(-)-JQ-1_1268524-71-5_DataSheet_MedChemExpress
IT-158BS TC 中等热转变温度(Tg)多功能填充环氧树脂和酚醛固化无铅层压板和预浸料说明书

IT-158BS/IT-158TCMultifunctional Filled Epoxy Resin and Phenolic-Cured Lead Free Laminate & PrepregIT-158 is a medium Tg (>150℃ by DSC) multifunctional filled epoxy with high thermal reliability and CAF resistance. It’s suitable for industrial PCB, automobile and can pass 260℃ Lead free assembly.Key Features =============================== Advanced Resin TechnologyIndustrial standard material with medium Tg (150℃ by DSC) multifunctional filled epoxy resin and excellent thermal reliability.Lead-Free Assembly CompatibleRoHS compliant and suitable for high thermal reliability needs, and Lead free assemblies with a maximum reflow temperature of 260℃. Friendly Processing and CAF ResistanceFriendly to PCB process that users can easily handle the process by current equipment and chemical.CAF ResistanceExcellent thermal reliability and CAF resistance providing long-term reliability for industrial boards and automobile application.Available in Variety of ConstructionsAvailable in a various of constructions, copper weights and glass styles, including standard(HTE), RTF and VLP copper foil. ApplicationsPC and Notebook Memory ModuleGame PlayerMultilayer PCB AutomobileServers and Networking Telecommunications Heavy Copper ApplicationIndustrial Approval UL 94 V-0IPC-4101C Spec / 99 RoHS CompliantREV 09-14ITEQ Laminate/ Prepreg : IT-158TC / IT-158BSIPC-4101C Spec /99LAMINATE (IT-158TC)Thickness<0.50 mm[0.0197 in] Thickness≧0.50 mm[0.0197 in] Units T est MethodPropertyTypical Value Spec Typical Value SpecMetric(English)IPC-TM-650(or as noted)Peel Strength, minimumA. Low profile copper foil and very low profile copperfoil - all copper weights > 17µm [0.669 mil]B. Standard profile copper foil1.After Thermal Stress2.At 125°C [257 F]3.After Process Solutions 0.88 (5.0)1.58 (9.0)1.31 (7.5)1.14 (6.5)0.70 (4.00)0.80 (4.57)0.70 (4.00)0.55 (3.14)0.88 (5.0)1.66 (9.5)1.40 (8.0)1.23 (7.0)0.70 (4.00)1.05 (6.00)0.70 (4.00)0.80 (4.57)N/mm(lb/inch)2.4.82.4.8.22.4.8.3Volume Resistivity, minimumA. C-96/35/90B. After moisture resistanceC. At elevated temperature E-24/125 3.0x1010--5.0x1010106--103--5.0x10101.0x1010--104103MΩ-cm 2.5.17.1Surface Resistivity, minimumA. C-96/35/90B. After moisture resistanceC. At elevated temperature E-24/125 1.0x1010--5.0x1010104--103--1.0x10103.0x1010--104103MΩ 2.5.17.1Moisture Absorption, maximum -- -- 0.08 0.5 % 2.6.2.1 Dielectric Breakdown, minimum -- -- 60 40 kV 2.5.6 Permittivity (Dk, 50% resin content)(Laminate & Laminated Prepreg)A. 1MHzB. 1GHzC. 2GHzD. 5GHzE. 10GHz 4.34.34.24.14.05.44.44.34.24.14.05.4 --2.5.5.92.5.5.13Loss Tangent (Df, 50% resin content) (Laminate & Laminated Prepreg)A. 1MHzB. 1GHzC. 2GHzD. 5GHzE. 10GHz 0.0160.0160.0170.0180.0190.0350.0160.0160.0170.0180.0180.035 --2.5.5.92.5.5.13Flexural Strength, minimumA. Length directionB. Cross direction ----------------450-480(65,250-69,600)370-400(53,650-62,350)415(60,190)345(50,140)N/mm2(lb/in2)2.4.4Arc Resistance, minimum 125 60 125 60 s 2.5.1 Thermal Stress 10 s at 288°C [550.4F],minimumA. UnetchedB. Etched PassPassPass VisualPass VisualPassPassPass VisualPass VisualRating 2.4.13.1Electric Strength, minimum(Laminate & Laminated Prepreg)45 30 -- -- kV/mm 2.5.6.2 Flammability,(Laminate & Laminated Prepreg)V-0 V-0 V-0 V-0 Rating UL94 Glass Transition Temperature(DSC) 155 150 minimum 155 150 minimum ˚C 2.4.25Decomposition Temperature-- -- 345 325 minimum ˚C2.4.24.6 (5% wt loss)X/Y Axis CTE (40℃ to 125℃) -- -- 11-13 -- ppm/˚C 2.4.24 Z-Axis CTEA. Alpha 1B. Alpha 2C. 50 to 260 Degrees C ------------402403.360 maximum300 maximum3.5 maximumppm/˚Cppm/˚C%2.4.24Thermal ResistanceA. T260B. T288 -------->60>3030 minimum5 minimumMinutesMinutes2.4.24.1CAF Resistance -- -- Pass AABUS Pass/Fail 2.6.25The above data and fabrication guide provide designers and PCB shop for their reference. We believe that these information are accurate, however, the data may vary depend on the test methods and specification used. The actual sales of the product should be according to specification in the agreement between ITEQ and its customer. ITEQ reserves the right to revise its data at any time without notice and maintain the best information available to users.REV 09-14。
2X M5 HiPer SYBR Premix EsTaq plus (with Tli RNase

2X M5 HiPer SYBR Premix EsTaq plus (with Tli RNaseH)使用说明书产品名称单位货号2X M5 HiPer SYBR Premix EsTaq plus (with Tli RNaseH) 50 μl反应× 40 次MF788-T2X M5 HiPer SYBR Premix EsTaq plus (with Tli RNaseH) 50 μl反应× 200 次MF788-012X M5 HiPer SYBR Premix EsTaq plus (with Tli RNaseH) 50 μl反应× 400 次MF788-02【储存条件】长期保存,请置于-20˚C,有效期24个月。
经常使用,可置于4˚C保存至少六个月。
【产品简介】本产品采用Sybrgreen嵌合荧光法进行荧光定量的专用试剂。
制品中含有荧光定量反应的最适浓度Sybrgreen,是一种2×浓度的预混试剂,进行实验时,PCR反应液的配制十分方便简单。
制品中使用了antiTaq抗体的Hot Start法用DNA聚合酶,与荧光定量反应适合Buffer组合,可以有效抑制非特异性的PCR扩增,大大提高PCR的扩增效率,可以进行高灵敏度的荧光定量扩增反应。
本产品Buffer 经过改良,使反应特异性比SybrGreenPremix Ex Taq (withTli RNaseH)(货号MF787)更高。
另外,本产品中添加了Tli RNaseH (耐热性RNaseH),以cDNA作为模板进行PCR反应时,可以很好地抑制由于cDNA中残存mRNA 对PCR反应造成的阻害作用。
适合于快速荧光定量扩增反应,可以在宽广的定量区域内得到良好的标准曲线,对靶基因进行准确定量、检测,重复性好,可信度高。
【产品组份】MF788-T MF788-012x M5 HiPer SYBR Premix EsTaq plus (withTli RNaseH) * 1.0 ml 5x1mlROX Reference Dye(50×)** 40μl 200μlROX Reference Dye II(50×)** 40μl200μl*由以下组分预混而成:EsTaq plus,dNTP Mixture,Mg2+,Tli RNaseH,Sybrgreen。
ACCUSPIN系统-Histopaque 1077产品说明书

Technical BulletinACCUSPIN™ System – Histopaque ® 1077Catalog Numbers A6929, A7054, and A0561Product DescriptionACCUSPIN System-Histopaque -1077products are intended for use in the isolation of lymphocytes and other mononuclear cells. The separation medium, Histopaque-1077, is a sterile-filtered, endotoxin tested solution of polysucrose and sodium diatrizoate, adjusted to a density of 1.077 g/mL. The ACCUSPIN tube is specially designed with two chambers separated by a porous high density polyethylene barrier (frit).Separation of lymphocytes and other mononuclear cells from whole blood and bone marrow using density gradientseparation media is based on a published method.1 Histopaque-1077 is suitable for human lymphocyte antigen (HLA) typing 2 and as the initial isolation step prior toenumeration of T, B, and ‘null’ lymphocytes.3 It may also be employed in the preparation of pure lymphocyte suspensions for cell culture and cytotoxicity assays.4ACCUSPIN System-Histopaque-1077 products consist of radiation sterilized polypropylene tubes fitted with a highdensity polyethylene frit and aseptically filled with Histopaque-1077.Histopaque-1077 is a sterile-filtered solution of polysucrose, 57 g/L, and sodium diatrizoate, 90 g/L.Density: 1.076–1.078 g/mL Endotoxin: 0.3 EU/mL pH: 8.8–9.0ACCUSPIN System-Histopaque-1077Catalog No. A692940 × 3 mLEach tube contains 3 mL ofHistopaque 1077-1 and will separate 3-6 mL of anticoagulated blood Catalog No. A7054 12 × 15 mLCatalog No. A0561100 × 15 mLEach tube contains 15 mL ofHistopaque 1077-1 and will separate 15-30 mL of anticoagulated bloodReagents and Equipment Required but Not ProvidedCentrifuge (swinging bucket rotor)capable of generating 100 to 1,000 g Centrifuge tubes for washing mononuclear cellsIsotonic phosphate buffered saline solution or appropriate cell culture mediumPrecautions and DisclaimerFor R&D use only. Not for drug, household, or other uses. Please consult the Safety Data Sheet for information regarding hazards and safe handling practices.Preparation InstructionsSpecimen Collection - Collect blood in preservative-free anticoagulant (EDTA or heparin) or use defibrinated blood. For best results, blood should be processed within 2 hours.On occasion, it may be necessary to dilute the blood sample 3 to 5-fold, depending on absolute cell numbers. A similar volume of prediluted blood may be used or the blood sample may be diluted directly in upper chamber of the ACCUSPIN tube (seeProcedure, step 3). This is appropriate for specimens with hematocrits above normal.Storage/StabilityStore the products at 2–8 C.Histopaque-1077 has an expiration period of 3 years. Reagent label bears expiration date.ProcedureAnticoagulated blood can be added to the top chamber of the tube without risk of mixing with the Histopaque-1077 in the lowerchamber under the frit. On centrifugation the whole blood migrates through the frit to contact with the Histopaque-1077. The elements of greater density displace a volume of Histopaque-1077 above the frit giving a clear separation of the bloodcomponents. The erythrocytes aggregate and the granulocytes become slightly hypertonic, increasing their sedimentation rate, resulting in pelleting at the bottom of the ACCUSPIN Tube. Lymphocytes and other mononuclear cells, e.g., monocytes, remain at the plasma/Histopaque-1077 interface. This dense band of mononuclear cells may be collected by pouring off the contents of the upper chamber or by means of a pipette. Erythrocyte contamination is avoided due to the barrier between the chambers.Most extraneous platelets are removed by low speed centrifugation during the washing steps.1. Bring desired number of tubes to roomtemperature. If Histopaque-1077 isabove the frit prior to use, centrifuge at 1,000 g for 30 seconds at room temperature.Note: Failure to bring ACCUSPIN System-Histopaque-1077 to room temperature may cause limited recovery of mononuclear cells. 2. Label tube(s).3. Freely pour the blood sample into theupper chamber of each ACCUSPIN System-Histopaque-1077 tube.a. Use 3–6 mL of whole blood withACCUSPIN System-Histopaque-1077 tubes, Catalog No. A6929. b. Use 15–30 mL of whole blood withACCUSPIN System-Histopaque-1077 tubes, Catalog Nos. A7054 or A0561. Note: Use of volumes of prediluted or whole blood other than those recommended may result in decreased recovery.4. Centrifuge at 1,000 g for 10 minutes atroom temperature or centrifuge at 800 g for 15 minutes at roomtemperature. Centrifugation at lower temperatures, such as 4 C, may result in cell clumping and poor recovery.Note: If platelet contamination is a concern, add the mononuclear cells to a 4-20% sucrose gradient that has been layered over Histopaque-1077.Centrifuge at 1,000 × g for 10 minutes at room temperature. The platelets will pellet at the bottom, while themononuclear cells will migrate to the Histopaque-1077 layer.5. After centrifugation, carefully aspiratethe plasma layer with a Pasteur pipette to within 0.5 cm of the opaque interface containing mononuclear cells. Properly dispose of the plasma layer.Note: Failure to remove the excesssupernatant may result in contamination of the mononuclear band with plasma proteins.6. Carefully transfer the opaque interfacewith a Pasteur pipette into a clean conical centrifuge tube.Note: Removal of Histopaque-1077 with the mononuclear band increasesgranulocyte contamination from residual granulocytes, which may remain at the mononuclear interface.7. Wash the cells by adding 10 mL ofisotonic phosphate buffered saline solution or appropriate cell culture medium and mix by gently drawing in and out of a Pasteur pipette. 8. Centrifuge at 250 g for 10 minutes. 9. Aspirate the supernatant and discard. 10. Resuspend cell pellet with 5 mL ofisotonic phosphate buffered saline solution or appropriate cell culture medium and mix by gently drawing in and out of a Pasteur pipette.11. Centrifuge at 250 g for 10 minutes. 12. Repeat steps 9, 10, and 11, discardsupernatant and resuspend cell pellet in 0.5 mL of isotonic phosphate buffered saline solution or appropriate cell culture medium. Erythrocytes and granulocytes should pellet to the bottom of the ACCUSPIN tube. Mononuclear cells should band at the interface between the Histopaque-1077 and the plasma. If observed results vary from expected results, please contact Sigma-Aldrich Technical Service for assistance.References1. Boyum, A., Separation of leukocytesfrom blood and bone marrow. Scand. J. Clin. Lab. Invest ., 21 (Suppl 97), 77 (1968).2. Amos, D.B., and Pool, P., “HLA typing” inManual of Clinical Immunology, Rose, N.R., and Friedman, H., eds., American Society for Microbiology, (Washington, DC: 1976) pp. 797-804.3. Winchester, R.J., and Ross, G., “Methodsfor enumerating lymphocyte populations” in Manual of Clinical Immunology, Rose, N.R., and Friedman, H. eds., American Society for Microbiology, (Washington, DC: 1976) pp. 64-76.4. Thorsby, E., and Bratlie, A., “A rapidmethod for preparation of pure lymphocyte suspensions.”Histocompatibility Testing, Terasaki, P.I., ed., 665-666 (1970).The life science business of Merck operates as MilliporeSigma in the U.S. and Canada.Merck, Sigma-Aldrich, ACCUSPIN, and Histopaque are trademarks of Merck KGaA, Darmstadt, Germany or its affiliates. All other trademarks are the property of their respective owners. Detailed information on trademarks is available via publicly accessible resources.© 2022 Merck KGaA, Darmstadt, Germany and/or its affiliates. All Rights Reserved.NoticeWe provide information and advice to our customers on application technologies and regulatory matters to the best of our knowledge and ability, but without obligation or liability. Existing laws and regulations are to be observed in all cases by our customers. This also applies in respect to any rights of third parties. Our information and advice do not relieve our customers of their own responsibility for checking the suitability of our products for the envisaged purpose.The information in this document is subject to change without notice and should not be construed as a commitment by the manufacturing or selling entity, or an affiliate. We assume no responsibility for any errors that may appear in this document.Contact InformationFor the location of the office nearest you, go to /offices .Technical ServiceVisit the tech service page on our web site at /techservice .Standard WarrantyThe applicable warranty for the products listed in this publication may be found at /terms .A0561 Technical Bulletin Rev 06/2022。
KSZ8851-16MLL DEMO BOARD 48-PIN ETHERNET CONTROLLE

