Preparation of A2 reverse grouping cells from A2B red blood cells
血管紧张素Ⅱ(ANGⅡ)检测试剂盒 操作步骤

血管紧张素Ⅱ(ANGⅡ)检测试剂盒(酶联免疫吸附试验法)检测范围:具体详见说明书检测种属:人、大小鼠、猪、犬、羊、兔、牛、马铃薯等动植物液体类标本: 包括血清、血浆、尿液、胸腹水、脑脊液、细胞培养上清等血管紧张素Ⅱ(ANGⅡ)检测试剂盒价格:欢迎咨询在线客服QQ:21570372 QQ:865468944血管紧张素Ⅱ(ANGⅡ)检测试剂盒Elisa实验原理:抗原或抗体能以物理性地吸附于固相载体表面,可能是蛋白和聚苯乙烯表面间的疏水性部分相互吸附,并保持它的免疫学活性;血管紧张素Ⅱ(ANGⅡ)检测试剂盒操作步骤1.标准品的稀释:准备小试管6只,依次编好号码,先在各小试管中加入标准品稀释液100ul,然后取原浓度标准品100ul加入一只已编好号的试管中,充分混匀;再在该试管中取100ul加入第二支试管中,充分混匀;再在该试管中取100ul加入第三只试管中,充分混匀;再在该试管中取100ul加入第四只试管中,充分混匀;再在该试管中取100ul加入第五只试管中,充分混匀;然后在该试管中取100ul,弃掉。
第六只试管作为0号标准品。
稀释后各管浓度分别是:320 pg/ml,160 pg/ml,80 pg/ml,40 pg/ml,20pg/ml,0 pg/ml。
在酶标包被板上设标准品孔,依次加入不同浓度的标准品50ul(建议每个浓度做2个平行孔)。
2.加样:分别设空白孔(空白对照孔不加样品、酶标试剂及生物素标记的抗IL-6抗体,其余各步操作相同)、待测样品孔。
在酶标包被板上待测样品孔中先加样品40μl,然后再加生物素标记的抗IL-6抗体10μl。
加样将样品加于酶标板孔底部,尽量不触及孔壁,轻轻晃动混匀。
3.温育:用封板膜封板后置37℃温育30分钟。
4.配液:将30倍浓缩洗涤液用蒸馏水30倍稀释后备用。
5.洗涤:小心揭掉封板膜,弃去液体,甩干,每孔加满洗涤液,静置30秒后弃去,如此重复5次,拍干。
6.加酶:每孔加入酶标试剂50μl,空白孔除外。
小鼠Ⅰ型胶原α2 (COL1a2)-ELISA试剂盒说明书
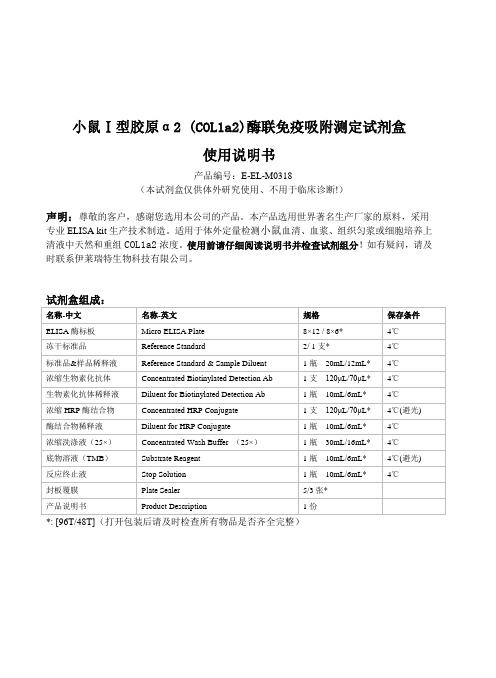
小鼠Ⅰ型胶原α2 (COL1a2)酶联免疫吸附测定试剂盒使用说明书产品编号:E-EL-M0318(本试剂盒仅供体外研究使用、不用于临床诊断!)声明:尊敬的客户,感谢您选用本公司的产品。
本产品选用世界著名生产厂家的原料,采用专业ELISA kit生产技术制造。
适用于体外定量检测小鼠血清、血浆、组织匀浆或细胞培养上清液中天然和重组COL1a2浓度。
使用前请仔细阅读说明书并检查试剂组分!如有疑问,请及时联系伊莱瑞特生物科技有限公司。
*: [96T/48T](打开包装后请及时检查所有物品是否齐全完整)检测原理:本试剂盒采用双抗体夹心ELISA法。
用抗小鼠COL1a2抗体包被于酶标板上,实验时标本或标准品中的COL1a2会与包被抗体结合,游离的成分被洗去。
依次加入生物素化的抗小鼠COL1a2抗体和辣根过氧化物酶标记的亲和素。
抗小鼠COL1a2抗体与结合在包被抗体上的小鼠COL1a2结合、生物素与亲和素特异性结合而形成免疫复合物,游离的成分被洗去。
加入显色底物(TMB),TMB 在辣根过氧化物酶的催化下现蓝色,加终止液后变黄。
用酶标仪在450nm波长处测OD值,COL1a2浓度与OD450值之间呈正比,通过绘制标准曲线求出标本中COL1a2的浓度。
标本收集:1.血清:全血标本于室温放置2小时或4℃过夜后于1000×g离心20分钟,取上清即可检测,收集血液的试管应为一次性的无热原,无内毒素试管。
2.血浆:抗凝剂推荐使用EDTA.Na2,标本采集后30分钟内于1000×g离心15分钟,取上清即可检测。
避免使用溶血,高血脂标本。
3.组织匀浆:用预冷的PBS (0.01M, pH=7.4)冲洗组织,以去除残留血液(匀浆中裂解的红细胞会影响测量结果),称重后将组织剪碎。
将剪碎的组织与对应体积的PBS(一般按1:9的重量体积比,比如1g的组织样本对应9mL的PBS,具体体积可根据实验需要适当调整,并做好记录。
3-Studies on the purification of__ peroxidase from horseradish roots using reverse micelles

Address reprint requests to Prof. Pyle, Biotechnology and Biochemical Engineering Group, University of Reading, Reading, Berks, RG6 2AP, United Kingdom Received 23 September 1994; revised 6 February 1995; accepted 15 February 1995
Keywords:Reverse micelles; protein purification; peroxidase purification; AOT; horseradish peroxidase Introduction
Reverse micelles can solubilize within their hydrophilic core water and macromolecules such as hydrophilic proteins, nucleic acids, and other entities, a-4 The surfactant shell layer generally protects enzymic proteins from denaturation by the organic phase, resulting in little or no damage to their catalytic activity. 5-9 It has been shown that on back extraction, the protein recovers near full activity1°-14; however, changes in conformation and activity of some proteins have been observed. 15-~8 A potential biotechnological application of reverse micelles is as an alternative solvent extraction technique in protein purification in particular, for prepurification and bulk reduction as one of the first steps in downstream processing. The concentration of a single protein using a continuous extraction and stripping process has been successfully achieved, 19 as have the separation of simple synthetic protein mixtures, 2° the purification of some enzymes from whole fermentation broths and crude preparations, 21'22 and the direct recovery of intracellular proteins from bacteria or yeasts. 23-26 This work was concerned with the purification of peroxidases. Peroxidase from soybean hulls has been purified by a combination of protein precipitation, diafiltration, and affinity-based reverse-micellar extraction. 27 In this study we explored the possibility of purification without the use of ligands. Here, we report the reverse-micellar purification of horseradish peroxidase (HRP) (E.C. 1.11.1.7) from a crude extract of horseradish roots (Armoracia rusticana). H R P occurs as multiple isoenzymes with a molecular weight of around 41.6 kDa28: The study focuses on the influence of the operational conditions on the selectivity of solubilization. The anionic surfactant AOT [aerosol-OT (sodium-bis[2ethylhexyl]sulphosuccinate)] was dissolved in 2,2,4-trimethylpentane (isooctane) to produce the reverse-micellar phase. A O T was chosen because it can solubilize positively
RACE步骤

1)RNA 提取采用试剂Trizol Reagent (购自invitrogen 公司)2)氯仿;异丙醇;70 % 乙醇;0.1 % DEPC;DEPC 处理的超纯水(2) 聚合酶链式反应(PCR)、核酸电泳试剂1) Tris-硼酸储存(5×TBE)缓冲液:54g Tris 碱,27.5 g 硼酸,20 ml 0.5 mol/L EDTA(pH 8.0)溶于1000 ml 水。
2) 溴化乙啶(10 mg/ml):100 ml 水中加入1 g 溴化乙啶,磁力搅拌数小时至溶解,避光保存3)点样缓冲液Loading buffer(10×): 0.25 %溴酚蓝,40 %甘油。
4)Ex Taq DNA Polymerase(TaKaRa),琼脂糖DNA 胶回收试剂盒(北京天根生物公司)5)DNA Makers:DL2000,DNA Maker Ⅲ6) 特异引物(上海生工合成)7) 5'RACE 试剂盒购置大连宝生物工程有限公司(3) 目的基因鉴定、培养基及培养液1)液体培养基:胰蛋白胨10.0g,酵母提取物5.0 g,NaCl 10.0g,调pH 至7.0,定容至1L,高压灭菌。
2)固体培养基:加琼脂15g/L,高压灭菌。
3)氨苄50μg/mL;IPTG 25 mg/mL;X-Gal 25 mg/mL 溶于DMF 中4)5×KCM 溶液3.73 g KCl,2.21 g CaCl2,5.08 g MgCl2,定容至100 ml。
总RNA 的提取实验用品的处理:离心管、枪头等塑料制品用0.1 % DEPC于37 ℃浸泡2h,和0.1 %的DEPC 水高压灭菌30min,研体、剪刀、镊子、容量瓶等180℃烘烤6h。
1)称取100 mg小麦叶片,液氮研磨至粉末状,迅速转入含1mL Trizol的1.5 ml离心管中,混匀,室温下静置5 min。
2)加0.2 ml氯仿,剧烈振摇15 sec,20 ℃静置3 min。
七氟醚下调TRPV4

前期研究发现,机械通气可上调花生四烯酸(AA )代谢途径关键限速酶胞质型磷脂酶A2(C-PLA2)活性使肺内AA 及其致炎性代谢产物生成增加继而引起VILI ,而七氟醚可通过阻断上述病理过程发挥其抗VILI 保护作用[1,2],但机械通气及七氟醚调控C-PLA2的具体机制尚未完全阐明。
由于细胞内钙离子浓度的增加是C-PLA2活化必不可少的条件[3],而TRPV4作为一种位于细胞膜上的钙离子通道,可被机械力直接激活[4],活化的TRPV4可上调C-PLA2表达继而引起高血压小鼠动脉内皮细胞收缩[5]。
体外实验研究发现,机械通气可激活TRPV4造成肺屏障功能破坏[6],而七氟醚可通过阻断瞬时受体电位钙离子通道发挥其心肌保护作用[7]。
据此我们推测机械通气所产生的机械力激活C-PLA2的具体机制与TRPV4有关,而七氟醚可通过阻断TRPV4/C-PLA2信号通路发挥其抗VILI 保护作用。
Sevoflurane alleviates ventilator-induced lung injury in rats by down-regulating the TRPV4/C-PLA2signaling pathwayWANG Wenfa 1,YANG Yong 2,WANG LI 3,GUO Xin 3,TIAN Lingfang 1,WANG He 1,HU Yuzhen 1,LIU Rui 31Department of Anesthesiology,Chuxiong Yi Autonomous Prefecture People's Hospital,Chuxiong 675000,China;2Experimental Center of Medical Function,Kunming Medical University,Kunming 650500,China;3Department of Anesthesiology,First People's Hospital of Yunnan Province/Affiliated Hospital of Kunming University of Science and Technology,Kunming 650032,China摘要:目的探讨七氟醚抗呼吸机诱导的肺损伤(VILI )的保护作用机制。
Appl Microbiol Biotechnol(2006)71833–839

