第十一章-基于靶点结构的药物分子设计PPT课件
计算机辅助药物设计ppt课件

3D-QSAR的评价
• 3D-QSAR从微观即从分子和原子的水平上揭示 了药物分子与受体相互作用的空间特征和在空 间结合的理化本质。 • 2D-QSAR则从药效学(包括药代动力学)的宏 观作用上考察构效关系,可作为预测同源物的 生物活性,是优化设计的工具。 • 2D-QSAR可以大批量地处理多达一二百个化合 物的数据组,但3D-QSAR目前难以办到。
14
3. 药物的化学结构与生物活性的关系 (SAR)
构效关系:药物的化学结构与生理活性的 关系。 目的:获得药物的生理活性与其结构间依 赖关系的规律,以便认识、解析药物的 作用方式和作用机理,预测某一化合物 的生物活性。
17
基于受体分子的组成、性状及空间结构还未 深入了解,对药物进行构效关系研究可间接 地阐明药物的作用机理。 研究内容:药效、药动及毒性的构效关系。 得到药物的药效基团、药动基团和毒性基团 高效、低毒、安全
O Cl
(4). 胆酸
• 肝细胞中含有胆酸系统,对胆酸有较强 亲和力。药物与胆酸偶联后,常具肝细 胞靶向特征。
OH COOH
O 苯丁氮芥 格仑伐他汀 N H
29
(CH2)n
O
OH
(5). 改变药动团的方法
a.改变亲脂性。例:增加烷基链长度,提高整个 分子的亲脂性。
b.改变电性。例:苯环对位引入氟原子,减低苯 环在体内被代谢羟基化的速率和程度。 c.改变立体性。例:引入体积较大基团,以保护 代谢敏感或易受攻击的基团。
42
2).Free-Wilson模型
• 它不需要Hansch方法那些理化参数,只需要将 药物的化学结构进行组合。该模型假定分子的
活性是母体化合物与取代基活性贡献之和,不
论其他位置取代基变换与否。 • 常用于同源系列化合物的设计。但结构参数缺 乏物理意义,难以指导新化合物设计而受到限 制。
药物设计的基本原理和方法 PPT

先导化合物发现的方法和途径
以现有的药物作为先导化合物
1.由药物副作用发现先导化合物 基于抗结核药异烟肼的副作用,发展了单胺氧化酶抑制剂类抗抑郁
药,如异丙烟肼。
二、新药设计与开发
Drug design and development
先导化合物发现的方法和途径
二、新药设计与开发
Drug design and developmentΒιβλιοθήκη 先导化合物发现的方法和途径
利用计算机进行靶向筛选得到先导化合物
以生物靶点为基础,利用计算机软件对化合物进行靶向合理筛选和从 头设计已成为发现先导化合物的一个重要手段。
二、新药设计与开发
Drug design and development
二、新药设计与开发
Drug design and development
先导化合物发现的方法和途径
用活性内源物质作为先导化合物
根据对生理病理的了解来研究新药,通常是针对与该生理活动有关的 酶或受体来设计药物,被称为合理药物设计。内源性神经递质、受体或酶 的底物就是初始的先导化合物。例如以炎症介质5-羟色胺为先导化合物研 发了抗炎药吲哚美辛。
高通量筛选(High-throughput screening)是以随机筛选和广泛筛选 为基础的。高通量筛选是利用近二、三十年来生物化学、分子生物学、分 子药理学和生物技术的研究成果,将已阐明影响生命过程的一些环节的酶、 受体、离子通道等作为药物作用的靶标进行分离、纯化和鉴定,由此建立 分子、细胞水平的高特异性的体外筛选模型,具有灵敏度高、特异性强、 需用药量少、快速筛选的特点。
二、新药设计与开发
Drug design and development
107863-药物设计-03基于靶点的药物设计

