DuraniMagliery2012ProteinExprPurif[1]
博来霉素气管多次给药诱导小鼠肺纤维化模型

博来霉素气管多次给药诱导小鼠肺纤维化模型作者:陈孟毅林帅杜朋李程程孟爱民来源:《中国医药导报》2017年第02期[摘要] 目的建立小鼠博来霉素气管多次给药肺纤维化模型。
方法 SPF级雄性C57BL/6J小鼠45只,按体重随机分为未处理组(5只)和处理组(40只),未处理组小鼠不经处理,处理组小鼠博来霉素气管给药(0.4 mg/mL),2周给药1次,共8次。
处理组小鼠在初次给药后2周、1个月、2个月、4个月时分别取出肺组织称重计算肺系数、固定做HE染色和Masson染色,制备冰冻切片进行细胞衰老β-半乳糖苷酶(SA-β-Gal)染色。
结果与未处理组小鼠比较,处理组小鼠在给药2周时肺系数显著升高(P < 0.05);同时,肺组织病理观察结果显示,给药2周时,小鼠肺泡腔内渗出和血管周围炎细胞浸润较为严重,病理评分与未处理组小鼠比较显著升高(P < 0.05)。
此外,处理组小鼠在给药2周、2个月与4个月时肺泡间隔增宽相对稳定,与未处理组小鼠比较,病理评分显著升高(P < 0.05),但胞浆渗出逐渐减少、炎细胞浸润减轻,评分接近未处理组小鼠水平;胶原染色结果显示处理组小鼠在给药2周时出现蓝绿色的胶原纤维,并在1、2、4个月时胶原纤维持续存在;SA-β-Gal染色结果表明,与未处理组小鼠比较,博来霉素气管给药4个月时小鼠的肺组织中衰老细胞明显增多。
结论博来霉素气管多次给药可以建立小鼠肺纤维化模型,且模型小鼠具有肺纤维化进展不可逆、炎性反应明显减少两个重要特点,并提示肺纤维化与衰老细胞有一定关系。
[关键词] 博来霉素;纤维化;肺;模型[中图分类号] R563.02;R363 [文献标识码] A [文章编号] 1673-7210(2017)01(b)-0008-04特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是一种原因不明、以弥漫性肺泡炎和肺泡结构紊乱最终导致肺间质纤维化为特征的慢性疾病[1-2]。
已知的miRNA的ID中英文对照

aae Aedes aegypti 埃及伊蚊aca Anolis carolinensis 安乐蜥aga Anopheles gambiae 冈比亚按蚊aly Arabidopsis lyrata 琴叶拟南芥ame Apis mellifera 意蜂api Acyrthosiphon pisum 豌豆蚜ata Aegilops tauschii 山羊草/节节麦ath Arabidopsis thaliana 拟南芥atr Amborella trichopoda 无油樟bdi Brachypodium distachyon 二穗短柄草blv Bovine leukemia virus 牛白血病病毒bma Brugia malayi 马来丝虫bmo Bombyx mori 家蚕bna Brassica napus 油菜bra Brassica rapa 白菜/芜菁bta Bos T aurus (普通)牛cbn Caenorhabditis brenneri 线虫brennericbr Caenorhabditis briggsae 线虫briggsaecel Caenorhabditis elegans 秀丽隐杆线虫cfa Canis familiaris 家犬chi Capra hircus 山羊cin Ciona intestinalis 玻璃海鞘cqu Culex quinquefasciatus 致倦库蚊cre Chlamydomonas reinhardtii 莱茵衣藻crm Caenorhabditis remanei 线虫remanei csa Ciona savignyi 玻璃海鞘savignyicte Capitella teleta 小头虫teletadan Drosophila ananassae 果蝇ananassaeddi Dictyostelium discoideum 盘基网柄菌der Drosophila erecta 果蝇万寿菊dgr Drosophila grimshawi 果蝇grimshawi dme Drosophila melanogaster 黑腹果蝇dmo Drosophila mojavensis 果蝇mojavensis dpe Drosophila persimilis 果蝇螨dps Drosophila pseudoobscura 果蝇pseudoobscura dpu Daphnia pulex 蚤状溞dre Danio rerio 斑马鱼dse Drosophila sechellia 果蝇sechelliadsi Drosophila simulans 果蝇simulansdvi Drosophila virilis 果蝇virlisdwi Drosophila willistoni 果蝇willistonidya Drosophila yakuba 果蝇yakubaebv Epstein Barr virus EB病毒eca Equus caballus 马efu Eptesicus fuscus 大棕蝠/大棕鲇egr Echinococcus granulosus 细粒棘球绦虫emu Echinococcus multilocularis 多房棘球绦虫esi Ectocarpus siliculosus 水云siliculosusfru Fugu rubripes 红鳍东方鲀gga Gallus gallus 原鸡ggo Gorilla gorilla 大猩猩gma Glycine max 大豆gra Gossypium raimondii 雷蒙德氏棉hbr Hevea brasiliensis 橡胶树hbv Herpes B virus 乙型肝炎病毒hcmv Human cytomegalovirus 人巨细胞病毒hme Heliconius melpomene 红带袖蝶hsa Homo sapiens 智人isc Ixodes scapularis 肩突硬蜱kshv Kaposi sarcoma-associated herpesvirus 卡波济肉瘤相关疱疹病毒lgi Lottia gigantean 帽贝lja Lotus japonicas 百脉根mdm Malus domestica 苹果mdo Monodelphis domestica 短尾负鼠mdv1 Mareks disease virus 马立克氏病病毒mes Manihot esculenta 木薯星虫mghv Mouse gammaherpesvirus 68 小鼠γ疱疹病毒mml Macaca mulatta 猕猴mmu Mus musculus 小家鼠mse Manduca sexta 烟草天蛾mtr Medicago truncatula 蒺藜苜蓿nve Nematostella vectensis海葵nvi Nasonia vitripennis 金小蜂oan Ornithorhynchus anatinus 鸭嘴兽oar Ovis aries 绵羊ocu Oryctolagus cuniculus 家兔oha Ophiophagus Hannah 眼镜王蛇ola Oryzias latipes 青鳉osa Oryza sativa 水稻pma Petromyzon marinus 海七鳃鳗ppc Pristionchus pacificus 蛔虫ppe Prunus persica 桃ppt Physcomitrella patens 小立碗藓ppy Pongo pygmaeus 猩猩prd Panagrellus redivivus 全齿复活线虫/腐生线虫ptc Populus trichocarpa 毛果杨ptr Pan troglodytes 黑猩猩pxy Plutella xylostella 小菜蛾rco Ricinus communis 蓖麻rno Rattus norvegicus 褐家鼠rrv Rhesus monkey rhadinovirus 猕猴Rhadino病毒sbi Sorghum bicolor 高粱sha Sarcophilus harrisii 袋獾sja Schistosoma japonicum 日本血吸虫sko Saccoglossus kowalevskii 橡子蠕虫sly Solanum lycopersicum 番茄sma Schistosoma mansoni 曼氏血吸虫sme Schmidtea mediterranea 淡水涡虫spu Strongylocentrotus purpuratus 紫色球海胆ssc Sus scrofa 野猪str Strongyloides ratti 鼠类圆线虫stu Solanum tuberosum 马铃薯tca Tribolium castaneum 赤拟谷盗tch Tupaia chinensis 树鼩tgu T aeniopygia guttata 斑胸草雀tni Tetraodon nigroviridis 金娃娃vvi Vitis vinifera 葡萄xtr Xenopus tropicalis 热带爪蟾zma Zea mays 玉米。
常见细菌中英文对照、菌组、菌属及代码(革兰阴性部分)

产碱杆菌属
axy
-
Alcaligenes xylosoxidans ss. xylosoxidans
木糖氧化无色杆菌木糖氧化亚种
NFR
无色杆菌属
b01
—
Shigella boydii serotype 1
鲍氏志贺菌血清1型
EBC
志贺菌属
b02
-
Shigella boydii serotype 2
鲍氏志贺菌血清2型
EBC
志贺菌属
b03
-
Shigella boydii serotype 3
鲍氏志贺菌血清3型
EBC
志贺菌属
b04
-
Shigella boydii serotype 4
鲍氏志贺菌血清4型
EBC
志贺菌属
b05
—
Shigella boydii serotype 5
鲍氏志贺菌血清5型
EBC
Pasteurella ureae
脲放线杆菌
GNCB
放线杆菌属
ave
-
Aeromonas veronii
维龙气单胞菌
FERM
气单胞菌属
avv
—
Aeromonas veronii biovar veronii
维龙气单胞菌
FERM
气单胞菌属
axy
-
Alcaligenes xylosoxidans
木糖氧化无色杆菌木糖氧化亚种
bud
-
Budvicia sp。
布戴约维采菌属
EBC
布戴约维采菌属
buk
-
Burkholderia sp.