SD13
SD7 40
CPU_D14 3
6
SD14
SD8 39
CPU_D15 4
5
SD15
SD9 36
SD10 35
33
SD11 34
CPU_CMD
33
SD12 33
R10
SD13 32
CPU_CSN
33
SD14 31
R12
SD15 30
CPU_WRN
33
CMD
11
R14
CPU_RDN
33
CSN
12
R16
5 6 7 8
TANT
C27
R28 10uF
470pF 2.49K
Power 3.3V 0.1uF (red LED)
CSN CMD
4.7K R27 4.7K R29
GBLC03C_0 D3
GND 2 GND
VR 5 3.3VA
INTRN 4.7K R30
VOUT = 1.24 X [ 1 + ( 2.49k/ 1.5K ) ]
5
4
3
KSZ8851-16MLL (48-pin) Demo Board Black Diagram
D
Headers 20x2
RJ45
LAN1 T
KSZ8851-16MLL
Reset
Power
+1.8V
+2.5V
+3.3V
STATUS LEDs
OSC
EEPROM
C
MIC5209YM
25 MHz
AT93C46
x2
2
1
DATE:
韩国LG化学公司INR18650 MH1 3200mAh可重充电锂电池说明书

PRODUCT SPECIFICATIONCONFIDENTIALPrepared Document No. Date Rev LGC MED/BDC Lee, Yong Seok LRB-PS-CY3200_MH1_Tentative2014-03-06 0Approved Checked DescriptionLGC MED/BDC Kim, Dong Myung Lithium Ion INR18650 MH1 3200mAhPRODUCT SPECIFICATIONRechargeable Lithium Ion BatteryModel : INR18650 MH1 3200mAh20 YOIDO-DONG YOUNGDUNGPO-GU,SEOUL 150-721, KOREAContents (3)1. General Information (4)1.1 Scope1.2 Application1.3 Product Classification1.4 Model Name2. Nominal Specification (5)2.1 Capacity2.2 Nominal Voltage2.3 Standard Charge2.4 Max. Charge Current2.5 Standard Discharge2.6 Max. Discharge Current2.7 Weight2.8 Operating Temperature2.9 Storage Temperature (for shipping state)3. Appearance and Dimension (6)3.1 Appearance3.2 Dimension4. Performance Specification (7)4.1 Standard Test Condition4.2 Electrical Specification4.3 Environmental Specification4.4 Mechanical Specification4.5 Safety Specification5. Cautions and Prohibitions in Handling (8)1. General Information1.1 ScopeThis product specification defines the requirements of the rechargeable lithium ion battery to be supplied to the Customer by LG Chem.1.2 Product classification: Cylindrical rechargeable lithium ion battery1.3 Model name: INR18650 MH12. Nominal Specification3. Appearance and Dimension3.1 AppearanceThere shall be no such defects as deep scratch, crack, rust, discoloration or leakage, which mayadversely affect the commercial value of the cell.3.2 DimensionDiameter : 18.39 ± 0.11 mmDiameter is defined as the largest data value measured on the "A" area of a cylindrical cellHeight : ≤65.15 mm.4. Performance Specification4.1 Standard test condition4.1.1 Standard ChargeUnless otherwise specified, “Standard Charge” shall consist of charging at constant current of 0.5C. Thecell shall then be charged at constant voltage of 4.20V while tapering the charge current. Charging shallbe terminated when the charging current has tapered to 50mA. For test purposes, charging shall beperformed at 23ºC ± 2ºC.4.1.2 Standard Discharge“Standard Discharge” shall consist of discharging at a constant current of 0.2C to 2.50V. Discharging isto be performed at 23 ºC ± 2 ºC unless otherwise noted (such as capacity versus temperature).4.1.3 High Drain rate Charge/discharge conditionCells shall be charged at constant current of 0.5C to 4.20V with end current of 50mA. Cells shall be discharged at constant current of 0.5C to 2.50V. Cells are to rest 10 minutes after charge and 20 minutes after discharge.5/104.2 Electrical Specification4.3 Environmental specification.* Remaining Capacity : After storage, cells shall be discharged with Std. condition(4.1.2) to measure the remaining capacity.** Recovery Capacity : After storage, cells shall be discharged with fast discharge condition(4.1.3), and then cells shall be charged with std. charge condition(4.1.1), and then discharged with Std. condition(4.1.2). This charge / discharge cycle shall be repeated three times to measure the recovery capacity.4.4 Mechanical Specification4.5 Safety Specification5. CautionWarning for using the lithium ion rechargeable battery. Mishandling of the battery may cause heat, fire and deterioration in performance. Be sure to observe the following.5.1 Cautions for Use and Handling•When using the application equipped with the battery, refer to the user’s manual before usage.•Please read the specific charger manual before charging.•Charge time should not be longer than specified in the manual.•When the cell is not charged after long exposure to the charger, discontinue charging.•Battery must be charged at operating temperature range 0 ~ 45℃.•Battery must be discharged at operating temperature range -20 ~ 60℃.•Please check the positive(+) and negative(-) direction before packing.•When a lead plate or wire is connected to the cell for packing, check out insulation not to short-circuit.•Battery must be stored separately.•Battery must be stored in a dry area with low temperature for long-term storage.•Do not place the battery in direct sunlight or heat.•Do not use the battery in high static energy environment where the protection device can be damaged.•When rust or smell is detected on first use, please return the product to the seller immediately.•The battery must be away from children or pets•When cell life span shortens after long usage, please exchange to new cells.5.2 Prohibitions•Do not use different charger. Do not use cigarette jacks (in cars) for charging.•Do not charge with constant current more than maximum charge current.•Do not disassemble or reconstruct the battery.•Do not throw or cause impact.•Do not pierce a hole in the battery with sharp things. (such as nail, knife, pencil, drill)•Do not use with other batteries or cells.•Do not solder on battery directly.•Do not press the battery with overload in manufacturing process, especially ultrasonic welding.•Do not use old and new cells together for packing.•Do not expose the battery to high heat. (such as fire)•Do not put the battery into a microwave or high pressure container.•Do not use the battery reversed.•Do not connect positive(+) and negative(-) with conductive materials (such as metal, wire)•Do not allow the battery to be immerged in or wetted with water or sea-water.5.3 Caution for the battery and the packPack shall meet under condition to maintain battery safety and last long performance of the lithium rechargeable cells.5.3.1 Installing the battery into the pack-. The cell should be inspected visually before battery assembly into the pack.-. Damaged cell should not be used. (damaged surface, can-distortion, electrolyte-smell)-. Different Lot Number cells should not be packaged into the same pack.-. Different types of cells, or same types but different cell maker’s should not be used together.5.3.2 Design of battery pack-. The battery pack should not be connected easily to any charger other than the dedicated charger.-. The battery pack has funcion not to cause external short cut easily.5.3.3 Charge-. Charging method is Constant Current-Constant Voltage (CC/CV).-. Charging should be operating under maximum charge voltage and current which is specified in the product specification. (Article. 2.4, 2.5)-. The battery should be charged under operating temperature specified in the product specification. (Article. 2.9) 5.3.4 Discharge-. Discharging method is Constant Current (CC).(In case of using the battery for mobile equipment, discharging mode could be Constant Power.)-. Discharging should be operating under maximum discharge current which is specified in the product specification. (Article. 2.7)-. Discharging should be done by cut off voltage which is specified in the product specification. (Article. 2.6)-. The battery should be discharged under operating temperature specified in the product specification.(Article. 2.9)5.3.5 Protection Circuit-. The protection circuit should be installed in the battery pack, charger.-. Charger or pack should have voltage sensing system to control over charge or discharge in order to maintain the battery’s normal operating mode and protect cell imbalance.-. Charger or pack should have warning system for over temperature, over voltage and over current.6. EXCLUSION OF LIABILITYTHE WARRANTY SHALL NOT COVER DEFECTS CAUSED BY NORMAL WEAR AND TEAR, INADEQUATE MAINTENANCE, HANDLING, STORAGER FAULTY REPAIR, MODIFICATION TO THE BATTERY OR PACK BY A THIRD PARTY OTHER THAN LGC OR LGC’S AGENT APPROVED BY LGC, FAILURE TO OBSERVE THE PRODUCT SPECIFICATION PROVIDED HEREIN OR IMPROPER USE OR INSTALLATION, INCLUDING BUT NOT LIMITED TO, THE FOLLOWING:-. DAMAGE DURING TRANSPORT OR STORAGE-. INCORRECT INSTALLATION OF BATTERY INTO PACK OR MAINTENANCE-. USE OF BATTERY OR PACK IN INAPPROPRIATE ENVIRONMENT-. IMPROPER, INADEQUATE, OR INCORRECT CHARGE, DISCHARGE OR PRODUCTION CIRCUIT OTHER THAN STIPULATED HEREIN-. INCORRECT USE OR INAPPROPRIATE USE-. INSUFFICIENT VENTILATION-. IGNORING APPLICABLE SAFETY WARNINGS AND INSTRUCTIONS-. ALTERING OR ATTEMPTED REPAIRS BY UNAUTHORIZED PERSONNEL-. IN CASE OF FORCE MAJEURE (EX. LIGHTENING, STORM, FLOOD, FIRE, EARTHQUAKE, ETC.)THERE ARE NO WARRANTIES – IMPLIED OR EXPRESS – OTHER THAN THOSE STIPULATED HEREIN. LG CHEM SHALL NOT BE LIABLE FOR ANY CONSEQUENTIAL OR INDIRECT DAMAGES ARISING OR IN CONNECTION WITH THE PRODUCT SPECIFICATION, BATTERY OR PACK.。
XMD8-87_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:XMD8–87 is a potent TNK2 inhibitor with IC 50 values of 38 and 113 nM for the D163E and R806Q mutations, respectively.IC50 & Target: IC50: 38 nM (TNK2, D163E mutation), 113 nM (TNK2, R806Q mutation)[1]In Vitro: XMD8–87 potently inhibits the growth of the TNK2 mutant expressing cell lines while having little or no effect on the control cells out to the highest tested concentrations (1,000 nM). XMD8–87 has IC 50s of 38 nM and 113 nM for the D163E and R806Q mutations. The effects of XMD8–87 on TNK2 cell lines are largely due to on–target effects on TNK2. Auto–phosphorylation of overexpressed TNK2 mutants could be blocked with TNK2 inhibitor XMD8–87[1].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[1]Kinase targets are tested with biochemical enzymatic kinase assays using the SelectScreen Kinase Profiling Service to determine IC 50 values. The compounds (XMD8–87) are assayed at 10 concentrations (3–fold serial dilutions starting from 1 μM) at an ATP concentration equal to the ATP Km [1].Cell Assay:[1]Cells are treated with the following inhibitors for 72 hours: dasatinib, AIM–100, XMD8–87 and XMD16–5. Cell viability is measured using a methanethiosulfonate (MTS)–based assay and absorbance (490 nm) is read at 1 and 3 hours after adding reagent [1].References:[1]. Maxson JE, et al. Identification and Characterization of Tyrosine Kinase Nonreceptor 2 Mutations in Leukemia through Integration of Kinase Inhibitor Screening and Genomic Analysis.Product Name:XMD8–87Cat. No.:HY-15811CAS No.:1234480-46-6Molecular Formula:C 24H 27N 7O 2Molecular Weight:445.52Target:Tyrosinase Pathway:Metabolic Enzyme/Protease Solubility:DMSO: ≥ 26 mg/mLCaution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@ Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
JQEZ5_SDS_MedChemExpress