Appl Microbiol Biotechnol(2006)71:833–839DOI10.1007/s00253-005-0207-3BIOTECHNOLOGICALLY RELEVANT ENZYMES AND PROTEINSJingxue Wang.Haijin MouXiaolu Jiang.Huashi GuanCharacterization of a novelβ-agarase from marine Alteromonas sp.SY37–12and its degrading productsReceived:22August2005/Revised:25September2005/Accepted:28September2005/Published online:24November2005 #Springer-Verlag2005Abstract The phenotypic and agarolytic features of anunidentified marine bacteria isolated from the southernocean of China was studied.The strain was gram-negative,aerobic,and polarly flagellated.It was identified as thegenus Alteromonas according to its morphological andphysiological characterization.In solid agar,the isolateproduced a diffusible agarase that caused agar softeningaround the colonies.An extracellular agarase was purifiedby the procedure of ammonium sulfate precipitation,gelfiltration on Sephacryl S-100HR,and ion-exchange chro-matography on diethylaminoethyl-Sepharose.The purifiedprotein exhibited a single band on SDS-PAGE with amolecular mass of39.5kDa.The enzyme hydrolyzed the β-1,4-glycosidic linkages of agar,yielding neoagarote-traose and neoagarohexaose as the main products.Theoptimum reaction temperature of the agarase was35°C,with a narrow range from30to45°C.The enzyme activityreached the maximum at pH7.0and in the presence of2%NaCl.Molecular mass and degrading products showed thatthe agarase from Alteromonas sp.SY37-12was muchdifferent from those previously reported.IntroductionAgar,as an important food additive and bacterial culturemedium,is one of the most well-known marine poly-saccharides.Additionally,various potential physiologicalactivities of polysaccharides and oligosaccharides derivedfrom agar have been reported,such as antivirus(Takemoto1966),antitumor(Fernandez et al.1989),immune en-hancement(Yoshizawa et al.1993),antioxidation(Wang et al.2004;Zhang et al.2003),and elicitor activity on plant (Weinberger et al.2001).Agar has been determined to have a linear chain structure composed of alternating residues[O-3,6-α-anhydro-L-galac-topyranosyl(1!3)O-β-D-galactopyranose]combined byβ-1,4bonds(Hamer et al.1977).It is confirmed that agar oligosaccharide can be attained by many methods,including chemical degradation and enzyme hydrolysis.A special enzyme hydrolyzing the agar,agarase(agarose4-glycanohy-drolase,E.C.3.2.1.81),has been found in certain marine mollusks(Usov and Miroshnikova1975).However,the most was reported from several bacterial genera,including Cytophaga (Van der Meulen et al.1974),Vibrio(Aoki et al.1990), Actinomyces(Stanier1942),Alteromonas(Leon et al. 1992),Pseudoalteromonas(Vera et al.1998),and Pseu-domonas(Ha et al.1997),etc.Most of these bacteria were isolated from marine environments,while a few species isolated from rivers(Agbo and Moss1979),hot spring (Shieh and Jean1998),soil(Sampietro and Vattuone de Sampletro1971),and sewage(van Hofsten and Malmqvist 1975)have also been described.Agarase-producing bacteria can be classified into two groups according to the mode of action on agar:α-agarases cleave theα-1,3linkage of agarose(Young et al.1978)and β-agarases cleave theβ-1,4linkage of agarose(Duckworthand Turvey1969).Some of the agarase have been purified and characterized in the past years.In our laboratory,we have isolated a marine bacterium,Alteromonas sp.strain37-12, which has the high ability to decompose the agar from the southern ocean of China.We describe here the identification of this strain,the characterization of its extracellularβ-agarase,and the composition of its degrading products.Materials and methodsIsolation of strains and cell growthThe agarase-producing strain SY37-12was isolated in this laboratory from the surface of rotted red algae in the South China Sea coast in Sanya,Hainan Island.The screeningJ.Wang.H.Mou(*). X.Jiang.H.GuanOcean University of China, Qingdao,266003,PR Chinae-mail:mousun@ Tel.:+86-532-82032290 Fax:+86-532-82894024was carried out on agar plates in a medium containing2.0% NaCl,0.1%K2HPO4,0.1%NH4Cl,0.05%MgSO4,0.01% CaCl2,and1.5%agar.The plates were incubated at33°C for48h.Colonies that formed pits or clearing zones on agar were picked up and purified further by the same plating method.For liquid culture,0.2%agar was added before sterilization.The agar used here was commercial agar powder extracted from Gracilaria verrucosa(Mingfu Sea-weed Industry Co.,Fujian Province,China).Alteromonas sp.strain37-12is stored now in the China Center for Type Culture Collection(CCTCC),with strain number M204009.Evaluation of agarase activityAgarase activity was determined by measuring the increase in the concentration of reducing sugar as described by von Borel et al.(1952).The isolated colonies were inoculated into a medium of the same composition as that of isolation medium,except that the agar concentration was lowered to 0.2%.The cells were incubated in an orbital shaker at 150rpm and33°C for24h.After centrifuging at7,000×g for15min to remove bacterial cells and gel residues,a1-ml culture supernatant was added to20ml pH7.0phosphate buffered saline solution(PBS)with0.5%agar substrate and incubated at33°C for30min.Then the1-ml reaction solution was mixed with1.5ml3,5-dinitrosalicylic acid (DNS)reagent.After being heated at100°C for5min and then cooled,the mixture was diluted to25ml with deionized water.Optical density was read at520nm,and values for reducing sugars were expressed as D-galactose equivalents.One unit of agarase activity was defined as the amount of enzyme that released1μmol reducing sugar (measured as D-galactose)from agar per minute under the above conditions.Purification of agaraseUnless specified otherwise,all operations were done at 4°C.An overnight culture of strain SY37-12was prepared in the medium described above and transferred to250ml of fresh medium containing0.2%agar.Incubation was carried out in an orbital shaker at150rpm and33°C for24h until the stationary phase,followed by centrifugation at7,000×g for15min.The supernatant was brought to80%saturation with solid ammonium sulfate overnight.The collected enzyme protein was resuspended in20ml of50mM Tris–HCl(pH7.5),sealed in dialysis bags(cut-off value, 12,000–14,000MW),and dialyzed three times against the same buffer at4°C for2days.The dialyzate was loaded onto a diethylaminoethyl(DEAE)-Sepharose column (1.5cm×20cm,Pharmacia)equilibrated with50mM Tris–HCl(pH7.5).The flow rate was adjusted to0.5ml/ min.The protein was eluted from the column with the same buffer containing a linear NaCl gradient(0∼1.0M),and the elute fractions were collected.The eluates were monitored continuously at280nm,and fractions were assayed for activity against agar.Fractions containing agarase activity were pooled and concentrated by polyethylene glycol20,000,then applied for gel filtration chromatography on Sephacryl S-100HR (1.2cm×92cm,Pharmacia)equilibrated with50mM Tris–HCl(pH7.8).The flow rate was adjusted to0.7ml/min. Fractions were assayed for protein and agarase activity. The enzyme was lyophilized and stored at−20°C and was stable for more than2months.The purification of the fractions was assessed by sodium dodecyl sulfate polyacrylamide gel electrophoresis(SDS-PAGE).SDS-PAGE was performed in a0.75-mm slab gel consisting of a stacking gel(5%polyacrylamide)and a separating gel(12.5%polyacrylamide)with25mM Tris–HCl buffer,pH7.8.Proteins were stained with Coomassie brilliant blue R-250.Molecular mass standards were phosphorylase (97.4kDa),bovine serum albumin(66.2kDa),ovalbumin (42.7kDa),carbonic anhydrase(31kDa),soybean trypsin inhibitor(21.5kDa),and lysosyme(14.4kDa).Effects of temperature and pH on enzymeactivity and stabilityThe optimum temperature of agarase was determined at different temperatures(from20to50°C).Temperature stability was determined by measuring the residual ac-tivities after incubation at different temperatures.These studies were carried out in sealed Eppendorf tubes that were completely immersed in a water bath at the required temperatures.Samples(1ml)were removed after incuba-tion at each of the indicated time periods,chilled on ice and then assayed for enzyme activity.The optimum pH was determined with agar as substrate dissolved in KH2PO4–NaOH buffer with different pH values(5.0–9.0).The optimum NaCl concentration in the reaction solution was determined in PBS solution(pH7.0) with different NaCl concentration(0–5.0%).Preparation of agar oligosaccharidesTen grams of agar powder was scattered in500ml deionized water.A50-ml agarase solution(total agarase activity was90U)extracted from strain SY37-12was added to the agar solution.The reaction was carried out at 33°C for12h,and then was stopped by heating the solution in boiling water for10min.Twofold ethanol was added to the reaction mixture to remove the high-molecular-mass polysaccharides.After centrifugation and concentration, the depolymerized end-products were produced by re-peated ethanol fractionation.Structural analysis of agar oligosaccharidesThe molecular mass distribution of the agar oligosaccha-rides was determined by matrix-assisted laser desorption834ionization time-of-flight mass spectrometry(MALDI-TOF-MS)using LDI-1700instrument(Linear Scientific, Inc.,USA).The instrument was fitted with a pulsed nitrogen laser at337nm with3ns pulse duration.2,5-Dihydroxybenzoic acid(DHB)was used as the matrix. Infrared spectroscopy was performed using a Nicolet Nexus470spectrophotometer(Thermo Nicolet,Madison, WI,USA).For13C-NMR spectroscopy analysis,the samples were taken up in2H2O and processed at30°C. Spectra were recorded on a JNM-ECP600SCM spec-trometer(JEOL,Tokyo,Japan).ResultsStrain properties and identificationBacteria isolated from different areas of the China Sea were screened for a stable and effective agar-decomposingenzyme.A total of216strains producing agar-decompos-ing enzyme were isolated from the soil or seaweed surface, including Vibrio,Alteromonas,and Cytophaga.Strain SY37–12,isolated from the red alga Gracilaria verrucosa collected on Hainan Island of China,produced a large quantity of extracellular agarase when incubated in basal salt medium.The strain SY37-12produced soft pits on the agar surface with clear haloes around the bacteria colonies, which was regarded as an obvious mark indicating the high ability of degrading agar.Electron micrograph of the strain showed a polarly flagellated and straight-rod-shaped cell, 0.5–0.6μm×1.2–1.5μm.It was gram-negative and oxidase-and catalase-positive.It requires sodium ion for growth,has an oxidative metabolism,and does not ac-cumulate poly-β-hydroxybutyrate(PHB)as an intracellu-lar reserve product.The preliminary identification results showed that the morphologic and physiological character-istics of this strain were in accordance with Alteromonas, according to Bergey’s Manual of Systematic Bacteriology (Holt et al.1994)(Table1).Agarase produced by SY37-12was shown to be an induced enzyme.In the presence of crude agar,the strain produced extracellular agarase.No detectable agarase activity was found in the culture medium without agar. Furthermore,the addition of carrageenan,alginate,starch, galactose,lactose,and glucose in the absence of agar showed no effects on the production of agarase of SY37-12.When the agar was used as the sole carbon source,theTable1Morphologic and physiological characteristics of Altero-monas sp.SY37–12Characteristics tested Results Cell shape Straightrod Microcysts or endospores-Polar flagellum+ Production of pigments-Growth at4°C-Growth at35°C+ Growth at40°C-Anaerobic growth-Organic growth factors required-Oxidase reaction+ Catalase reaction+Argin1ine dihydrolase-Accumulation of PHB-O/F test O Production of H2S-Requirement of sodium for growth+ Hydrolysis of agar,starch,carrageenan,and gelatin+ Hydrolysis of alginate,chitin,and cellulose-Utilization of D-glucose,D-galactose,D-fructose,sucrose, cellobiose,and D-mannose+ Utilization of L-arabinose,dulcitol,raffinose,andD-ribose-Culture time (h)Agaraseactivity(U/ml)Biomass(OD6)Agarase activityFig.1Growth and agarase activity of Alteromonas sp.SY37–12. Fermentation liquids were monitored at600nm for biomass(□)and assayed for agarase activity(◊)by measuring the increase in the concentration of reducingsugar01836547290108Elution time (min)OD28Agaraseactivity(U/ml)Fig.2Ion-exchange chromatography of agarase on DEAE-Sepha-rose.The Tris buffer containing gradient NaCl rising from0to 1.0M at a flow-rate of0.5ml/min was used to wash out the sample. Fractions were monitored continuously at280nm for protein content(♦)and assayed for agarase activity(□)by measuring the increase in the concentration of reducing sugar835agarase activity of fermentation medium of SY37-12reached a maximum of 1.8U/ml after cultivation in conventional batch culture for 20h,whereas the biomass peaked at 24h (Fig.1).Purification of agaraseAccording to the pattern of ion-exchange chromatography on DEAE-Sepharose,several protein peaks were contained in the agarase sample (Fig.2).Additional purification of the fractions with agarase activity was achieved by gel filtration on Sephacryl S-100(Fig.3).Three protein peaks were shown in the chromatography,which was in ac-cordance with the result of SDS-PAGE (Fig.4).Thepurified enzyme had a molecular mass of 39.5kDa,as determined by a comparison with the mobility of protein standards.The purification steps and recovery rate of the agarase is summarized in the Table 2.Enzyme propertiesThe optimum reaction temperature of the agarase was 35°C,with a narrow range from 30to 45°C.At 20°C,the enzyme activity was only 30%in comparison with the maximum.The agarase was heat-labile,with rapid loss of activity when treated at 50°C for 15min or at 70°C for 1min (Fig.5).It even lost above 80%of its original activity after incubation at 90°C for 20s.The effect of pH on