持有的态度——工作中出现一些困难是正常的,也是难免的,但是只要有坚忍不拔的毅力、良好的合作精神以及事前周密而充分的准备,任何困难都是可以克服。
19、如果录用了你,你将怎样开展工作?回答提示:①如果应聘者对于应聘的职位缺乏足够的了解,最好不要直接说出自己开展工作的具体办法。
②可以尝试采用迂回战术来回答,如“首先听取领导的指示和要求,然后就有关情况进行了解和熟悉,接下来制定一份近期的工作计划并报领导批准,最后根据计划开展工作。
”。
分析:这个问题的主要目的也是了解应聘者的工作能力和计划性、条理性,而且重点想要知道细节。
如果向思路中所讲的迂回战术,面试官会认为回避问题,如果引导了几次仍然是回避的话,此人绝对不会录用了。
20、你希望与什么样的上级共事?回答提示:①通过应聘者对上级的“希望”可以判断出应聘者对自我要求的意识,这既上一个陷阱,又是一次机会。
②最好回避对上级具体的希望,多谈对自己的要求。
③如“做为刚步入社会的新人,我应该多要求自己尽快熟悉环境、适应环境,而不应该对环境提出什么要求,只要能发挥我的专长就可以了。
分析:这个问题比较好的回答是,希望我的上级能够在工作中对我多指导,对我工作中的错误能够立即指出。
总之,从上级指导这个方面谈,不会有大的纰漏。
21、与上级意见不一时,你将怎么办?回答提示:①一般可以这样回答“我会给上级以必要的解释和提醒,在这种情况下,我会服从上级的意见。
”②如果面试你的是总经理,而你所应聘的职位另有一位经理,且这位经理当时不在场,可以这样回答:“对于非原则性问题,我会服从上级的意见,对于涉及公司利益的重大问题,我希望能向更高层领导反映。