质粒DNA提取、酶切及琼脂糖凝胶电泳
质粒DNA的提取与电泳鉴定_百替生物

质粒DNA的提取与电泳鉴定质粒DNA的提取是从事基因工程工作中的一项基本实验技术,但提取方法有很多种,以下介绍一种最常用的方法:碱裂解法:此方法适用于小量质粒DNA的提取,提取的质粒DNA可直接用于酶切、PCR扩增、银染序列分析。
方法如下:1、接1%含质粒的大肠杆菌细胞于2ml LB培养基。
2、37℃振荡培养过夜。
3、取1.5ml菌体于Ep管,以4000rpm离心3min,弃上清液。
4、加0.lml溶液I(1%葡萄糖,50mM/L EDTA pH8.0,25mM/L Tris-HCl pH8.0)充分混合。
5、加入0.2ml溶液II(0.2 mM/L NaOH,1%SDS),轻轻翻转混匀,置于冰浴5 min 。
6、加入0.15m1预冷溶液III(5 mol/L KAc,pH4.8),轻轻翻转混匀,置于冰浴5 min 。
7、以10,000rpm离心20min,取上清液于另一新Ep管8、加入等体积的异戊醇,混匀后于?0℃静置10min。
9、再以10,000rpm离心20min,弃上清。
10、用70%乙醇0.5ml洗涤一次,抽干所有液体。
11、待沉淀干燥后,溶于0.05mlTE缓冲液中煮沸法1、将1.5ml培养液倒入eppendorf管中,4℃下12000g离心30秒。
2、弃上清,将管倒置于卫生纸上几分钟,使液体流尽。
3、将菌体沉淀悬浮于120ml STET溶液中, 涡旋混匀。
4、加入10ml新配制的溶菌酶溶液(10mg/ml), 涡旋振荡3秒钟。
5、将eppendorf管放入沸水浴中,50秒后立即取出。
6、用微量离心机4℃下12000g离心10分钟。
7、用无菌牙签从eppendorf管中去除细菌碎片。
8、取20ml进行电泳检查。
质粒DNA的大量提取和纯化在制作酶谱、测定序列、制备探针等实验中需要高纯度、高浓度的质粒DNA,为此需要大量提取质粒DNA。
大量提取的质粒DNA一般需进一步纯化,常用柱层析法和氯化绝梯度离心法。
实验八质粒DNA的提取及琼脂糖凝胶电泳检测

当加入pH4.8的乙酸钾高盐缓冲液恢复 pH至中性时,共价闭合环状的质粒DNA的两 条互补链仍保持在一起,因此复性迅速而准 确,而线性的染色体DNA的两条互补链彼此 已完全分开,复性就不会那么迅速而准确, 它们缠绕形成网状结构,通过离心,染色体 DNA与不稳定的大分子RNA,蛋白质-SDS复 合物等一起沉淀下来而被除去。
2.简要叙述酚氯仿抽提DNA体系后出现的现象及其成 因?
3.沉淀DNA时为什么要用无水乙醇及在高盐、低温条 件下进行?
质粒3种构型
1.超螺旋的共价闭合环状质粒DNA(covalelltly closed circular DNA,简称cccDNA); 2.开环质粒DNA,即共价闭合环状质粒DNA的一条链 断裂,(open circular DNA,简称ocDNA); 3.线形质粒DNA,即共价闭合环状质粒DNA 两条链发 生断裂(linear DNA,简称LDNA)。
2. 琼脂糖凝胶电泳检测DNA
一、实验目的
通过本实验学习琼脂糖凝胶电泳检测 DNA的方法和技术。
二、实验原理
DNA分子在琼脂糖凝胶中泳动时有电荷效应和分子 筛效应。DNA分子在高于等电点的pH 溶液中带负 电荷,在电场中向正极移动。由于糖—磷酸骨架在 结构上的重复性质,相同数量的双链DNA几乎具有 等量的净电荷,因此它们能以同样的速度向正极方 向移动。在一定的电场强度下,DNA分子的迁移速 度取决于分子筛效应,即DNA分子本身的大小和构 型。具有不同的相对分子质量的DNA片段泳动速度 不一样,可进行分离。DNA分子的迁移速度与相对 分子质量的对数值成反比关系。
2.取1.4 ml培养液至Eppendorf管中,12000rpm离心30 秒,弃上清,取沉淀。
3.将细菌沉淀悬浮于100µl预冷溶液Ⅰ中,振荡混匀。 4.加入200µl溶液Ⅱ,缓慢颠倒离心管,混匀(勿剧烈
质粒dna的提取及电泳检测实验报告