the enzyme activity was determined at 33°C in the pH range 5.0–9.0.The agarase exhibited maximum activity at pH 7.0.Since strain Alteromonas sp.SY37-12camefromElution time (min)O D 2800.20.40.60.811.21.4A g a r a s e a c t i v i t y (U /m l )Fig.3Gel filtration chromatography of agarase on Sephacryl S-100.The Tris buffer at a flow-rate of 0.7ml/min was used to wash out the sample.Fractions were monitored continuously at 280nm for protein content (♦)and assayed for agarase activity (□)by measuring the increase in the concentration of reducingsugarFig.4SDS-PAGE of purified ne 1molecular mass standards,lane 2purified agarase by Sephacryl S-100(only one band with a molecular mass of 39.5kDa),lane 3partly purified agarase by DEAE-Sepharose (three bands exist in the sample)Table 2Purification of agarase from Alteromonas sp.SY37-12StepV olume (ml)Agarase (U)Protein (mg)Specific activity (U/mg)Purification (-fold)Yield (%)Cell-free medium 1,0001,818606.2 3.0110080%(NH 4)2SO 4precipitate 45727157.84.61.540.0DEAE-Sepharose 20064217.137.512.535.3Sephacryl S-100HR2002593.183.527.814.2ab510152025Time (min)106050403020Time (s)R e l a t i v e a c t i v i t y (%)R e l a t i v e a c t i v i t y (%)Fig.5Effect of temperature on stability of agarase836the ocean,high NaCl concentration was necessary for its growth and enzyme production.When enzyme activity was measured in the presence of salt,a significant elevation could be observed.The enzyme activity reached the maximum in the presence of 2%NaCl,and then declined when the NaCl concentration continued to increase.MALDI-TOF-MS of the agarooligosaccharidesAfter sufficient hydrolysis and repeated fractionation by ethanol precipitation,the main depolymerized end-prod-ucts were collected.The MALDI-TOF mass spectrum of the sample was shown in Fig.6.According to the mass spectrum,the sample was an oligosaccharide mixture with polymerization degree from 4to 10.No sulfate group was linked in the sugar ring according to the molecular mass assignment.This speculation could be confirmed further by IR spectrum analysis and chemical determination (data not shown).The main composition was tetrasaccharide (MM-630Da)containing two galactopyranose residues (G-units)and two 3,6-anhydrogalaxtopyranose residues (An-units),hexasaccharide (MM-936Da)containing three G-units and three An-units,and octosaccharide (MM-1242Da)con-taining four G-units and four An-units.Furthermore,several minor peaks could be found in the mass spectrum,which were attributed to pentasaccharide (MM-792Da)containing three G-units and two An-units,heptasaccharide (MM-998Da)containing four G-units and three An-units,and decasaccharide (MM-1548Da)containing five G-units and five An-units.13C-NMR of agarooligosaccharidesMALDI-TOF mass analysis of the hydrolysis products of agar generated by agarase from Alteromonas sp.SY 37-12showed the presence of agarotetraose and agarohexaose as the main products.These products were further analyzed by NMR to determine the specificity of the cleavage.Anomeric carbons usually give downfield signals in 13C-NMR spectrum.The configuration of anomeric carbons can be determined according to the downfield shifts.The 13C-NMR spectrum of the oligosaccharide mixture showed a typical pattern for neoagarooligosaccharide (Fig.7).The neoagarooligosaccharide series is typically produced by the cleavage of β-(1,4)linkages by β-agarase.Resonances at about 97and 93ppm are characteristic for the βand αanomeric forms,respectively,of galactose residues at the reducing end of the neoagarooligosaccharides,indicating that the cleavage occurs at the β-(1,4)linkages.No peaks were present at around 90.4ppm corresponding to hydrolyzed α-(1,3)linkages.Thus,the 13C-NMR spectrum confirmed that agarase from strain SY 37-12is a β-agarase,specifically hydrolyzing the β-(1,4)glycosidic linkage between D -galactose and 3,6-anhydro-L-galactose.Fig.713C-NMR spectrum of the hydrolysis products of agar produced by agarase from Alteromonas sp.SY37–12837DiscussionWe describe here the characterization of a new agarolytic bacterium isolated from the southern ocean of China.This strain was identified as Alteromonas sp.according to its morphologic and physiological characteristics.The purified agarase extracted from Alteromonas sp.SY37-12has a molecular mass of39.5kDa,as indicated by SDS-PAGE. This value is close to those reported forβ-agarase from Pseudoalteromonas sp.N-1(33kDa)(Vera et al.1998), Pseudomonasatlantica(32kDa)(Morrice et al.1983),and Vibrio sp.AP-2(34kDa)(Aoki et al.1990).This enzyme acts as an endoenzyme,which decreased rapidly the viscosity of agar substrate at the initial reaction stage. Agarases(E.C.3.2.1.81)are classified into two groups depending upon their specificity of the degradation on the agar:α-agarases cleave theα-1,3linkage of agar,yielding oligosaccharides with3,6-anhydro-L-galactose at the re-ducing end and D-galactose at the nonreducing end,and β-agarase cleave theβ-1,4linkage of agar,yielding oligo-saccharides with D-galactose at the reducing end and3,6-anhydro-L-galactose at the nonreducing end(Vera et al. 1998).From the experiment result on analyzing the oligosaccharides by13C-NMR,it could be deduced that the agarase from the Alteromonas sp.SY37-12wasβ-agarase,which produced the3,6-anhydro-L-galactose as the nonreducing end and D-galactose as the reducing end. According to the assignment of MALDI-TOF-MS,the main composition of hydrolytic products was neotetrasac-charide(An-G-An-G),neohexasaccharide(An-G-An-G-An-G),and neooctosaccharide(An-G-An-G-An-G-An-G). In addition,pentasaccharide(G-An-G-An-G),heptasac-charide(G-An-G-An-G-An-G),and decasaccharide(An-G-An-G-An-G-An-G-An-G)could also be found in the products.It did not release detectable amounts of agaro-biose and agarotriose.With future experiments,the enzyme was confirmed to be not capable of hydrolyzing agarote-trose,agaropentose,agarohexose,and agaroheptose.It could be speculated that the minimal polymer degree of the agar substrate depolymerized by agarase from Alteromonas sp.SY37-12is eight.The agarase produce mainly agarooligosaccharides with even degree of polymerization, with D-galactose as the reducing end.As an endoenzyme,it cleaves theβ-1,4linkage of agar at random and produces a large quantity of low-molecular-mass fragments during the hydrolysis.Therefore,at the end of the hydrolytic reaction, the oligosaccharides with odd degree of polymerization,Table3Molecular mass and depolymerized products of characterized agarasesGroup Strain Molecular mass(kDa)Products Referenceα-Agarase Alteromonas GJIB360(bipolymer)Agarotetraose Potin et al.1993 Vibrio JI010784(bipolymer)Agaropentaose,agarotriose,agarobiose,3,6-anhydro-galactose and D-galactoseSugano et al.1994 Bacillus MK03320(octamer)Agaropentaose,agarotriose,3,6-anhydro-galactose and D-galactoseSuzuki et al.2002β-agarase Cytophaga flevensis26Neoagarotetraose,neoagarobiose Van der Meulen et al.1974 Pseudoalteromonas N-133Neoagarotetraose,neoagarohexaose Vera et al.1998Pseudomonas-like I a:210Neoagarohexaose,neoagarotetraose,neoagarobioseMalmqvist1978II:63NeoagarotetraosePseudomonas atlantica I:32Neoagarotetraose,neoagarobiose Morrice et al.1983II b:N/A NeoagarobioseVibrio AP-2I:34Neoagarotetraose Aoki et al.1990II:20NeoagarobioseIII:18NeoagarotetraoseVibrio JT0107I:107Neoagarotetraose,neoagarobiose Sugano et al.1993,1995II:72Neoagarotetraose,neoagarobioseVibrio PO-303I:87.5Neoagarohexaose,neoagarotetraose Araki et al.1998II:115NeoagarobioseIII:57Neoagarooctaose,neoagarodecaoseBacillus MK0392Neoagarotetraose Suzuki et al.2003Bacillus cereusASK20290Neoagarobiose Kim et al.1999 Alteromonas E-l82Neoagarobiose Kirimura et al.1999Alteromonas C-152Neoagarotetraose Leon et al.1992Alteromonas SY37-1239.5Neoagarotetraose,neoagarohexaose This worka There are several fractions with agarase activity produced by the strainb Data not available838especially pentasaccharide and heptasaccharide,are un-avoidably formed in the products.Molecular mass and depolymerized products of char-acterized agarases were given in Table3,which showed that the molecular mass of agarase from Alteromonas sp. SY37-12is different from the others,especially that from Alteromonas sp.E-1(Kirimura et al.1999)and Alteromo-nas sp.C-1(Leon et al.1992).The main depolymerized products produced by this enzyme are unique in compar-ison with the reports.Therefore,a conclusion was drawn here that the agarase produced by Alteromonas sp.SY37-12might be a novel enzyme.Further characterization of this enzyme will be discussed in the near future. ReferencesAgbo JAC,Moss MO(1979)The isolation and characterization of agarolytic bacteria from a low land river.J Gen Microbiol 113:355–368Aoki T,Araki T,Kitamikado M(1990)Purification and character-ization of a novelβ-agarase from Vibrio sp.AP-2.Eur J Biochem187:461–465Araki T,Hayakawa H,Lu Z,Karita S,Morishita T(1998) Purification and characterization of agarases from a marine bacterium,Vibrio sp.PO-303.J Mar Biotechnol6:260–265 Duckworth M,Turvey JR(1969)The action of a bacterial agarase on agarose,porphyran and alkali treated porphyran.Biochem J 113:687–692Fernandez LE,Valiente OG,Mainardi V,Bello JL,Velez H,Rosado A(1989)Isolation and characterization of an antitumor active agar-type polysaccharide of Gracilaria dominguensis.Carbo-hydr Res190:77–83Ha J,Kim GT,Kim SK,Oh TK,Yu JH,Kong IS(1997)β-Agarase from Pseudomonas sp.W7:purification of the recombinant enzyme from Escherichia coli and the effects of salt in its activity.Biotechnol Appl Biochem26:1–6Hamer GK,Bhattacharjee SS,Yaphe W(1977)Analysis of the enzymic hydrolysis products of agarose by13C-n.m.r.spec-troscopy.Carbohydr Res54:C7-C10Holt JG,Krieg NR,Sneath PHA,Staley JT,Williams ST(1994) Bergey’s manual of determinative microbiology,9th edn.Williams and Wilkins,BaltimoreKim BJ,Kim HJ,Duck Ha S,Hee Hwang S,Seok Byun D,Ho Lee T,Yul Kong J(1999)Purification and characterization ofβ-agarase from marine bacterium Bacillus cereus ASK202.Biotechnol Lett21:1011–1015Kirimura K,Masuda N,Iwasaki Y,Nakagawa H,Kobayashi R, Usami S(1999)Purification and characterization of a novelβ-agarase from an alkalophilic bacterium,Alteromonas sp.E-1.J Biosci Bioeng87:436–441Leon O,Quintana L,Peruzzo G,Slebe JC(1992)Purification and properties of an extracellular agarase from Alteromonas sp.strain C-1.Appl Environ Microbiol58:4060–4063 Malmqvist M(1978)Purification and characterization of two different agarose-degrading enzymes.Biochim Biophys Acta 537:31–43Morrice LM,McLean MW,Williamson FB,Long WF(1983)β-Agarases I and II from Pseudomonas atlantica.Purifications and some properties.Eur J Biochem135:553–558Potin P,Richard C,Rochas C,Kloareg B(1993)Purification and characterization of the alpha-agarase from Alteromonas agar-lyticus(Cataldi)comb.nov.,strain GJ1B.Eur J Biochem 214:599–607Sampietro AR,Vattuone de Sampletro MA(1971)Characterization of the agarolytic system of agarobacterium pastinator.Biochim Biophys Acta244:65–76Shieh WY,Jean WD(1998)Alterococcus agarolyticu s,gen.nov., sp.nov.,a halophilic thermophilic bacterium capable of agar degradation.Can J Microbiol44:637–645Stanier RY(1942)Agar-decomposing strains of the Actinomyces coelicolor species group.J Bacteriol44:555–570Sugano Y,Terada I,Arita M,Noma M,Matsumoto T(1993) Purification and characterization of a new agarase from a marine bacterium,Vibrio sp.strain JT0107.Appl Environ Microbiol59:1549–1554Sugano Y,Kodama H,Terada I,Yamazaki Y,Noma M(1994) Purification and characterization of a novel enzyme,alpha-neoagarooligosaccharide hydrolase,from a marine bacterium, Vibrio sp.strain JT0107.J Bacteriol176(22):6812–6818 Sugano Y,Nagae H,Inagaki K,Yamamoto T,Terada I,Yamazaki Y (1995)Production and characteristics of some newβ-agarases from a marine bacterium,Vibrio sp.strain JT0107.J Ferment Bioeng79:549–554Suzuki H,Sawai Y,Suzuki T,Kawai K(2002)Purification and characterization of an extracellularα-neoagarooligosaccharide hydrolase from Bacillus sp.MK03.J Biosci Bioeng93:456–463Suzuki H,Sawai Y,Suzuki T,Kawai K(2003)Purification and characterization of an extracellularβ-agarase from Bacillus sp.MK03.J Biosci Bioeng95:328–334Takemoto KK(1966)Plaque mutants of animal viruses.Prog Med Virol8:314–348Usov AI,Miroshnikova LI(1975)Isolation of agarase from Littorinamandshurica by affinity chromatography on BiogelA.Carbohydr Res43(1):204–212Van der Meulen HJ,Harder W,Vedkamp H(1974)Isolation and characterization of Cytophaga flevensis sp.nov.,a new agarolytic flexibacterium.Antonie Van Leeuwenhoek40:329–346van Hofsten B,Malmqvist M(1975)Degradation of agar by a Gram-negative bacterium.J Gen Microbiol87:150–158Vera J,Alvarez R,Murano E,Slebe JC,Leon O(1998)Identifi-cation of a marine agarolytic Pseudoalteromonas and char-acterization of its extracellular agarase.Appl Environ Microbiol 64:4378–4383von Borel E,Hostettler F,Deuel H(1952)Fasciculus I-15.Quantitative zuckerbestimmung mit3,5-dinitrosalicylsaure and phenol.Helv Chim Acta35:115–120Wang JX,Jiang XL,Mou HJ,Guan HS(2004)Anti-oxidation of agar oligosaccharides produced by agarase from a marine bacterium.J Appl Phycol16:333–340Weinberger F,Richard C,Kloareg B,Kashman Y,Hoppe H, Friedlander M(2001)Structure-activity relationships of oligoagar elicitors towards Gracilaria conferta.J Phycol 37:418–426Yoshizawa Y,Enomoto A,Todoh H,Ametani A,Kaminogawa S (1993)Activation of murine macrophages by polysaccharide fractions from marine algae(Porphyra yezoensis).Biosci Biotechnol Biochem57:1862–1866Young KS,Bhattacharjee SS,Yaphe W(1978)Enzymic cleavage of theα-linkages in agarose,to yield agaro-oligosaccharides.Carbohydr Res66:207–211Zhang QB,Li N,Zhou GF,Lu XL,Xu ZH,Li Z(2003)In vivo antioxidant activity of polysaccharide fraction from Porphyra haitanesis(Rhodephyta)in aging mice.Pharmacol Res48(2):151–155839。
ABO-Rh正反定型血型定型操作规程