伯克霍尔德菌属
支气管肺泡灌洗液G 试验联合GM 试验在

医学食疗与健康 2022年9月中第20卷第26期·影像学及诊断检验·支气管肺泡灌洗液G试验联合GM试验在侵袭性肺部真菌感染诊断中的效能研究黎志勤…张妙芬…张霆(东莞市人民医院检验科,广东 东莞 523000)【摘要】目的:探讨支气管肺泡灌洗液(BALF)(1,3)β-D葡聚糖检测(G试验)联合半乳甘露聚糖检测(GM)试验在侵袭性肺部真菌感染(IPFI)患者中的诊断效果及效能。
方法:选择2018年5月至2021年2月疑似IPFI患者76例为对象,所有患者入院后均行支气管肺泡灌洗液G试验,GM试验检查,并以病原学检测结果作为“金标准”,绘制ROC曲线,分析G试验与GM试验在侵袭性肺部真菌感染中的诊断价值及效能。
结果:76例疑似IPFI患者经“金标准”检查确诊51例。
G试验检查确诊43例,GM试验检查确诊46例,两种方法联合检查确诊49例,与“金标准”诊断符合率为89.47(68/76),诊断灵敏度为90.20%(46/51)、特异度为88.00%(22/25),阳性预测值为93.88%(46/49)、阴性预测值为81.48%(22/27);ROC曲线结果表明:支气管肺泡灌洗液G试验,GM试验联合检测在IPFI 患者中的诊断灵敏度和特异度高于单一支气管肺泡灌洗液G试验,GM试验(P<0.05)。
结论:支气管肺泡灌洗液G试验,GM试验用于IPFI患者中中具有较高的检出率,且二者联合检查诊断灵敏度和特异度较高,可指导临床…诊疗。
【关键词】支气管肺泡灌洗液G试验;半乳甘露聚糖检测;侵袭性肺部真菌感染;诊断效能【中图分类号】R562…【文献标识码】A…【文章编号】2096-5249(2022)26-0128-04Efficacy of bronchoalveolar lavage fluid G test combined with GM test in the diagnosis of invasive pulmonary fungal infectionLi Zhi-qin, Zhang Miao-fen, Zhang TingClinical Laboratory of Dongguan People's Hospital, Dongguan 523000, Guangdong, China【Abstract】Objective: To investigate the effect of bronchoalveolar lavage fluid(BALF)(1, 3)β-D glucan test(G test)combined with galactomannan test(GM)test in invasive pulmonary fungal infection(IPFI)diagnostic efficacy and efficacy in patients. Methods: A total of 76 patients with suspected IPFI from May 2018 to February 2021 were selected as the subjects. All patients underwent bronchoalveolar lavage fluid G test and GM test after admission, and the etiological test results were used as the "gold standard". The ROC curve was drawn to analyze the diagnostic value and efficacy of G test and GM test in invasive pulmonary fungal infection. Results: Of the 76 suspected IPFI patients, 51 were confirmed by the "gold standard" examination. 43 cases were confirmed by G test, 46 cases were confirmed by GM test, and 49 cases were confirmed by combined examination of the two methods. The coincidence rate with the "gold standard" diagnosis was 89.47(68/76), and the diagnostic sensitivity was 90.20%(46/51). The specificity was 88.00%(22/25), the positive predictive value was 93.88%(46/49), and the negative predictive value was 81.48%(22/27); the ROC curve results showed that: bronchoalveolar lavage fluid G test, GM test The diagnostic sensitivity and specificity of combined detection in IPFI patients were higher than those of single bronchoalveolar lavage fluid G test and GM test (P<0.05). Conclusion: Bronchoalveolar lavage fluid G test and GM test have high detection rate in IPFI patients, and the combined detection of the two has high diagnostic sensitivity and specificity, which can guide clinical diagnosis 作者简介:黎志勤(1984.07—),男,本科,主管技师,研究方向:微生物学。
麻保沙星欧洲标准

14、菲利平Trade name:FeilipingActive ingredient:Lincomycin hydrochloride , Spectinomycin sulfate .Attending functions:Long-term broad-spectrum antibacterial. Can prevent diseases of porcine proliferative enteropathy(PPE),mycoplasma hyopneumoniae, diarrhea and so on.Usage and Dosage:For pig:a ton of feed to add this product 1000g, Continuous use 10-15 days. Doubling dose in serious condition .Specification:1000g:Fincomycin hydrochloride 22g (2200 million units) + spectinomycin sulfate 22 g(2200 million units)Packaging:100 0g/bagAvailable period:2 yearNote:5-day withdrawal period for pigStore in a cool and dry place.【商品名】:菲利平【有效成分】:盐酸林可霉素硫酸大观霉素【功能主治】:广谱抗菌药,可预防增生性肠炎、猪肺炎支原体、猪痢疾及常见细菌感染。
【用法用量】猪每吨饲料添加本品1000g,连用10~15天。
病情严重时剂量加倍。
【规格】1000g:林可霉素22g(2200万单位)+大观霉素22g(2200万单位)【包装】 1000g/袋【有效期】 2 年【注意事项】1.停药期:猪5天;2.存放于阴凉干燥处。
“魔法子弹”ADC药物——乳腺癌的克星

100多年前,免疫学之父Paul Ehrlich (如图1所示)首次提出“魔法子弹”的设想,即选择性地向目标细胞输送毒性药物,而避免伤害人体正常细胞的概念。
之后,“魔法子弹”ADC (Antibody -Drug Conjugate )药物开创于生物技术和药物研发领域,是一种结合抗体、毒素和连接子的复合物,用于精确靶向肿瘤细胞并释放药物。
它的历史可以追溯到20世纪80年代,当时科学家开始尝试将抗体与毒素结合,以增强抗肿瘤治疗的效果。
图1 免疫学之父Paul Ehrlich乳腺癌是最常见的女性恶性肿瘤之一,具有很强的异质性。
传统的化疗、内分泌治疗和放疗对于不同类型的乳腺癌患者效果各自受限。
ADC 药物利用其高度特异性的抗体识别癌细胞表面的靶点,并通过连接药物分子的化学链将细胞毒性药物传递到癌细胞内部。
一些ADC 药物已经在乳腺癌治疗中进行了临床试验,并取得一些出色的治疗效果。
最早的ADC 药物是通过化学手段将毒素与抗体连接而成,但这种方法存在许多限制和挑战。
随着科技的进步和对ADC 技术的深入研究,研究人员开始采用更精确的方法来制造ADC 药物。
其中一个重要的突破是使用基因工程技术生产抗体。
这些抗体可以特异性地结合到肿瘤细胞表面的抗原上,从而实现更准确的靶向治疗。
另一个关键的进展是连接子的开发。
连接子是将抗体与毒素连接在一起的分子,它能够在抗体与毒素之间建立稳定的化学键。
这种连接子需要具备足够的稳定性,以便在血液循环中保持连接,又能在抗体与肿瘤细胞结合后迅速释放毒素。
“魔法子弹”ADC 药物的进一步发展还受益于对肿瘤细胞生物学和信号传导途径的深入了解。
研究人员可以根据不同肿瘤类型的特征选择适当的抗原作为靶点,并设计相应的ADC 药物。
“魔法子弹”ADC 药物的起源可以追溯到对抗体、毒素和连接子的深入研究,以及对肿瘤生物学的进一步认识。
这些技术和知识的不断发展促使ADC 药物在肿瘤治疗领域取得显著进展,并成为一种重要的治疗策略之一。
蛋白聚合原因-方式及其分析Protein Aggregation Pathways, Induction Factors and Analysis

Protein Aggregation:Pathways,Induction Factorsand AnalysisHANNS-CHRISTIAN MAHLER,1WOLFGANG FRIESS,2ULLA GRAUSCHOPF,1SYLVIA KIESE11Formulation R&D Biologics,Pharmaceutical and Analytical R&D,F.Hoffmann-La Roche Ltd.,Basel,Switzerland2Department of Pharmacy,Pharmaceutical Technology and Biopharmaceutics,Ludwig-Maximilians-Universitaet Muenchen,Munich,GermanyReceived27March2008;revised1August2008;accepted5August2008Published online29September2008in Wiley InterScience().DOI10.1002/jps.21566ABSTRACT:Control and analysis of protein aggregation is an increasing challengeto pharmaceutical research and development.Due to the nature of protein in-teractions,protein aggregation may occur at various points throughout the lifetimeof a protein and may be of different quantity and quality such as size,shape,morphology.It is therefore important to understand the interactions,causes and analyses of suchaggregates in order to control protein aggregation to enable successful products.Thisreview gives a short outline of currently discussed pathways and induction methodsfor protein aggregation and describes currently employed set of analytical techniquesand emerging technologies for aggregate detection,characterization and quantification.A major challenge for the analysis of protein aggregates is that no single analyticalmethod exists to cover the entire size range or type of aggregates which mayappear.Each analytical method not only shows its specific advantages but also hasits limitations.The limits of detection and the possibility of creating artifacts throughsample preparation by inducing or destroying aggregates need to be consideredwith each method used.Therefore,it may also be advisable to carefully compareanalytical results of orthogonal methods for similar size ranges to evaluate methodperformance.ß2008Wiley-Liss,Inc.and the American Pharmacists Association J Pharm Sci98:2909–2934,2009Keywords:protein aggregation;protein analysis;protein formulation;molecularweight determination;particle sizing;size exclusion chromatography;analytical ultra-centrifugation;light scattering;SDS–PAGE;orthogonal methodsINTRODUCTIONThe breakthrough of recombinant DNA technol-ogy in the mid1970s has allowed the development of many recombinant therapeutic proteins and thus has resulted in many protein-based products to reach the market.1,2The control and analysis of protein aggregation during production of a bio-therapeutic drug is an increasing challenge to many pharmaceutical research and development groups and companies.Aggregation is potentially encountered during various steps of the manu-facturing process of biopharmaceuticals,which include fermentation,purification,formulation and during storage.Biopharmaceuticals forH.-C.Mahler and S.Kiese contributed equally to this study. Correspondence to:Hanns-Christian Mahler(Telephone: 41-61-68-83174;Fax:41-61-68-88689;E-mail:hanns-christian.mahler@)Journal of Pharmaceutical Sciences,Vol.98,2909–2934(2009)Pharmacists AssociationJOURNAL OF PHARMACEUTICAL SCIENCES,VOL.98,NO.9,SEPTEMBER20092909clinical trials require full characterization includ-ing accurate quantification of protein aggregates to meet the drug product specifications.Protein aggregates potentially cause adverse effects,such as an immune response,3,4which may cause neutralization of the endogenous protein with essential biological functions leading to a life-threatening situation for the patient and aggre-gates may also potentially impact the drug’s efficacy.5The scientific fact base to clearly link specific types and sizes of aggregates to immune responses is however currently still under inves-tigation.A potential increase in immune res-ponses caused by aggregates has been reported previously,3whereas in contrast no enhanced immunogenicity was shown for example in the case of aggregated rFVIII.6There are monographs and acceptance criteria/ limits in the pharmacopoeias for visible and subvisible particles(i.e.,insoluble proteins aggre-gates)—for example,United States Pharmaco-poeia(USP)<788>,7European Pharmacopoeia (Ph.Eur.) 2.9.198and Ph.Eur 2.9.209—for parenteral products.However,limits for soluble aggregates have to be set case-by-case as there are no predefined limits laid down in general for biopharmaceuticals within regulatory documents. In order to control protein aggregation to enable safe and successful products,it is important to understand the origin of protein aggregates,and the analytical techniques for characterizing their full size range.This review article aims to collate and discuss available literature on the major causes of aggre-gation and the analytical methods/techniques to characterize protein aggregates.PATHWAYS AND INDUCTION FACTORSDefinition and Mechanism of Protein Aggregation The term‘‘protein aggregation’’has been given many definitions and terminologies within the literature.10,11The authors define‘‘protein aggre-gates’’as a summary of protein