O
N
O
H3C O
HN
Cl
N
N
吉非替尼
口服有效
疗效比较
progression free survival (PFS)
H3C CH3
H3 C
O
HO O CH
O NH O H3 C
3
CH3 OH
O OH
CH3
H
O
HO AcO
多西他赛 BzO
O 双联疗法
H3N O
Pt
H3 N O
卡铂 O
美国政府的笑柄
• 1971年,美国总统尼克松:美国政府将 “向癌症开战”,并预期10年内将可找到 “完全治愈”癌症的治疗方法。
胃酸中和与胃粘膜保护
中和胃酸
氢氧化铝
主要有效成分之一
胃粘膜保护 硫糖铝
不溶于水,不被吸收;酸性 条件下解离出硫酸蔗糖复合 离子,形成不溶性带负电荷 的胶体,与溃疡处带正电荷 的蛋白质渗出物结合,形成 保护膜,促进溃疡愈合
RO O RO
OR
O OR
OR
O
OR
OR
OR
R=SO3[Al2(OH)5]
枸橼酸铋钾 易溶于水,在溃疡表面形成坚
本科生《药物设计学》专业课
基于靶点的药物设计
Target Based Drug Design
引子:一个百年老药的诞生
H3C
CO
OH
OH
O
苯酚
Y1842 动物组织与木材防腐 Y1862 创口消毒,强腐蚀性
柳树树皮提取物
COOH
水杨酸
Y1860 柯伯尔合成法 杀菌剂,腐蚀性降低
氧化 苷键断裂
OH
CH2OH O O HO
最新基于配体的药物和设计主题讲座课件
量体裁衣
量衣裁衣
基于配体的药物设计:在生物靶点未知的情况下, 通过研究与靶点具有特异性结合配体的结构性信息, 发现先导化合物的方法。
基于配体的药物设计流程
定量构效关系 QSAR
基于配体的药物 设计
5.用三维等势线系数图显示QSAR方程,体现结构和活性关系。
图中各取代基性质及方位变化对活性的影响用不同颜色表示。用户可进 一步设计新的化合物,并预测其活性。
国外应用CADD成功的实例
国内药物设计成功的实例
比较分子相似因子分析法(CoMSIA)
氢键
CoMSIA作为SYBBYL一个模板已经实现商业化,预计今后 的应用将更为广泛。
QSAR与药效团比较
共同点:
两者均研究具有同类活性的一系列化合物与靶点相互作用,并认定 其活性部位是一致的。
不同点:
定量构效关系QSAR
药效团
研究的是基于同一母核(或骨架)
的系列化合物,侧重于对先导化合 物的优化
研究不同结构类型的多种先导化合 物的构效关系,更体现了活性配体 分子的抽象特征。涵盖了设计新的 配体分子所需要的三维结构信息, 为我们发现先导化合物新结构类型 特供有效途径。
Hansch模型的提出标志着药物定量构效关系研究的开始, 也被认为从盲目药物设计过渡到合理药物设计的重要标志。
Hansch模型揭开了经典QSAR研究的篇章,成为QSAR发 展历史中的里程碑。
表示化合物上取代基对化学反应的影响。
指示变量
指示变量:常用于线性自由性相关分析中描述某些不能用连 续性变量说明的某种特征。
在Hansch方法的指导下,人们成功地设计了诺氟沙星等喹 诺酮类抗菌药。
《分子分析》课件

利用生物信息学的方法和技术,对分子数据 进行处理、分析和挖掘,揭示生命活动的规 律和机制。
生物信息学与分子分析的 交叉融合
通过生物信息学的手段,对分子数据进行深 入挖掘和多维度分析,为疾病诊断、药物研
发和个性化医疗提供有力支持。
纳米技术与分子分析的结合
纳米技术在分子分析中的应用
02
分子分析的基本原理
分子结构与性质
分子结构
分子中的原子通过化学键相互连接,形成特定的空间排列,决定 了分子的性质。
分子性质
分子的性质由其化学键和分子构型决定,包括稳定性、反应活性、 物理性质和化学性质等。
分子结构与性质的关系
了解分子的结构有助于预测其性质,从而为分子设计和合成提供指 导。
分子光谱分析
基因组学与蛋白质组学研究
总结词
分子分析技术是基因组学和蛋白质组学研究的重要手段,有助于深入了解生物体的生命 活动和疾病发生机制。
详细描述
基因组学和蛋白质组学研究需要大规模地分析基因和蛋白质的表达、结构和功能。分子 分析技术如高通量测序、质谱分析等,能够快速、准确地获取这些信息,为生物医学研
究提供有力支持。
05
分子分析的未来发展
高通量与高灵敏度分析技术
高通量分析技术
通过自动化和高效率的检测系统,实现 大规模样本的同时检测和分析,提高分 析速度和效率。
VS
高灵敏度分析技术
利用新型的信号放大技术和高灵敏度检测 器,实现对低浓度样本的精准检测,有助 于发现早期病变和微量污染。
生物信息学与分子分析的结合
利用纳米材料的独特性质和功能,开发新型的分子检 测技术和器件,提高检测的灵敏度和特异性。
纳米技术与分子分析的交叉融合
药物作用靶点(课堂PPT)