Inhibitors, Agonists, Screening LibrariesSafety Data Sheet Revision Date:Jan.-22-2018Print Date:Jan.-22-20181. PRODUCT AND COMPANY IDENTIFICATION1.1 Product identifierProduct name :JQEZ5Catalog No. :HY-100846CAS No. :1913252-04-61.2 Relevant identified uses of the substance or mixture and uses advised againstIdentified uses :Laboratory chemicals, manufacture of substances.1.3 Details of the supplier of the safety data sheetCompany:MedChemExpress USATel:609-228-6898Fax:609-228-5909E-mail:sales@1.4 Emergency telephone numberEmergency Phone #:609-228-68982. HAZARDS IDENTIFICATION2.1 Classification of the substance or mixtureNot a hazardous substance or mixture.2.2 GHS Label elements, including precautionary statementsNot a hazardous substance or mixture.2.3 Other hazardsNone.3. COMPOSITION/INFORMATION ON INGREDIENTS3.1 SubstancesSynonyms:NoneFormula:C30H38N8O2Molecular Weight:542.68CAS No. :1913252-04-64. FIRST AID MEASURES4.1 Description of first aid measuresEye contactRemove any contact lenses, locate eye-wash station, and flush eyes immediately with large amounts of water. Separate eyelids with fingers to ensure adequate flushing. Promptly call a physician.Skin contactRinse skin thoroughly with large amounts of water. Remove contaminated clothing and shoes and call a physician.InhalationImmediately relocate self or casualty to fresh air. If breathing is difficult, give cardiopulmonary resuscitation (CPR). Avoid mouth-to-mouth resuscitation.IngestionWash out mouth with water; Do NOT induce vomiting; call a physician.4.2 Most important symptoms and effects, both acute and delayedThe most important known symptoms and effects are described in the labelling (see section 2.2).4.3 Indication of any immediate medical attention and special treatment neededTreat symptomatically.5. FIRE FIGHTING MEASURES5.1 Extinguishing mediaSuitable extinguishing mediaUse water spray, dry chemical, foam, and carbon dioxide fire extinguisher.5.2 Special hazards arising from the substance or mixtureDuring combustion, may emit irritant fumes.5.3 Advice for firefightersWear self-contained breathing apparatus and protective clothing.6. ACCIDENTAL RELEASE MEASURES6.1 Personal precautions, protective equipment and emergency proceduresUse full personal protective equipment. Avoid breathing vapors, mist, dust or gas. Ensure adequate ventilation. Evacuate personnel to safe areas.Refer to protective measures listed in sections 8.6.2 Environmental precautionsTry to prevent further leakage or spillage. Keep the product away from drains or water courses.6.3 Methods and materials for containment and cleaning upAbsorb solutions with finely-powdered liquid-binding material (diatomite, universal binders); Decontaminate surfaces and equipment by scrubbing with alcohol; Dispose of contaminated material according to Section 13.7. HANDLING AND STORAGE7.1 Precautions for safe handlingAvoid inhalation, contact with eyes and skin. Avoid dust and aerosol formation. Use only in areas with appropriate exhaust ventilation.7.2 Conditions for safe storage, including any incompatibilitiesKeep container tightly sealed in cool, well-ventilated area. Keep away from direct sunlight and sources of ignition.Recommended storage temperature:Powder-20°C 3 years4°C 2 yearsIn solvent-80°C 6 months-20°C 1 monthShipping at room temperature if less than 2 weeks.7.3 Specific end use(s)No data available.8. EXPOSURE CONTROLS/PERSONAL PROTECTION8.1 Control parametersComponents with workplace control parametersThis product contains no substances with occupational exposure limit values.8.2 Exposure controlsEngineering controlsEnsure adequate ventilation. Provide accessible safety shower and eye wash station.Personal protective equipmentEye protection Safety goggles with side-shields.Hand protection Protective gloves.Skin and body protection Impervious clothing.Respiratory protection Suitable respirator.Environmental exposure controls Keep the product away from drains, water courses or the soil. Cleanspillages in a safe way as soon as possible.9. PHYSICAL AND CHEMICAL PROPERTIES9.1 Information on basic physical and chemical propertiesAppearance Light yellow to yellow (Solid)Odor No data availableOdor threshold No data availablepH No data availableMelting/freezing point No data availableBoiling point/range No data availableFlash point No data availableEvaporation rate No data availableFlammability (solid, gas)No data availableUpper/lower flammability or explosive limits No data availableVapor pressure No data availableVapor density No data availableRelative density No data availableWater Solubility No data availablePartition coefficient No data availableAuto-ignition temperature No data availableDecomposition temperature No data availableViscosity No data availableExplosive properties No data availableOxidizing properties No data available9.2 Other safety informationNo data available.10. STABILITY AND REACTIVITY10.1 ReactivityNo data available.10.2 Chemical stabilityStable under recommended storage conditions.10.3 Possibility of hazardous reactionsNo data available.10.4 Conditions to avoidNo data available.10.5 Incompatible materialsStrong acids/alkalis, strong oxidising/reducing agents.10.6 Hazardous decomposition productsUnder fire conditions, may decompose and emit toxic fumes.Other decomposition products - no data available.11.TOXICOLOGICAL INFORMATION11.1 Information on toxicological effectsAcute toxicityClassified based on available data. For more details, see section 2Skin corrosion/irritationClassified based on available data. For more details, see section 2Serious eye damage/irritationClassified based on available data. For more details, see section 2Respiratory or skin sensitizationClassified based on available data. For more details, see section 2Germ cell mutagenicityClassified based on available data. For more details, see section 2CarcinogenicityIARC: No component of this product present at a level equal to or greater than 0.1% is identified as probable, possible or confirmed human carcinogen by IARC.ACGIH: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by ACGIH.NTP: No component of this product present at a level equal to or greater than 0.1% is identified as a anticipated or confirmed carcinogen by NTP.OSHA: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by OSHA.Reproductive toxicityClassified based on available data. For more details, see section 2Specific target organ toxicity - single exposureClassified based on available data. For more details, see section 2Specific target organ toxicity - repeated exposureClassified based on available data. For more details, see section 2Aspiration hazardClassified based on available data. For more details, see section 212. ECOLOGICAL INFORMATION12.1 ToxicityNo data available.12.2 Persistence and degradabilityNo data available.12.3 Bioaccumlative potentialNo data available.12.4 Mobility in soilNo data available.12.5 Results of PBT and vPvB assessmentPBT/vPvB assessment unavailable as chemical safety assessment not required or not conducted.12.6 Other adverse effectsNo data available.13. DISPOSAL CONSIDERATIONS13.1 Waste treatment methodsProductDispose substance in accordance with prevailing country, federal, state and local regulations.Contaminated packagingConduct recycling or disposal in accordance with prevailing country, federal, state and local regulations.14. TRANSPORT INFORMATIONDOT (US)This substance is considered to be non-hazardous for transport.IMDGThis substance is considered to be non-hazardous for transport.IATAThis substance is considered to be non-hazardous for transport.15. REGULATORY INFORMATIONSARA 302 Components:No chemicals in this material are subject to the reporting requirements of SARA Title III, Section 302.SARA 313 Components:This material does not contain any chemical components with known CAS numbers that exceed the threshold (De Minimis) reporting levels established by SARA Title III, Section 313.SARA 311/312 Hazards:No SARA Hazards.Massachusetts Right To Know Components:No components are subject to the Massachusetts Right to Know Act.Pennsylvania Right To Know Components:No components are subject to the Pennsylvania Right to Know Act.New Jersey Right To Know Components:No components are subject to the New Jersey Right to Know Act.California Prop. 65 Components:This product does not contain any chemicals known to State of California to cause cancer, birth defects, or anyother reproductive harm.16. OTHER INFORMATIONCopyright 2017 MedChemExpress. The above information is correct to the best of our present knowledge but does not purport to be all inclusive and should be used only as a guide. The product is for research use only and for experienced personnel. It must only be handled by suitably qualified experienced scientists in appropriately equipped and authorized facilities. The burden of safe use of this material rests entirely with the user. MedChemExpress disclaims all liability for any damage resulting from handling or from contact with this product.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
agilent-5067-5585_chinese.pdf-高灵敏度d1000试剂安全技术说明书

High Sensitivity D1000 Reagents, Part Number 5067-5585*************(24小时)化学品安全技术说明书GHS product identifier 应急咨询电话(带值班时间)::供应商/ 制造商:安捷伦科技贸易(上海)有限公司中国(上海)外高桥自由贸易试验区英伦路412号(邮编:200131)电话号码: 800-820-3278传真号码: 0086 (21) 5048 2818High Sensitivity D1000 Reagents, Part Number 5067-5585化学品的推荐用途和限制用途High Sensitivity D1000 Sample Buffer 5190-6504High Sensitivity D1000 Ladder 5067-5587部件号:部件号(化学品试剂盒):5067-5585安全技术说明书根据 GB/ T 16483-2008 和 GB/ T 17519-2013GHS化学品标识:高灵敏度 D1000 试剂, 部件号 5067-5585推荐用途限制用途High Sensitivity D1000Sample Buffer1 x 300 µl 样品瓶5067-5587High Sensitivity D1000Ladder1 x 20 µl 样品瓶::有关环境保护措施,请参阅第 12 节。
物质或混合物的分类根据 GB13690-2009 和 GB30000-2013紧急情况概述High Sensitivity D1000 Sample Buffer液体。
High Sensitivity D1000 Ladder 液体。
High Sensitivity D1000 Sample Buffer无色。
High Sensitivity D1000 Ladder 无色。
High Sensitivity D1000 Sample Buffer无气味的。
RayBio