species of higher molecular weight such as‘‘oligomers’’or‘‘multi-mers’’instead of the desired defined species(e.g.,a monomer).Aggregates are thus a universal term for all kinds of not further defined multimeric species that are formed by covalent bonds or noncovalent interactions.Different mechanisms that may lead to forma-tion of various types of aggregates are currently under discussion.There is no single protein aggregation pathway but a variety of pathways, which may differ between proteins12and may result in different end states.A protein may undergo various aggregation pathways depending on the environmental conditions,including dif-ferent types of applied stress.Also,the initial state of a protein that is prone for subsequent aggregation may differ.It may be constituted by the native structure,13by a degraded14or modified structure,15by a partially unfolded structure15,16 or by the fully unfolded state.12The aggregation process in general may lead to soluble and/or insoluble aggregates which may precipitate.13,17–19The morphology of these inso-luble aggregates may be in the form of amorphous orfibrillar material which is dependent on the protein and its environment.Noncovalent aggre-gates are formed solely via weak forces such as Van der Waals interactions,hydrogen bonding, hydrophobic and electrostatic interactions20 whereas covalent aggregates may for example form via disulfide bond linkages through free thiol groups11,21,22or by nondisulfide cross-linking pathways such as dityrosine formation.23Aggre-gation may be reversible24or irreversible where the irreversible aggregates could be permanently eliminated by preparative separation processes such asfiltration techniques.25The formation of reversible aggregates is often considered to be caused by the self-assembly of protein molecules, which could be induced by changes in pH or ionic strength of the protein solution.26–30One model that has been applied to describe irreversible protein aggregation is the Lumry-Eyring two state model.31According to this model the native protein undergoesfirst a reversible conformational change to an aggregation-prone state,which subsequently assembles irreversibly to the aggregated state.In this model protein aggregation is thereby controlled by conforma-tional and colloidal mechanisms.18,25In many cases,aggregation was described to follow a nucleation–propagation polymerization mechanism,whereby the nucleus can be formed by an altered monomeric structure or by a multimeric species.32New reports also suggest the appearance of heterogenous nucleation which is induced by micro-and nanoparticles of foreign matter,which for example could be shed from the equipment during processing.33,34Much insight in protein aggregation pathways is obtained from research in thefield of amyloidfiber formation35 and sickle cell hemoglobin.36In the area ofJOURNAL OF PHARMACEUTICAL SCIENCES,VOL.98,NO.9,SEPTEMBER2009DOI10.1002/jps 2910MAHLER ET AL.pharmaceutically relevant proteins such as mono-clonal antibodies,the published reports on aggre-gation pathways are still very limited.Protein aggregates have been categorized pre-viously11,37based on different aspects.However, since the term‘‘protein aggregation’’has often been used lacking adequate definition,the authors suggest classifying‘‘protein aggregates’’based on the above considerations into the following categories:(a)by type of bond:nonconvalent aggregates(bound by weak electrostatic forces)38versus covalent aggregates(e.g.,causedby disulfide bridges);11,21(b)by reversibility:reversible26,29,30versusirreversible27aggregates;(c)by size:small soluble aggregates(oligo-mers)such as dimers,trimers,tetramers,etc.versus large 10-mer oligomers versusaggregates in the diameter range of someapprox.20nm to approx.1m m versus inso-luble particles in the1–25m m range versuslarger insoluble particles visible to the eyeunder defined inspection conditions;17,18,39(d)by protein conformation:aggregates withpredominantly native structure40versusaggregates with predominantly nonnativestructure(i.e.,partially unfolded multi-meric species,39,41,42fibrillar aggregates43–45).Induction Factors Causing Protein Aggregation Aggregation can be induced by a wide variety of conditions,including temperature,mechanical stress such as shaking and stirring,pumping, freezing and/or thawing and formulation.Also, because partially unfolded protein molecules are part of the native state ensemble,aggregation can occur under nonstress conditions where the native state is highly favored.Since processes that may cause stress upon a protein are commonly utilized during manufacturing of biopharmaceuticals including fermentation,purification,formulation,filling,shipment and storage,it is important to understand their effect on the induction of protein aggregates,both their influence on the aggre-gation rate as well as the type of aggregate potentially induced.Furthermore,it is generally acknowledged that formulation parameters including the protein concentration itself and other parameters such as pH,the qualitative and quantitative composition and formulation/packa-ging interactions play a major role in the control of protein aggregation.The following section outlines in general a few of the processes and conditions which have been reported to be involved in the formation of protein aggregation. TemperatureAn increase in temperature accelerates chemical reactions such as oxidation and deamidation of biopharmaceuticals,which could in turn lead to higher aggregate levels.46Higher temperature also has a direct effect on the conformation of polypeptide chains on the level of its quaternary, tertiary,and secondary structure,and can lead to temperature-induced unfolding that promotes in many cases aggregation.A measure for thermal stability of a protein is the melting temperature (T m),at which50%of protein molecules are unfolded during a thermal unfolding transition. Melting temperatures vary among proteins,and lie usually in a range between40and808C.39It is generally important to store biopharmaceuticals well below their T m,usually at2–88C,and to avoid processing temperatures for example during fermentation,purification and manufacturing that go above the T m.During accelerated stability studies of biopharmaceuticals high temperature storage conditions,such as408C are used to gain stability data within comparably short times, but since aggregation processes do not necessarily follow Arrhenius behavior,the extrapolation to predict aggregation at lower storage tempera-tures remains challenging.47Freezing and ThawingFreezing introduces complex physical and chemi-cal changes including creation of new ice/solution interfaces,48–50adsorption to container surfaces,50 cryoconcentration of the protein and solutes,51,52 and pH changes due to crystallization of buffer components.53These effects are regarded as possible causes of freezing-induced protein dena-turation and aggregation.The freezing rate as well as the method and control of thawing has been previously reported to influence the rate of protein aggregation.52,54The freezing/thawing container and thefill volume play a major role in the extent of induced protein aggregation.55 This poses a significant challenge during formula-tion development since freeze/thaw stability test-ing is suggested to be tested at scale but may have to be carried out at small scale due to limitedDOI10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.98,NO.9,SEPTEMBER2009PROTEIN AGGREGATION2911availability of protein material in early stages, potentially not correlating to the freezing/thawing behavior and stability at large scale.Bhatnagar et al.56have collated numerous reports on freeze/ thaw experiments in terms of rate of freezing and thawing as well as freeze–thaw cycles. Agitation StressAgitation stress such as stirring,pumping,and shaking during manufacturing and transport has been described to cause aggregation.13,18,57,58 These types of stress could induce shearing, interfacial effects,cavitation,local thermal effects and rapid transportation of either aggregated or adsorbed species from the interface into solu-tion.13Although agitation stress is sometimes referred to as shear stress,several studies59–61 suggest that shear alone does not cause protein aggregation.Agitation has been described to potentially cause cavitation62where cavitation is described as the rapid formation of voids or bubbles within the liquid which rapidly collapse thus producing shock waves,highly turbulent flow conditions,extreme pressures and tempera-ture which may result in the generation of hydroxyl and hydrogen radicals thus leading to the formation of protein aggregates.63,64Mechan-ical stress testing in lab experiments could be performed under controlled conditions using,for example,horizontal or vertical shakers,13,18stir-red reactors65and pumps,11,66rheometers such as concentric-cylinder shear devices or cone-plate, rotating-disk reactors,57,58to mimic‘‘real-life’’mechanical stresses which proteins may experi-ence.Yet it is difficult to estimate the level of agitation stress that a protein experiences during processing,shipment,etc.Thus,the conditions applied in stress studies do not necessarily reflect the real life situation.It needs to be noted that the above mentioned different conditions may