17
2. 上市药物靶点的类别
• 生物化学分类: 上市小分子和生化药靶点, 50%以上属4 个关键生化类别: G-蛋白偶联受体(26.8%),核受体(13%),配体门控性离子通道(7.9%)及电 压门控离子通道(5.5%) 。其他包括青霉素结合蛋白,髓过氧化酶,神经递 质,DNA拓扑异构酶,纤维结合蛋白,细胞色素P450等。
试剂特异结合并产生临床疗效的分子结构。不包括药理学和生物化学研究工 具以及相关技术。得出的结论:目前的分子药物靶点数目为218个。 • Overington 等(2006)提出有价值的评估: 药物数量减至1357种。所有药物通 过324个靶标起作用,其中266个为人类基因衍生的,其余为病原体靶标。 • 小分子药物作用于248种蛋白,其中207个靶标由人体基因组编码;口服小分子 药物靶标227个,其中186个为人类基因靶标。
14
药物研发需要生物学,化学,电脑 物理,仪器分析等多个领域合作
15
五. 靶点药物开发概况
• 现代药物开发是基于特定靶寻找先导化合物,进而筛选候选药物的过程。 • 人类基因组研究结果预测: 有600种小分子靶点,1800多蛋白质靶点,及
2100种基因治疗和siRNA治疗靶点。 • 失败教训: 近20年对高选择性靶点的集中研发,候选新药经临床试验后
ad靶向治疗药物38葡萄糖苷酶小肠内水解糖苷键生成单糖葡萄糖激酶gk醛糖还原酶ar二肽肽激酶dpp蛋白激酶cpkc蛋白酪氨酸激酶ptk和蛋白酪氨酸磷酸酶ptp一氧化氮合酶nos血管紧张素转换酶ace肉碱酯酰转移酶cpt糖代谢酶蛋白磷酸化酶39天然靶点药物研究模式已知确立大量提取物的筛选几百种可组合活性比较找出作用强的几中细胞组织动物实验可委托临床试验酶活性测定40天然药物提取分离与制备提取分离新方法
药物分子设计

药物分子设计近些年来各种各样的新型疾病依次出现。
因此寻找可以治愈这些疾病的药物对人们来说至关重要。
随着分子生物学和药物化学的发展药物设计进入了理性阶段其中药物分子设计是目前新药发现的主要方向。
它是依据生物化学、酶学、分子生物学以及遗传学等生命科学的研究成果针对这些基础研究中所揭示的包括酶、受体、离子通道及核酸等潜在的药物设计靶点并参考其它类源性配体或天然产物的化学结构特征设计出合理的药物分子[1]。
本文主要就药物分子设计研究策略中的选择性与基于受体-配体之间的结合力作进一步详细介绍。
药物设计开始由定性进入定量研究阶段为定量药物设计奠定理论和实践基础[2]。
药物设计逐渐形成一门独立的分支学科。
70年代以后药物设计开始综合运用药物化学、分子生物学、量子化学、统计数学基础理论和当代科学技术以及电子计算机等手段开辟了药物设计新局面。
随着分子生物学的进展对酶与受体的理解更趋深入对有些酶的性质、酶反应历程、药物与酶复合物的精细结构得到阐明模拟与受体相结合的药物活性构象的计算机分子图像技术在新药研究中已取得可喜的成果。
运用这些新技术从生化和受体两方面进行药物设计是新药设计的趋向[3]。
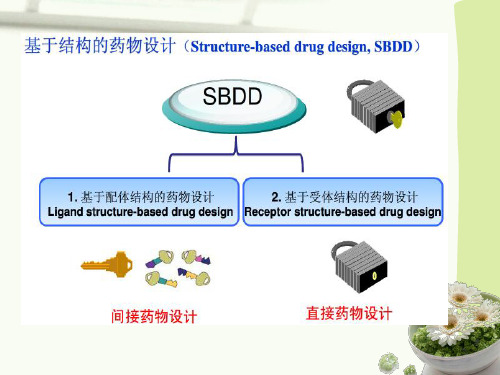
计算机辅助药物分子设计大致可分为直接药物设计(基于结构的药物设计)、间接药物设计(基于受体的药物设计)和基于结构的药物设计与组合技术相结合的策略[4]。
直接法是从已知受体的三维结构设计配体或药物分子,前提条件是受体的三维立体结构已知[5]。
间接法则是从一系列作用于同一受体并显效的药物分子中找出共同的基本结构,根据结构的相似性与性质的相似性之间的关系,推导出新的先导化合物。
间接法的优点是绕开了确定受体立体结构这一难点。
直接药物设计方法设计过程大体分为三步:分析受体的构象,确定受体的活性区域,在活性区域搜索可能的结合位点;寻找与受体结合位点相匹配的配体分子,得到候选化合物;最后是对候选化合物进行评价,有人形象地称之为对分子打分[6]。
生物技术药物