ABO-Rh正反定型血型定型操作规程一、项目名称、检验方法名称1.项目名称:ABO-Rh正反定型血型定型2.检验方法名称:柱凝集二、方法学原理该试剂卡是基于柱凝集的原理而设计。
正常具有抗原的人红细胞将与相应的抗体发生凝集反应,BioVue血型诊断试剂卡的微柱中含有玻璃珠介质和试剂。
加入红细胞并将试剂卡离心后,发生凝集的红细胞会被阻挡在玻璃珠的上面,而未凝集的游离红细胞则会在离心力的作用下通过玻璃珠之间的间隙到达微柱的底部。
三、试剂卡品牌、代号、包装规格、内含物1.试剂卡品牌:美国强生奥索(Ortho-Clinical Diagnostica,Inc)2.代号:BioVue -ABO/Rh-Reverse3.包装规格:400卡/箱试剂卡(707100);100卡/箱试剂卡(707155)4.试剂成分:BioVue ABO-Rh正反定型血型诊断试剂卡有6根微柱组成,每一柱中含有缓冲液,其成分为牛血清蛋白,大分子增强剂以及0.1%叠氮钠,0.01M的EDTA.成分描述第一柱:抗-A试剂抗-A鼠单克隆(lgM)抗体混和;蓝色染料第二柱:抗-B试剂抗-B鼠单克隆(lgM)抗体混和;黄色染料第三柱:抗-D试剂(抗RH)抗-D人单克隆(lgM)抗体第四柱:质控优化增强剂,用于血型检测的质控对照第五,第六柱:反定型稀释液优化增强剂,用于反定型血型检测5.试剂的储存与效期:储存条件: 2~25℃环境中保存。
请勿将试剂存放在自动除霜的冰箱中,也不要冷冻保存。
并且不要放在离热源(如孵育箱、辐射器、大型设备、冰箱、冰柜等)很近或阳光直射的地方。
效期:试剂盒上有记载四、仪器品牌、型号BioVue专用离心机;BioVue专用孵育器五、样本的采集与准备:在采集标本前对病人/献血者无特殊要求,血液采集应符合相关的技术标准及管理规范。
含有或不含有抗凝剂的标本均可使用。
标本采集后应尽快进行检测。
如果实验不能立即进行,应将标本置于2-8℃保存。
阿加曲班与替罗非班治疗超溶栓时间窗急性进展性脑梗死的随机对照研究