induce different species of aggregates,qualitatively and quantitatively.13Protein ConcentrationThe increase in protein concentration has been reported to enhance the formation of protein aggregates for many proteins under quiescent storage.67–70The formation of aggregates is occurring by at least bimolecular interaction of protein molecules and thus,this reaction is per se considered to be concentration-dependent.68At high protein concentrations,macromolecular crowding occurs,a term used to describe the effect of high total volume occupancy by macro-molecular solutes upon the behavior of each macromolecular species in that solution may appear.According to this excluded volume theory, self-assembly and thus potentially aggregation may be favored,40but at the same time unfolding that is a prerequisite for many aggregation reactions may be reduced.40,71An increase in protein concentration has been shown to increase the size of aggregates,as in the case of Beta-lactoglobulin.72Quiescent storage showed the acceleration of aggregation formation in higher protein concentrated formulations however upon agitation stress more aggregation was seen in low protein concentration samples.68On the other hand,a decrease in protein concentration via dilution(e.g.,during preparation of drugs for clinical administration or sample preparation) has been shown to affect the aggregate content as aggregates formed by weak reversible interaction can dissociate as the protein concentration decreases.73Solvent and Surface EffectsChanges to the solution environment of a protein, for example,pH,ionic strength,buffer species, excipients and contact materials,could induce the formation of protein aggregates.A change in pH has a strong influence on the aggregation rate as the pH determines the electrostatic interactions through charge distribution on the protein sur-face.74Under acidic conditions protein cleavage may occur,whereas under neutral to alkaline conditions deamidation and oxidation are favored. Such modifications depend on the primary sequence as well as structure75and may lead to increased aggregation.Different protein aggrega-tion behavior has been shown within different buffer systems with equivalent pH.76,77For interferon-tau,a high protein aggregation rate in phosphate buffer was observed while the process was much slower in Tris and histidine buffer.76The quantity of excipients in the solvent may also have an impact on the aggregation behavior.The ability of the surfactants such as polysorbates(PS)to stabilize a protein against aggregation has been shown to depend on the protein to surfactant ratio.13,18Such a change in the concentration of an excipient/stabilizer is also observed when a drug formulation is added to infusion bags and this concentration change may have marked influence on aggregate levels. Furthermore,contact materials such as glass,JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.98,NO.9,SEPTEMBER2009DOI10.1002/jps 2912MAHLER ET AL.steel,silicone,plastic,rubbers,etc.may influence aggregation78where histidine formulated bulk of an IgG1including sodium chloride resulted in high aggregate levels when stored in a stainless steel tank.79Chemical Modifications of the Protein Aggregation reactions can occur following a chemical modification of a protein.Chemical modifications can include reactions such as deamidation,isomerization,hydrolysis,and oxi-dation.39Modifications of amino acid side chains by for example deamidation or isomerization may distort the conformation of proteins80potentially leading to aggregation or self-association.Oxida-tion reactions such as disulfide bond formation or methionine oxidation may be promoted by light exposure,peroxide contaminations of excipients, or simply the presence of oxygen during the manufacturing process.Light exposure may lead to photolytic degradation of the protein through photo-oxidation of the side-chains of certain amino acids such as Met,Tyr,Trp,His,Cys, and Phe81–83and has been shown to induce protein aggregation.82The quality in terms of impurity levels of an excipients such as the nonionic surfactants PS20and PS80,in a protein formulation may result in oxidation reactions. Polysorbates undergo auto-oxidation which results in hydroperoxide formation.84The impur-ity levels of hydroperoxide within PS may vary from lot-to-lot as well as from manufacturer-to-manufacturer.85The levels of hydroperoxide may also depend on storage conditions and storage time of the PS and thus leading to potential differences in oxidation and also potentially in subsequent protein aggregation.84PROTEIN AGGREGATION:ANALYTICAL TECHNIQUESProteins are therapeutically used in a wide range of indications.Before these biopharmaceuticals enter clinical R&D programs to evaluate their therapeutic potential,they need to be extensively characterized and adequately monitored during and after manufacturing and storage with regard to structural and biological integrity,process and product related impurities,and molecular and biological properties.Recent technological progress has significantly amplified the speed of characterizing proteins and with these advances, various analytical methods are now available to better characterize biopharmaceuticals.Molecu-lar weight,conformation,size and shape,and state and extent of aggregation are a few of the physico-chemical properties studied.Several methods are available for the quanti-fication and size estimations or the characteriza-tion of protein aggregates(Tab.1).However,the inherent differences in what is being measured and the requirements of most of these methods for data evaluation may result in inconsistencies between the methods in the reported mean size, size distributions,and quantity of an aggregate species for a given sample.One of the major challenges with the analysis of protein aggregates is that currently no single analytical method exists to cover the entire size range in which aggregates may appear,especially a routine method to quantify submicron particles.The protein aggre-gates may constitute only a minute fraction of the total protein mass and may be particularly of interest due to their potential role in immuno-genicity.3Therefore different analytical methods have to be employed in order to detect these minute aggregate fractions as well as to cover the size range from a few nanometers to hundred micrometers to large visible particles(Fig.1). Additionally,analytical methods used for asses-sing protein aggregates need to be closely looked at with regard to their performance and limita-tions,such as their specific limit of detection as well as the possibility to create artifacts,such as either inducing or destroying aggregates during sample preparation(dilution or increasing the concentration)thus potentially shifting the aggre-gation equilibrium,or the loss of aggregates by adsorption onto column material or membranes during analysis.Therefore,it may be advisable to carefully compare the analytical results obtained from various methods,that is,the use of orthogonal methods and to assess any data differences on a case-by-case basis with regards to method set-up and parameters.The authors define the use of orthogonal methods as‘‘the use of a combination or a variety of different analytical methods,each having its own characteristic measuring principle,for example,by size,quan-tification or structure,etc.’’The use of such orthogonal methods is also suggested in the current European Medicines Agency(EMEA) draft guideline on‘‘production and quality control of monoclonal antibodies and related substances.’’86DOI10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.98,NO.9,SEPTEMBER2009PROTEIN AGGREGATION2913The following sections shall discuss various analytical methods available to measure protein aggregation,their pitfalls,as well as the advan-tages in comparison to other techniques.Size Exclusion ChromatographySince its introduction by Porath and Flodin in the late1950s87the conventional size exclusion chromatography(SEC)or gelfiltration has become an essential tool for the analysis and purification of proteins.SEC is one of the most used analytical methods for the detection and quantification of protein aggregates.SEC analysis allows both for sizing of aggregates,and their quantification.Utilizing various column materials in combina-tion with high performance liquid chromatogra-phy(HPLC)results in the selective and rapid separation of macromolecules based on their shape and size(hydrodynamic radius)in a mole-cular weight range of roughly5–1000kDa.88This fractionation range of the column is based on the fact that oligomers that are too large to penetrate the pores of the matrix are excluded from the packing pore volume and elute with the void volume of the column.89The aggregation size determination may vary between different SEC methods,suggesting that the upper size range of an aggregate which escapes the SEC determina-tion needs to be assessed case by case.However, insoluble aggregates are not considered to be measurable by SEC due to potential removal via filtration by the column or precolumn or by the sample preparation for SEC(e.g.,centrifugation). Factors such as protein shape,protein glycosyla-tion or pegylation90could affect the accuracy if the molecular weight of protein species is determined based on a