RayBio® Mouse PAI-I IQLEISA KitCatalog #: IQM-PAIIUser ManualLast revised April 6, 2023Caution:Extraordinarily useful information enclosedISO 13485 Certified3607 Parkway Lane, Suite 200Peachtree Corners, GA 30092 Tel: 1-888-494-8555 (Toll Free) or 770-729-2992, Fax: 770-206-2393Web: , Email: *******************RayBiotech, Inc.________________________________________RayBio® Mouse PAI-I IQLEISA Kit ProtocolTable of ContentsSection Page # I.Introduction3II.Reagents3III.Storage3IV.Additional Materials Required4V.Reagent Preparation4VI.Assay Procedure5VII.Assay Procedure Summary8VIII.Calculation of ResultsA. Typical DataB. Sensitivity and Recovery 8 9 9IX.Troubleshooting Guide10I. INTRODUCTIONThe RayBio®I mmuno Q uantitative E nzyme L inked I mumuno S orbent A ssay (IQELISA) is an innovative new assay that combines the specificity and ease of use of an ELISA with the sensitivity of real-time PCR. This results in an assay that is simultaneously familiar and cutting edge and enables the use of lower sample volumes while also providing more sensitivity. The RayBio® Mouse PAI-I IQLEISA Kit is a modified ELISA assay with high sensitivity qPCR readout for the quantitative measurement of Mouse PAI-I in serum, plasma, and cell culture supernatants. This assay employs an antibody specific for Mouse PAI-I coated on a 96-well PCR plate. Standards and samples are pipetted into the wells and PAI-I present in a sample is bound to the wells by the immobilized antibody. The wells are washed and a detection affinity molecule is added to the plates. After washing away unbound detection affinity molecule, primers and a PCR master mix are added to the wells and data is collected using qPCR. C t values obtained from the qPCR are then used to calculate the amount of antigen contained in each sample, where lower C t values indicate a higher concentration of antigen.II. REAGENTS1.PAI-I Microplate (Item A)**: 96 well PCR plate coated with anti-Mouse PAI-I.2.Wash Buffer I Concentrate (20x) (Item B): 25 ml of 20x concentrated solution.3.Standards (Item C): 2 vials of recombinant Mouse PAI-I.4.Assay Diluent B (Item E): 15 ml of 5x concentrated buffer.5.Detection Affinity Reagent for PAI-I (Item F): 2 vials of a 4x concentrated solution of anti-Mouse PAI-I affinity reagent.6.IQELISA Detection Reagent (Item G): 1 mL of a 10x concentrated stock.7.Primer Solution (Item I): 1.5 mL vial.8.PCR Master Mix (Item J): 1.4 mL vial.9.PCR Preparation buffer (Item K): 1mL vial of 10x concentrated buffer.10.Final Wash Buffer (Item L): 10 mL vial of 10x concentrated buffer.**The PCR plate used is a 0.2 mL, non-skirted 96-well plate (ThermoFisher, cat. no.:AB0600). Please ensure compatibility with your PCR machine prior to purchase. For additional information contact technical support (**************************).III. STORAGEMay be stored for up to 6 months at 2°to 8°C from the date of shipment. Standard (recombinant protein) should be stored at -20°C or -80°C (recommended at -80°C) after reconstitution. Opened PCR plate or reagents may be stored for up to 1 month at 2° to 8°C. Note: the kit can be used within one year if the whole kit is stored at -20°C. Avoid repeated freeze-thaw cycles.IV. ADDITIONAL MATERIALS REQUIRED1.Real-time PCR instrument, Bio-Rad recommended2.Precision pipettes to deliver 2 µl to 1 mL volumes.3.Adjustable 1-25 mL pipettes for reagent preparation.4.100 mL and 1 L graduated cylinders.5.Absorbent paper.6.Distilled or deionized water.7.Log-log graph paper or computer and software for data analysis.8.Tubes to prepare standard or sample dilutions.9.Heating block or water bath capable of 80°CV. REAGENT PREPARATION1.Bring wash buffer, samples, assay diluents, and PCR plate to room temperature (18 -25°C) before use. PCR master mix and Primer solution should be kept at 4°C at alltimes.2.Sample dilution: If your samples need to be diluted, 1x Assay Diluent B should be usedfor dilution of serum/plasma samples.Suggested dilution for normal serum/plasma: 2 -20 fold*.*Please note that levels of the target protein may vary between different specimens.Optimal dilution factors for each sample must be determined by the investigator.3.Assay Diluent B should be diluted 5-fold with deionized water.4.Briefly spin the Detection Antibody vial before use. Add 25 µL of 1x Assay Diluent B intothe vial to prepare a detection antibody concentrate. Pipette up and down to mix gently (the concentrate can be stored at 4°C for 5 days). This concentrate should be diluted 80-fold with 1x Assay Diluent B and used in step 4 of the Assay Procedure.5.PCR preparation buffer should be transferred to a 15 mL tube and diluted with 9 mL ofdeionized or distilled water before use.6.Final Wash Buffer should be transferred to a 15 mL tube and diluted with 9 mL ofdeionized or distilled water for every 1 mL of 10x concentrate used before use.7.Preparation of standard: Preparation of standard: Briefly spin a vial of Standards. Add400 µl 1X Assay Diluent B into Standards vial to prepare a 100 ng/ml standard solution.Dissolve the powder thoroughly by a gentle mix. Add 100 µl of the PAI-1 standard from the vial of Standards, into a tube with 400 µl 1X Assay Diluent B to prepare a 20,000pg/ml standard solution. Pipette 200 µl 1X Assay Diluent B into each tube. Use the 20,000 pg/ml standard solution to produce a dilution series (shown below). Mix eachtube thoroughly before the next transfer. 1X Assay Diluent B serves as the zero standard (0 pg/ml).400.00µL+ 100 µL100 µL + 200 µL 100 µL + 200 µL 100 µL + 200 µL 100 µL + 200 µL 100 µL+ 200 µL100 µL+ 200 µL20000pg/ml 6666.667pg/ml 2222.222pg/ml 740.741pg/ml 246.914pg/ml 82.305pg/ml 27.435pg/ml 0pg/ml8.If the Wash Buffer Concentrate (20x) contains visible crystals, warm to room temperature and mix gently until dissolved. Dilute 20 mL of Wash Buffer Concentrate into deionized or distilled water to yield 400 mL of 1x Wash Buffer.9.Prepare the IQELISA detection reagent by calculating how much will be needed. This may be accomplished by multiplying the number of wells to be assayed by the volume you plan to use per well. Once the volume of IQELISA detection reagent is known,prepare the reagent by diluting it 1:10 with deionized water and mixing thoroughly.VI. ASSAY PROCEDUREOptional Visual Aid: IQELISA [Good Laboratory Practice Guide]1.Bring all reagents and samples to room temperature (18 - 25°C) before use. It isrecommended that all standards and samples be run in triplicate. Partial plate runs may be accomplished by cutting the PCR plate into the desired number of strips using a pair of sturdy scissors, wire cutters, or shears. The remainder may be saved and used for a later date. If this is done, the PCR Plate Film should also be cut to a suitable size.2.Add 10-25 µL of each standard (see Reagent Preparation step 2) and sample intoappropriate wells. Volumes should be consistent between all wells, samples, andstandards. As little as 10 µL can be used if sample volume is limited, however thisincreases the chance of technical error. Ensure there are no bubbles present at thebottom of the wells. Dislodge any bubbles with gentle tapping or with a pipette tip being careful not to contact the sides or bottom of the well. Cover well and incubate for 1.5 -2.5 hours at room temperature.3.Discard the solution and wash 4 times with 1x Wash Solution. Wash by filling each wellwith Wash Buffer (100 µL) using a multi-channel Pipette or autowasher. Completeremoval of liquid at each step is essential to good performance. After the last wash,remove any remaining Wash Buffer by aspirating or decanting. Invert the plate and blot it against clean paper towels.4.Add 25 µL of prepared Detection Antibody (Reagent Preparation step 4) to each well.Incubate for 1 hour at room temperature with gentle shaking.5.Discard the solution. Repeat the wash as in step 3.6.Add 50 µL of prepared IQELISA detection reagent and incubate 1 hour with rocking(Reagent Preparation step 9)7.Discard the solution. Repeat the wash as in step 3, for a total of 6 washes.8.Add 75 µL of Final wash buffer to each well and incubate for 4 minutes with rocking.Remove the solution from each well and blot against paper towels.9.Add 75 µL of 1x PCR preparation buffer to each well and incubate for 10 seconds beforeremoving the buffer. Blot the plate after the buffer is removed to ensure completeremoval of the buffer.10.Add 10 µL of the Primer solution to each well of the plate. At this stage the plate can becovered and stored at -20°C for use the next day if needed.11.Add 10 µL of PCR Master Mix to each well and pipette thoroughly to mix the well (atleast 3x up and down).12.Cover the plate with the supplied PCR Plate Film, taking care to insure the film iscompletely and even pressed onto the plate, creating an air tight seal around each well of the plate.Optional Visual Aid: Sealing the plate [qPCR]13.Place the plate into a real-time PCR instrument using a FITC compatible wave length fordetection with the following settings for cycling1.2 minute activation at 95°C2.15 seconds 95°C denaturation3.25 seconds 60°C annealing/extension4.Repeat steps 2 and 3 34x**Optional: Include a melt curve to view potential plate contamination that can causehigh background and lower the sensitivity. This can be seen in the visual aid onYouTube.VII. ASSAY PROCEDURE SUMMARY1.Prepare all reagents, samples and standards as instructed.2.Add 25 µL standard or sample to each well. Incubate 1.5 - 2.5 hours at roomtemperature.3.Add 25 µL Detection Antibody to each well. Incubate 1 hour at room temperature.4.Add 50 µL of IQELISA Detection Reagent to each well. Incubate 1 hour5.Add 10 µL Primer solution and 10 µL of PCR master mix to each well6.Run real-time PCRVIII. CALCULATION OF RESULTSThe primary data output of the IQELISA kit is C t values. These values represent the number of cycles required for a sample to pass a fluorescence threshold. As the DNA is amplified additional fluorescent signal is produced, with each cycle resulting in an approximate doubling of the DNA. Therefore, higher levels of DNA (directly related to the amount of antigen in the sample) result in lower C t values.Calculate the mean C t for each set of triplicate standards, controls and samples. Subtract the C t value of each sample from the control to obtain the difference between the control and sample (Delta C t). Plot the values of the standards on a graph using a log scale for concentration on the x axis. This graph is the quickest way to visualize results, although not the most accurate. If this method is used the concentration of unknown samples can be estimated using a logarithmic line of best fit.The line of best fit will have an equation y = mln(x)+b, where y is the Delta C t value and x is the concentration. It may be helpful to use 5 significant figures for m and b to minimize rounding errors. To calculate the concentration of unknown sample this can be entered into Excel in the following format=EXP((y-b)/m))Where y is the Delta C t obtained during the assay, and b and m are obtained from the line of best fit.Alternatively, for a more accurate representation linear regression may be used. Both the Delta C t and Concentration can be transformed using a log base of 10, plotted on a graph as described above, along with a line of best fit (using a linear model). The equation of this line may be used to calculate the antigen concentration of unknown samples. This is the method used for the analysis spreadsheet for IQELISA available online.A. TYPICAL DATAThese data are for demonstration only. A standard curve must be run with each assay.B. SENSITIVITY and RECOVERYThe minimum quantifiable dose of PAI-I is typically 78.125 pg/ml, however levels as lower than 78.125 pg/ml may be detected outside of the quantification range.Serum spike tests show recovery is 75% with a range from 67% to 80%.Intraplate CV is below 10% for all samples and Interplate CV is below 15%.X. TROUBLESHOOTING GUIDEThis product is for research use only.©2023 RayBiotech, Inc11。
胆碱酯酶(ChE)检测试剂盒(羟胺氯化铁比色法)
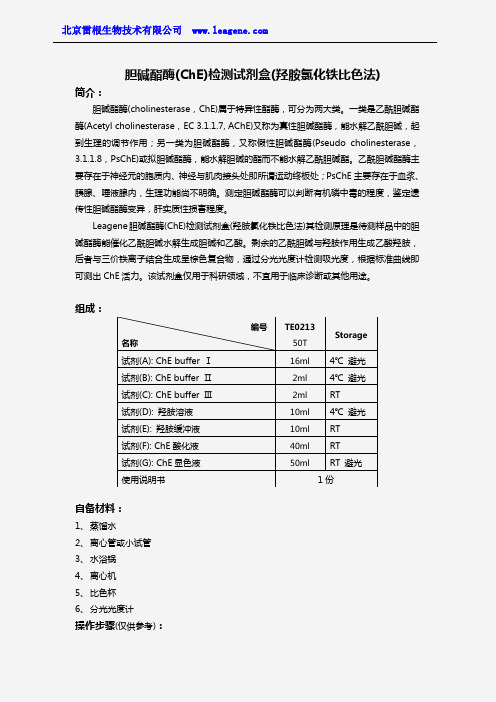
胆碱酯酶(ChE)检测试剂盒(羟胺氯化铁比色法)简介:胆碱酯酶(cholinesterase,ChE)属于特异性酯酶,可分为两大类。
一类是乙酰胆碱酯酶(Acetyl cholinesterase,EC 3.1.1.7, AChE)又称为真性胆碱酯酶,能水解乙酰胆碱,起到生理的调节作用;另一类为胆碱酯酶,又称假性胆碱酯酶(Pseudo cholinesterase,3.1.1.8,PsChE)或拟胆碱酯酶,能水解胆碱的酯而不能水解乙酰胆碱酯。
乙酰胆碱酯酶主要存在于神经元的胞质内、神经与肌肉接头处即所谓运动终板处;PsChE主要存在于血浆、胰腺、唾液腺内,生理功能尚不明确。
测定胆碱酯酶可以判断有机磷中毒的程度,鉴定遗传性胆碱酯酶变异,肝实质性损害程度。
Leagene胆碱酯酶(ChE)检测试剂盒(羟胺氯化铁比色法)其检测原理是待测样品中的胆碱酯酶能催化乙酰胆碱水解生成胆碱和乙酸。
剩余的乙酰胆碱与羟胺作用生成乙酸羟胺,后者与三价铁离子结合生成呈棕色复合物,通过分光光度计检测吸光度,根据标准曲线即可测出ChE活力。
该试剂盒仅用于科研领域,不宜用于临床诊断或其他用途。
组成:自备材料:1、蒸馏水2、离心管或小试管3、水浴锅4、离心机5、比色杯6、分光光度计操作步骤(仅供参考):编号名称TE021350TStorage试剂(A): ChE buffer Ⅰ16ml 4℃避光试剂(B): ChE buffer Ⅱ2ml 4℃避光试剂(C): ChE buffer Ⅲ2ml RT试剂(D): 羟胺溶液10ml 4℃避光试剂(E): 羟胺缓冲液10ml RT试剂(F): ChE酸化液40ml RT试剂(G): ChE显色液50ml RT 避光使用说明书1份1、准备样品:①血浆、血清样品:血浆、血清按照常规方法制备,可以直接用于本试剂盒的测定,-70℃冻存,用于ChE的检测。
②细胞或组织样品:取恰当细胞或组织进行匀浆,低速离心取上清,-70℃冻存,用于ChE的检测。
核苷类药物知识