汇报人: 202X-01-05
• 生物技术药物概述 • 生物技术药物的种类 • 生物技术药物的研发与应用 • 生物技术药物的法规与监管 • 生物技术药物的未来发展 • 案例研究:生物技术药物的研发与
应用
01
生物技术药物概述
定义与分类
定义
生物技术药物是指利用生物技术生产 的药物,包括基因工程药物、抗体药 物、细胞治疗药物等。
法,为治疗癌症、遗传性疾病等提供更多有效手段。
基因编辑技术的药物研发应用
基因治疗
利用基因编辑技术修复或替换缺陷基因,治疗遗传性疾病和罕见 病。
细胞免疫治疗
通过基因编辑技术改造免疫细胞,增强其抗肿瘤能力,为癌症治 疗提供新的策略。
疫苗研发
利用基因编辑技术设计和优化新型疫苗,提高疫苗的免疫效果和 安全性。
基因治疗药物
总结词
基因治疗药物是通过将正常基因导入病变细胞,纠正或补偿 缺陷基因引起的疾病。
详细描述
基因治疗药物在罕见病、遗传性疾病等领域取得显著成果, 如用于治疗囊性纤维化的基因疗法。
细胞治疗药物
总结词
细胞治疗药物是通过体外培养或修饰 人体细胞来治疗疾病,包括干细胞治 疗和免疫细胞治疗等。
详细描述
。
案例二
要点一
总结词
基因治疗药物为罕见病提供了全新的治疗策略,但仍面临 技术挑战和伦理问题。
要点二
详细描述
基因治疗是通过向患者体内导入功能正常的基因,纠正或 补偿缺陷基因引起的疾病。近年来,基因治疗药物在罕见 病治疗中取得了重要进展,如囊性纤维化、血友病等。这 些药物通过基因工程技术,将正常基因导入患者体内,实 现了对疾病的根治。然而,基因治疗药物仍面临技术挑战 和伦理问题,如基因编辑的安全性和伦理审查等。
药物分子设计

高含量紫杉醇的红豆杉适宜的生态环境:
①寒冷地区:冬天-20℃左右,夏天20℃以下; ②林中蔽阴散生,或阴坡上避免阳光直射; ②富含有机质的酸性土壤,5<pH<7
红豆杉中紫杉醇的提取
①溶剂萃取:乙酸乙酯-丙酮(1;1); ②色谱技术分离; ③膜分离; ④离子交换树脂
紫杉醇分子中含有6-8-6-4环系,含氧四元环对维持活 性有重要作用,是个必需的药效团。
先导化合物(1ead compound),或称原形物 (prototype):具有某种生物活性的结构,但未必 是可实用的药物。
先导化合物可以是天然生物活性物质,或已 知活性的药物或化合物结构中,提取出决定 生物活性的部分结构。
一、先导化合物的产生
1、天然生物活性物质——次级代谢产物
其产生的原因尚不清楚,但比较普遍的认识是,次级代 谢产物是生物体为保护自己和繁衍物种而产生的防御性 或引诱性的物质。
moleculardrugdesign药物分子设计由多学科相互穿插交替进行药物设计学分子生物学结构生物学分子生物学结构生物学基因组生物信息学基因组生物信息学数学统计学数学统计学药物化学有机药物化学药物化学有机药物化学计算机科学计算化学计算机科学计算化学分子药理学一般药理学分子药理学一般药理学研究与开发新药的化学过程
Desipramine
推测:其它抗抑郁药的侧链和胺基N-去甲基化,是否也活化?
N 阿米替林
N H 去甲替林
5、幸运发现先导化合物
例1:青霉素的发现 例2:苯并二氮卓的发现 苯并二氮卓(Librium)是最早的安定药,也是偶然发现的。
例3:顺铂Cisplastin的发现
H2N H2N
Cl Pt
Cl
二、先导化合物的优化
• 剖裂(dissection) • 拼合(association) • 局部修饰(local manipulation)
靶向药物PPT课件

1. 单克隆抗体药物
西妥昔单抗(Cetuximab)(德国默克雪兰诺生产 爱必妥) 和帕尼单抗(Panitumumab)都是被 FDA 批准应用的抗 EGFR 单克隆抗体药物。
这两种单克隆抗体都能直接封闭 EGFR 的胞外配基结合 位点:Cetuximab(嵌合抗体)于 2004 年被 FDA 批准用于 晚期结肠癌的治疗,临床试验显示,Cetuximab 虽然不能延 长患者的存活期,但可以减小某些患者体内的肿瘤体积并减 缓肿瘤的生长速度,同时也能有效治疗头颈鳞状细胞癌和非 小细胞肺癌(NSCLC),但 Cetuximab 属于人鼠嵌合抗体, 反复用药会产生人抗鼠抗体反应(HAMA),具有局限性。
肿瘤分子靶向治疗药物分类
早期的靶向治疗主要是利用抗体直接封闭肿瘤 细胞膜表面相关抗原,如 CD20、CD33、CD52 等, 但这些抗原在正常淋巴细胞中也有表达,所以抗 CD20 抗体等药物会严重影响机体的免疫功能。
针对实体肿瘤治疗的分子靶点分为: 一、抗血管内皮素生长因子(VEGF) 二、抗人表皮生长因子受体 2(HER-2)/neu 三、抗表皮生长因子受体(EGFR)
埃罗替尼(Erlotinib)是另一种有效、可逆的选择性 HER-1/EGFR TKI,于 2004 年获得 FDA 批准上市,应用 于晚期或转移性 NSCLC 的治疗。临床研究表明该药在难治 性晚期 NSCLC 的治疗中具有较高价值,最常见的不良反应 有皮疹(75%)和腹泻(56%),其中最严重的不良反应是 间质性肺病(ILD),甚至可危及生命,一旦确诊为 ILD, 应立刻中断治疗并采取相应的治疗措施。
一、抗表皮生长因子受体(EGFR)治疗药物
EGFR 在不同类型的肿瘤组织中都呈异常高表达,是肿 瘤分子靶向治疗的一个理想靶点。EGFR 是相对分子质量为 170 000 的细胞膜糖蛋白,含有一个胞外配基结合位点、一 个跨膜亲脂部分和一个胞内蛋白酶结合位点。当信号分子与 胞外配基结合后,EGFR 出现二聚体现象,使配基的亲和力 增强,激活内部酪氨酸激酶活性,诱导酪氨酸磷酸化,导致 一系列生化和生理反应,促使肿瘤细胞增殖分裂和永生化。 目前,已有多个针对 EGFR 的靶向治疗药物被批准上市并应 用于临床。
第四章-基于结构的药物设计PPT优质课件