calibration curve using calibration standards.91Well characterized,water-soluble and globular proteins are used as calibration standards,which may differ in their elution properties in comparison with the protein of interest.It has been reported that basing the molecular weight solely on the elution volume has resulted in incorrectly identifying peaks as dimers.37,91,92It was also shown that modification of the mobile phase,such as the inclusion of arginine suppresses protein adsorption to the column matrix,93,94and that additional peaks may be induced due to a high salt content in theTable1.Frequently Used Methods for the Analysis of Protein AggregationCategory Method ApplicationQuantification and/or size estimation SE-HPLC Size estimation and quantification(soluble aggregates)RP-HPLC Size estimation and quantification(soluble aggregates) SDS–PAGE Size estimation and to distinguish reducible covalentfrom noncovalent aggregatesCapillary electrophoresis Size estimation and quantification(soluble aggregates) Fieldflow fraction(e.g.,AF4)Size estimation and quantification(soluble aggregates) Microscopic methods(e.g.,Light,electron,atomic force microscopy)Size and shape estimationStatic light scattering Size and shape estimationDynamic light scattering Size distributionAnalytical ultracentrifugation Size,shape estimation and quantificationLight obscuration Size and quantification(insoluble aggregates)Coulter counter Size and number quantification(insoluble aggregates) Visible inspection Absence or presence of visible aggregatesUV–Vis spectroscopy,turbidity/opalescence/clarity(visuallyor instrumentally)Soluble and insoluble aggregates;solution property(no quantification possible)Characterization Circular dichroism Structural analysisFluorescence spectroscopy Structural analysis(FT-)infrared spectroscopy Structural analysisRaman spectroscopy Structured analysisNuclear magnetic resonancespectroscopyStructural analysisJOURNAL OF PHARMACEUTICAL SCIENCES,VOL.98,NO.9,SEPTEMBER2009DOI10.1002/jps 2914MAHLER ET AL.mobile phase.73Dilution,occurring during the chromatography process or sample preparation,may lead to the dissociation of reversibly formed aggregates as the concentration decreases.73SEC cannot only be used in combination with UV or fluorescence detectors,but also with other detec-tors such as light scattering detectors (e.g.,multi-angle laser light scattering (MALLS)),to take advantage of the light scattering technique in combination with the separation technique of soluble aggregates such as to increase accuracy in molecular weight determination.Further details of light scattering are to be discussed in Light Scattering Section.Sodium Dodecyl (lauryl)Sulfate–Polyacrylamide Gel Electrophoresis ChromatographyGel electrophoresis has been used since the 1960s 95,96and has become a commonly usedversatile analytical tool for estimating protein size,identifying proteins,determining sample purity and evaluating presence of disulfide bonds to name a few applications.The detection size is limited to proteins/aggregates with a weight range between ca.5and 500kDa with the possibility to extend the weight range of an electrophoresis gel by various techniques such as gradient gels or particular buffer systems.97The use of the anionic detergent sodium dodecyl (lauryl)sulfate (SDS)in the separation of proteins into fractions has been known for over 70years 98and the combination with gel electrophoresis has become a very commonly used system for mole-cular weight determination which is also called the Laemmli system.99SDS–PAGE has the ability to detect covalently linked aggregates,or SDS nondissociable aggregates,however noncovalent associated proteins species are separated into their constituent polypeptide chains.69As an anionic detergent,SDS denatures proteinsandFigure 1.Schematic representation of the approximate range of detectable protein sizes (diameter)of various analytical methods.DOI 10.1002/jpsJOURNAL OF PHARMACEUTICAL SCIENCES,VOL.98,NO.9,SEPTEMBER 2009PROTEIN AGGREGATION 2915。
2012年美国FDA批准的新分子实体

但是它却能够与促红 和 疗 效 作 了评价 ,其 中一 项 患 者 年龄 为 1 2 岁 及 以 细胞生成素没有任何的相关性 ,
上 ,另外 一项 患 者 年 龄 为 6 - 1 1 岁 。两 项 研 究结 果 细胞生成素受体结合 ,从而促进红细胞的生成。临床 显 示 ,受试 者 经 I v a c a f t o r 治 疗 后 ,患者 的肺 功 能 研究表明,在健康志愿者以及慢性肾功能疾病的患者
相 比较 轻 ,对 眼 睛 表 面 的 损 害 较 小 ,患 者 使 用 时 结 合 ,并 且 产 生 一 个 正 电子 信 号 ,通 过 正 电子 发
的舒适性和依从性得到明显改善 。本次批准是 依 射断 层造影( P E T )扫描技术可 测定脑内淀粉样蛋 据来 自在9 0 5 例患者的五项对照临床研究疗效和安 白水 平 ,进而 对患者 的病 情进 行诊 断 。P ET 扫
遗 传性疾病 ,影 响肺和其它器官 ,最终 导致过早
死亡 。它 是 由于 一 种 基 因突 变( 缺 陷) 所 致 ,该 基 因 7 芦西纳坦 ,L u c i n a c t a n t
编码 的C F T R蛋白在体内调节离子( 如氯离子) 和水
是一种无菌 ,无热原肺表面活性剂 ,仅为气管
的半 衰期 ,故每 月给药 一次便 足以达 到治疗 目的 该
6 他 氟前列素,T a l f u p r o s t
药能够增加 心肌梗死 、静脉血栓 栓塞 、肿瘤 进展或 复
是 一种新 型P G F 2 a 衍 生物 ,对 虹膜 睫状 体F P受 发的风险,其最常见的不良反应为呼吸困难、腹泻、
V i s mo d e g i b的安全性和有效性进行了评估,在接受 全结果 。他氟前列素滴眼液被显示有强有力的降眼 V i s mo d e g i b 治疗的转移性基底细胞癌患者 中,部分 内压作用。在长达2 年时 间的临床研究 中,他氟前 缓解率为3 0 %;在局部晚期基底细胞癌患者 中,完 列素滴眼液在 傍晚每天给药1 次 ,分别在3  ̄ 1 1 6 个月 全加部分缓解率为4 3 %。该药主要的不 良反应为味 时将眼内压从基线压2 3 - 2 6 mmHg 降至6 - 8 mmHg 和
水飞蓟素通过调控Nrf2抗氧化通路抑制NLRP3炎症小体改善非酒精性脂肪性肝病

网络出版时间:2020-6-514:08 网络出版地址:http://kns.cnki.net/kcms/detail/34.1086.r.20200604.1026.032.html水飞蓟素通过调控Nrf2抗氧化通路抑制NLRP3炎症小体改善非酒精性脂肪性肝病胡 俊,饶紫兰,陈江木,房太勇(福建医科大学附属第二医院消化内科,福建泉州 362000)doi:10.3969/j.issn.1001-1978.2020.07.016文献标志码:A文章编号:1001-1978(2020)07-0971-07中国图书分类号:R 332;R322.47;R364.5;R349.1;R575.5;R977.6摘要:目的 探究水飞蓟素(Silymarin)影响非酒精性脂肪性肝病(NAFLD)病变进展的机制。
方法 溶剂、(PA+LPS)、(PA+LPS+Silymarin)3种方式干预人肝癌HepG2细胞后,分别用Westernblot、qPCR及荧光探针法检测Nrf2、SIRT2、NLRP3以及其下游Casp1P45、Casp1P20蛋白的表达水平以及Nrf2、HO 1、NQO 1的细胞mRNA表达水平以及ROS代谢水平的影响。
(PA+LPS+siNC)、(PA+LPS+Silymarin+siNrf2)、(PA+LPS+Silymarin+siNC)、(PA+LPS+siNrf2)四种方式干预HepG2细胞后,再次检测上述蛋白和mRNA等指标。
溶剂、HFD和(HFD+Silymarin)3种方式干预小鼠后,对肝脏HE病理切片进行评估,IHC检测肝脏石蜡包埋切片的NLRP3蛋白的组织表达水平的改变。
结果 Silyma rin可明显升高HepG2细胞的Nrf2和SIRT2表达水平,降低NLRP3、Casp1P20和ROS表达水平,以及降低小鼠肝脏的NLRP3表达水平。
用Nrf2 siRNA特异性抑制Nrf2之后,Si lymarin对于NLRP3、Casp1P20和ROS表达水平的下调受到明显抑制。
用于治疗结缔组织疾患的来自菠萝蛋白酶的蛋白水解提取物[发明专利]
![用于治疗结缔组织疾患的来自菠萝蛋白酶的蛋白水解提取物[发明专利]](https://img.taocdn.com/s3/m/5ef400c04793daef5ef7ba0d4a7302768e996fc3.png)
(10)申请公布号 (43)申请公布日 2014.03.26C N 103687607A (21)申请号 201280035497.X(22)申请日 2012.07.1961/509,612 2011.07.20 USA61K 36/88(2006.01)A61K 38/48(2006.01)A61P 19/02(2006.01)A61P 19/04(2006.01)(71)申请人麦迪伍德有限公司地址以色列亚夫恩(72)发明人利奥尔·罗森伯格 盖伊·鲁宾艾伦·阿斯库莱(74)专利代理机构北京安信方达知识产权代理有限公司 11262代理人高瑜 郑霞(54)发明名称用于治疗结缔组织疾患的来自菠萝蛋白酶的蛋白水解提取物(57)摘要本发明涉及从菠萝蛋白酶获得的用于治疗结缔组织疾病的蛋白水解提取物。
特别地,本发明涉及包含从菠萝蛋白酶获得的用于治疗疾病例如掌腱膜挛缩症和阴茎硬结症的蛋白水解提取物的药物组合物。
(30)优先权数据(85)PCT国际申请进入国家阶段日2014.01.17(86)PCT国际申请的申请数据PCT/IL2012/050261 2012.07.19(87)PCT国际申请的公布数据WO2013/011514 EN 2013.01.24(51)Int.Cl.权利要求书2页 说明书12页 附图8页(19)中华人民共和国国家知识产权局(12)发明专利申请权利要求书2页 说明书12页 附图8页(10)申请公布号CN 103687607 A1.一种治疗结缔组织疾病的方法,其包括对需要这种治疗的受试者施用包含治疗有效量的从菠萝蛋白酶获得的蛋白水解提取物和药学上可接受的载体的药物组合物,其中所述蛋白水解提取物包含选自由茎菠萝蛋白酶和凤梨蛋白酶组成的组的至少一种半胱氨酸蛋白酶,并且其中所述结缔组织疾病与过度胶原沉积相关联。
2.根据权利要求1所述的方法,其中所述结缔组织疾病选自由掌腱膜挛缩症、阴茎硬结症、肩周炎和Ledderhose疾病组成的组。
硫酸铵是一种中性盐
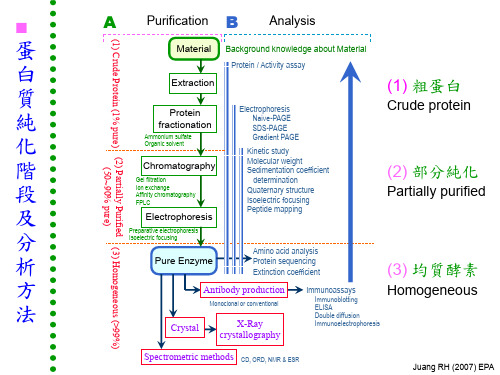
0 28 56 86 117 148 181 214 249 285 323 402 488 30 0 28 57 87 118 151 184 218 254 291 369 453 35 0 29 58 89 120 153 187 222 258 335 418 40 0 29 59 90 123 156 190 226 302 383 45 0 30 60 92 125 159 194 268 348 50 0 30 61 93 127 161 235 313 55 0 31 62 95 129 201 279 60 0 31 63 97 168 244 65 0 32 65 134 209 70
■ 硫酸銨 (ammonium sulfate) 的使用法
(1) 硫酸銨容易吸收空氣中的水分而結塊,使用前最好先把硫 酸銨磨碎,平鋪在烤箱 (約 60℃) 內烘過,切勿過熱。 (2) 添加硫酸銨時,要在冰浴中進行,不可一次把硫酸銨倒入, 而以小量分多次慢慢溶入,並不時攪拌,以免造成局部濃度過 高。 硫酸銨全加完後,再攪拌約 10 - 30 min,使溶解完全平 衡,然後進行離心,注意所要的是沉澱或上清,要弄清楚! (3) 最後所得的沉澱溶解在最少量的緩衝液中,或者以沉澱形 式保存,均 相當安定;但要記得其中所含的硫酸銨,是否對 下一步檢定或純化有影響,可以透析法除去之。
Juang RH (2007) EPA
■ 鹽影響蛋白質溶解度 Salt effects protein solubility
● 鹽溶 Salting-in:
加鹽使蛋白質溶入水溶液中
● 鹽析 Salting-out:
加鹽使蛋白質由水溶液中沉澱出來
Juang RH (2007) EPA
■ 鹽溶 Salting-in effect
Durapore

滤膜类型Durapore®亲水性滤膜•0.45 µm 内置预过滤器•0.45 µm 无内置预过滤器过滤器的类型•OptiScale®工艺开发过滤工具•Millipak®小型囊式过滤器和Millidisk®小型筒式过滤器•Opticap® XL即用型囊式过滤器•Opticap® XLT即用型囊式过滤器•筒式过滤器2符合法规亲水性 Durapore ®0.45µm 过滤器的设计、开发、和生产过程均符合ISO®9001质量体系标准所要求的质量管理体系,每个Durapore ®过滤器出厂时均附有质量证书。
每个Millipak®囊式过滤器、Opticap® XL 囊式过滤器和筒式滤芯在生产过程中均经过100%完整性测试且具有符合法规要求的验证指南支持。
为了产品的追溯和易于辨识,每个过滤器上均标有产品名称和识别特征。
OptiScale®工艺开发过滤工具OptiScale®即用型囊式过滤器为工艺筛选和放大提供了便利的小规模解决方案。
此类“内置式”过滤器适用于生物药品的评估。
OptiScale®囊式过滤器为高效地开发化合物和生物治疗药物提供了加速上市策略。
OptiScale®即用型囊式过滤器非常适用于工艺开发和筛选。
OptiScale®囊式过滤器的安装比常规的25 mm 和47 mm 滤片更加快速、简便。
Millipak®小型囊式过滤器和Millidisk®小型筒式过滤器Millidisk®筒式过滤器和Millipak®即用型囊式过滤器为层叠式圆盘式过滤器,用于去除液体和气体中的颗粒物和微生物。
Millipak®和Millidisk®过滤器采用不带支撑网的叠片式设计,能最大限度减少残留体积以及颗粒脱落现象。
perilipin1简介

perilipin1简介摘要:I.引言- 介绍perilipin1- 阐述其在生物体内的作用II.perilipin1 的结构和功能- 描述perilipin1 的结构- 详述perilipin1 在生物体内的功能III.perilipin1 与疾病的关系- 阐述perilipin1 与肥胖的关系- 介绍perilipin1 在代谢性疾病中的作用IV.perilipin1 的研究进展- 介绍当前perilipin1 的研究领域- 讨论perilipin1 作为药物靶点的可能性V.结论- 总结perilipin1 的重要性和研究意义- 展望perilipin1 未来的研究方向正文:perilipin1,全称为脂肪细胞Perilipin 1,是一种在生物体内发挥重要作用的蛋白质。
它主要存在于脂肪细胞中,并在脂肪的储存和释放过程中起着关键作用。
近年来,随着研究的深入,perilipin1 与肥胖以及代谢性疾病的关系日益受到科学家们的关注。
perilipin1 的结构相对复杂,由多个结构域组成,包括N-末端脂肪酰基辅酶A 结合域、中央α螺旋结构域和C-末端Perilipin 结构域。
这些结构域使得perilipin1 具有多种生物学功能。
在脂肪细胞中,perilipin1 能够结合脂肪酰基辅酶A,从而抑制脂肪的分解,有助于脂肪的储存。
同时,perilipin1 还能够结合细胞内的其他蛋白质,参与细胞信号传导过程。
研究表明,perilipin1 与肥胖及代谢性疾病密切相关。
在肥胖患者体内,脂肪细胞中的perilipin1 水平上升,导致脂肪的储存增加,从而引发肥胖。
此外,perilipin1 在糖尿病、高脂血症等代谢性疾病中也发挥着重要作用。
因此,研究perilipin1 在脂肪代谢中的作用机制,有助于揭示肥胖及代谢性疾病的发病机制,为治疗这些疾病提供新的思路。
近年来,perilipin1 的研究取得了重要进展。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