核苷类药物知识核苷类药物的综述,免费下载的,大家给好评吧!O(∩_∩)O~1. 前言核苷和脱氧核苷是由核苷碱基分别和核糖或脱氧核糖以苷键形式而构成的,它们是组成核糖核酸(RNA)和脱氧核糖核酸(DNA)的基本元件,是遗传基因的基础。
核苷和脱氧核苷系列衍生物具有多种生物活性物质,可以直接或间接地作为药物使用,在治疗多种重大的疾病方面起到极其重要的作用,国外已经研究开发出系列化药物并商品化,国内研究与开发较晚,发展前景非常广阔.1。
1 核苷类药物的合成与生产从20世纪40年代末期,国外就开始核苷及其系列药物的合成与开发。
目前世界排名前25位制药大公司都有自己的核苷衍生物生产或加工厂,并且均有持有专利的核苷类药物上市,并且从20世纪90年代起投入巨资用于基因药物的研究。
据国外有关资料预计,2003年基因药物的市场价值将超过30亿美元.在亚洲,日本是最早开发核苷类药物和基因药物的国家,如武田、住友、味之素等公司均有相关的中间体开发机构和生产基地。
另外韩国、印度在20世纪90年代初开始投入这类产品的开发与生产.中国在核苷及其衍生物方面的开发研究与生产始于20世纪90年代末期,但是核苷及其中间体品种少,部分原料依赖进口,与目前快速发展的生命科学及相关药物研究不相适应。
1。
2 核苷类药物的应用核苷与脱氧核苷系列化合物主要用于医药领域,用途广泛,而且新产品层出不穷,应用范围不断扩大.(一)抗病毒药物。
核苷类抗病毒药物品种繁多,结构多样,主要以破坏病毒转录,干扰或终止病毒核酸的合成为目的,用于抗疱疹病毒、HIV、HBV、以及流感和呼吸系统病毒等DNA和RNA病毒。
目前在这方面应用最多,而且新出现的药物主要集中于治疗上述疾病。
(二)抗肿瘤药物。
目前用于临床和正在研究的核苷类抗肿瘤药物有数十种,它们的主要作用是干扰肿瘤的DNA合成,或者影响核酸的转录过程,抑制蛋白质的合成,从而达到治疗肿瘤的效果。
(三)抗真菌类药物.具有这方面作用的核苷类化合物已经有多种用于临床应用,其中有部分产品对多种真菌具有抑制作用,而且对哺乳动物几乎无毒性。
德州仪器(Texas Instruments)DRV8871 3.6A 刷式直流电机驱动器说明书

Copyright © 2017, Texas Instruments IncorporatedProduct Folder Order Now Technical Documents Tools &SoftwareSupport &CommunityDRV8871ZHCSE26B –AUGUST 2015–REVISED JULY 2016具有内部电流感测功能的DRV88713.6A 刷式直流电机驱动器(PWM 控制)1特性•H 桥电机驱动器–驱动一个直流电机、一个步进电机的绕组或其他负载• 6.5V 至45V 宽工作电压范围•565m Ω(典型值)R DS(on)(HS +LS)• 3.6A 峰值电流驱动能力•PWM 控制接口•无需感测电阻即可实现电流调节•低功耗休眠模式•小型封装尺寸–8引脚HSOP 封装,带有PowerPAD™– 4.9mm ×6mm •集成保护特性–VM 欠压闭锁(UVLO)–过流保护(OCP)–热关断(TSD)–自动故障恢复2应用•打印机•电器•工业设备•其他机电应用3说明DRV8871器件是一款刷式直流电机驱动器,适用于打印机、电器、工业设备以及其他小型机器。
两个逻辑输入控制H 桥驱动器,该驱动器由四个N 沟道金属氧化物半导体场效应晶体管(MOSFET)组成,能够以高达3.6A 的峰值电流双向控制电机。
利用电流衰减模式,可通过对输入进行脉宽调制(PWM)来控制电机转速。
如果将两个输入均置为低电平,则电机驱动器将进入低功耗休眠模式。
DRV8871器件具有高级电流调节电路,该电路不使用模拟电压基准或外部感应电阻器。
这种新型解决方案采用标准的低成本、低功耗电阻来设置电流阈值。
该器件能够将电流限制在某一已知水平,这可显著降低系统功耗要求,并且无需大容量电容来维持稳定电压,尤其是在电机启动和停转时。
该器件针对故障和短路问题提供了全面保护,包括欠压锁定(UVLO)、过流保护(OCP)和过热保护(TSD)。
抗核抗体检测试剂盒(磁条码免疫荧光法)产品技术要求丽珠

1性能指标
1.1外观
试剂盒各组份应齐全、完整,标签清晰,易识别;液体无渗漏。
1.2准确度
对准确度参考品进行检测,双链DNA(dsDNA)IgG抗体的相对偏差应在±20%范围内。
1.3最低检测限
对检测限参考品进行检测,dsDNA IgG抗体的检测限应不高于10 IU/mL;其它定性指标对应的各指标的L1应为阳性、L2应为阳性或阴性、L3应为阴性。
1.4线性
dsDNA IgG抗体在10~ 600 IU/mL范围内,其线性相关系数(r)r2应不小于0.950。
1.5阴性参考品符合率
对10份阴性参考品进行检测,对应指标的阴性参考品符合率应为100%。
1.6阳性参考品符合率
对12份阳性参考品进行检测,对应指标的阳性参考品符合率应为100%。
1.7重复性
对重复性参考品检测10次,同一指标的变异系数(CV)应不大于15%。
1.8批间差
用三个批号试剂盒检测同一份重复性参考品,批间差应不大于20%。
1.9dsDNA 校准品均匀性
1.9.1瓶内均匀性
校准品的瓶内均匀性(变异系数,CV)应不大于15%。
1.9.2瓶间均匀性
校准品的瓶间均匀性(变异系数,CV)应不大于15%。
艾滋病检测试剂技术参数和要求

艾滋病检测试剂技术参数和要求一、人类免疫缺陷病毒抗原抗体诊断试剂盒(酶联免疫法)参数和要求▲1、原理:用双抗原夹心法检测标本中的HlV抗体,同时用双抗体夹心法检测标本中的HIVp24抗原;2、方法:酶联免疫试验(ELISA);3、有效期:到用户指定地点有效期为8个月(含)以上;★4、2017-2019年在全国艾滋病病毒抗体诊断试剂临床质量评估中敏感性均达到100%,特异性不低于99.6%,功效性不低于99.7%;提供评估报告复印件5、获得食品药品监督管理局颁发的医疗器械注册证(提供复印件);6、HIV-1/2抗体国家阳性参考品(+/+)为20/20;HIV-1P24抗原国家阳性参考品(+/+)为10/10;▲7.可检测样品:人血清或血浆二、HIV/HCV/HBV/梅毒四合一快检试剂参数和要求1、单人份包装;★2、试剂可同时检测血液中的梅毒抗体、艾滋抗体、乙肝表面抗原和丙肝抗体★3、统一加样,全血标本加样总量不超过150μL;4、加样后15分钟内出结果;5、有效期≥24个月,到货后有效期≥15个月;★6、可以检测血清、血浆和全血样品;7、试剂性能可靠,近三年参加过全国HlV抗体诊断试剂临床质量评估:敏感度、特异性、功效率均≥99.0%(须提供报告)★8、具有中国国家食品药品监督管理部门颁发的医疗器械注册证;注:★为必须达到三、人类免疫缺陷病毒I型尿液抗体检测试剂盒(胶体金法).产品要求:经国家药品监督管理局批准,获得产品注册证;派2.预期用途:可用于体外定性检测人尿液样本中的HIV-1抗体,可用于消费者自检。
(以注册证具体描述为准)3.检测原理:间接法。
▲4.检测方法:胶体金法;5.储存条件及有效期:2・30。
C干造处保存,有效期不少于(可包含)12个月;▲6.包装规格:1人份/盒,单人份包装▲7.判定时间:15min内可以进行结果判定;(以说明书为主)8.灵敏度和特异度:均大于98%(参考说明书临床试验结果)注▲为关键条款,※为必须项。
BYD Microelectronics Co., Ltd. BF6971A Datasheet说明

BYDBF6971ABF6971AS14规格书通用MCU控制器产品概述日期:2013年01月06日目录目录 (2)BF6971AS14 (4)1、规格 (4)1.1特性 (4)1.2 应用 (4)2、介绍 (5)3、引脚 (5)3.1 引脚图 (5)3.2 引脚描述 (6)4、电气特性 (7)4.1 AC特性 (7)4.2 DC特性 (7)4.3极限参数 (7)5、GPIO端口 (8)5.1 PA端口 (8)5.2 PB端口 (8)5.3 PC端口 (9)6、寄存器 (9)6.1 特殊功能寄存器总表(SFR) (10)7、WDT (11)8、复位 (12)8.1上电、掉电复位 (12)8.2 FLASH编程复位 (12)8.3软件复位 (13)8.4看门狗定时器溢出复位 (13)8.5 PC指针溢出复位 (13)9、空闲模式 (13)10、定时器 (14)10.1 定时器0和定时器1 (14)10.2 定时器时钟控制 (17)11、中断 (18)11.1 中断源及入口地址 (18)11.2 中断SFR (19)11.3 中断响应 (21)11.4 中断优先级 (21)11.5中断采样 (22)11.6中断等待 (22)12、PWM输出模块 (22)13、UART0 (23)13.1 UART0中的SFR (24)13.2 UART0工作方式 (25)13.3 多机通信 (27)14、IIC通信 (27)14.1 IIC中的SFR (28)14.2 IIC说明 (30)15、应用电路 (34)15.1 BF6971AS14应用电路 (34)16、封装 (35)I——SOP20 (35)免责声明 (36)BF6971AS141、规格1.1特性●IO数目——17个GPIO——GPIOB支持40mA灌电流●LP(低功率)模式——低功率模式:空闲模式●通信模式——IIC(支持标准模式100K或快速模式400K)——UART●供电电压:2.7~5.5V●PWM模块:16bit PWM输出●ROM——8K FLASH●RAM——512字节SRAM●封装型号:SOP201.2 应用●白色家电2、介绍BF6971AS14最多可以支持17路GPIO 的芯片,其中8路GPIO 支持40mA 的灌电流,在驱动LED 时无需三极管等电流放大器件。
Bioanalytical Method ValidationGuidance for Indust