阿加曲班与替罗非班治疗超溶栓时间窗急性进展性脑梗死的随机对照研究孙丽霞,许自强,韩 蕊,宋晓锋,尤书德摘要 目的:探讨应用阿加曲班和替罗非班治疗超溶栓时间窗急性进展性脑梗死(APCI )的疗效与安全性㊂方法:选取2019年10月 2022年5月河南科技大学附属许昌市中心医院神经内科收治的超溶栓时间窗且拒绝行全脑血管造影及血管内介入治疗的APCI 病人217例,采用随机数字表法分为观察A 组(73例)㊁观察B 组(72例)和对照组(72例)㊂观察A 组采用阿加曲班+常规治疗,观察B 组采用替罗非班+常规治疗,对照组采用阿司匹林和氯吡格雷双联抗血小板聚集+常规治疗㊂3组病人均于治疗前及治疗后24h ㊁14d 和90d 采用美国国立卫生研究院卒中量表(NIHSS )评估病人神经功能,采用功能独立性评定(FIM )量表评定病人日常生活自理能力㊂治疗过程中监测病人血常规㊁凝血功能及皮肤黏膜㊁大小便等情况㊂结果:3组治疗前NIHSS 评分及FIM 量表评分比较差异无统计学意义(P >0.05);治疗后24h ,3组病人NIHSS 评分及FIM 量表评分比较差异有统计学意义(P <0.05),观察A组和观察B 组NIHSS 评分及FIM 量表评分比较差异无统计学意义(P >0.05)㊂治疗后14d 和90d ,3组病人NIHSS 评分及FIM 量表评分比较差异有统计学意义(P <0.01),观察A 组和观察B 组NIHSS 评分及FIM 量表评分比较差异有统计学意义(P <0.05)㊂3组病人治疗过程中均未出现严重出血等不良反应㊂结论:阿加曲班或替罗非班治疗超溶栓时间窗急性进展性脑梗死疗效均优于双联抗血小板聚集,阿加曲班效果更佳㊂关键词 急性进展性脑梗死;阿加曲班;替罗非班;神经功能;日常生活自理能力d o i :10.12102/j.i s s n .1672-1349.2023.05.034 急性进展性脑梗死(acute progressive cerebral infarction ,APCI )是一种严重而难治的脑梗死临床亚型,占急性脑梗死的26%~43%[1]㊂在我国,APCI 是指发病后虽经积极治疗,神经功能仍进展恶化的脑梗死,一般在发病后6h 至1周内进展,大多在发病后24h 内进展[2]㊂有研究显示,APCI 总死亡率㊁总致残率较高,3个月不良预后发生率也较高,预后较差[3]㊂APCI 发病机制复杂,溶栓仍是目前首选方案,但受溶栓时间窗的限制,目前国际及国内的溶栓率均较低㊂针对APCI 病人,目前临床仍以抗血小板治疗为主,但效果并不理想[4],因而APCI 的治疗成为临床一大难题㊂国内外学者曾进行了低分子肝素㊁巴曲酶等大量积极探索,也有学者应用阿加曲班㊁替罗非班等抗凝及抗血小板药物[5-6],但均因效果不肯定㊁证据不充分等因素未得到指南认可[7]㊂积极探索可早期控制病情㊁促进神经功能缺损恢复的有效治疗方案对降低APCI 病人死亡率㊁致残率及致残程度,减轻家庭及社会负担至关重要㊂2019年以来,我院采用阿加曲班和替罗非班治疗超溶栓时间窗APCI ,取得较好的临床疗效㊂现报道如下㊂基金项目 2019年度河南省医学科技攻关计划联合共建项目(No.LHGJ20191384)作者单位 河南科技大学附属许昌市中心医院(河南许昌461000)通讯作者 尤书德,E -mail :***************引用信息 孙丽霞,许自强,韩蕊,等.阿加曲班与替罗非班治疗超溶栓时间窗急性进展性脑梗死的随机对照研究[J ].中西医结合心脑血管病杂志,2023,21(5):926-930.1 资料与方法1.1 临床资料 选取2019年10月 2022年5月我院神经内科收治的超溶栓时间窗且拒绝行全脑血管造影及血管内介入治疗的APCI 病人217例,采用随机数字表法分为观察A 组(73例)㊁观察B 组(72例)和对照组(72例)㊂观察A 组研究过程中因合并严重肺部感染死亡1例,治疗过程中监测活化部分凝血活酶时间(APTT )达危急值停药退出2例,后期失访1例,最终入组69例,男39例,女30例;年龄28~79(56.99ʃ13.13)岁;病程6~157(54.01ʃ37.70)h ㊂观察B 组因病情危重家属放弃治疗1例,后期失访1例,最终入组70例,男40例,女30例;年龄29~78(57.23ʃ11.57)岁;病程7~150(56.19ʃ41.94)h ㊂对照组后期失访1例,最终入组71例,男43例,女28例;年龄27~78(55.54ʃ13.55)岁;病程7~152(51.59ʃ37.78)h ㊂3组病人在性别㊁年龄㊁病程方面比较,差异均无统计学意义(P >0.05),具有可比性㊂1.2 诊断标准 经头颅磁共振成像(MRI )及CT 明确诊断为急性脑梗死㊂入院1周内任意1次美国国立卫生研究院卒中量表(NIHSS )评分增加ȡ2分为APCI ,且经头部CT/MRI 除外脑出血㊁梗死后出血㊁短暂性脑缺血发作㊁严重感染㊁心功能不全等所致病情进展㊂1.3 纳入标准 ①首次发病或既往有神经系统疾病史但不影响神经功能评分的病人;②年龄25~80岁;③血压ɤ180/110mmHg (1mmHg =0.133kPa );④发病6~168h ;⑤NIHSS 评分3~20分;⑥头颅影像学排除脑出血;⑦经本院伦理委员会批准,且病人或其家属签署知情同意书㊂1.4排除标准 ①发病48h内使用抗凝药物者;②发病前半年内有溶栓治疗史者,发病后接受静脉溶栓或血管内治疗者;③合并严重内外科系统疾病者;④1年内有活动性内脏出血㊁重要部位手术及合并凝血功能障碍者;⑤合并智力或精神障碍不能配合检查及治疗者;⑥依从性较差,无法很好地配合治疗及调查者㊂1.5病例脱落标准①经筛选合格进入研究者中途退出,没有完成治疗周期者;②病人依从性差,自行加减药及随访失访者㊂1.6治疗方法1.6.1观察A组发病小于48h的APCI病人,前48 h给予阿加曲班60mg加入生理盐水持续静脉泵入; 48h后改用阿加曲班10mg加入生理盐水静脉泵入3h,每天2次,总疗程7d㊂同时给予常规治疗(他汀类药物㊁丁苯酞氯化钠注射液㊁改善循环㊁针灸㊁康复等㊂7d 后在常规治疗基础上加用双抗治疗(阿司匹林100 mg㊁氯吡格雷片75mg)㊂发病48h至7d的病人,给予阿加曲班10mg加入生理盐水持续静脉泵入3h,每天2次,总疗程10d,同时给予常规治疗,10d后在常规治疗基础上加用双抗治疗,总疗程14d㊂1.6.2观察B组给予替罗非班治疗,起始30min内按0.4μg/(kg㊃min)静脉输注,后按0.1μg/(kg㊃min)速度维持72h,同时给予常规治疗,3d后常规治疗基础上加用双抗治疗,总疗程14d㊂1.6.3对照组给予常规治疗基础上加用双抗治疗,总疗程14d㊂1.7观察指标1.7.1疗效指标治疗前和治疗后24h㊁14d及90d,分别用NIHSS评定病人的神经功能,评分越高表示病人的神经功能缺损程度越严重;采用功能独立性评定(FIM)量表评定病人日常生活自理能力,该量表由6个维度㊁18个条目组成,总分18~126分,评分越高表明日常生活活动能力越强㊂两个量表均由神经内科主治医师以上人员进行评定,研究开始前统一进行人员培训以确保评定的准确性,尽量减少量表评定不一致导致的结果偏倚㊂在治疗后24h判断病人超早期疗效,治疗后14d判断病人近期疗效,治疗后90d判定病人远期疗效㊂参考脑卒中病人临床神经功能缺损程度评分标准(1995)[8]进行效果评价㊂痊愈:临床症状完全消失,NIHSS评分下降>90%;显效:临床症状大部分改善,NIHSS评分下降50%~90%;有效:临床症状有所缓解,NIHSS评分下降20%~49%;无效:临床症状未改善,NIHSS评分下降<20%或有上升趋势㊂总有效率=(痊愈例数+显效例数+有效例数)ː总例数ˑ100%㊂1.7.2安全性指标观察病人用药期间有无皮肤黏膜㊁牙龈㊁颅内㊁消化道等出血表现㊂用药前完善血尿粪常规㊁凝血功能㊁血液生化等检查㊂观察A组于用药后2h㊁24h㊁3d㊁10d复查凝血功能,重点观察APTT,观察B组和对照组于用药后3d㊁10d复查血常规及凝血功能,重点观察血小板计数(PLT)㊂1.8统计学处理采用SPSS23.0统计软件进行数据处理㊂定性资料用频数㊁百分比(%)表示,采用χ2检验;符合正态分布的定量资料以均数ʃ标准差(xʃs)表示,多个时间点比较采用重复测量方差分析(若Mauchly检验结果P<0.05以多变量检验结果为准;若P>0.05以主体内效应检验结果为准),多组间比较采用方差分析,两组间比较采用t检验㊂以P<0.05为差异有统计学意义㊂2结果2.13组近期疗效比较治疗14d后,观察A组总有效率为95.65%,观察B组总有效率为94.29%,对照组总有效率88.73%,3组近期疗效比较差异有统计学意义(P<0.01),观察A组与观察B组比较差异无统计学意义(P>0.05)㊂详见表1㊂表13组近期疗效比较组别例数痊愈(例)显效(例)有效(例)无效(例)总有效率(%)观察A组697536395.65观察B组7074514494.29对照组7121546888.73注:3组总有效率比较,χ2=67.596,P<0.01㊂2.23组远期疗效比较治疗90d后,观察A组总有效率为98.55%,观察B组总有效率为95.71%,对照组总有效率91.55%,3组远期疗效比较差异有统计学意义(P<0.01),观察A组与观察B组比较差异无统计学意义(P>0.05)㊂详见表2㊂表23组病人远期疗效比较组别例数痊愈(例)显效(例)有效(例)无效(例)总有效率(%)观察A组6910535198.55观察B组708509395.71对照组7142437691.55注:3组总有效率比较,χ2=53.802,P<0.01㊂2.33组NIHSS评分比较治疗前,3组病人NIHSS 评分比较差异无统计学意义(P>0.05)㊂治疗后24h,3组NIHSS评分比较差异有统计学意义(P<0.05),观察A组和观察B组NIHSS评分比较差异无统计学意义(P>0.05)㊂治疗后14d及90d,3组NIHSS评分比较差异均有统计学意义(P<0.01),观察A组和观察B组NIHSS评分比较差异有统计学意义(P<0.05)㊂详见表3㊂表33组NIHSS评分比较(xʃs)单位:分组别例数治疗前治疗后24h治疗后14d治疗后90d 观察A组698.43ʃ2.788.07ʃ2.49 3.20ʃ1.94 2.23ʃ1.64观察B组708.44ʃ2.708.10ʃ2.19 3.84ʃ1.85① 2.86ʃ1.53①对照组718.37ʃ2.798.97ʃ2.63① 5.32ʃ2.22① 4.03ʃ1.99①F值0.016 3.08620.58719.385P0.9840.048<0.001<0.001注:F时间=685.504,P<0.001;F组间=5.350,P=0.005;F交互=9.487,P<0.001㊂与观察A组同时间比较,①P<0.01㊂2.4两组FIM评分比较治疗前,3组FIM评分比较差异无统计学意义(P>0.05)㊂治疗后24h,3组FIM 评分比较差异有统计学意义(P<0.05),观察A组和观察B组比较差异无统计学意义(P>0.05)㊂治疗后14d及90d,3组FIM评分比较差异均有统计学意义(P<0.01),观察A组和观察B组FIM评分比较差异有统计学意义(P<0.05)㊂详见表4㊂表43组FIM评分比较(xʃs)单位:分组别例数治疗前治疗后24h治疗后14d治疗后90d 观察A组6963.06ʃ21.0265.93ʃ18.97107.93ʃ14.80114.55ʃ12.60观察B组7063.51ʃ22.5566.00ʃ20.11100.17ʃ15.20①107.63ʃ13.10①对照组7163.18ʃ18.5859.11ʃ17.21①87.94ʃ16.45①97.94ʃ16.57①F值0.009 3.12429.61224.112P0.9910.046<0.001<0.001注:F时间=568.492,P<0.001;F组间=8.769,P<0.001;F交互=14.419,P<0.001㊂与观察A组同时间比较,①P<0.01㊂2.53组APTT比较治疗前,3组APTT比较差异无统计学意义(P>0.05)㊂治疗后3d,观察A组APTT 明显延长,观察B组和对照组仅轻度延长,观察A组与治疗前比较差异有统计学意义(P<0.01),观察B 组和对照组与治疗前比较差异无统计学意义(P>0.05), 3组治疗后3d APTT比较差异有统计学意义(P< 0.01)㊂详见表5㊂表53组APTT比较(xʃs)单位:s 组别例数治疗前治疗后3d治疗后10d 观察A组6924.13ʃ2.8328.59ʃ5.43①24.23ʃ2.77观察B组7024.16ʃ2.8724.35ʃ2.7724.19ʃ2.59对照组7123.69ʃ2.6223.81ʃ2.6723.75ʃ2.53F值0.64632.5890.725P0.525<0.0010.485注:F时间=47.999,P<0.001;F组间=8.160,P<0.001;F交互=28.183,P<0.001㊂与同组治疗前比较,①P<0.01㊂2.63组PLT比较治疗前,3组PLT比较差异无统计学意义(P>0.05)㊂治疗后3d及10d,3组病人PLT比较差异无统计学意义(P>0.05),3组治疗后3d 及10d与治疗前比较差异均无统计学意义(P> 0.05)㊂详见表6㊂表63组PLT比较(xʃs)单位:ˑ109/L 组别例数治疗前治疗后3d治疗后10d 观察A组69204.30ʃ55.61203.74ʃ49.59206.17ʃ52.71观察B组70197.79ʃ54.80197.01ʃ48.19201.41ʃ56.17对照组71208.72ʃ55.82207.59ʃ50.68207.24ʃ53.37 F值0.6940.8120.231P0.5010.4450.794注:F时间=1.844,P=0.161;F组间=0.552,P=0.577;F交互=0.822,P=0.512㊂3讨论APCI是神经内科危急重症,以进展快㊁破坏性强为特点,治疗不及时或治疗不当易导致病人死亡率及致残率增加㊂关于脑梗死进展机制,目前并不明确,多数认为与血栓进展㊁侧支循环建立障碍㊁脑灌注压降低和血流动力学异常等有关[9-11]㊂目前,发病6h内的APCI仍以溶栓治疗为主,但对于超溶栓时间窗且无血管内治疗指征及拒绝血管内治疗的病人仍缺乏明确有效的内科治疗手段㊂为提高其临床疗效,降低死亡率及致残率,国内外进行了大量的研究,然而研究结论不一㊂我院神经内科多年来在进展性脑梗死的治疗过程中亦进行了大量积极的探索,发现阿加曲班和替罗非班的疗效明显,但两种药物何种疗效更佳目前尚缺乏客观有力的证据,探索比较两种药物的疗效及安全性对进展性脑梗死的更优化治疗可提供有利的依据㊂研究发现,急性缺血性脑血管病人的凝血酶活性普遍增强[12],凝血酶参与了缺血性脑血管病的发生及进展,加速脑组织的死亡[13]㊂因此,凝血酶可能成为预防和治疗该类疾病的新靶点[14]㊂阿加曲班为新型直接凝血酶抑制剂,相对传统抗凝药物,具有更高的选择性,能与凝血酶完全㊁快速㊁可逆地结合,进入血栓内部,从而阻止病情的发生和发展㊂阿加曲班可抑制游离的凝血酶及已形成血凝块的凝血酶[15],且出血风险较低㊂由于分子量小,阿加曲班容易穿透血管内皮和细胞屏障㊂因此,它对包括动脉粥样硬化血栓形成在内的微血管疾病的治疗是有效的[16],且可作为抗血小板药物抑制动脉粥样硬化斑块的增长[17]㊂血小板聚集与血栓形成密切相关,早期抑制血小板聚集的连锁反应可有效阻止血栓进展,是治疗APCI 的关键㊂但在临床实践中发现,对于APCI病人,即使早期积极给予双抗治疗,神经功能进行性恶化的病人仍较多,这与彭斌[18]报道的对于超溶栓时间窗APCI 病人,双抗联合他汀治疗的显效率不高一致㊂替罗非班可直接抑制血栓形成的关键和唯一通路,抑制血小板聚集,拮抗血栓形成[19]㊂且该药起效迅速,作用持续3~8h,与阿司匹林㊁氯吡格雷联用可产生协同作用㊂本研究对发病6h至1周的超溶栓时间窗且不具备血管内治疗指征及拒绝行血管内治疗的APCI病人,对阿加曲班㊁替罗非班及双抗加常规治疗3种方案进行对比观察,结果发现,阿加曲班及替罗非班对无论发病48h以内病人,还是发病大于48h且小于1周的APCI病人均能有效阻止病情进展,疗效均明显优于双抗治疗,这与文献报道结果[18]相一致㊂本研究发现,阿加曲班及替罗非班在应用后的24h 内即可有效阻止神经功能缺损恶化,促进病情恢复,与双抗治疗比较差异有统计学意义㊂探讨其机制发现,阿加曲班分子质量仅527Da,该药起效快,作用时间短,正常机体仅需1~3h即可达到稳态血药浓度[20],可能是阿加曲班早期使用即可获效的原因㊂而文献报道替罗非班半衰期亦比较短,起效快,给药5min血小板抑制率即可达96%,且可溶解微血管内的微血栓或者通过改善血栓的结构,使血栓发生内源性溶栓过程,清除或抑制急性血栓形成而实现血管再通[21]㊂本研究发现,观察A组与观察B组的近期及远期疗效比较差异均无统计学意义(P>0.05),但治疗后14d及90d的NIHSS评分及FIM量表评分比较差异均有统计学意义(P<0.01),这说明虽然两组病人的总体疗效相近,但阿加曲班在改善神经功能缺损及日常生活自理能力方面仍优于替罗非班㊂目前国内外对阿加曲班及替罗非班治疗急性脑梗死均进行了大量的研究,但将两种药物进行对比研究的不多,杨斌等[22]研究发现,阿加曲班治疗急性缺血性脑卒中的临床总有效率优于应用替罗非班治疗的病人㊂而陈宏涛等[23]的研究发现,阿加曲班治疗心源性栓塞型的APCI更佳,替罗非班则更适宜于小动脉闭塞型的病人,两者治疗大动脉粥样硬化病人的效果相当㊂本研究采用NIHSS评分评价病人神经功能缺损情况,采用FIM量表评价病人日常生活自理能力㊂FIM量表是目前国际上运用较多的一种功能评价量表,不仅可准确地评估病人的运动功能,而且可充分评估病人的理解㊁表达㊁社交㊁问题解决和记忆功能[24]㊂FIM量表与运动㊁认知分量表都具有较高的一致性及良好的信度㊁效度[25],在入院和出院评估时与Barthel 指数量表具有较高的一致性[26-27]㊂本研究发现,观察A组2例病人治疗后3d因APTT达危急值停药,临床未见出血症状,停药4h后复查APTT已明显下降,第2日复查APTT已恢复正常㊂这与文献报道[28]该药停药后APTT在2~4h内即可恢复正常一致,故在应用过程中需监测凝血功能,尤其是APTT,以确保用药的安全性㊂应用阿加曲班治疗过程中6例病人出现牙龈出血或者皮肤瘀斑,应用替罗非班治疗过程中5例病人出现牙龈出血或者皮肤瘀斑,使用双抗治疗有8例出现牙龈出血或者皮肤瘀斑,但继续用药均未发现出血增加及其他部位出血等不良反应㊂虽然观察A组在治疗后3d复查APTT 较治疗前明显升高,3组比较差异有统计学意义,但临床未见严重出血事件发生,说明3种治疗方案均具有良好的安全性㊂本研究仅观察了3种不同治疗方案对APCI病人治疗后24h的超早期疗效,以及治疗后14d及90d 的近期㊁远期疗效,未就脑梗死TOAST分型进一步进行更细化地研究,今后将针对APCI病人的不同分型进行更加细化的研究以期探讨阿加曲班和替罗非班对不同分型病人疗效是否存在差异,为今后临床的进一步精准治疗提供更加有力的证据㊂参考文献:[1]秦洁行,苗玲.进展性脑梗死的相关因素[J].中国临床康复,2005,9(33):114-116.[2]JOHNSTON K C,BRUNO A,PAULS Q,et al.Intensive vsstandard treatment of hyperglycemia and functional outcome inpatients with acute ischemic stroke:the SHINE randomizedclinical trial[J].JAMA,2019,322(4):326-335.[3]WANG P E,YAN G Z,TAN B Y Q,et al.Bilateral watershedinfarcts in infective endocarditis[J].QJM:an International Journalof Medicine,2019,112(5):375-376.[4]SENERS P,HURFORD R,TISSERAND M,et al.Is unexplainedearly neurological deterioration after intravenous thrombolysisassociated with thrombus extension[J].Stroke,2017,48(2):348-352.[5]胡欣慧,张建斌,郑东梅.阿加曲班联合阿司匹林治疗急性脑梗死的研究进展[J].中国实用神经疾病杂志,2021,24(14):1277-1282.[6]NIU J L,DING Y L,ZHAI T T,et al.The efficacy and safety oftirofiban for patients with acute ischemic stroke:a protocol forsystematic review and a meta-analysis[J].Medicine,2019,98(9):e14673.[7]中华医学会神经病学分会,中华医学会神经病学分会脑血管病学组.中国急性缺血性脑卒中诊治指南2018[J].中华神经科杂志,2018,51(9):666-682.[8]全国第四届脑血管病学术会议.脑卒中患者临床神经功能缺损程度评分标准(1995)及各类脑血管疾病诊断要点[J].中华神经科杂志,1996,29(6):379-383.[9]李博,李飞,冯秀龙,等.进展性脑梗死与头颈CT血管成像血管狭窄及斑块特征的相关性研究[J].实用放射学杂志,2022,38(1):14-17;46.[10]陈晓娟.进展性脑梗死的相关危险因素分析[J].中外医学研究,2021,19(2):17-20.[11]高欣欣,张彬,王红霞,等.进展性缺血性脑卒中相关危险因素的研究进展[J].当代医药论丛,2021,19(7):12-15.[12]MCCONNELL J P,CHERYK L A,DUROCHER A,et al.Urinary11-dehydro-thromboxane B2and coagulation activation markersmeasured within24h of human acute ischemic stroke[J].Neuroscience Letters,2001,313(1):88-92.[13]CHEN L,CAO S S,YANG J X.Argatroban plus aspirin versusaspirin in acute ischemic stroke[J].Neurological Research,2018,40(10):862-867.[14]PLEŞERU A M,MIHAILǍR G.The role of thrombin in centralnervous system activity and stroke[J].Clujul Medical,2018,91(4):368-371.[15]KATHIRESAN S,SHIOMURA J,JANG I K.Argatroban[J].Journalof Thrombosis and Thrombolysis,2002,13(1):41-47. [16]UEDA M.Pathology of Athero Thrombos IS(ATIS)[J].Drugs,2010,70(Suppl1):3-8.[17]OGURO H,MITAKI S,TAKAYOSHI H,et al.Retrospective analysisof argatroban in353patients with acute noncardioembolic stroke[J].Journal of Stroke and Cerebrovascular Diseases,2018,27(8):2175-2181.[18]彭斌.急性缺血性脑卒中的诊治[J].中华神经科杂志,2020,53(2):122-126.[19]黄志宝,何忠莲,汤红薇,等.替罗非班联合丁苯酞注射液治疗进展性脑梗死[J].血栓与止血学,2018,24(1):10-12.[20]YEH R W,J ANG I K.Argatroban:update[J].American HeartJournal,2006,151(6):1131-1138.[21]杜大勇,赵莲花,李博,等.替罗非班治疗超时间窗急性脑梗死的临床研究[J].中华神经医学杂志,2020,19(5):470-476. [22]杨斌,石秋艳,屈征,等.阿加曲班和替罗非班治疗急性缺血性脑卒中的有效性和安全性对比[J].中国老年学杂志,2021,41(11):2261-2263.[23]陈宏涛,杨伟民.阿加曲班与替罗非班治疗进展性缺血性脑卒中的效果比较[J].临床医学研究与实践,2021,6(18):37-39. [24]薛凯文,刘翔翔,张泽宇,等.脑卒中患者日常生活活动能力评定量表反应性研究进展[J].康复学报,2022,32(4):374-380. [25]HONG I,WOO H S,SHIM S,et al.Equating activities of daily livingoutcome measures:the Functional Independence Measure andthe Korean version of Modified Barthel Index[J].Disability andRehabilitation,2018,40(2):217-224.[26]LINACRE J M,HEINEMANN A W,WRIGHT B D,et al.Thestructure and stability of the Functional Independence Measure[J].Archives of Physical Medicine and Rehabilitation,1994,75(2):127-132.[27]GOSMAN-HEDSTRÖM G,SVENSSON E.Parallel reliability of theFunctional Independence Measure and the Barthel ADL index[J].Disability and Rehabilitation,2000,22(16):702-715. [28]北京神经科学学会血管神经病学专业委员会,阿加曲班治疗急性缺血性卒中中国专家共识组.阿加曲班治疗急性缺血性卒中中国专家共识2021[J].中国卒中杂志,2021,16(9):946-953.(收稿日期:2022-11-22)(本文编辑郭怀印)。
开启片剂完整性的窗户(中英文对照)