肾上腺素能激动α- 和β- 肾上腺素受体 而异丙肾上腺素仅能激.动β-肾上腺素受体
立体因素的影响
药物多种对应异构体中只有一个能与受体发生 特异性结合。
β-OH的立体结构对活性影响显著,β-碳为R构型的肾上 腺素左旋体是β-碳为S构型的右旋体的12倍。
.
一个对映体完全占据受体结合部位
.
另一个对映体仅能部分匹配
: 药效构象 药物分子与受体结合时所采取的
实际构象,并不一定采取它的优势构象,实际构 象为药效构象。
.
药效构象(pharmacophoric conformation)
.
1.3 基于结构的药物设计
间接药物设计
.
直接药物设计
第二节 计算机辅助药物设计 Computer-Aided Drug Design
力场总能量,即分子总能量= 键合作用+ 非键作用
.
键合能的构成:
.
力场的总能量,即分子总能量Etotal: Etotal = Ec + Eb + Et + Ev + Eh + Ee + Ed
Ec 键的伸缩能(compression energy) Eb 键角的弯曲能(bending energy) Et 键的二面角扭转能(torsional energy) Ev 范德华作用能(van de Waals energy) Eh 氢键作用能( hydrogen bonding energy) Ee 静电作用能(electrostatic energy) Ed 偶极作用能(dipole energy)
量子力学——研究微观粒子(电子、原子、分 子)运动规律的理论。它用波函数描写粒子的 运动状态,以薜定谔方程确定波函数的变化规 律并对各物理量进行计算。
农药分子设计PPT课件

N H O CH3
咪唑喹啉酸
2、类同合成法
类同合成也称为衍生合成,这种方法是从某个已经开 发的新药分子结构出发,进行结构变化和修饰,谋求开发 出同一系列的衍生物,或以该化合物为先导,衍生出二级 先导化合物,从而开发出结构不同的新品种。有跟着走的 意思,也称为me too。
有关应用这一方法开发新药的例子很多,几乎每出现 一类新型结构的光导化合物,世界各地都有很多药物研究 者采用类同合成法来发现新的药物,而且基本都能找到不 同特点的药物。例如,超高效磺酰脲类除草剂的开发研究 过程就充分体现了这一方法特点。自从杜邦公司报道了第 一个超高效磺酰脲类除草剂氯磺隆以来,经过近20年的 发展,世界各国农药研究部门已经合成了数十万个磺酰脲 类化合物,并开发出数十个各具特点的新型除草剂品种。
图17-4
其分子中的酯基水解后形成相应的酸再一次显示出较好 的除草活性,从而引起了以化合物6为先导化合物的研究,最 终开发出了新型的咪唑啉酮类超高效ALS抑制剂。
CH3OCH2
O
OH
N
N
CH3 CH3
N H
O CH3
甲氧咪草烟
H3C
H O CH3
咪唑乙烟酸
O
OH
N
N CH3 CH3
总之,该方法最大的优点是方向明确,有一个样板, 可以少走弯路,省力省钱,缺点是难以脱开别人专利的保 护范围。这一方法也是我国发现新药用得最多、最快、最 省钱的途径。当然,该法也是世界各国采用较多的开发新 药的方法。较高层次地进行类同合成则是对巳开发的分子 结构进行较大改变,包括骨架改变,以谋求二次先导化合 物,进而开发出新型化学结构的品种,如采用生物等排原 理。
图17-5 人们对这一先导进行了结构与活性关系的研究,并相继开 发出系列沙蚕毒素类杀虫剂,如杀虫双、杀虫环都是很好的水 稻田杀虫剂,它们的化学结构见图17-6。
新型药物靶点的分子设计与合成