ReviewSimplifying protein expression with ligation-free,traceless and tag-switching plasmidsVenuka Durani a ,Brandon J.Sullivan b ,Thomas J.Magliery a ,c ,⇑aDepartment of Chemistry,The Ohio State University,Columbus,OH 43210,USAbOhio State Biochemistry Program,The Ohio State University,Columbus,OH 43210,USA cDepartment of Biochemistry,The Ohio State University,Columbus,OH 43210,USAa r t i c l e i n f o Article history:Received 17March 2012and in revised form 1June 2012Available online 21June 2012Keywords:PlasmidProtein expression Co-expression Protease cleavageLigation independent cloning Traceless tagginga b s t r a c tSynthetic biology and genome-scale protein work both require rapid and efficient cloning,expression and purification.Tools for co-expression of multiple proteins and production of fusion proteins with purifica-tion and solubility tags are often desirable.Here we present a survey of plasmid vectors that provide for some of these features with a focus on tools for rapid cloning and traceless tagging –a setup that facil-itates removal of fusion tags post-purification leaving behind no ‘scar’on the final construct.Key features are reviewed,including plasmid replication origins and resistance markers,transcriptional promoters,cloning methods,and fusion tags and their removal by proteolysis.We describe a vector system called pHLIC,which assembles features for simple cloning,overexpression,facile purification,and traceless cleavage,as well as flexibility in modifying the vector to exchange fusion tags.Ó2012Elsevier Inc.All rights reserved.ContentsIntroduction.............................................................................................................9Origin of replication .....................................................................................................10Antibiotic resistance .....................................................................................................10Cloning regions .........................................................................................................10Transcription promoters..................................................................................................12Fusion tags.............................................................................................................13Protease cleavage sites ...................................................................................................13Seamless cloning and traceless tagging.. (13)Vectors for traceless tagging –some examples............................................................................13pHLIC vectors ......................................................................................................14Conclusion.............................................................................................................15Appendix A.Supplementary data . (15)References (15)IntroductionHigh-throughput approaches in modern protein science –including structural genomics and proteomics –have necessitated the development of high-throughput methods for cloning,protein expression,and purification [1–5].Even for laboratories studying a single protein target,such as designed proteins,these steps are often expensive and time-consuming prerequisites,particularly for beginning researchers.Solutions to these problems have emerged [6–9]:recombinogenic (e.g.,Gateway [10])cloning and ligation-independent cloning (LIC)1[11,12];vectors with very1046-5928/$-see front matter Ó2012Elsevier Inc.All rights reserved./10.1016/j.pep.2012.06.007⇑Corresponding author at:Department of Chemistry,The Ohio State University,Columbus,OH 43210,USA.Fax:+16142921685.E-mail addresses:vdurani@ (V.Durani),bsulliva@ (B.J.Sullivan),magliery.1@ (T.J.Magliery).1Abbreviations used:MBP,maltose binding protein;GST,glutathione S-transferase;CBP,calmodulin binding protein;Trx,thioredoxin;PSI–MR,protein structure initiative–materials repository;LIC,ligation independent cloning;SLiCE,seamless ligation cloning extract;IPTG,isopropyl b -D -1-thiogalactopyranoside;IMAC,immo-bilized metal-ion affinity chromatography;SUMO,small ubiquitin-like modifier;Ub,ubiquitin;DUBs,deubiquitylating enzymes;TEV,tobacco etch virus;TIM,triose-phosphate isomerase.strong promoters such as T7and cspA ;and the use of fusion tags such as hexahistidine (6ÂHis)[13],maltose binding protein (MBP)[14,15],glutathione S-transferase (GST)[16,17],calmodu-lin-binding peptide (CBP)[18,19]and thioredoxin (Trx)[20]for solubility and/or purification.Many useful vectors are available through the Protein Structure Initiative Material Repository (PSI–MR),and information about these vectors is available in a search-able database called DNASU [7].While affinity tagging results in convenient purification,many applications demand removal of the fused tag post-purification,typically by proteolysis.However,the recognition sequences of restriction enzymes for cloning and of proteases for freeing the fi-nal protein typically put constraints on one or more amino acids in the protein,leaving a ‘scar’that may not be desirable.This problem has initiated work on seamless cloning and traceless tagging strat-egies,and plasmid vectors that simplify such manipulations are desirable.Plasmid vectors require various components to carry out their functions (Fig.1).The origin of replication determines the copy number of a plasmid;antibiotic resistance provides a selection marker;promoter regions facilitate gene transcription;and careful engineering of cloning sites can provide for features like ligation-independent cloning and fusion tags to facilitate solu-bility and purification of the expressed protein.Vectors with un-ique combinations of these functionalities are especially suited for specific applications in structural genomics and synthetic biol-ogy.Here we present a brief survey of plasmid vectors useful for protein expression with a focus on vectors that facilitate quick cloning and traceless tagging of proteins.Origin of replicationThe origin of replication is the smallest region of the plasmid that is able to replicate on its own and is also the site where plas-mid replication is initiated [21].The replicator region of a plasmidthat includes the origin of replication determines its copy ually for large scale protein purification we turn to high copy number plasmids that can maintain tens to hundreds of copies of plasmid per cell.Production of recombinant proteins in Escherichia coli often results in degradation,aggregation or misfolding of pro-teins,and co-expression [22]of molecular chaperones can provide a solution to this problem [23–25].Co-expression of proteins is also desirable for purification of multi-subunit complexes and studying interacting proteins [26–28].However,plasmid incom-patibility,a mechanism that prevents the stable co-existence of two similar plasmids in the same bacterial cell,can cause problems for these kinds of applications,and having access to plasmids with different origins of replication that are compatible with each other is important.The best developed class of plasmid vectors for many manipulations including protein expression is based on the ColE1/pMB1[29]replicons including the pBR322[30,31],pUC [32,33]and pET (Novagen)vector systems.Vectors containing ColE1/pMB1de-rived origins of replication fall into the same incompatibility group [34,35].On the other hand,plasmids like pACYC177and pACYC184[36,37]with origins of replication derived from p15A [38],although incompatible with other vectors with p15A derived ori-gins,are compatible with ColE1/pMB1derived plasmids.These are two compatible vector groups that are commonly used for co-expression of proteins.Since ColE1based plasmids are very common for production of target proteins,several chaperone-encoding plasmids are available on vectors with p15A derived rep-licons [23].For experiments in which the population of three or more different plasmids needs to be maintained in a cell,other plasmids with pSC101[39],CloDF13[40,41],ColA [42,43],RF1030[44]and pEC [45]replicons can be used [22].Antibiotic resistanceIn order to ensure that a plasmid vector is taken up and main-tained in a cell,genes encoding resistance to antibiotics are ex-pressed from plasmids.Moreover,if populations of multiple vectors are to be maintained in a cell,then not only do their repli-cator regions need to be compatible,their antibiotic resistance markers also need to be orthogonal to ensure co-transformation.The most common antibiotic resistance markers are ampicillin,kanamycin,chloramphenicol and tetracycline.Table 1has exam-ples of some vectors with spectinomycin,zeocin and erythromycin resistance.Ampicillin is especially susceptible to degradation when present the bacterial culture and this effect can be observed as ‘satellite’colonies on agar plates.This effect can be alleviated to some extent by using carbenicillin [46],a less degradation-susceptible analog of ampicillin.Cloning regionsMost plasmid vectors that are commercially available have multiple unique restriction enzyme sites arranged in tandem in a polylinker cloning region.These can be used for traditional liga-tion-dependent cloning methods (Fig.2a)where a target gene is amplified by PCR using primers containing unique restriction sites,followed by restriction enzyme digest and ligation into a vector containing compatible cohesive ends.These