开启片剂完整性的窗户日本东芝公司,剑桥大学摘要:由日本东芝公司和剑桥大学合作成立的公司向《医药技术》解释了FDA支持的技术如何在不损坏片剂的情况下测定其完整性。
太赫脉冲成像的一个应用是检查肠溶制剂的完整性,以确保它们在到达肠溶之前不会溶解。
关键词:片剂完整性,太赫脉冲成像。
能够检测片剂的结构完整性和化学成分而无需将它们打碎的一种技术,已经通过了概念验证阶段,正在进行法规申请。
由英国私募Teraview公司研发并且以太赫光(介于无线电波和光波之间)为基础。
该成像技术为配方研发和质量控制中的湿溶出试验提供了一个更好的选择。
该技术还可以缩短新产品的研发时间,并且根据厂商的情况,随时间推移甚至可能发展成为一个用于制药生产线的实时片剂检测系统。
TPI技术通过发射太赫射线绘制出片剂和涂层厚度的三维差异图谱,在有结构或化学变化时太赫射线被反射回。
反射脉冲的时间延迟累加成该片剂的三维图像。
该系统使用太赫发射极,采用一个机器臂捡起片剂并且使其通过太赫光束,用一个扫描仪收集反射光并且建成三维图像(见图)。
技术研发太赫技术发源于二十世纪九十年代中期13本东芝公司位于英国的东芝欧洲研究中心,该中心与剑桥大学的物理学系有着密切的联系。
日本东芝公司当时正在研究新一代的半导体,研究的副产品是发现了这些半导体实际上是太赫光非常好的发射源和检测器。
二十世纪九十年代后期,日本东芝公司授权研究小组寻求该技术可能的应用,包括成像和化学传感光谱学,并与葛兰素史克和辉瑞以及其它公司建立了关系,以探讨其在制药业的应用。
虽然早期的结果表明该技术有前景,但日本东芝公司却不愿深入研究下去,原因是此应用与日本东芝公司在消费电子行业的任何业务兴趣都没有交叉。
这一决定的结果是研究中心的首席执行官DonArnone和剑桥桥大学物理学系的教授Michael Pepper先生于2001年成立了Teraview公司一作为研究中心的子公司。
TPI imaga 2000是第一个商品化太赫成像系统,该系统经优化用于成品片剂及其核心完整性和性能的无破坏检测。
Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and st