新型药物靶点的分子设计与合成随着现代科学技术的不断进步,药物研发领域也迎来了新的机遇和挑战。
在药物研发过程中,分子设计与合成是一个关键的环节,它直接影响到药物的疗效和安全性。
本文将对新型药物靶点的分子设计与合成进行探讨,以期提高药物的治疗效果,为疾病的治疗带来新希望。
一、药物靶点研究的意义药物靶点是指在疾病发生过程中起关键作用的分子结构,通过与之相互作用,药物能够实现对疾病的干预和治疗。
药物靶点研究的意义在于找到药物的作用位点,从而更好地设计和合成药物分子,提高药物的治疗效果。
二、分子设计的原理和方法分子设计是指通过计算机辅助药物设计和模拟技术,根据药物靶点的结构特点和作用机制,设计出具有高亲和力和选择性的化合物。
分子设计的原理和方法主要包括构效关系研究、药效团分析、3D-QSAR模型建立等。
1. 构效关系研究构效关系研究是一种通过拆解和修改分子结构,分析结构与活性之间的关系的方法。
通过这种方法,可以确定分子中活性团的位置和作用方式,从而指导分子的设计和合成。
2. 药效团分析药效团分析是一种通过研究不同化合物的共同药效团,确定药物靶点的活性位点及其与药物之间的相互作用方式。
通过药效团分析,可以帮助设计出具有更高亲和力和选择性的药物分子。
3. 3D-QSAR模型建立3D-QSAR模型是一种通过分子结构的三维信息和活性数据进行建模和预测的方法。
通过3D-QSAR模型,可以准确地预测新化合物的活性,为药物的设计和合成提供指导。
三、分子合成的原理和方法分子合成是指通过一系列化学反应,将原始物质转化为目标化合物的过程。
在药物研发中,分子合成是制备药物的关键环节,合成策略和方法的选择直接影响到药物的产率和纯度。
1. 合成策略的选择合成策略是指在分子设计的基础上,选择适当的方法和反应路径进行合成。
常用的合成策略包括逆合成法、正向合成法和中间体合成法等。
通过合理选择合成策略,可以提高合成的效率和产率。
2. 反应条件的控制反应条件的控制是合成过程中非常重要的一环。
药物设计学 11第十一章 基于片段的药物分子设计

对靶蛋白要求低,无需确定结构,无需进行放射性核素标记,避开对靶蛋白分子量的限制。 片段类药性三原则(RO3) 分子量<300(160~250)、氢键供体或受体数目≤3、脂水分配系数log P ≤ 3
➢ 这种方法得到片段与报告配体结合到药靶相同区域, 避免检测到与药靶非功能区域结合的片段,有效降 低假阳性。
19
2. 检测受体的筛选 TDBS
2.1 先决条件
I. 已知靶蛋白的结构 II. 对靶蛋白进行15N标记
2.2 原理
➢ 当小分子与靶蛋白结合后,会改变结合位点局部的 化学环境,通过15N标记蛋白的二维15N-HSQC谱图, 可以找到各酰胺信号15N或1H的化学位移变化。
合的药物发现新方法。
➢ 首先筛选得到低分子量和低亲和力的片段,
然后基于药靶结构信息将片段进行优化或连
接,得到与药靶亲和力高且类药性强的新分 子。
3
第一节 基于片段的分子设计原理
一、发展历程和基本理论
1.发展背景—高通量筛选的缺陷
I. 盲目性大,命中率很低。理论计算,含有30个重原 子的化合物有1060种,而高通量筛选的化合物数即使 以百万计(106),筛选也只占很少部分。
选技术平台、片段库的结晶筛选、结果分析和片段
不能获得靶蛋白结合位点的信息以及片段在结合位点的取向。 2 实例—发现细菌U1061A功能域配体
检对测靶配 蛋体白➢的进筛行选15NL片D标B记S 段库——含溴片段(利于测定、易于衍生化)
这种方法得到片段与报告配体结合到药靶相同区域,避免检测到与药靶非功能区域结合的片段,有效降低假阳性。
药物及生物活性小分子发现与分子设计PPT课件

通过对药物及生物活性小分子的研究, 可以发现新的药物先导物,加速新药 的研发进程。
02
药物及生物活性小分子的 发现
基于疾病靶点的药物筛选
总结词
基于疾病靶点的药物筛选是一种利用已知疾病靶点特征筛选小分子药物的方法。
详细描述
该方法首先确定与特定疾病相关的靶点,然后通过筛选与靶点相互作用的小分 子来寻找潜在的药物候选者。这种方法有助于提高药物发现的效率和特异性。
物质转运
一些药物及生物活性小分子可以作 为载体或通道,影响细胞内外的物 质转运,调节细胞内环境稳定。
药物及生物活性小分子的研究意义
疾病治疗
药物及生物活性小分子是疾病治疗的 重要手段之一,通过调节细胞功能和 代谢,达到治疗疾病的目的。
药物研发
生命科学研究
药物及生物活性小分子是研究生命活 动规律的重要工具,可以用于研究细 胞信号转导、基因表达调控等生命过 程。
合理药物设计
合理药物设计(Rational Drug Design)是一种综合利用基于结构的药 物设计、计算机辅助药物设计和药效团模型等手段,对已知活性分子进 行结构改造或对靶点蛋白进行理性设计的方法。
Rational Drug Design旨在通过理性设计手段,预测和优化小分子药物 的理化性质、药效团特征以及与靶点蛋白的结合模式,以提高药物的疗
结构生物学的发展为SBDD提供了重要 SBDD的关键步骤包括:小分子配体的
的基础,通过解析靶点蛋白的三维结构, 对接与优化、药效团模型的建立、虚拟
可以深入理解其功能机制,从而为药物
筛选以及先导化合物的优化等。
设计提供指导。
计算机辅助药物设计
计算机辅助药物设计(CADD)利用计算机技术对药物分子的理化性质、药效团特征等进行 预测和优化,以加速药物发现的过程。
第十一章基于靶点结构的药物分子设计