质粒dna的提取及电泳检测实验报告一、实验目的本实验的主要目的是学习质粒DNA的提取方法和电泳检测技术,并通过实验观察质粒DNA的提取情况和电泳检测结果。
二、实验原理质粒DNA的提取方法主要采用碱裂解法,通过将细胞裂解后,用酸性物质中和碱性物质,使DNA分离出来,最后用酒精沉淀纯化。
电泳检测是通过将DNA 样品置于琼脂糖凝胶上,加上电场后,DNA在凝胶中移动,通过电泳带电的原理,将DNA分离出来,从而观察DNA的大小和形状。
三、实验步骤1.准备样品:将含有目标DNA的菌落挑出放入EP管中,加入100μl TE缓冲液(10mmol/L Tris-HCl,1.0mmol/L EDTA,pH8.0),用微量移液器均匀混合。
2.制备裂解液:取10μl 0.25mol/L NaOH加入菌液中,轻轻摇匀,并立即加入200μl 1%SDS(十二烷基硫酸钠)液,翻转混匀。
3.裂解DNA:加入150μl NaOH-SDS溶液,翻转10次,置于80℃水浴中裂解细胞壁和细胞膜,20min后取出,加入150μl冷KAc/EDTA溶液(3mol/L KAc,pH5.2,0.5mol/L EDTA),翻转混匀,冰上静置5min。
4.离心沉淀:10000r/min离心10min,将上清液移至新的96孔板中,加入0.6倍体积的冰凉乙醇,反复翻转,置于-20℃上进行冷却,离心12000r/min 10min,去掉上清液,用95%乙醇洗涤沉淀物,最后用300μl TE缓冲液溶解沉淀DNA。
5.电泳检测:取相同浓度的DNA样品,加入琼脂糖,混合后加入电泳槽中,通电检测10min。
四、实验结果经过实验操作后,取出DNA样品,通过电泳检测,观察到有一条长度合适的条带,证明质粒DNA已经成功提取,并且检测结果与对照组差异不大,说明实验操作规范,结果可信。
五、实验结论本次实验通过碱裂解法成功提取了质粒DNA,并通过电泳检测技术检测到了DNA条带,证明提取质粒DNA的方法可行,同时也说明电泳检测是一种可靠的分子生物学技术,对于DNA分离和检测有着重要的应用价值。
质粒的酶切—琼脂糖凝胶电泳分离DNAppt课件

1. 实验目的 2. 实验原理 3. 实验仪器、材料与试剂 4. 实验步骤 5. 实验结果及讨论
实验目的
限制性内切酶切割出目的DNA
了解琼脂糖凝胶电泳的原理
掌握琼脂糖凝胶的制作及进行电泳的方 法
实验原理
利用限制性内切酶具有特定的识别及切割位 点,定点切割环状质粒DNA。
Tris 242g 冰醋酸 57.1ml 0.5mol/L EDTA(pH8.0) 100ml 3. 溴化乙锭溶液 10mg/ml水溶液,室温,棕色瓶或铝 铂纸包装保存。 4. 10×DN切体系
调制酶切体系如下: (共 20μL)
无菌水 6μL,
质粒
10 μL
Buffer K 2μL,
EcoR I 1μL,
BamH I 1 μL
37℃酶切1.5小时。
琼脂糖凝胶制备
1. 洗净电泳槽、制胶槽、梳子、挡板,晾干。 2. 插好挡板、梳子。 3. 根据实验所需称取0.4克琼脂糖,放入制胶
的容器中(一般用三角瓶),然后加入40ml电 泳缓冲液(1×TAE)。 4. 加热煮沸,使琼脂糖完全溶解。 5. 溶液冷至约60℃时,加入溴化乙锭4 μL (终浓 度为0.1 g/l),轻轻摇匀,倒入制胶槽上,检查 有无气泡。
纯度;DNA的甲基化程度;温度;缓冲液成分 (DDT,BSA)等。
使用3 mg的DNA Marker DL 2,000(500 ng/条带)进行1% 的琼脂糖凝胶电泳后,各DNA条带自上至下分别为2kb,1Kb, 750bp,500bp,250bp,100bp。
9.用移液器吸起离心管中溶液,移入凝胶的梳孔中。
10.插上导线,打开电泳仪电源,按照需要调节 电压至120V,电泳开始,观察电流情况或电泳 槽中负极的铂金丝是否有气泡出现。
质粒DNA酶切及琼脂糖凝胶电泳分离鉴定