traditional methods often produce ‘background’due to incomplete digestion and re-ligation of the digested vector and require unique restriction en-zyme sites in the vector that do not occur in the DNA to be cloned.These limitations and others such as difficulty to manipulate very large inserts have led to research in recombinogenic cloning meth-ods especially for applications in structural genomics.Homologous recombination occurs through stretches of DNA shared by the two molecules.Because the sequence of the homology regions canbe1.Schematic plasmid map (not drawn to scale)showing various important regions of plasmid vectors that are discussed in this review.and Purification 85(2012)9–17chosen freely,any position on a target molecule can be specifically altered(for reviews on recombinogenic cloning see references [47–49]).Gateway cloning(Fig.2b)vectors have several features required for high-efficiency and accurate cloning using recombino-genic methods[10].Once an insert is cloned into an entry vector for Gateway cloning,it can be easily swapped into other Gateway vectors using clonase activity.This makes Gateway cloning very useful for applications where the target construct is required in various contexts like different solubility tags or different origins of replication.Ligation-independent cloning(Fig.2c)[50]addsflanking se-quence extensions to the target gene that are shorter than those used in recombinogenic methods while still avoiding some of the drawbacks of traditional cloning methods[51].By taking advan-tage of the controlled exonuclease activity of T4DNA polymerase, the LIC procedure creates long single-stranded overhangs of DNA in the vector and the insert.These overhangs hybridize on mixing the vector and insert together.When transformed into E.coli,the nicks are sealed by the cellular machinery.Hence no use of ligase is required,and since the vector has two fairly large overhangs which can be designed to be completely non-complementary to each other,the chances of re-sealing the vector are very small. Moreover,since this scheme does not require digestion of the in-sert with any restriction enzymes,there are no complications regarding multiple occurrences of the preferred cloning enzymes within the sequence to be cloned.A new cloning method calledTable1List of plasmid vectors.Vector name Fusion tags a Cleavage sites‘Scar’residues post-cleavage b Cloning method Promoter region Ori Res.Ref.Origins of replication and antibiotic resistance markers:pET11a T7None13amino acids Ligation T7ColE1Amp[99] pURI2-Ery6ÂHis EK protease M LIC lpp ColE1Ery[100] pACYCDuet-16ÂHis None13amino acids Ligation T7p15A Cmp[101] pT7LICK6ÂHis(Ct)None DHHHHHH(Ct)LIC T7pSC101Kan[12] pEC None N.A.N.A.Ligation P BAD pEC Kan[45] pMCSG216ÂHis TEV protease SNA LIC T7Clo-DF13Spec[102] pCOLADuet-16ÂHis None14amino acids Ligation T7ColA Kan[43] Cloning methods:pET32a Trx-6ÂHis EK protease AMA Ligation T7ColE1Amp[99] pVP168ÂHis-MBP TEV protease S Gateway T5ColE1Amp[8] pNIC28-Bsa46ÂHis TEV protease SM LIC T7ColE1Kan[4] pBL None N.A.N.A.SLiCE T7ColE1Amp[52] pCR-BluntII-None N.A.N.A.TOPO T7,SP6ColE1Kan,[55,56] TOPO ZeoTranscription promoters:pVP33A8ÂHis-MBP3C and TEV AIA Ligation T5ColE1Amp[8] pURI26ÂHis EK protease M LIC lpp ColE1Amp[13] pURI2-TEV6ÂHis TEV protease GM LIC lpp ColE1Amp[100] pURI36ÂHis EK protease M LIC T7ColE1Amp[13] pBAD/His6ÂHis EK protease DRWGSELE Ligation P BAD ColE1Amp[103] pCKSP6None N.A.N.A.Ligation SP6ColE1Amp[104] PinPoint Xa1Biotin Factor Xa None Ligation tac ColE1Amp[105] pCOLD I6ÂHis Factor Xa HM Ligation cspA ColE1Amp[72] Tag-switching plasmids:pHLIC6ÂHis TEV protease None LIC T7ColE1Amp[98] pHLIC-MBP6ÂHis-MBP TEV protease None LIC T7ColE1AmppMCSG76ÂHis TEV protease SNA LIC T7ColE1Amp[11] pMCSG96ÂHis-MBP TEV protease SNA LIC T7ColE1Amp[95] pMCSG106ÂHis-GST TEV protease SNA LIC T7ColE1Amp[102] pMCSG116ÂHis TEV protease SNA LIC T7p15A Cmp[102] pMCSG136ÂHis-MBP TEV protease SNA LIC T7p15A Cmp[97] pMCSG146ÂHis-GST TEV protease SNA LIC T7p15A Cmp[97] pLIC-His6ÂHis TEV protease GAAS LIC T7ColE1Amp[2] pLIC-MBP6ÂHis-MBP TEV protease GAAS LIC T7ColE1Amp[2] pLIC-GST6ÂHis-GST TEV protease GAAS LIC T7ColE1Amp[2] pOPINF6ÂHis3C protease GP LIC T7ColE1Amp[1] pOPINM6ÂHis-MBP3C protease GP LIC T7ColE1Amp[1] pOPINJ6ÂHis-GST3C protease GP LIC T7ColE1Amp[1] Protease cleavage sites:pMCSG19MBP-6ÂHis TVMV and TEV SNA LIC T7ColE1Amp[95] pVP33K8ÂHis-MBP3C and TEV AIA Ligation T5ColE1Kan[8] pCAL-n-EK CBP EK protease None LIC T7ColE1Amp[19] pHUE6ÂHis-Ub Usp2-cc None Ligation T7ColE1Amp[77] pHLIC-smt36ÂHis-SUMO SUMO protease None LIC T7ColE1AmpC-terminal His-tags:pT7LICB6ÂHis(Ct)None DHHHHHH(Ct)LIC T7ColE1Amp[12] pT7LICC6ÂHis(Ct)None DHHHHHH(Ct)LIC T7p15A Cmp[12] pT7LICK6ÂHis(Ct)None DHHHHHH(Ct)LIC T7pSC101Kan[12] pURI2-Cter6ÂHis(Ct)None HHHHHH(Ct)LIC lpp ColE1Amp[100]Ori,origin of replication;Res,antibiotic resistance marker;Ref,reference;Amp,ampicillin;His,histidine tag;EK,enterokinase;LIC,ligation independent coning;Ery, erythromycin;Cmp,chloramphenicol;Ct,C-terminal;Kan,kanamycin;N.A.,not applicable;TEV,Tobacco Etch Virus;Spec,spectinomycin;Trx,thioredoxin;MBP,maltose binding protein;Zeo,zeocin;3C,human rhinovirus3C protease site;GST,glutathione S-transferase;SUMO,Small Ubiquitin-like Modifier;TVMV,tobacco vein mottling virus protease;CBP,calmodulin binding protein.a All the fusion tags are on the N-terminus unless otherwise specified as Ct(C-terminus).b Extra N-terminal(unless otherwise specified)residues that remain after protease cleavage due to the design of the cloning site are listed in one letter amino acid codes.In some cases more residues may be required based on efficiency of protease cleavage.For vectors that have a tag but no cleavage site,the length of the tag is mentioned.V.Durani et al./Protein Expression and Purification85(2012)9–1711SLiCE(Seamless Ligation Cloning Extract)relies on in vitro recom-bination in bacterial cell extract(Fig.2d)[52].Extracts from the two recAÀstrains DH10B and JM109yielded the highest cloning efficiencies indicating that SLiCE is facilitated by a RecA-indepen-dent mechanism.SLiCE facilitates seamless cloning by recombining short end homologies(>15bp)with or withoutflanking heterolo-gous sequences.Seamless cloning using SLiCE has been demon-strated using plasmid vector pBL[52].Other recently-reported methods of cloning that fall into this category include Gibson clon-ing(enzymatic assembly of DNA molecules using T5exonuclease, Phusion polymerase and Taq ligase)[53]and CPEC(Circular Poly-merase Extension Cloning that involves extension of the overlap between single stranded vector and insert to form a complete plas-mid)[54].TOPO cloning(Fig.2e)is a method of cloning where a vector and insert with single nucleotide overhangs are ligated together by tak-ing advantage of topoisomerase activity[55].The single nucleotide overhang in the insert is created upon amplification by Taq poly-merase and this technique,like LIC,negates the use of restriction enzyme digest for the insert.The vector pCR-BluntII-TOPO(Invitro-gen)uses TOPO cloning in conjunction with the ccdB(control of cell death)gene for zero background cloning[56].The CcdB protein causes cell death by poisoning bacterial DNA gyrase,which leads to degradation of the host chromosome[57,58].Ligation of an in-sert into the vector disrupts the ccdB gene and enables the recom-binant colonies to grow.This method eliminates background from self-ligation of the vector.Transcription promotersPromoter regions affect the level of gene expression in E.coli,and the suitability of a promoter for protein expression depends on three main factors.First,the promoter must be strong and have the capacity to over-produce proteins to the extent that they be-come a significant percentage of the total expressed protein in the cell.Second,it is desirable to have a highly tunable(or at least highly repressible)promoter particularly for proteins that have adverse ef-fects on the growth of the host cell.Third,the promoter should be easily inducible typically by chemical or thermal means.Several promoter systems used for protein production in E.coli,their regu-lation mechanisms,and methods of induction can be found in refer-ences[59,60].Vectors with SP6(phage promoter,mainly used for cell free expression[61,62]),T5(phage promoter,induced by IPTG, very high level of expression,tight regulation possible[63]),hybrid lpp(E.coli promoter,induced by IPTG,very high level ofexpression Fig.2.Cloning schemes for(a)ligation-based cloning;(b)ligation-independent cloning;(c)recombinogenic cloning(Gateway);(d)SLiCE cloning;and(e)TOPO cloning.[64]),P BAD(E.coli promoter,induced by L-arabinose,variable level of expression from low to high,tight regulation possible[65]),tac (E.coli hybrid promoter,induced by IPTG,moderately high level of expression,high basal level of expression[66]),cspA(E.coli pro-moter,cold shock induction at temperatures below20°C,low level of expression[67])and T7(phage promoter,induced by IPTG,very high level of expression,tight regulation possible[68])promoters are listed in Table1.The pET vector system that has the promoter system from bacteriophage T7has been very widely used due to the orthogonality of this system to E.coli promoters,leading to ease of cloning toxic genes and also its capacity to overproduce proteins at very high levels constituting up to50%of the total cell protein [69–71].The T7promoter,along with some of the other promoters listed above,is indirectly induced by IPTG in that T7RNA polymer-ase is expressed from the lac promoter-operator.One of the limitations of the T7promoter system,however,is that rapid over-expression of relatively ill-behaved proteins can lead to pro-tein misfolding,aggregation and segregation into inclusion bodies. Although using a cold shock promoter like cspA[67](pCold vectors by Takara Bio Inc.