第十一章基于靶点结构的药物分子设计药物设计是药物发现研究的重要组成部分。
药物设计的主要目标是设计出具有良好疗效、能直接作用于治疗靶点的药物分子。
基于靶点结构的药物分子设计是药物设计领域的一个重要研究方向,它通过对靶点结构的认识,设计出更精准、更有效、更具选择性和更安全的药物分子。
一、基于靶点结构的药物设计的原理基于靶点结构的药物设计的原理是先确定治疗的靶点分子结构,然后利用计算机分子模拟等技术,设计出具有较高亲和性、良好选择性和稳定性的药物分子。
通过分析药物分子与靶点分子相互作用的方式、结构和特点,预测药物的生物效应并寻找具有良好效应的化合物。
基于靶点结构的药物设计可以有效地减少药物筛选的时间和成本,提高药物的成功率。
二、基于靶点结构的药物分子设计的步骤(一)靶点制备和结构测定制备给定的靶点,并确定结晶条件,测定靶点的晶体结构。
(二)分析靶点结构特征对靶点的结构进行分析,包括小分子结构、大分子的结构和空间结构。
通过分析靶点的结构特征,确定药物分子在靶点的作用模式和优化结构。
(三)药物筛选利用计算机分子模拟等技术,筛选靶点的药物分子。
药物分子的筛选包括确定药物与靶点的结合位点、寻找靶点结构中能与药物分子作用的区域、设计药物分子的基本结构等。
(四)药物分子的优化设计通过计算机分子模拟等技术,预测药物分子与靶点分子相互作用的方式和结构特点,优化药物分子的化学结构,提高药物分子与靶点分子的的亲和力和选择性。
(五)药物分子的合成和生物测试根据药物分子的结构,设计具有活性的分子,并合成实验室中的物质,进行生物活性测试。
分析分子的适应性和毒副作用。
三、基于靶点结构的药物分子设计应用案例基于靶点结构的药物设计已经成功地应用于药物研究与开发。
以下是本领域标志性的案例:(一)利用靶点结构设计抗HIV药物研究人员通过计算机模拟,预测了HIV-1逆转录酶(RT)的结构,成功地设计出了一些抑制剂。
其中多个抑制剂以不同方式与RT结合,从而抑制了病毒的复制。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
全新药物设计
靶点-配体复合物 晶体结构
结合位点 分析
配体设计 片段药物设计
配体合成
片段设计
配体设计
活性测试
数据库搜索 分子对接
购买少量 化合物
基于生物大分子靶点结构的药物设计方法
第一节 靶蛋白结构的预测
• 蛋白质结构与功能研究已成为后基因组时代最具挑战 性的研究课题。
• 当前测定蛋白质结构的主要方法仍然是X-射线晶体学 方法和多维核磁共振技术。
药物设计学
第十一章 基于靶点结构的
药物分子设计
【学习要求】
1. 掌握基于靶点结构的药物设计、全新药物设计、计算机 虚拟筛选、基于片段药物设计的基本概念。
2. 熟悉蛋白质三维结构预测法、分子对接方法及分类、基 于片段药物设计的基本思路、基于片段药物设计的优点; 片段筛选的主要检测技术;片段优化的常用方法。
• 代表性的活性位点分析方法的软件有GRID、MCSS和 HINT等相关程序。
GRID
• GRID程序由Goodford研究小组开发,其基本原理是将 靶点蛋白的活性部位划分为有规则的网格,应用分子力 场的方法计算探针分子(水分子或甲基等)在不同的格 点上与受体活性部位的相互作用能,以此解析探针分子 与靶点活性部位的相互作用情况,发现最佳作用位点。
• Adlington等应用MCSS对前列腺特异性免疫抗原 (PSA)的活性位点进行了详细分析,以此对已有的 PSA抑制剂进行结构优化,从而得到了迄今为止活性 最高的PSA抑制剂,其IC50为(226±10)nmol/L。
HINT
• HINT(hydrophobic interaction)是Kellogg等研究 的计算分子脂水分配系数及评价的程序,目前已商 业化并已有SYBYL和InsightⅡ下的版本。在SYBYL 最新版本中,HINT已作为一个正式模块推出,并能 够进一步计算和显示疏水场及两分子间的疏水相互 作用,并为CoMFA计算提供疏水场值。