Guidance for Industry Bioanalytical Method ValidationU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Veterinary Medicine (CVM)May 2001BPGuidance for Industry Bioanalytical Method ValidationAdditional copies are available from:Drug Information Branch (HFD-210)Center for Drug Evaluation and Research (CDER)5600 Fishers Lane, Rockville, MD 20857 (Tel) 301-827-4573Internet at /cder/guidance/index.htmorCommunications Staff (HFV-12)Center for Veterinary Medicine (CVM)7500 Standish Place, Rockville, MD 20855 (Tel) 301–594-1755Internet at /cvmU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Veterinary Medicine (CVM)May 2001BPTable of ContentsI.INTRODUCTION (1)II.BACKGROUND (1)A.F ULL V ALIDATION (2)B.P ARTIAL V ALIDATION (2)C.C ROSS-V ALIDATION (3)III.REFERENCE STANDARD (4)IV.METHOD DEVELOPMENT: CHEMICAL ASSAY (4)A.S ELECTIVITY (4)B.A CCURACY, P RECISION, AND R ECOVERY (5)C.C ALIBRATION/S TANDARD C URVE (5)D.S TABILITY (6)E.P RINCIPLES OF B IOANALYTICAL M ETHOD V ALIDATION AND E STABLISHMENT (8)F.S PECIFIC R ECOMMENDATIONS FOR M ETHOD V ALIDATION (10)V.METHOD DEVELOPMENT: MICROBIOLOGICAL AND LIGAND-BINDING ASSAYS (11)A.S ELECTIVITY I SSUES (11)B.Q UANTIFICATION I SSUES (12)VI.APPLICATION OF VALIDATED METHOD TO ROUTINE DRUG ANALYSIS (13)A CCEPTANCE C RITERIA FOR THE R UN (15)VII.DOCUMENTATION (16)A.S UMMARY I NFORMATION (16)B.D OCUMENTATION FOR M ETHOD E STABLISHMENT (17)C.A PPLICATION TO R OUTINE D RUG A NALYSIS (17)D.O THER I NFORMATION (19)GLOSSARY (20)GUIDANCE FOR INDUSTRY1Bioanalytical Method ValidationI.INTRODUCTIONThis guidance provides assistance to sponsors of investigational new drug applications (INDs), new drug applications (NDAs), abbreviated new drug applications (ANDAs), and supplements in developing bioanalytical method validation information used in human clinical pharmacology, bioavailability (BA), and bioequivalence (BE) studies requiring pharmacokinetic (PK) evaluation. This guidance also applies to bioanalytical methods used for non-human pharmacology/toxicology studies and preclinical studies. For studies related to the veterinary drug approval process, this guidance applies only to blood and urine BA, BE, and PK studies.The information in this guidance generally applies to bioanalytical procedures such as gas chromatography (GC), high-pressure liquid chromatography (LC), combined GC and LC mass spectrometric (MS) procedures such as LC-MS, LC-MS-MS, GC-MS, and GC-MS-MS performed for the quantitative determination of drugs and/or metabolites in biological matricessuch as blood, serum, plasma, or urine. This guidance also applies to other bioanalytical methods, such as immunological and microbiological procedures, and to other biological matrices, such as tissue and skin samples.This guidance provides general recommendations for bioanalytical method validation. The recommendations can be adjusted or modified depending on the specific type of analytical method used. II.BACKGROUND1 This guidance has been prepared by the Biopharmaceutics Coordinating Committee in the Center for Drug Evaluation and Research (CDER) in cooperation with the Center for Veterinary Medicine (CVM) at the Food and Drug Administration.This guidance has been developed based on the deliberations of two workshops: (1) Analytical Methods Validation: Bioavailability, Bioequivalence, and Pharmacokinetic Studies (held on December 3B5, 19902 ) and (2) Bioanalytical Methods Validation C A Revisit With a Decade of Progress (held on January 12B14, 20003).Selective and sensitive analytical methods for the quantitative evaluation of drugs and their metabolites (analytes) are critical for the successful conduct of preclinical and/or biopharmaceutics and clinical pharmacology studies. Bioanalytical method validation includes all of the procedures that demonstrate that a particular method used for quantitative measurement of analytes in a given biological matrix, such as blood, plasma, serum, or urine, is reliable and reproducible for the intended use. The fundamental parameters for this validation include (1) accuracy, (2) precision, (3) selectivity, (4) sensitivity, (5) reproducibility, and (6) stability. Validation involves documenting, through the use of specific laboratory investigations, that the performance characteristics of the method are suitable and reliable for the intended analytical applications. The acceptability of analytical data corresponds directly to the criteria used to validate the method.Published methods of analysis are often modified to suit the requirements of the laboratory performing the assay. These modifications should be validated to ensure suitable performance of the analytical method. When changes are made to a previously validated method, the analyst should exercise judgment as to how much additional validation is needed. During the course of a typical drug development program, a defined bioanalytical method undergoes many modifications. The evolutionary changes to support specific studies and different levels of validation demonstrate the validity of an assay’s performance. Different types and levels of validation are defined and characterized as follows:A.Full Validation•Full validation is important when developing and implementing a bioanalytical method for the first time.•Full validation is important for a new drug entity.• A full validation of the revised assay is important if metabolites are added to an existing assay for quantification.B.Partial ValidationPartial validations are modifications of already validated bioanalytical methods. Partial validation can range from as little as one intra-assay accuracy and precision determination to a nearly full2 Workshop Report: Shah, V.P. et al., Pharmaceutical Research: 1992; 9:588-592.3 Workshop Report: Shah, V.P. et al., Pharmaceutical Research: 2000; 17:in press.validation. Typical bioanalytical method changes that fall into this category include, but are not limited to:•Bioanalytical method transfers between laboratories or analysts•Change in analytical methodology (e.g., change in detection systems)•Change in anticoagulant in harvesting biological fluid•Change in matrix within species (e.g., human plasma to human urine)•Change in sample processing procedures•Change in species within matrix (e.g., rat plasma to mouse plasma)•Change in relevant concentration range•Changes in instruments and/or software platforms•Limited sample volume (e.g., pediatric study)•Rare matrices•Selectivity demonstration of an analyte in the presence of concomitant medications•Selectivity demonstration of an analyte in the presence of specific metabolitesC.Cross-ValidationCross-validation is a comparison of validation parameters when two or more bioanalytical methods are used to generate data within the same study or across different studies. An example of cross-validation would be a situation where an original validated bioanalytical method serves as thereference and the revised bioanalytical method is the comparator. The comparisons should be done both ways.When sample analyses within a single study are conducted at more than one site or more than one laboratory, cross-validation with spiked matrix standards and subject samples should be conducted at each site or laboratory to establish interlaboratory reliability. Cross-validation should also be considered when data generated using different analytical techniques (e.g., LC-MS-MS vs.ELISA4) in different studies are included in a regulatory submission.All modifications should be assessed to determine the recommended degree of validation. The analytical laboratory conducting pharmacology/toxicology and other preclinical studies for regulatory submissions should adhere to FDA=s Good Laboratory Practices (GLPs)5 (21 CFR part 58) and to sound principles of quality assurance throughout the testing process. The bioanalytical method for human BA, BE, PK, and drug interaction studies must meet the criteria in 21 CFR 320.29. The analytical laboratory should have a written set of standard operating procedures (SOPs) to ensure a complete system of quality control and assurance. The SOPs should cover all aspects of analysis from the time the sample is collected and reaches the laboratory until the results of the analysis are reported. The SOPs also should include record keeping, security and chain of sample custody4 Enzyme linked immune sorbent assay5 For the Center for Veterinary Medicine, all bioequivalence studies are subject to Good Laboratory Practices.(accountability systems that ensure integrity of test articles), sample preparation, and analytical tools such as methods, reagents, equipment, instrumentation, and procedures for quality control and verification of results.The process by which a specific bioanalytical method is developed, validated, and used in routine sample analysis can be divided into (1) reference standard preparation, (2) bioanalytical method development and establishment of assay procedure, and (3) application of validated bioanalytical method to routine drug analysis and acceptance criteria for the analytical run and/or batch. These three processes are described in the following sections of this guidance.III.REFERENCE STANDARDAnalysis of drugs and their metabolites in a biological matrix is carried out using samples spiked with calibration (reference) standards and using quality control (QC) samples. The purity of the reference standard used to prepare spiked samples can affect study data. For this reason, an authenticated analytical reference standard of known identity and purity should be used to prepare solutions of known concentrations. If possible, the reference standard should be identical to the analyte. When this is not possible, an established chemical form (free base or acid, salt or ester) of known purity can be used. Three types of reference standards are usually used: (1) certified reference standards (e.g., USP compendial standards); (2) commercially supplied reference standards obtained from a reputable commercial source; and/or (3) other materials of documented purity custom-synthesized by an analytical laboratory or other noncommercial establishment. The source and lot number, expiration date, certificates of analyses when available, and/or internally or externally generated evidence of identity and purity should be furnished for each reference standard.IV.METHOD DEVELOPMENT: CHEMICAL ASSAYThe method development and establishment phase defines the chemical assay. The fundamental parameters for a bioanalytical method validation are accuracy, precision, selectivity, sensitivity, reproducibility, and stability. Measurements for each analyte in the biological matrix should be validated. In addition, the stability of the analyte in spiked samples should be determined. Typical method development and establishment for a bioanalytical method include determination of (1) selectivity, (2) accuracy, precision, recovery, (3) calibration curve, and (4) stability of analyte in spiked samples.A.SelectivitySelectivity is the ability of an analytical method to differentiate and quantify the analyte in thepresence of other components in the sample. For selectivity, analyses of blank samples of theappropriate biological matrix (plasma, urine, or other matrix) should be obtained from at leastsix sources. Each blank sample should be tested for interference, and selectivity should be ensured at the lower limit of quantification (LLOQ).Potential interfering substances in a biological matrix include endogenous matrix components, metabolites, decomposition products, and in the actual study, concomitant medication and other exogenous xenobiotics. If the method is intended to quantify more than one analyte, each analyte should be tested to ensure that there is no interference.B.Accuracy, Precision, and RecoveryThe accuracy of an analytical method describes the closeness of mean test results obtained by the method to the true value (concentration) of the analyte. Accuracy is determined by replicate analysis of samples containing known amounts of the analyte. Accuracy should be measured using a minimum of five determinations per concentration. A minimum of three concentrations in the range of expected concentrations is recommended. The mean value should be within 15% of the actual value except at LLOQ, where it should not deviate by more than 20%. The deviation of the mean from the true value serves as the measure of accuracy.The precision of an analytical method describes the closeness of individual measures of an analyte when the procedure is applied repeatedly to multiple aliquots of a single homogeneous volume of biological matrix. Precision should be measured using a minimum of five determinations per concentration. A minimum of three concentrations in the range of expected concentrations is recommended. The precision determined at each concentration level should not exceed 15% of the coefficient of variation (CV) except for the LLOQ, where it should not exceed 20% of the CV. Precision is further subdivided into within-run, intra-batch precision or repeatability, which assesses precision during a single analytical run, and between-run, inter-batch precision or repeatability, which measures precision with time, and may involve different analysts, equipment, reagents, and laboratories.The recovery of an analyte in an assay is the detector response obtained from an amount of the analyte added to and extracted from the biological matrix, compared to the detector response obtained for the true concentration of the pure authentic standard. Recovery pertains to the extraction efficiency of an analytical method within the limits of variability. Recovery of the analyte need not be 100%, but the extent of recovery of an analyte and of the internal standard should be consistent, precise, and reproducible. Recovery experiments should be performed by comparing the analytical results for extracted samples at three concentrations (low, medium, and high) with unextracted standards that represent 100% recovery.C.Calibration/Standard CurveA calibration (standard) curve is the relationship between instrument response and known concentrations of the analyte. A calibration curve should be generated for each analyte in thesample. A sufficient number of standards should be used to adequately define the relationship between concentration and response. A calibration curve should be prepared in the same biological matrix as the samples in the intended study by spiking the matrix with known concentrations of the analyte. The number of standards used in constructing a calibration curve will be a function of the anticipated range of analytical values and the nature of theanalyte/response relationship. Concentrations of standards should be chosen on the basis of the concentration range expected in a particular study. A calibration curve should consist of a blank sample (matrix sample processed without internal standard), a zero sample (matrix sample processed with internal standard), and six to eight non-zero samples covering the expected range, including LLOQ.1.Lower Limit of Quantification (LLOQ)The lowest standard on the calibration curve should be accepted as the limit ofquantification if the following conditions are met:C The analyte response at the LLOQ should be at least 5 times the responsecompared to blank response.C Analyte peak (response) should be identifiable, discrete, and reproducible witha precision of 20% and accuracy of 80-120%.2.Calibration Curve/Standard Curve/Concentration-ResponseThe simplest model that adequately describes the concentration-response relationshipshould be used. Selection of weighting and use of a complex regression equation should be justified. The following conditions should be met in developing a calibration curve:C#20% deviation of the LLOQ from nominal concentrationC#15% deviation of standards other than LLOQ from nominal concentrationAt least four out of six non-zero standards should meet the above criteria, including the LLOQ and the calibration standard at the highest concentration. Excluding thestandards should not change the model used.D.StabilityDrug stability in a biological fluid is a function of the storage conditions, the chemical properties of the drug, the matrix, and the container system. The stability of an analyte in a particular matrix and container system is relevant only to that matrix and container system and should not be extrapolated to other matrices and container systems. Stability procedures should evaluate the stability of the analytes during sample collection and handling, after long-term (frozen at theintended storage temperature) and short-term (bench top, room temperature) storage, and after going through freeze and thaw cycles and the analytical process. Conditions used in stability experiments should reflect situations likely to be encountered during actual sample handling and analysis. The procedure should also include an evaluation of analyte stability in stock solution.All stability determinations should use a set of samples prepared from a freshly made stock solution of the analyte in the appropriate analyte-free, interference-free biological matrix. Stock solutions of the analyte for stability evaluation should be prepared in an appropriate solvent at known concentrations.1.Freeze and Thaw StabilityAnalyte stability should be determined after three freeze and thaw cycles. At least three aliquots at each of the low and high concentrations should be stored at the intendedstorage temperature for 24 hours and thawed unassisted at room temperature. Whencompletely thawed, the samples should be refrozen for 12 to 24 hours under the sameconditions. The freeze–thaw cycle should be repeated two more times, then analyzedon the third cycle. If an analyte is unstable at the intended storage temperature, thestability sample should be frozen at -700C during the three freeze and thaw cycles.2.Short-Term Temperature StabilityThree aliquots of each of the low and high concentrations should be thawed at roomtemperature and kept at this temperature from 4 to 24 hours (based on the expectedduration that samples will be maintained at room temperature in the intended study) and analyzed.3.Long-Term StabilityThe storage time in a long-term stability evaluation should exceed the time between the date of first sample collection and the date of last sample analysis. Long-term stabilityshould be determined by storing at least three aliquots of each of the low and highconcentrations under the same conditions as the study samples. The volume of samples should be sufficient for analysis on three separate occasions. The concentrations of allthe stability samples should be compared to the mean of back-calculated values for the standards at the appropriate concentrations from the first day of long-term stabilitytesting.4.Stock Solution StabilityThe stability of stock solutions of drug and the internal standard should be evaluated at room temperature for at least 6 hours. If the stock solutions are refrigerated or frozenfor the relevant period, the stability should be documented. After completion of thedesired storage time, the stability should be tested by comparing the instrumentresponse with that of freshly prepared solutions.5.Post-Preparative StabilityThe stability of processed samples, including the resident time in the autosampler, should be determined. The stability of the drug and the internal standard should be assessedover the anticipated run time for the batch size in validation samples by determiningconcentrations on the basis of original calibration standards.Although the traditional approach of comparing analytical results for stored samples with those for freshly prepared samples has been referred to in this guidance, other statistical approaches based on confidence limits for evaluation of an analyte=s stability in abiological matrix can be used. SOPs should clearly describe the statistical method andrules used. Additional validation may include investigation of samples from dosedsubjects.E.Principles of Bioanalytical Method Validation and Establishment•The fundamental parameters to ensure the acceptability of the performance of a bioanalytical method validation are accuracy, precision, selectivity, sensitivity,reproducibility, and stability.• A specific, detailed description of the bioanalytical method should be written. This can be in the form of a protocol, study plan, report, and/or SOP.•Each step in the method should be investigated to determine the extent to which environmental, matrix, material, or procedural variables can affect the estimation of analyte in the matrix from the time of collection of the material up to and including the time ofanalysis.•It may be important to consider the variability of the matrix due to the physiological nature of the sample. In the case of LC-MS-MS-based procedures, appropriate steps should be taken to ensure the lack of matrix effects throughout the application of the method,especially if the nature of the matrix changes from the matrix used during method validation.• A bioanalytical method should be validated for the intended use or application. All experiments used to make claims or draw conclusions about the validity of the methodshould be presented in a report (method validation report).•Whenever possible, the same biological matrix as the matrix in the intended samples should be used for validation purposes. (For tissues of limited availability, such as bone marrow, physiologically appropriate proxy matrices can be substituted.)•The stability of the analyte (drug and/or metabolite) in the matrix during the collection process and the sample storage period should be assessed, preferably prior to sampleanalysis.•For compounds with potentially labile metabolites, the stability of analyte in matrix from dosed subjects (or species) should be confirmed.•The accuracy, precision, reproducibility, response function, and selectivity of the method for endogenous substances, metabolites, and known degradation products should beestablished for the biological matrix. For selectivity, there should be evidence that thesubstance being quantified is the intended analyte.•The concentration range over which the analyte will be determined should be defined in the bioanalytical method, based on evaluation of actual standard samples over the range,including their statistical variation. This defines the standard curve.• A sufficient number of standards should be used to adequately define the relationship between concentration and response. The relationship between response and concentration should be demonstrated to be continuous and reproducible. The number of standards used should be a function of the dynamic range and nature of the concentration-responserelationship. In many cases, six to eight concentrations (excluding blank values) can define the standard curve. More standard concentrations may be recommended for nonlinear than for linear relationships.•The ability to dilute samples originally above the upper limit of the standard curve should be demonstrated by accuracy and precision parameters in the validation.•In consideration of high throughput analyses, including but not limited to multiplexing, multicolumn, and parallel systems, sufficient QC samples should be used to ensure control of the assay. The number of QC samples to ensure proper control of the assay should be determined based on the run size. The placement of QC samples should be judiciously considered in the run.•For a bioanalytical method to be considered valid, specific acceptance criteria should be set in advance and achieved for accuracy and precision for the validation of QC samples over the range of the standards.F.Specific Recommendations for Method Validation•The matrix-based standard curve should consist of a minimum of six standard points, excluding blanks, using single or replicate samples. The standard curve should cover the entire range of expected concentrations.•Standard curve fitting is determined by applying the simplest model that adequately describes the concentration-response relationship using appropriate weighting and statistical tests for goodness of fit.•LLOQ is the lowest concentration of the standard curve that can be measured with acceptable accuracy and precision. The LLOQ should be established using at least five samples independent of standards and determining the coefficient of variation and/orappropriate confidence interval. The LLOQ should serve as the lowest concentration on the standard curve and should not be confused with the limit of detection and/or the low QC sample. The highest standard will define the upper limit of quantification (ULOQ) of an analytical method.•For validation of the bioanalytical method, accuracy and precision should be determined using a minimum of five determinations per concentration level (excluding blank samples).The mean value should be within ±15% of the theoretical value, except at LLOQ, where it should not deviate by more than ±20%. The precision around the mean value should not exceed 15% of the CV, except for LLOQ, where it should not exceed 20% of the CV.Other methods of assessing accuracy and precision that meet these limits may be equally acceptable.•The accuracy and precision with which known concentrations of analyte in biological matrix can be determined should be demonstrated. This can be accomplished by analysis ofreplicate sets of analyte samples of known concentrations C QC samples C from anequivalent biological matrix. At a minimum, three concentrations representing the entire range of the standard curve should be studied: one within 3x the lower limit of quantification (LLOQ) (low QC sample), one near the center (middle QC), and one near the upperboundary of the standard curve (high QC).•Reported method validation data and the determination of accuracy and precision should include all outliers; however, calculations of accuracy and precision excluding values that are statistically determined as outliers can also be reported.•The stability of the analyte in biological matrix at intended storage temperatures should be established. The influence of freeze-thaw cycles (a minimum of three cycles at twoconcentrations in triplicate) should be studied.•The stability of the analyte in matrix at ambient temperature should be evaluated over a time period equal to the typical sample preparation, sample handling, and analytical run times.•Reinjection reproducibility should be evaluated to determine if an analytical run could be reanalyzed in the case of instrument failure.•The specificity of the assay methodology should be established using a minimum of six independent sources of the same matrix. For hyphenated mass spectrometry-basedmethods, however, testing six independent matrices for interference may not be important.In the case of LC-MS and LC-MS-MS-based procedures, matrix effects should beinvestigated to ensure that precision, selectivity, and sensitivity will not be compromised.Method selectivity should be evaluated during method development and throughout methodvalidation and can continue throughout application of the method to actual study samples.•Acceptance/rejection criteria for spiked, matrix-based calibration standards and validation QC samples should be based on the nominal (theoretical) concentration of analytes.Specific criteria can be set up in advance and achieved for accuracy and precision over therange of the standards, if so desired.V.METHOD DEVELOPMENT: MICROBIOLOGICAL AND LIGAND-BINDING ASSAYSMany of the bioanalytical validation parameters and principles discussed above are also applicable to microbiological and ligand-binding assays. However, these assays possess some unique characteristics that should be considered during method validation.A.Selectivity IssuesAs with chromatographic methods, microbiological and ligand-binding assays should be shown to be selective for the analyte. The following recommendations for dealing with two selectivity issues should be considered:1.Interference From Substances Physiochemically Similar to the Analyte•Cross-reactivity of metabolites, concomitant medications, or endogenouscompounds should be evaluated individually and in combination with the analyteof interest.•When possible, the immunoassay should be compared with a validated reference method (such as LC-MS) using incurred samples and predetermined criteria foragreement of accuracy of immunoassay and reference method.。
74LVC1G157单片2输入多路选择器产品数据手册说明书