DIFFERENT SYNAPTIC AND SUBSYNAPTIC LOCALIZATION OF ADENOSINE A2A RECEPTORS IN THE HIPPOCAMPUS AND STRIATUM OF THE RATN.REBOLA,P.M.CANAS,C.R.OLIVEIRAAND R.A.CUNHA*Center for Neurosciences of Coimbra,Institute of Biochemistry,Fac-ulty of Medicine,University of Coimbra,3004-504Coimbra,PortugalAbstract—Adenosine A2Areceptors are most abundant in thestriatum where they control the striatopallidal pathway thuscontrolling locomotion.Extra-striatal A2Areceptors are con-siderably less abundant but their blockade confers robustneuroprotection.We now have investigated if striatal andextra-striatal A2Areceptors have a different neuronal locationto understand their different functions.The binding densityof the A2Aantagonist,[3H]-7-(2-phenylethyl)-5-amino-2-(2-furyl)pyrazolo[4,3e][1,2,4]triazolo[1,5-c]pyrimidine([3H]SCH58261),was enriched in nerve terminals membranes(Bmax؍103؎12fmol/mg protein)compared with total mem-branes(Bmax؍29؎4fmol/mg protein)from the hippocampus,the same occurring with A2Areceptor immunoreactivity.Incontrast,there was no enrichment of[3H]SCH58261binding or A2Areceptor immunoreactivity in synaptosomalcompared with total membranes from the striatum.Furthersubcellular fractionation of hippocampal nerve terminals re-vealed that A2Areceptor immunoreactivity was enriched inthe active zone of presynaptic nerve terminals,whereas itwas predominantly located in the postsynaptic density in thestriatum,although a minority of striatal A2Areceptors werelocated in the presynaptic active zone.These results indicatethat A2Areceptors in the striatum are not enriched in syn-apses in agreement with the preponderant role of A2Arecep-tors in signal processing in striatopallidal neurons.In con-trast,hippocampal A2Areceptors are enriched in synapses,mainly in the active zone,in accordance with their role incontrolling neurotransmitter release.This regional variationin the neuronal distribution of A2Areceptors reinforces thecare required to extrapolate our knowledge from striatal A2Areceptors to other brain preparations.©2005IBRO.Pub-lished by Elsevier Ltd.All rights reserved.Key words:A2Areceptor,adenosine,hippocampus,striatum,nerve terminals.Adenosine is a neuromodulator in the nervous systemwhere it mainly inhibits neurotransmitter release throughactivation of A1receptors(Dunwiddie and Masino,2001).Adenosine can also activate facilitatory receptorsof the A2A subtype(Fredholm et al.,2003).These A2Areceptors were initially thought to be exclusively locatedin the basal ganglia in particular in the medium spinalneurons of the indirect pathway(reviewed in Svenningssonet al.,1999).Subsequent molecular biology,autoradiog-raphy and membrane binding,immunological,neuro-chemical and electrophysiological studies provided evi-dence for the presence of A2Areceptors in the limbic andneocortex(reviewed in Cunha,2001),albeit with a con-siderably lower density than in the striatum(see Lopeset al.,2004).However,some studies consistently founddifferences between the pharmacological properties(e.g.Johansson and Fredholm,1995;Cunha et al.,1996;Lindström et al.,1996;but see Rebola et al.,2002)and transducing systems(e.g.Cunha and Ribeiro,2000;Cunha et al.,1999;Rebola et al.,2003b;but seeGubitz et al.,1996;Okada et al.,2001)operated by theprototypical A2Areceptors in the striatum and the pur-ported A2Areceptors of the cortex or hippocampus.Thisis accompanied by an essentially different role fulfilledby striatal and extra-striatal A2Areceptors.In fact,themain role of striatal A2Areceptors is to postsynapticallycontrol the signaling in striatopallidal neurons throughcAMP and/or DARPP-32phosphorylation(reviewed inFerréet al.,2003;Fredholm et al.,2003)and,accord-ingly,antagonists of A2Areceptors are undergoing clin-ical trials as anti-parkinsonian drugs(see Schwarzschildet al.,2002).In contrast to the predominant postsynapticrole of striatal A2Areceptors,the main function ascribedto extra-striatal A2Areceptors has been as a presysnap-tic modulatory system facilitating the evoked release ofneurotransmitters(reviewed in Cunha,2001).These differences between striatal and extra-striatalA2Areceptors may be due to a different subcellularlocalization of A2Areceptors in striatal and extra-striataltissue.A possible different subcellular localization of A2Areceptors in striatal and extra-striatal tissues may alsoshed light on the surprising impact of A2Areceptors incontrolling neuronal viability.In fact,either antagonistsof A2Areceptors or genetic disruption of A2Areceptorsconfer a robust neuroprotection against cortical damage(e.g.Behan and Stone,2002;Chen et al.,1999;Monopoli et al.,1998),which is greater than that ob-served in striatal tissue(e.g.Chen et al.,1999;Melani etal.,2003),where A2Areceptors are far more abundant.Thus,we now investigated if the subcellular and thesubsynaptic localization of A2Areceptors are different inthe striatum and in the hippocampus.*Corresponding author.Tel:ϩ351-239-820190;fax:ϩ351-239-822776.E-mail address:racunha@clix.pt(R.A.Cunha).Abbreviations:PBS,phosphate buffer saline;TBS-T,Tris-buffered salinewith0.1%Tween20;XAC,8-{4-[(2-aminoethyl)amino]carbonylmethyl-oxyphenyl}xanthine;[3H]SCH58261,7-(2-phenylethyl)-5-amino-2-(2-furyl)pyrazolo[4,3e][1,2,4]triazolo[1,5-c]pyrimidine.Neuroscience132(2005)893–9030306-4522/05$30.00ϩ0.00©2005IBRO.Published by Elsevier Ltd.All rights reserved. doi:10.1016/j.neuroscience.2005.01.014893EXPERIMENTAL PROCEDURESMale Wistar rats (6–8weeks old,140–160g;obtained from Harlan Ibérica,Barcelona,Spain)were used throughout this study.All experiments were carried out according to the local guidelines on the ethical use of experimental animals,based on the EU guidelines for use of experimental animals (86/609/EEC),with care to minimize the number of animals used and their suffering,the rats being anesthetized under halothane atmo-sphere before being killed by decapitation.Preparation of total and synaptosomal membranesTotal membranes from the striatum or hippocampus or from Percoll-purified striatal or hippocampal synaptosomes were prepared as previously described (Cunha et al.,1996).Briefly,the two striata or hippocampi from one rat were homogenized at 4°C in sucrose solution (0.32M)containing 50mM Tris–HCl,2mM EGTA and 1mM dithiothreitol,pH 7.6.The resulting homogenates were centrifuged at 3000ϫg for 10min at 4°C,the supernatants collected and centri-fuged at 14,000ϫg for 20min at 4°C.The pellets of one of the samples of striatal or hippocampal tissue were taken as the total membrane fraction and were either resuspended in an incubation buffer containing 25mM Tris,2mM MgCl 2,pH 7.4for binding studies or in SDS-PAGE buffer (see below)for Western blot analysis.The pellet of the other sample was resuspended in 1ml of a 45%(v/v)Percoll solution made up in a Krebs solution (composition 140mM NaCl,5mM KCl,25mM HEPES,1mM EDTA,10mM glucose,pH 7.4).After centrifugation at 14,000ϫg for 2min at 4°C,the top layer was removed (synaptosomal fraction),washed in 1ml Krebs solution and resuspended in 10ml of incubation buffer.This mixture was centrifuged at 14,000ϫg for 20min at 4°C and the pellet corre-sponded to the nerve terminal membranes and was either resus-pended in the incubation buffer for binding studies or in SDS-PAGE buffer (see below)for Western blot analysis.Membrane binding experimentsBoth total membranes and nerve terminal membranes (from either hippocampus or striatum)were first incubated with 2U/ml aden-osine deaminase for 30min at 37°C,to remove endogenous adenosine.The mixture was then centrifuged at 14,000ϫg for 10min at 4°C and the pellets resuspended in the incubation solution.Binding of [3H]-7-(2-phenylethyl)-5-amino-2-(2-furyl)pyr-azolo[4,3e][1,2,4]triazolo[1,5-c]pyrimidine ([3H]SCH 58261)was for 1h at room temperature (23–25°C)with 213–304g of hippocampal or 23–41g of striatal membrane protein in a final volume of 300l in the incubation solution containing 4U/ml adenosine deaminase,as previously described (Alfaro et al.,2004;Lopes et al.,2004).Specific binding was determined by subtraction of the nonspecific binding,which was measured in the presence of 1M 8-{4-[(2-aminoethyl)amino]carbonylmethyl-oxyphenyl}xanthine (XAC),a mixed A 1/A 2receptor antagonist.All binding assays were performed in duplicate.The binding reactions were stopped by vacuum filtration through glass fiber filters (GF/C filters)using a 24well Brandel harvester.The filters were then placed in scintillation vials and 4ml of scintillation liquid (Ready Safe,Pharmacia,Lisboa,Portugal)added.Radioactivity was de-termined after at least 12h with a counting efficiency of 55–60%.To ensure an error lower than 5%of counts,the samples were counted for 10min.The protein concentration was determined using the Bio-Rad protein assay based on Bradford dye-binding procedure (see Cunha et al.,1996).The specific binding from saturation experiments was fitted by non-linear regression to one binding site equation using the Raphson-Newton method,performed with a commercial software (GraphPad,San Diego,CA,USA)to determine the binding pa-rameters (dissociation constant,K d ,and maximal number of bind-ing sites,B max ).Western blot analysisThe analysis of adenosine A 2A receptor immunoreactivity was carried out in total membranes and in membranes from a Percoll-purified synaptosomal fraction of the striatum or hippocampus,as previously described (Rebola et al.,2002).Briefly,after determin-ing the amount of protein,each sample was diluted with two volumes of SDS-PAGE buffer containing 8M urea,100mM dithiothreitol,2%(w/v)sodium dodecyl sulfate and 375mM Tris–HCl pH 6.8and incubated for 2h at 37°C.These diluted samples and the pre-stained molecular weight markers (Amersham)were separated by SDS-PAGE (10%with a 4%concentrating gel)under reducing conditions and electro-transferred to polyvinyli-dene difluoride membranes (0.45m,from Amersham).After blocking for 2h at room temperature with 5%milk in Tris-buffered saline,pH 7.6containing 0.1%Tween 20(TBS-T),the mem-branes were incubated overnight at 4°C with either a goat anti-adenosine A 2A receptor antibody (raised against the C-terminus of the receptor,1:500dilution)or with a mouse anti-adenosine A 2A receptor antibody (raised against the third intracellular loop of the receptor,1:1000dilution).After four washing periods for 10min with TBS-T containing 0.5%milk,the membranes were incubated with the alkaline phosphatase-conjugated anti-goat or anti-mouse secondary antibody (1:2000dilution;from Calbiochem)in TBS-T containing 1%milk during 90min at room temperature.After five 10min-washes in TBS-T with 0.5%milk the membranes were incubated with enhanced chemi-fluorescence during 5min and then analyzed with a VersaDoc 3000(Biorad).The membranes were then re-probed and tested for tubulin immunoreactivity to confirm that similar amounts of protein were applied to the gels.Briefly,the membranes were incubated for 1h at room temperature with a 0.1M glycine (pH 7.2)solution then blocked as previously described before incubation with the anti-tubulin antibody (1:1000).The membranes were then washed and incubated with alkaline phosphatases-conjugated secondary an-tibody as previously described.Subsynaptic fractionationThe separation of the presynaptic active zone,postsynaptic den-sity and non-synaptic fractions from striatal or hippocampal nerve terminals was carried out as initially described by Phillips et al.(2001).Briefly,synaptosomes were diluted 1:10in cold 0.1mM CaCl 2and an equal volume of 2ϫsolubilization buffer (2%Triton X-100,40mM Tris,pH 6.0)was added to the suspension.The membranes were incubated for 30min on ice with mild agitation and the insoluble material (synaptic junctions)pelleted (40,000ϫg for 30min at 4°C).The supernatant (extrasynaptic fraction)was decanted and proteins precipitated with six volumes acetone at Ϫ20°C and recovered by centrifugation (18,000ϫg for 30min at Ϫ15°C).The synaptic junctions pellet was washed in solubiliza-tion buffer (pH 6.0)and resuspended in 10volumes of a second solubilization buffer (1%Triton X-100,20mM Tris but at pH 8.0).This increase in pH allows the dissociation of the extracellular matrix that maintains the presynaptic active zone tightly bound to the postsynaptic density (see Phillips et al.,2001).Hence,the active zone is solubilized whereas the postsynaptic density is essentially preserved because the amount of detergent is not enough for its solubilization (Phillips et al.,2001).After incubation for 30min on ice with mild agitation,the mixture was centrifuged and the supernatant (presynaptic fraction)processed as described for the extrasynaptic fraction.The protease inhibitor,phenylmethylsulfonyl fluoride (1mM;from Sigma Ibérica)was added to the suspension in all extraction steps.The pellets from the supernatants and the final insoluble pellet (postsynaptic frac-tion)were solubilized in 5%SDS,the protein concentration deter-mined by the bicinchoninic acid protein assay (Pierce)and the samples were added to an equal volume of 2ϫSDS-PAGE sam-ple buffer prior to freezing at Ϫ20°C.N.Rebola et al./Neuroscience 132(2005)893–903894As previously reported (Pinheiro et al.,2003;Rebola et al.,2003a ),this fractionation procedure allows an effective separation (over 90%efficiency)of markers of the presynaptic (e.g.syntaxin or SNAP25),postsynaptic (e.g.PSD-95or NMDA receptor sub-units)and non-synaptic (e.g.synaptophysin)fractions,and can be used to access the subsynaptic distribution of adenosine recep-tors by Western blot analysis (Rebola et al.,2003a ).Immunohistochemistry experiments in rat hippocampal or striatal neuronal culturesTo obtain hippocampal neurons,neurons were dissociated from hippocampi of E17Wistar rat embryos,after treatment with trypsin (2.0mg/ml,15min,37°C)and deoxyribonuclease I (0.15mg/ml)in Ca 2ϩ-and Mg 2ϩ-free Hanks’balanced solution (137mM NaCl,5.36mM KCl,0.44mM KH 2PO 4,0.34mM Na 2HPO 4,4.16mM NaHCO 3,5mM glucose,1mM sodium pyruvate,10mM HEPES,pH 7.4)The cells were cultured in serum-free Neurobasal medium,supplemented with B27(GIBCO),glutamate (25M),glutamine (0.5mM)and gentamicin (0.12mg/ml),as described previously (Silva et al.,2001).Cultures were kept at 37°C in a humidified incubator in 5%CO 2/95%air,for 7–8days,the time required for maturation of hippocampal neurons.The cultured striatal neurons were obtained following a pre-viously described methodology (Mena et al.,1997).Neurons were dissociated from striata of P2rat pups and plated at 80,000cells/well over a glial monolayer.This glial monolayer was prepared by dissociation of cerebral cortical tissue from P2-3rat pups in a solution containing 20U/ml papain in 1.0mM cysteine,0.001%Phenol Red,116mM NaCl,5.4mM KCl,26mM NaHCO 3,2mM NaH 2PO 4,1mM MgSO 4,500M EDTA and 25mM glucose at pH 7.3.Cells were plated at a density of 150,000cells/well in a medium containing 90%minimal essential medium,10%calf serum,0.33%glucose,500M glutamine,12U/ml penicillin and 12g/ml streptomycin.After 2weeks,the glial growth medium was removed and serum free neuronal medium (neurobasal me-dium supplemented with B27,25M glutamate,500M glu-tamine,500M kynurenate and 0.12mg/ml gentamicin)was added for 48h before plating the striatal neurons.Cultures were kept at 37°C in a humidified incubator in 5%CO 2/95%air,for at least 4–5days,until use.Immunocytochemistry in the coverslip-mounted neurons was carried out as previously described (Silva et al.,2001).Briefly,the cultures were washed twice with 1ml phosphate buffer saline (PBS;140mM NaCl,3mM KCl,20mM Na 2HPO 4, 1.5mM KH 2PO 4)kept at 37°C during 10min,and fixed with 4%parafor-maldehyde with 4%sucrose for 30min at 37°C.Coverslip-mounted cells were then permeabilized with 0.2%Triton X-100for 2min at room temperature and non-specific binding subsequently blocked with 3%bovine serum albumin for 30min at room tem-perature.Cells were then incubated for 1h at room temperature either with goat anti-adenosine A 2A receptor antibody (1:500dilu-tion)and with mouse anti-synaptophysin antibody (1:200dilution)or with a mouse anti-adenosine A 2A receptor antibody (1:1000dilution)and a rabbit anti-synaptophysin antibody (1:500dilution).After being washed three times with 200l PBS for 5min,incu-bation was conducted for 1h at room temperature with the sec-ondary antibodies,either a mixture of an AlexaFluor-488-(green)labeled donkey anti-goat IgG antibody (Molecular Probes;1mg/ml;1:200dilution)and AlexaFluor-598-(red)labeled goat anti-mouse IgG antibody (Molecular Probes;1mg/ml;1:200dilution)or a mixture of an AlexaFluor-488-(green)labeled donkey anti-mouse IgG antibody (Molecular Probes;1mg/ml;1:200dilution)and AlexaFluor-598-(red)labeled goat anti-rabbit IgG antibody (Molecular Probes;1mg/ml;1:200dilution).After three washing periods of 5min with 200l PBS,the cells were mounted using a Prolong Antifade kit (Amersham)and,after drying,were visualized in a Zeiss Axiovert 200fluorescence microscope equipped with acooled CCD camera or with a Biorad 600confocal microscope and analyzed with MetaFluor 4.0software.Drugs and solutions[3H]SCH 58261(specific activity 77Ci/mmol)was prepared by Amersham (Buckinghamshire,UK)and was a generous gift of Dr.Ennio Ongini (Shering-Plough,Milan,Italy),adenosine deami-nase (calf intestine suspension,2000U/ml,EC 3.5.4.4)was from Sigma (Reagente 5;Oporto,Portugal),XAC was from Research Biochemical International (Reagente 5;Oporto,Portugal),goat anti-adenosine A 2A receptor antibody (200g/ml,raised against the C-terminus of the rat A 2A receptor)was from Santa Cruz Biotechnology-Europe (Freelab;Lisbon,Portugal),mouse anti-adenosine A 2A receptor antibody (1mg/ml,raised against the third intracellular loop of the receptor)and mouse anti-PSD-95antibody (1mg/ml)were from Upstate Biotechology (Lake Placid,NY,USA),mouse anti-␣tubulin antibody (0.5mg/ml)and rabbit anti-synaptophysin antibody (0.5mg/ml)were from Zymed Laborato-ries (Alfredo Cavalheiro;Lisbon,Portugal)and mouse anti-syntaxin antibody (0.5mg/ml)and mouse anti-synaptophysin antibody (100g/ml)were from Sigma (Sintra,Portugal).[3H]SCH 58261and adenosine deaminase were prepared directly into the incubation solution each day and [3H]SCH 58261was made up as a 5mM stock in dimethylsulphoxide.All cell culture solutions were purchased from GIBCO BRL (Life Technologies,Scotland,UK).All other reagents were of the highest purity available.StatisticsData are mean ϮS.E.M.values or mean (95%confidence interval)of n experiments.Significance was considered at P Յ0.05using the Student’s t -test.RESULTSDifferent subcellular localization of A 2A receptors in the striatum and hippocampusTo investigate the synaptic localization of striatal and extra-striatal A 2A receptors,we started to test if there was an enrichment of A 2A receptors in nerve terminals in either the hippocampus or striatum.Thus,we compared the bind-ing density of the selective A 2A receptor antagonist,[3H]SCH 58261,in total membranes from the striatum and hippocampus and in membranes of synaptosomes,the best purified model to study presynaptic mechanisms (e.g.Cunha,1998).We first confirmed that the synaptosomal membranes indeed corresponded to an enriched fraction of nerve terminals.In fact,we now observed that there was an over 450%enrichment of synaptophysin immunoreac-tivity (a protein located in synaptic vesicles)in synaptoso-mal membranes compared with total membranes in both the striatum and hippocampus,as evaluated by Western blot analysis.In the striatum,the A 2A receptor antagonist,[3H]SCH 58261,bound to total membranes with a K d of 0.96nM (95%confidence interval:0.65–1.23nM,n ϭ5)and a B max of 997Ϯ27fmol/mg protein (n ϭ5;Fig.1A),as previously ob-served (Alfaro et al.,2004).[3H]SCH 58261binding in syn-aptosomal membranes from the rat striatum was similar to that found in total striatal membranes (Fig.1A),both in terms of affinity (K d ϭ0.98,95%confidence interval:0.63–1.33nM,n ϭ6)and of density (B max ϭ940Ϯ67fmol/mg protein,n ϭ6).Thus,there was no significant (P Ͼ0.05)enrichment ofN.Rebola et al./Neuroscience 132(2005)893–903895[3H]SCH 58261binding in nerve terminals from the rat striatum.The situation was different in the hippocampus.[3H]SCH 58261,bound to total hippocampal membranes with a K d of 1.01nM (95%confidence interval:0.54–1.48nM,n ϭ6)and a B max of 29.1Ϯ3.7fmol/mg protein (n ϭ6;Fig.1B).Thus,the binding affinity of [3H]SCH 58261was similar in the striatum and hippocampus,but the density of [3H]SCH 58261binding was nearly 20times lower in the hippocampus than in the striatum.And,in contrast with what was observed in the striatum,there was a marked (P Ͻ0.05)enrichment of [3H]SCH 58261binding in nerve terminals of the rat hip-pocampus (Fig.1B).In fact,[3H]SCH 58261bound to hippocampal synaptosomal membranes with a B max of 103Ϯ12fmol/mg protein (n ϭ6)and a K d of 1.04nM (95%confidence interval:0.75–1.33nM,n ϭ6).This indicates that A 2A receptors are mostly located in nerve terminals in the hippocampus,in contrast to what occurs in the striatum.Since there was a large variability in results in the binding experiments in hippocampal tissue due to the low signal-to-noise ratio,we used a second experimental ap-proach to confirm this different location in nerve terminals of striatal and extra-striatal A 2A receptors,now based on the use of antibodies rather than of a selective ligand for A 2A receptors.We have previously validated the selectivity of a goat antibody against the C-terminus of A 2A receptors for extra-striatal A 2A receptors,based on the similar mo-lecular weight of the band recognized by the antibody in striatal and cortical membranes of rat and mouse and to the disappearance of this single 44kDa band in cortical membranes of A 2A receptor knockout mice (see Lopes et al.,2004),the same occurring for striatal membranes (L.O.Porciu ´ncula and R.A.Cunha,unpublished obser-vations).We also verified in immunohistochemical exper-iments that this A 2A receptor antibody stains rat brain sections in a manner similar to radiolabeled A 2A receptor ligands,i.e.predominantly in the basal ganglia (unpub-lished observations).The identification of A 2A receptors by Western blot was also confirmed with another A 2A receptor antibody,a mouse anti-A 2A receptor raised against the third intracellular loop of the A 2A receptor (see Hettinger et al.,2001;i.e.a different epitope from that recognized by the goat anti-A 2A receptor antibody).When used at a dilu-tion of 1:1000,this second antibody also recognized a single band of 44–45kDa both in striatal and hippocampal tissue derived from either rat and mouse (the technical sheet of this antibody already provides data showing the lack of signal using this dilution of the antibody in striatal tissue derived from A 2A receptor knockout mice).Thus,we now compared by Western blot analysis the density of immunoreactivity found in total membranes and synapto-somal membranes from the hippocampus and striatum.As illustrated in Fig.2A and C using the goat anti-A 2A receptor antibody,there was a consistently lower A 2A recep-tor immunoreactivity in synaptosomal membranes compared with total membranes from the striatum.This again confirms that A 2A receptors are mostly located outside nerve terminals in the striatum.The opposite was observed in the hippocam-pus.In fact,as illustrated in Fig.2B and D,there was a consistent and significant (P Ͻ0.05)enrichment of A 2A recep-tor immunoreactivity in synaptosomal membranes compared with total membranes of the hippocampus.These results confirm the data obtain in binding studies,indicating that A 2A receptors are enriched in nerve terminals in the hippocampus but not in the striatum.It should be noted that considerably higher amounts of protein from hippocampal compared with striatal tissue had to be applied to the gels to observe A 2A receptor immunoreactivity,again indicating that the density of A 2A receptors is considerably lower in the hippocampus than in the striatum.Different subcellular localization of A 2A receptors in the striatal and hippocampal neuronsWe next decided to use a different preparation that allows visualizing intact neurons to confirm the different relative localization of A 2A receptors in nerve terminals of the hip-pocampus or striatum.Thus,we compared the patternofFig. 1.Saturation binding curves of the selective A 2A receptor antagonist,[3H]SCH 58261,to total membranes (filled circles)and synaptosomal membranes (open circles)of rat striatum (A)and hip-pocampus (B).The ordinates represent the specific binding of [3H]SCH 58261,obtained upon subtraction of the non-specific binding determined with 1M XAC from total binding,which represented 11–19%of the total binding in the striatum and 42–67%of the total binding in the hippocampus.The Scatchard plots derived from these saturation curves were linear (not shown for the sake of clarity).The data presented in the figures are mean ϮS.E.M.of five to six experi-ments.Note that the density of binding of [3H]SCH 58261to striatal membranes is nearly 20-fold greater than that observed in hippocam-pal membranes.N.Rebola et al./Neuroscience 132(2005)893–903896A 2A receptor immunoreactivity in cultured hippocampal neurons and striatal neurons obtained from rat pups.As illustrated in Fig.3A using the goat anti-A 2A recep-tor antibody,we found that A 2A receptor immunoreactivity in cultured hippocampal neurons was present in the cell bodies (see Lee et al.,2003)but displayed the most in-tense labeling in a punctuate manner away from cell bod-ies.The double labeling of cultured hippocampal neurons with antibodies against A 2A receptors and against synap-tophysin (a marker of nerve terminals),revealed an exten-sive co-localization of A 2A receptor and synaptophysin immunoreactivity in these cultured hippocampal neurons (Fig.3C).This reinforces our previous conclusion that A 2A receptors are indeed mainly located in nerve terminals in the hippocampus since this was observed both in cultured neurons that were derived from immature rats and in mem-brane preparations obtained from adult rats.Again,and as observed in native tissue from adult animals,the distribution of A 2A receptors in cultured striatal neurons differed from that found in hippocampal neurons.In fact,it was observed that A 2A receptor immunoreactivity was mostly found in the cell bodies of striatal neurons,although there was also a less intense A 2A receptor immu-noreactivity in branched filaments emerging from cell bodies (Fig.3D).The double labeling of cultured striatal neurons with antibodies against A 2A receptors and against synaptophysin revealed a minor co-localization of A 2A re-ceptor and synaptophysin immunoreactivity,since most of the A 2A receptor immunoreactivity was not co-located with synaptophysin in these cultured striatal neurons (Fig.3F).This confirms our previous conclusion that A 2A receptors are mostly located outside nerve terminals in striatal tis-sues,which does not obviously exclude a minor localiza-tion of A 2A receptors in nerve terminals in striatal neurons.As was carried out for the Western blot analysis,we also repeated the characterization of A 2A receptor immu-noreactivity in cultured neurons from either the hippocam-pus or the striatum with the two different antibodies against different epitopes of the A 2A receptor.We confirmed that the relative distribution of A 2A receptor immunoreactivityinFig.2.A 2A Receptor immunoreactivity in total membranes and synaptosomal membranes from the rat striatum (A,C)and hippocampus (B,D).A and B show representative Western blots comparing the A 2A receptor immunoreactivity,corresponding to the 44kDa band,when applying different amount of protein (indicated below each line)of total membranes (total,first three lanes from the left in each panel)and synaptosomal membranes (synaptosomal,first three lanes from the right in each panel)from the rat striatum (A)and hippocampus (B).A re-probing of the membranes with an anti-tubulin antibody,shown below each gel in each panel,confirmed that similar amounts of protein were added for each concentration of total and synaptosomal membranes.C and D show the average relative immunoreactivity of A 2A receptors in total membranes (filled squares)and synapto-somal membranes (open circles)in the striatum (C)and hippocampus (D)according to the amount of protein applied to gel in three different Western blot analysis using membranes from different animals.*P Ͻ0.05comparing A 2A receptor immunoreactivity in total and synaptosomal membranes.The blots presented were obtained using the goat anti-A 2A receptor antibody and similar results were obtained in two experiments using the mouse anti-A 2A receptor antibody (data not shown).N.Rebola et al./Neuroscience 132(2005)893–903897。
EP09 A3 Measurement Procedure Comparison and Bias Estimation Using Patient Samples