质粒DNA酶切及琼脂糖凝胶电泳分离鉴定组员:孙慜、孙苏、谢拓燕、郑倩倩、王佳玲【实验目的】1、了解DNA的限制性内切酶酶切的基本原理2、学习琼脂糖凝胶电泳检测DNA的方法和技术【实验原理】①DNA的限制性内切酶酶切原理(1)限制性内切酶能特异地结合于一段被称为限制性酶识别序列的DNA序列之内或其附近的特异位点上,并切割双链DNA,但不能切单链DNA。
它可分为三类:I类和III类酶在同一蛋白质分子中兼有切割和修饰(甲基化)作用且依赖于ATP的存在。
III类酶结合于识别位点并随机的切割识别位点不远处的DNA,而Ⅲ类酶在识别位点上切割DNA分子,然后从底物上解离。
Ⅱ类由两种酶组成: 一种为限制性内切核酸酶(限制酶),它切割某一特异的核苷酸序列; 另一种为独立的甲基化酶,它修饰同一识别序列。
Ⅱ类中的限制性内切酶在分子克隆中得到了广泛应用,它们是重组DNA的基础。
绝大多数Ⅱ类限制酶识别长度为4至6个核苷酸的回文对称特异核苷酸序列(如EcoRⅠ识别六个核苷酸序列:5‘- G↓AATTC-3‘), 有少数酶识别更长的序列或简并序列。
Ⅱ类酶切割位点在识别序列中, 有的在对称轴处切割, 产生平末端的DNA片段(如SmaⅠ:5‘-CCC↓GGG-3‘); 有的切割位点在对称轴一侧, 产生带有单链突出末端的DNA片段称粘性未端, 如EcoRⅠ切割识别序列后产生两个互补的粘性末端。
5‘…G↓AATTC…3‘→5‘… G AATTC…3‘3‘…CTTAA↑G …5‘→3‘… CTTAA G…5‘DNA限制性内切酶酶切图谱又称DNA的物理图谱,它由一系列位置确定的多种限制性内切酶酶切位点组成,以直线或环状图式表示。
在DNA序列分析、基因组的功能图谱绘制、DNA的无性繁殖、基因文库的构建等工作中,建立限制性内切酶图谱都是不可缺少的环节,近年来发展起来的RFLP(限制性片段长度多态性)技术更是建立在它的基础上。
②琼脂糖凝胶电泳条带的观察:(2)通过观察示踪染料的迁移距离可以判断DNA的迁移距离。
质粒DNA的提取与琼脂糖凝胶电泳鉴定

实验题目质粒DNA的提取与琼脂糖凝胶电泳鉴定实验目的掌握质粒提取的原理与各种试剂的作用和琼脂糖凝胶电泳原理和操作实验原理质粒存在于细菌,线粒体等,独立于染色体之外,能够稳定遗传并自我复制,是一个双链环装DNA。
对数期大肠埃希菌含有质粒。
溶液一可使细菌充分重悬,溶液二使细菌裂解,释放质粒和基因组DNA.溶液三起到中和作用,使得质粒DNA迅速复性,而基因组DNA因为分子巨大,复性时间长。
离心后,质粒DNA留在上清液,而基因组DNA和细胞碎片沉淀在离心管。
琼脂糖凝胶电泳蛋白质和核酸会根据pH不同带有不同电荷,在电场中受力大小不同,因此泳动的速度不同,根据这个原理可将其分开。
电泳缓冲液的pH在6~9之间,离子强度0.02~0.05为最适。
常用1%的琼脂糖作为电泳支持物。
用各种浓度的琼脂糖凝胶可以分离长度为200bp至近50bp的DNA。
此外,直接用低浓度的核酸染料进行染色,可以确定DNA 在凝胶中的位置。
实验主要步骤质粒DNA提取1.取4ml细菌平均分两份加入离心管,以12000r/min离心1min,去除上清液2.加100ul用冰预冷溶液I,移液枪打散细菌沉淀使其成为悬浮液3.加入200ul溶液II,盖紧盖口,翻转五次,充分混合,避免振荡,置于冰上4.加入150ul冰预冷溶液III,盖紧盖口,翻转离心管5次,温和摇匀直到粘稠的细菌裂解物出现,放于冰上5min5.离心管以12000r/min离心5min6.取上清液加入等量的酚:氯仿混合液,轻轻摇匀,以12000r/min离心7min7.取上清液加入2倍体积100%乙醇沉淀DNA,轻轻摇匀,12000r/min离心5min,去除上清液,倒置在滤纸上干燥8.用1ml70%乙醇洗涤DNA沉淀,按照步骤7去除上清液,空气干燥10min9.用50ul无菌水溶解质粒DNA琼脂糖凝胶电泳:1.制胶。
将电泳缓冲液和琼脂糖在微波炉中熔化,混匀,冷却至55°℃,加入EB染料,倒入已封好的凝胶灌制平台上,插上样品梳。
质粒DNA的限制性内切酶酶切及琼脂糖凝胶电泳分析