74LVC1G157Single 2-input multiplexerRev. 9 — 8 October 2019Product data sheet1. General descriptionThe 74LVC1G157 is a single 2-input multiplexer which select data from two data inputs (I0 and I1)under control of a common data select input (S). The state of the common data select inputdetermines the particular register from which the data comes. The output (Y) presents the selecteddata in the true (non-inverted) form.Inputs can be driven from either 3.3 V or 5 V devices. This feature allows the use of these devicesas translators in mixed 3.3 V and 5 V applications.This device is fully specified for partial power-down applications using I OFF. The I OFF circuitrydisables the output, preventing the damaging backflow current through the device when it ispowered down.Schmitt-trigger action at all inputs makes the circuit highly tolerant to slower input rise andfall times.2. Features and benefits•Wide supply voltage range from 1.65 V to 5.5 V•High noise immunity•Complies with JEDEC standard:•JESD8-7 (1.65 V to 1.95 V)•JESD8-5 (2.3 V to 2.7 V)•JESD8B/JESD36 (2.7 V to 3.6 V)•±24 mA output drive (V CC = 3.0 V)•CMOS low power consumption•Latch-up performance exceeds 250 mA•Direct interface with TTL levels•Inputs accept voltages up to 5 V•ESD protection:•HBM JESD22-A114F exceeds 2000 V•MM JESD22-A115-A exceeds 200 V•Multiple package options•Specified from -40 °C to +85 °C and -40 °C to +125 °C3. Ordering information4. Marking[1]The pin 1 indicator is located on the lower left corner of the device, below the marking code.5. Functional diagram6. Pinning information6.1. Pinning74LVC1G157I1SGND I0Y001aac6561236V CC54Fig. 5.Pin configuration SOT363 (SC-88) and SOT457 (SC-74)74LVC1G157GND 001aac657I1I0V CCSYT ransparent top view231546Fig. 6.Pin configuration SOT886 (XSON6)74LVC1G157GND 001aaf545I1I0V CC S YT ransparent top view231546Fig. 7.Pin configurationSOT891, SOT1115 and SOT1202 (XSON6)6.2. Pin description7. Functional descriptionTable 4. Function tableH = HIGH voltage level; L = LOW voltage level; X = don’t care.8. Limiting valuesTable 5. Limiting valuesIn accordance with the Absolute Maximum Rating System (IEC 60134). Voltages are referenced to GND (ground = 0 V).[1]The input and output voltage ratings may be exceeded if the input and output current ratings are observed.[2]For SOT363 (TSSOP6) packages: P tot derates linearly with 3.7 mW/K above 83 °C.For SOT457 (TSOP6) packages: P tot derates linearly with 4.1 mW/K above 89 °C.For SOT886 (XSON6) packages: P tot derates linearly with 3.3 mW/K above 74 °C.For SOT891 (XSON6) packages: P tot derates linearly with 3.3 mW/K above 74 °C.For SOT1115 (XSON6) packages: P tot derates linearly with 3.2 mW/K above 71 °C.For SOT1202 (XSON6) packages: P tot derates linearly with 3.3 mW/K above 74 °C.9. Recommended operating conditions10. Static characteristicsTable 7. Static characteristicsAt recommended operating conditions. Voltages are referenced to GND (ground = 0 V).[1]All typical values are measured at T amb = 25 °C.11. Dynamic characteristicsTable 8. Dynamic characteristicsVoltages are referenced to GND (ground = 0 V); for test circuit see Fig. 9.[1]Typical values are measured at T amb = 25 °C and V CC = 1.8 V, 2.5 V, 2.7 V, 3.3 V and 5.0 V respectively.[2]t pd is the same as t PLH and t PHL.[3]C PD is used to determine the dynamic power dissipation (P D in μW).P D = C PD x V CC2 x f i x N + Σ(C L x V CC2 x f o) where:f i = input frequency in MHz;f o = output frequency in MHz;C L = output load capacitance in pF;V CC = supply voltage in Volts;N = number of inputs switching;Σ(C L x V CC2 x f o) = sum of the outputs.11.1. Waveforms and test circuit12. Package outline13. Abbreviations14. Revision history15. Legal informationData sheet status[1]Please consult the most recently issued document before initiating orcompleting a design.[2]The term 'short data sheet' is explained in section "Definitions".[3]The product status of device(s) described in this document may havechanged since this document was published and may differ in case ofmultiple devices. The latest product status information is available onthe internet at https://.DefinitionsDraft — The document is a draft version only. The content is still under internal review and subject to formal approval, which may result in modifications or additions. Nexperia does not give any representations or warranties as to the accuracy or completeness of information included herein and shall have no liability for the consequences of use of such information. Short data sheet — A short data sheet is an extract from a full data sheet with the same product type number(s) and title. A short data sheet is intended for quick reference only and should not be relied upon to contain detailed and full information. For detailed and full information see the relevant full data sheet, which is available on request via the local Nexperia sales office. In case of any inconsistency or conflict with the short data sheet, the full data sheet shall prevail.Product specification — The information and data provided in a Product data sheet shall define the specification of the product as agreed between Nexperia and its customer, unless Nexperia and customer have explicitly agreed otherwise in writing. In no event however, shall an agreement be valid in which the Nexperia product is deemed to offer functions and qualities beyond those described in the Product data sheet.DisclaimersLimited warranty and liability — Information in this document is believedto be accurate and reliable. However, Nexperia does not give any representations or warranties, expressed or implied, as to the accuracyor completeness of such information and shall have no liability for the consequences of use of such information. Nexperia takes no responsibility for the content in this document if provided by an information source outside of Nexperia.In no event shall Nexperia be liable for any indirect, incidental, punitive, special or consequential damages (including - without limitation - lost profits, lost savings, business interruption, costs related to the removalor replacement of any products or rework charges) whether or not such damages are based on tort (including negligence), warranty, breach of contract or any other legal theory.Notwithstanding any damages that customer might incur for any reason whatsoever, Nexperia’s aggregate and cumulative liability towards customer for the products described herein shall be limited in accordance with the Terms and conditions of commercial sale of Nexperia.Right to make changes — Nexperia reserves the right to make changesto information published in this document, including without limitation specifications and product descriptions, at any time and without notice. This document supersedes and replaces all information supplied prior to the publication hereof.Suitability for use — Nexperia products are not designed, authorized or warranted to be suitable for use in life support, life-critical or safety-critical systems or equipment, nor in applications where failure or malfunctionof an Nexperia product can reasonably be expected to result in personal injury, death or severe property or environmental damage. Nexperia and its suppliers accept no liability for inclusion and/or use of Nexperia products in such equipment or applications and therefore such inclusion and/or use is at the customer’s own risk.Quick reference data — The Quick reference data is an extract of the product data given in the Limiting values and Characteristics sections of this document, and as such is not complete, exhaustive or legally binding. Applications — Applications that are described herein for any of these products are for illustrative purposes only. Nexperia makes no representation or warranty that such applications will be suitable for the specified use without further testing or modification.Customers are responsible for the design and operation of their applications and products using Nexperia products, and Nexperia accepts no liability for any assistance with applications or customer product design. It is customer’s sole responsibility to determine whether the Nexperia product is suitableand fit for the customer’s applications and products planned, as well asfor the planned application and use of customer’s third party customer(s). Customers should provide appropriate design and operating safeguards to minimize the risks associated with their applications and products. Nexperia does not accept any liability related to any default, damage, costs or problem which is based on any weakness or default in the customer’s applications or products, or the application or use by customer’s third party customer(s). Customer is responsible for doing all necessary testing for the customer’s applications and products using Nexperia products in order to avoid a default of the applications and the products or of the application or use by customer’s third party customer(s). Nexperia does not accept any liability in this respect.Limiting values — Stress above one or more limiting values (as defined in the Absolute Maximum Ratings System of IEC 60134) will cause permanent damage to the device. Limiting values are stress ratings only and (proper) operation of the device at these or any other conditions above thosegiven in the Recommended operating conditions section (if present) or the Characteristics sections of this document is not warranted. Constant or repeated exposure to limiting values will permanently and irreversibly affect the quality and reliability of the device.Terms and conditions of commercial sale — Nexperia products aresold subject to the general terms and conditions of commercial sale, as published at /profile/terms, unless otherwise agreed in a valid written individual agreement. In case an individual agreement is concluded only the terms and conditions of the respective agreement shall apply. Nexperia hereby expressly objects to applying the customer’s general terms and conditions with regard to the purchase of Nexperia products by customer.No offer to sell or license — Nothing in this document may be interpreted or construed as an offer to sell products that is open for acceptance or the grant, conveyance or implication of any license under any copyrights, patents or other industrial or intellectual property rights.Export control — This document as well as the item(s) described herein may be subject to export control regulations. Export might require a prior authorization from competent authorities.Non-automotive qualified products — Unless this data sheet expressly states that this specific Nexperia product is automotive qualified, the product is not suitable for automotive use. It is neither qualified nor tested in accordance with automotive testing or application requirements. Nexperia accepts no liability for inclusion and/or use of non-automotive qualified products in automotive equipment or applications.In the event that customer uses the product for design-in and use in automotive applications to automotive specifications and standards, customer (a) shall use the product without Nexperia’s warranty of the product for such automotive applications, use and specifications, and (b) whenever customer uses the product for automotive applications beyond Nexperia’s specifications such use shall be solely at customer’s own risk, and (c) customer fully indemnifies Nexperia for any liability, damages or failed product claims resulting from customer design and use of the product for automotive applications beyond Nexperia’s standard warranty and Nexperia’s product specifications.Translations — A non-English (translated) version of a document is for reference only. The English version shall prevail in case of any discrepancy between the translated and English versions.TrademarksNotice: All referenced brands, product names, service names and trademarks are the property of their respective owners.Contents1. General description (1)2. Features and benefits (1)3. Ordering information (2)4. Marking (2)5. Functional diagram (2)6. Pinning information (3)6.1. Pinning (3)6.2. Pin description (3)7. Functional description (3)8. Limiting values (4)9. Recommended operating conditions (4)10. Static characteristics (5)11. Dynamic characteristics (6)11.1. Waveforms and test circuit (7)12. Package outline (8)13. Abbreviations (14)14. Revision history (14)15. Legal information (15)© Nexperia B.V. 2019. All rights reservedFor more information, please visit: Forsalesofficeaddresses,pleasesendanemailto:*************************** Date of release: 8 October 2019Mouser ElectronicsAuthorized DistributorClick to View Pricing, Inventory, Delivery & Lifecycle Information:N experia:74LVC1G157GS,13274LVC1G157GN,13274LVC1G157GF,13274LVC1G157GM,11574LVC1G157GM,132 74LVC1G157GV,12574LVC1G157GW,125。
凯杰HIV说明书