2. Allowable random error (REa)
• Precision • Target SD values for routine QC
• • Medicare Part B, 2010: approx. 44,000,000 glucose blood tests
– • 2.2 million errors with 95% goal – • >132,000 errors with 99.7% goal
• +/- 2SD corresponds to 95% of data (2 sigma) • +/- 3SD corresponds to 99.7% of data ( 3 sigma) •+/- 6SD corresponds to 99.9993% of data ( 6 sigma)
– Best practices recommend measuring at least 3 replicates. – SEa is a fraction of the TEa.
Carol R Lee
1
Accuaracy, Bias and Trueness
4/26/2016
What error do you want?
• Analytical error:
– The amount of error that your lab test actually has
Performance Standards
Patient Results
10
TEa includes both bias and imprecision
• The CLIA Calibration Verification experiment assesses the difference (bias) from the measured value to the true value.
成组序贯实验原理 group sequential test-概述说明以及解释

成组序贯实验原理group sequential test-概述说明以及解释1.引言1.1 概述概述部分的内容如下:成组序贯实验是一种重要的临床试验设计方法,旨在提高临床试验效率和减少试验风险。
传统的临床试验通常是一次性地进行,无法及时根据中间结果进行调整。
然而,成组序贯实验通过在试验过程中多次进行中间分析,可以在保证试验可靠性的前提下,提前终止试验或选择优胜的治疗方案。
成组序贯实验的基本原理是通过不断积累来自试验治疗组和对照组的数据,根据预定的监测规则进行中间分析,以确定是否已达到预设的效果或是否存在显著差异。
与传统试验相比,成组序贯实验可以根据中间结果进行统计推断,而无须等待试验结束,从而加快了试验过程。
成组序贯实验具有许多优势。
首先,它可以在试验过程中及时掌握到试验治疗方案的效果,这对于优化治疗方案和提高临床效果非常重要。
其次,成组序贯实验可以在中间分析时终止试验,从而减少了试验时间和资源的消耗。
此外,它还可以降低试验过程中的风险,保护受试者的权益。
随着临床研究的不断发展,成组序贯实验的重要性逐渐凸显。
它不仅可以提高试验效率,减少试验成本,还可以为临床决策提供及时可靠的依据。
未来,随着统计学和数据分析方法的进一步改进,成组序贯实验在医学研究中的应用前景将更加广阔。
1.2文章结构文章结构部分的内容可以包括以下信息:文章结构部分旨在介绍整篇文章的组织架构,让读者能够清晰地了解文章的组织框架和各个部分的内容。
本文按照以下结构进行组织和呈现:第一部分为引言部分,主要包括概述、文章结构和目的。
在概述中,将简要介绍成组序贯实验原理及其重要性。
在文章结构部分,将详细列举并描述本文各个章节的内容,以便读者能够有一个清晰的整体概念。
最后,在目的部分,将明确阐述本文的目标和意义,以便读者对文章有一个明确的预期。
第二部分为正文部分,主要包括成组序贯实验的基本原理和其优势两个部分。
在基本原理部分,将详细介绍成组序贯实验的概念、基本原理和实施流程,以及与传统实验设计的区别和联系。
不规则抗体筛选试验标准操作规程(sop)

不规则抗体筛选试验标准操作规程一、目的为规范微柱凝胶抗球蛋白检测卡在红细胞不规则抗体筛选试验中的应用,保证实验结果的准确性和有效性。
二、适用范围输血科进行的微柱凝胶卡式不规则抗体试验。
三、原理红细胞血型抗原—抗体反应体系在微柱凝胶卡微孔上端反应室反应后,如红细胞血型抗原与相应特异性抗体发生凝集反应后,离心后被排阻在具有三维空间网状结构的凝胶表层或者颗粒间隙之中,不能通过凝胶筛网,而未发生抗原—特异性抗体凝集的红细胞则可以通过凝胶筛网沉降至微柱凝胶底部。
四、步骤和方法1、取微柱凝胶抗球蛋白检测卡放入专用离心机,3000r/min离心1min后撕开锡纸膜,正立放置待用。
2、在标记为I、II、III的各反应腔中分别加入对应的I、II、III号筛选红细胞稀释液(1滴细胞+3滴lis液)50微升,再在标记为I,II,III的各反应腔中分别加入对应的受检者血清(血浆)20微升,加入后轻轻震荡3次使其充分混合。
3、将检测卡在微柱凝胶专用孵育器中37℃孵育15min后,用专用离心机以1800r/min·2min,2500r/min·1min离心后取出判断结果。
5、结果判读:I、II、III型红细胞任何一种出现阳性反应,表明该血清(血浆)不规则抗体筛选阳性。
五、质量控制微柱凝胶试剂卡,试剂筛选红细胞在有效期内使用。
六、注意事项1、检测卡若在冰箱储存,使用前须放置实验室5—10min平衡至室温。
2、撕开微柱凝胶卡锡纸膜时,注意避免各微柱间特异性抗体试剂的交叉污染。
3、细菌污染,纤维蛋白析出及抗凝不佳的血,可产生假阳性结果,建议使用血清。
4、严格遵循离心力,离心时间要求,离心速度过快,离心时间过长均易产生假阴性结果。
5、由于试剂生产厂家,批号不同等因素,红细胞浓度,吸取样品的量及离心条件等环节要求有所差异,以试剂说明书为准。
α-淀粉酶菌株摇瓶发酵初筛、复筛及酶活力测定

初筛菌株α -淀粉酶活力计算记录表
初筛菌株α -淀粉酶活力计算记录表
班次: 组号: 反应时间 菌株编号 分 秒 分钟 稀释倍数 姓名: 酶活力单位 (U/mL)
备注说明
复筛菌株α -淀粉酶活力计算记录表
复筛菌株α -淀粉酶活力计算记录表
班次: 菌株编号: 重复瓶号 1 2 3 4 斜面直接接种 反应时间 分 秒 分钟 稀释倍数 酶活力单位 (U/mL) 备注说明 组号: 姓名:
实验室摇瓶机(摇床)的作用
使培养基一直保持均匀状态——菌体始终 接触到新的营养成分(相对来说);快速 稀释代谢废物(一般对细胞有毒害作用, 对人类来说可能有用) 增加培养基中溶解氧的浓度,提高菌种的 有氧代谢强度,加快物质的转化,有利于 代谢产物的合成和积累。
摇瓶机的种类
旋转式——转速相应较高(200转以上), 培养液贴壁旋转; 往复式——转速不易太高,一般120转/分 左右,培养液来回振荡,振荡液面较高; 往复式的溶氧效果较好(相对旋转式); 注意摇瓶机的振幅(偏心距)。
发酵培养基(8瓶/组) (100mL装于500mL三角瓶)
玉米淀粉(可溶性淀粉 ) 1g; 豆粕粉 3g; Na2HPO4 .12H2O 0.8g; (NH4)2SO4 0.4g; NH4Cl 0.15g; CaCO3 0.10g 水100mL 121.3℃灭菌20分钟 初筛和复筛所需要的一起准备配制。
本次实验基本流程
冷却后接种 准备发酵 灭菌 环接种 培养基 (1瓶/株) 恒温摇 瓶培养 发酵液棉 花过滤 测定酶 活力 确定1-2 株复发酵 菌株
一级液体 活化菌种
液体菌种接种 于发酵培养基 (3-5瓶/株) 灭菌 准备发酵 培养基
恒温摇 瓶培养
发酵液棉 花过滤
螺旋翻折法实操考试中的模拟题

螺旋翻折法实操考试中的模拟题(含答案)1. 请列出螺旋翻折法中一般的实验步骤。
答案:一般的实验步骤包括:蛋白质提取、沉淀、溶解、结晶、纯化和鉴定等步骤。
2. 在螺旋翻折法实验中,如何选择适当的缓冲液?答案:选择适当的缓冲液需要考虑多个因素,如蛋白质的等电点、缓冲液的pH值、离子强度、缓冲剂的种类等。
一般建议选择pH值接近蛋白质等电点的缓冲液,并根据具体情况进行调整。
3. 螺旋翻折法中,如何判断蛋白质结晶的质量?答案:判断蛋白质结晶的质量需要观察结晶的外观和形态,如是否呈现规则的形状和大小,是否有杂质等。
此外,还可以通过显微镜和X射线衍射等技术来进一步确定结晶的质量。
4. 在螺旋翻折法实验中,如何纯化蛋白质?答案:纯化蛋白质可以通过多种技术实现,如离子交换、凝胶过滤、亲和层析等。
具体选择哪种方法需要根据蛋白质的性质和特点进行判断。
5. 在螺旋翻折法实验中,如何鉴定蛋白质的纯度?答案:鉴定蛋白质的纯度可以通过多种技术实现,如SDS-PAGE、Western blot、质谱等。
具体选择哪种方法需要根据蛋白质的特点和实验需求进行判断。
6. 在螺旋翻折法实验中,如何调节缓冲液的pH值?答案:调节缓冲液的pH值需要使用一些常见的缓冲剂,如Tris-HCl、MES、HEPES等,并在适当的浓度下加入酸或碱来调节pH值。
注意,调节后需要使用电极等仪器检测pH值是否达到预期值。
7. 在螺旋翻折法实验中,如何去除杂质和不纯物质?答案:去除杂质和不纯物质需要使用一些常见的技术,如离心、过滤、洗涤等。
具体选择哪种方法需要根据实验需求和蛋白质的特点进行判断。
8. 在螺旋翻折法实验中,如何对蛋白质进行再结晶?答案:对蛋白质进行再结晶需要使用一些常见的技术,如改变结晶条件、添加结晶助剂等。
具体选择哪种方法需要根据实验需求和蛋白质的特点进行判断。
9. 在螺旋翻折法实验中,如何使用X射线衍射技术来确定蛋白质的晶体结构?答案:使用X射线衍射技术来确定蛋白质的晶体结构需要进行多个步骤,如收集X射线数据、解析晶体结构等。
rafls细胞实验记录

rafls细胞实验记录
选择类风湿关节炎关节成纤维样滑膜细胞的细胞MH7A和正常人关节成纤维样滑膜细胞HFLS用于实验研究.MH7A和HFLS细胞生长至汇合度80%左右时进行细胞传代,三代至六代细胞用于实验研究。
台盼蓝染色实验鉴定细胞活性。
利用Transwell细胞迁移实验和Transwell细胞侵袭实验检测并比较两种细胞的迁移、侵袭能力。
Trizol法分别提取两组细胞的总RNA,并进行RNA定量分析。
按照SYBR RPrimeScriptTM miRNA RT -PCRKit (Takara,RR716)说明书进行Poly(A)加尾反应和反转录成cDNA。
选定用于实验的miRNAs和内参,查找miRNAs数据库,分别找到选定miRNAs的原始序列,并按照引物设计原则进行引物设计,合成扩增miRNAs的引物。
进行实时定量聚合酶链式反应(Real-time PCR),并对实验数据进行统计学分析。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
©
SI
M
TI
Se
rv
Flow cytometry Flow cytometry analysis of native and enzymeconverted RBC was performed using an FACScan flow cytometer (Cytomics FC 500 Beckman Coulter, Brea, United States of America) with anti-B monoclonal antibodies (Changchun Brother Biotech Co., Ltd., Changchun, China) and fluorescein isothiocyanateconjugated affinity purified goat anti-mouse IgM (Jackson ImmunoResearch Laboratories, Inc., West Grove, United States of America). Briefly, 10 μ L cells were fixed overnight at room temperature under gentle agitation by the addition of 100 μ L of 2% paraformaldehyde (w/v, Sigma-Aldrich, St. Louis, United States of America) in PBS to prevent agglutination of antigen-positive cells. Next, 2 μL packed RBC were prewashed with 500 μL PBS twice and resuspended in 50 μL PBS. Then 50 μL of undiluted primary antibody was added and incubated for 60 minutes in the dark at 25 °C. After two washes and resuspension in 100 μL PBS, 2 μL of undiluted secondary antibody was added and incubated for 60 minutes in the dark at 25 °C. The cells were analysed after another two washes (as above) and resuspension in 500 μL PBS.
iz
ห้องสมุดไป่ตู้
iS
rl
305
Gao H-W et al
Results
The enzyme-treated RBC did not agglutinate with the three kinds of commercial anti-B monoclonal antibodies or with the anti-B serum from ten group A donors in multiple observations of visual microscope fields in different conditions. All the results were rechecked using anti-B antibody: native A2B showed a higher positive rate, 99.3%, but native A2 and enzymatically converted A2 RBC gave low positive rates of 0.3% and 0.7%, respectively (Figure 1). Moreover the enzymatically produced A2 RBC, just like the A2 control RBC, did not agglutinate with anti-A1 monoclonal antibodies or anti-A1 serum (Table I). These results indicate that we successfully obtained A2 RBC from A2B RBC through
1 2
Department of Blood Biochemistry and Molecular Biology, Beijing Institute of Transfusion Medicine, Beijing; Shaanxi Blood Center, Xi'an, People's Republic of China
Blood Transfus 2013; 11: 305-7 DOI 10.2450/2012.0020-12
© SIMTI Servizi Srl
Materials and methods
Enzymatic treatment of A2B red blood cells with B. fragilis -galactosidase Fresh human whole blood (blood group A2B) was obtained from Shaanxi Province Blood Centre (Xi'an, China), and the buffy coat was removed. Enzymatic conversion was performed in 1 mL reaction mixtures containing 200 mmol/L glycine and 3 mmol/L NaCl, at pH 6.8 (conversion buffer), with 30% packed RBC as described by Liu et al.9,10. Briefly, RBC were prewashed 1:1 and 1:4 (v/v) in conversion buffer before addition of -galactosidase (0.005 mg/mL packed RBC) and incubation for 60 min with gentle mixing at 26 °C, followed by four repeat washing cycles with 1:4 (v/v) phosphate-buffered saline (PBS) by centrifugation at 2,000 rpm for 5 min. The washed, enzyme-converted RBC were stored in monoammonium phosphate nutrient solution at 4 °C.
Introduction
Correct typing of donors' and recipients' blood group is of paramount importance in transfusion services. There are two distinct parts in ABO typing, forward and reverse typing, both of which are routinely carried out1. Reverse typing is obligatory, because it can help to reveal mistyping, weak A subgroups with anti-A1 and unexpected IgM antibodies. Any discrepancy between the results of the tests with serum or plasma and red cells should be investigated. Reverse grouping cells, including A, B, O and A 2 type red blood cells (RBC), are important to resolve ABO discrepancies. Anti-A and/ or anti-B antibodies can be easily detected by red blood cells with A and/or B blood group antigens. The presence of an anti-A1 should be confirmed by testing serum against A1, A2, and O red cells. This method necessitates A2 reverse grouping cells. Very few people in China have the A2 blood group. In the Xi'an area (north-western China) only about 0.0006% of the population have A2 RBC, while about 0.003% of the population have the A2B group are. The ratio of the prevalence of A2B:A2 is 5 (unpublished data). In the Shanghai area (eastern China), this same ratio is about 2.5. Thus, there are more people with A2B group RBC than there are with A2 RBC2,3. As it is known that -galactosidase, a kind of exoglycosidase, can remove the reducing end -galactose residues of group B antigen by hydrolysis and based on the characteristics of the group B epitopes and -galactosidase hydrolysis reaction, -galactosidase has been used in B to O RBC conversion study since the 1980s4. Our group has been researching in this area for more than 10 years5,6 and we recently obtained a novel -galactosidase from B. fragilis (which belong to CAZy GH110) with highly specific activity, greatly restricted substrate specificity and a neutral pH optimum7,8. Our research showed that this novel -galactosidase can convert B RBC to O RBC with high substrate specificity and at low cost7,8. We, therefore, started to study the A2B to A2 conversion using -galactosidase in order to broaden the source of A2 RBC.