质粒DNA的限制性内切酶酶切及琼脂糖凝胶电泳分析摘要限制性内切酶发现于某些大肠杆菌体内,能够“限制”噬菌体对其感染并帮助将已殖入的噬菌体序列移除,可以识别双链DNA上特异序列(限制性位点)并酶切。
限制酶极大地促进了分子生物学、基因工程与遗传工程领域的进展。
[1]本实验通过对大肠杆菌质粒DNA的酶切处理,经琼脂糖凝胶电泳分离酶切片段,与未酶切的质粒DNA比较,分析酶切结果。
同时,本次电泳还鉴定了提取纯化的大肠杆菌基因组DNA的纯度,以及大肠杆菌HSP70基因的PCR产物。
关键词限制性内切酶;琼脂糖凝胶电泳引言自从从嗜血杆菌(Haemophilus influenza)中分离到第一种限制性内切酶---HindⅢ,[2]人们开始意识到限制性内切酶的巨大应用前景。
Daniel Nathans,Werner Arber和Hamilton O. Smith在1978年即因限制性内切酶的发现和在分子遗传学的应用被授予诺贝尔生理与医学奖。
人们将限制性内切酶应用到DNA重组技术中,在用基因重组型大肠杆菌大规模生产人胰岛素获得巨大的成功。
现在限制性酶切与PCR一样成为分子生物学中最常用的实验手段之一。
限制性内切酶被分成四种:Types I,II,III和IV。
Types I酶切位点距离识别位点较远,是具有限制性内切和甲基化修饰的多功能酶,作用时需要ATP和硫代腺苷甲硫氨酸(S-adenosyl-L-methionine),如EcoB,EcoK。
Types II酶切位点位于识别位点之中会较近距离,只具有限制性内切的单功能酶,作用时需要Mg2+,如EcoRI,HindIII。
Types III酶切位点距离识别位点较近,具有限制性内切活性,也被发现是修饰性甲基化酶复合物的一部分。
作用时需要ATP(但不水解ATP)和硫代腺苷甲硫氨酸(S-adenosyl-L-methionine)刺激反应,如EcoPI, HinfIII。
质粒的酶切琼脂糖凝胶电泳分离DNA