• 蛋白质结构的测定速度却远远落后于基因组测序和氨 基酸序列的测定速度,无法满足蛋白组学及其相关的 学科需要。
同源模建的主要步骤 (1)目标序列与模板序列的比对; (2)根据同源蛋白的多重序列比对结果,确定同源蛋
白的结构保守区以及相应的框架结构; (3)目标蛋白质结构保守区的主链建模; (4)目标蛋白质结构变异区的主链建模; (5)侧链的安装和优化; (6)对模建结构进行优化和评估。
基于配体结构的药物设计
• 是从研究一系列药物分子对同一受体的活性出发, 比较它们的结构变化与生物活性之间的关系,找到 对该受体能发生结合并产生活性的最普遍的结构因 素,并根据此结构特征设计新的药物分子。
以靶点结构为主的药物设计可分为三大类
• 全新药物设计:根据靶点活性位点构建配体 • 分子对接:以靶点结构来搜寻配体 • 基于片段的药物设计:根据靶点活性位置来构建配
活性位点的分析方法
• 通过探针来探测简单的分子或碎片如何能够与生物大 分子的活性位点很好地结合。用于分析的探针可以是 一些简单的分子或碎片,例如水或苯环作为探针,通 过分析它们与活性位点的相互作用情况,可以找到这 些分子或碎片在活性部位中的可能结合位置。
• 活性位点分析法通常不能直接产生完整的配体分子,但 它得到的有关靶点结合的信息对后面的全新药物设计和 分子对接等都有很好的指导意义。
• 应用GRID程序研究流感病毒的重要靶点神经氨酸酶时, 以氨为探针分子搜寻神经氨酸酶结合位点时发现用胍基 取代抑制剂Neu5Ac2en的4-羟基,得到的化合物扎那米 韦(zanamivir)活性大为提高,现已作为抗A型感冒病 毒药物上市。
MCSS
• MCSS是Karplus课题组发展的一种活性位点分析方 法,其基本思路与GRID方法相似,但处理方式更为 细致、深入。例如GRID方法中仅考虑探针和蛋白质 的非键相互作用,而MCSS法进一步包括了探子分子 片段的构象能;GRID计算采用系统搜索法将探针分 子片段依次放在每个格点上,而MCSS法将探针分子 以多拷贝形式放置在活性口袋中,利用蒙特卡罗模拟 结合分子力学进行优化来寻找最佳作用位点。
从头预测(de novo prediction)
• 蛋白质结构从头预测是一个尚未成熟的研究领域,但 发展潜力十分巨大。因为该方法不需要知道任何一个 目标序列的同源蛋白质,仅从蛋白质的一级结构预测 其高级结构,一旦从头预测的方法获得重大突破,将 有助于人们理解蛋白质折叠的过程,影响蛋白质结构 稳定性的因素等基本问题。
第二节 分子对接与虚拟筛选
一、分子对接
• 分子对接(molecular docking)是通过研究小分子配 体与靶点生物大分子相互作用,预测其结合模式和亲 和力,进而实现基于结构的药物设计的一种重要方法。 根据配体与靶点作用的“锁钥原理”,分子对接可以 有效地确定与靶受体活性部位空间和电性特征互补匹 配的小分子化合物。
序列比对
• 序列比对是同源模建的关键,大多数的序列比对方法 都是以目标蛋白质和模板蛋白质序列之间的相似性为 基础的,其准确性可以通过进行多序列比对得到提高。 目前常用的序列比对程序有FASTA和BLAST等。许多 药物设计软件公司也开发了同源模建法预测蛋白的软 件模块,如Tripos公司的Composer、Accelyrs公司的 Homology等。
3. 了解全新药物设计的常用方法、磁共振检测技术的分类 和原理;SAR-by-NMR的原理和应用;Tether和二次 Tether技术的原理;结晶筛选的研究流程。
ቤተ መጻሕፍቲ ባይዱ
基于靶点结构的药物设计
• 是指一般应用由X射线衍射、磁共振或分子模拟 (同源建模法等)提供的蛋白质结构信息,来辅助 设计具有生物活性的化合物的过程。
折叠识别(fold recognition)
• 当目标蛋白质找不到已知结构的蛋白质作模板时,可 以采用蛋白质折叠识别方法进行三维结构预测。
• 折叠识别法就是总结出已知的蛋白质结构模式作为目 标蛋白质进行匹配的模式,然后经过现有的数据库的 观察,总结出可以区分正误结构的平均势函数作为判 别标准,来选择最佳的匹配方式。