检测限度 经采用 CLB(荷兰血液传输中心实验室)HIV RNA monitor (Cat.No. S2039,标定 HIV RNA 载量为 25,000 copies/ml)测试,灵敏度达到 5.0×102 IU/ml。
【参考值(参考范围)】
1. 结果分析条件设定 阈值(threshold)设定原则以阈值线刚好超过正常阴性对照曲线(无规
则的噪音线)的最高点为准。 基线(baseline)应选择该次实验指数扩增前所有样本荧光信号比
较稳定的一段区域(所有样本荧光信号无较大波动),起始点(循环 数)应避开荧光采集起始阶段的信号波动,终止点(循环数)应比 最早出现指数扩增的样本 Ct 值减少 1-2 个循环。 【注】具体设置方法请参照各仪器使用说明书。 2. 质控标准 2.1 将校准品 1-4 的浓度值输入,仪器将自动以定量标准品的浓度值为 横坐标,以其实际测得的外标 Ct 值为纵坐标给出标准曲线,标准曲 线的拟和度的绝对值应大于等于 0.980,否则视为定量结果无效。 2.2 阴性对照 HIV RNA 应为 0.0IU/ml(Ct 值应无数值显示); 强阳性对照 Ct 值≤25.0; 强阳性对照 Ct 值≤临界阳性对照 Ct 值≤30.0; 否则此次实验视为无效。所有实验从样本处理开始重做。
95oC:5sec, 60oC:30sec,40 个循环。
反应体系为 50μl (Rotor-Gene Q 为 25μl)。
使用 iCycler 荧光 PCR 检测仪时循环程序设置如下:
42oC:30 min;95oC:3min; 95oC:10sec, 55oC:30sec,72oC:60sec,5 个循环; 95oC:5sec, 60oC:30sec,42 个循环。 反应体系为 50μl。 使用 LC1.2/2.0 荧光 PCR 检测仪时循环程序设置如下: 42oC:30min;92oC:1min; 92oC:10sec, 52oC:20sec,72oC:30sec,5 个循环; 92oC:5sec, 60oC:30sec,40 个循环。 反应体系为 25μl。 4.2. 仪器检测通道选择 多荧光检测仪荧光信号收集时设定为 Fam 荧光素。荧光信号收集设 在 60 oC。 LC1.2/2.0 荧光 PCR 检测仪,仪器通道选择 F1 通道。荧光信号收 集设在 60 oC,收集方式设为 SINGLE。 其它荧光 PCR 检测仪的具体循环程序设置方法请参照以上设置方法 及各类仪器使用说明书和实际情况而适当调整。 4.3. 样本的设定: 在扩增开始前条件设定时,HIV-1 校准品设为“Standard”并输入 试剂盒内给定浓度值;待检样本和对照品设定为“Unknown”或 “Sample”。