[72])is sometimes a solution to this problem,a major disadvantage of that system is that it becomes repressed1–2h after lowering the temperature,and this time frame might not be sufficient for a high level accumulation of all proteins.Fusion tagsBesides cold shock,there are several approaches to the expres-sion of proteins that end up in the insoluble fraction,including the co-overexpression of chaperones[23–25],periplasmic expres-sion of proteins[73]and the use of solubilizing fusion tags.Protein fusions are efficient tools for protein purification,detection and immobilization.One of the most commonly used fusion tags for protein purification is the hexahistidine(6ÂHis)[13]tag that facil-itates purification by immobilized metal-ion affinity chromatogra-phy(IMAC)purification with Ni–NTA agarose.This method is inexpensive;the resin has very high capacity and can be reused many times.It is less expensive than other resins such as streptavi-din agarose used for purification of biotin-tagged proteins,and it has a higher capacity than the less-expensive amylose resin used to purify proteins with an MBP tag[59,74].Although fusion tags were originally developed for purification purposes,it was found that many tags lead to better solubilization of proteins fused at their C-termini.It is believed that the general mechanism for this effect involves the fast and efficient folding of the N-terminal fusion tag upon exiting the ribosome immediately after translation,thus pulling the C-terminally fused protein into solution.The most com-monly used fusion tags for solubility enhancement are maltose binding protein(MBP)[14,15],calmodulin-binding peptide(CBP) [18,19],glutathione S-transferase(GST)[16,17],and thioredoxin (Trx)[20].More recently it has been shown that the N-terminal fu-sion of SUMO protein[75,76],ubiquitin[77],and HaloTag7[78]also enhance expression levels and solubility of ill-behaved proteins.Several fusion tags can have multiple functionalities;for instance, MBP and HaloTag7can be used for both solubility enhancement and purification.Similarly,SUMO and ubiquitin(Ub)protein fusions can be used as both solubility tags and also protease cleavage tags.Some-timesfinding the optimum tag for an application requires some trial and error and also some applications may require the same protein with different tags.Hence,tag switching vectors where the same in-sert can be used to clone with various tags are desirable.Protease cleavage sitesEven though fusion tags often solubilize proteins and provide options for easy purification,it is often desirable to remove them after purifimon proteases for this purpose include TEV protease[79–81]and related3C protease[82],enterokinase [83–85],Factor Xa[86],thrombin[87,88]and SUMO protease [76].TEV protease,the capsid protease of the tobacco etch virus, is among the most robust of the proteases in common use[89]. It is more specific than enterokinase[85]or Factor Xa[90],and it is active under a wide range of conditions.It is commercially avail-able and can also be overexpressed and purified from E.coli using a standard protocol[91].The general form of its recognition se-quence is taken to be E-X-X-Y-X-Q-(G/S),with cleavage after the Gln residue;the most commonly used sequence is ENLYFQG.Re-cently,Kapust et al.demonstrated that the TEV protease will cleave with greater than90%efficiency with12different amino acids at the P10site(C-terminal to Gln)and greater than50%efficiency for all other amino acids except proline[92].Effectively,TEV pro-tease can be used to quantitatively produce mature proteins with any N-terminal amino acid except Pro under reasonable reaction conditions.However,proteins with structured N-terminal regions sometimes do not display very efficient cleavage with TEV protease presumably due to inaccessibility of the protease recognition and cleavage site.For such cases,having an alternative option is desir-able,such as Factor Xa,which cleaves I-(E/D)-G-R-X between R and X,where X can be any amino acid except proline[93].Another op-tion is the SUMO protein fusion.In this case,the100residue SUMO protein serves as the recognition site for SUMO protease,which cleaves after the C-terminal Gly of SUMO.It has been shown that recombinant SUMO–GFP fusions are efficiently cleaved when any amino acid except proline is in P1´position of the cleavage site [76].Similar to SUMO,the ubiquitin tag also serves as a recognition site for deubiquitylating enzymes(DUBs),however most DUBs that have been isolated from various species are relatively large and dif-ficult to express and purify.No stable DUB known with general activity against a range of fusion proteins was known.Recently,a mouse DUB,Usp2,was engineered to provide a minimal catalyti-cally active deubiquitylating domain(Usp2-cc)[77].It was ex-pressed and purified with a polyhistidine tag that allowed the in vitro cleavage of Ub from the desired protein as well as its selec-tive removal from the cleavage reaction.Seamless cloning and traceless taggingGeneration of the overhang for cloning into a vector requires restriction enzyme scission,which places a limitation on the se-quence of the overhang and usually results in a particular protein sequence at the site of fusion.A way to get around this problem and accomplish‘seamless’cloning[94]is to digest with a type IIs restriction enzyme which cuts outside of its recognition sequence and therefore in any sequence.(GeneArt by Invitrogen and CloneEZ by GenScript are kits that can be used for seamless cloning.)Even for routine protein expression and purification,it would be useful to have a veryflexible plasmid vector for high-level E.coli overex-pression of proteins using an N-terminal6ÂHis tag that could be removed tracelessly–which is to say,with no residues left behind and few or no constraints on the amino acid next to the scissile bond.To circumvent some issues with ligation-dependent cloning, a ligation-independent method is desirable,but with minimal ex-pense,and with theflexibility to use ligation-dependent methods, as well.Several vectors which nearlyfit these criteria are available, perhaps most notably the pMCSG7vector[11]and the pHLIC vec-tor system(described here).Vectors for traceless tagging–some examplesToday several plasmid vectors are available that have cloning sites designed to attach fusion tags and also provide for proteaseV.Durani et al./Protein Expression and Purification85(2012)9–1713cleavage sites to remove the fusion tags later.These vectors have combinations of different methods of cloning,different fusion tags and different protease cleavage sites (several examples are listed in Table 1).For instance,vectors pURI [13]and pCAL-n-EK [19]can be used for ligation-independent cloning,and use enterokinase,which is not as specific as TEV and also more expensive.Vectors like pOPINJ [1],pLIC [2],pNIC28[4],pVp [8]allow for TEV cleavage of 6ÂHis tag,optionally C-terminal)6ÂHis tag to a target protein cloned by LIC under the control of the T7promoter.Variants of this vector that allow for TEV cleavable MBP and GST fusions are also available and this series has some ColE1derived vectors and others with p15A derived replicons for co-expression [97].However,the LIC cloning scheme designed for these vectors requires that the cleaved N-terminal sequence begin with Ser-Asn-Ala.We have constructed a related vector in which there are no constraints on the N-terminal sequence,and in which the protease recognition se-quence can be removed or changed by a simple standard cloning step.We call the resulting vector pHLIC (plasmid for 6ÂHis tagging and Ligation Independent Cloning,Fig.3and Supplementary material ).To achieve high-level expression and simple,inexpensive purifi-cation,we selected a pET vector system with a T7promoter for overexpression in DE3-lysogenized cells,and we selected an N-ter-minal 6ÂHis-tag.It would be difficult to achieve ‘‘traceless’’scission with a C-terminal tag,since virtually all useful (highly specific and active)proteases have their recognition elements upstream of or overlapping the scissile bond.To truly make this traceless,we se-lected the robust and easy-to-prepare TEV protease,which cleaves Fig.3.Schematic maps of the pHLIC and pHLIC-MBP plasmids.The AlwNI site is not unique in the pHLIC-MBP vector.4.LIC scheme for pHLIC.BsaI digested pHLIC vector (a)is treated with T4DNA polymerase in the presence of dGTP to create large overhangs (b).The PCR product for insert (c)is treated with T4DNA polymerase in the presence of dCTP to create complementary overhangs (d).Then (b)and (d)are mixed together leading to the formation desired clone by hybridization of the overhangs of the vector and insert.This product has nicks,at positions indicated by arrows in (e),which are resolved in the cell upon transformation.and Purification 85(2012)9–17。