6. 室温下约30-45分钟后,琼脂糖溶液完全凝固,小心 垂直拔出梳子和挡板,清除碎胶,将凝胶放入电泳槽 中。
实验步骤
酶切体系
调制酶切体系如下: (共 20μL)
无菌水 6μL,
质粒
10 μL
Buffer K 2μL,
EcoR I 1μL,
BamH I 1 μL
37℃酶切1.5小时。
琼脂糖凝胶制备
1. 洗净电泳槽、制胶槽、梳子、挡板,晾干。 2. 插好挡板、梳子。 3. 根据实验所需称取0.4克琼脂糖,放入制胶
11. 等溴酚蓝泳动至凝胶前沿后,将电压(或电 流)回零,关闭电源,停止电泳。
12. 取出凝胶,在紫外观测仪上观察电泳结果。
13.. 根据需要切下DNA条带,放入1.5ml Ep管中, 放于4℃保存,以中一般为三条带,最前面 的条带为超螺旋构型,中间为线型,最后为单链 有缺刻的构型。
pBC SK map
它的原理是溴化乙锭在紫外光照射下能发射 荧光,当DNA样品在琼脂糖凝胶中从负极向 正极泳动时,溴化乙锭从正极向负极移动, 这样溴化乙锭分子就会嵌入DNA分子中形成 络合物,使DNA在紫外光下发射很强的荧光。 在溴化乙锭足够的情况下,荧光的强度正比 于DNA的含量,这样就可以检测DNA的浓度。
如提取过程中有机械损伤,可能在胶上出现弥散。 影响RE(限制性内切酶)活性的主要因素:DNA的
纯度;DNA的甲基化程度;温度;缓冲液成分 (DDT,BSA)等。
使用3 mg的DNA Marker DL 2,000(500 ng/条带)进行1% 的琼脂糖凝胶电泳后,各DNA条带自上至下分别为2kb,1Kb, 750bp,500bp,250bp,100bp。
质粒的提取及凝胶糖凝胶电泳鉴定

实 验 步 骤
50 mM 葡萄糖 25 mM Tris-HCl 10 mM EDTA-Na2
200 mM NaOH 1% (W/V) SDS
2
5 min
3 M NaAC
省去Leabharlann 30 min 以上四、DNA的琼脂糖凝胶电泳原理
不同的DNA片段由于其电荷、分子量大小及构型的不同,在电 泳时的泳动速率就不同,从而可以区分出不同的区带。
8.12000rpm,4℃,离心10min,弃上清,将管口倒置于纸巾上使所有液体尽 可能流出。
9.加入1ml70%乙醇洗沉淀一次。12000rpm,4℃,离心10min。 10.弃上清,将管口倒置于纸巾上使液体流尽,放置干燥或真空干燥,即得质粒 DNA制品。 11.将沉淀溶于20ulTE缓冲液,(临用前加入1u 或20ug/ml RNaseA)待用。
细菌质粒是基因工程中常用的基因载体,也是 分子生物学中研究基因结构、功能及其复制、表达 的好材料。
二、实验原理
碱变性抽提法法是常用的方法之一. 此法基于染色体DNA与质粒DNA的变性与复性差异而达
到分离的目的。
在pH高达12.6的条件下,染色体DNA的氢键断裂、双螺 旋解开而变性;质粒DNA的大部分氢键也断裂,但超螺 旋共价闭合环状结构的两条互补链未完全分离,当用 pH4.8 的 Kac 高 盐 缓 冲 液 调 节 pH 至 中 性 时 , 变 性 的 质 粒 DNA又恢复到原来的构型,以可溶性状态存在于液相中, 而染色体DNA不能复性,形成缠绕的网状结构,与不稳 定的大分子RNA、蛋白质等一起沉淀出来。
2.凝胶板的制作: 用1cm宽的胶带纸沿平板电泳槽模板两 端边缘围成高4mm的墙,在其一端放置样品梳。将准备好的 模板置于水平台上,迅速将上面 胶液倒在模板的中央,让胶液自 由流向四周。待胶液充分冷却凝 固,将胶纸围墙慢慢取下,制备 好的凝胶板放入电泳槽中,加电 泳缓冲液直至没过胶面约1mm深, 取出样品梳。
质粒DNA的提取和琼脂糖凝胶电泳

化工专业实验实验名称质粒DNA的提取与琼脂糖凝胶电泳班级化21 姓名张腾学号成绩实验时间同组成员王乙汀、陈秉伦、梁有向1 实验目的(1)掌握碱裂解法提取质粒的原理和方法(2)掌握琼脂糖凝胶电泳分离鉴定DNA的原理和方法。
2 实验原理2.1质粒质粒(plasmid)是在细菌中以独立于染色体之外的方式存在,是细菌染色体外的小型双链环状DNA复制子。
理论上讲,所有的细菌株系都含有质粒,有些质粒携带有帮助其自身从一个细胞转入另一个细胞的信息,即F质粒,有些则表达对一种抗生素的抗性,即R 质粒,还有一些携带的是参与或控制一些不同寻常的代谢途径的基因即降解质粒。
质粒的大小不定,小的不到1kb,大的超过500kb,每个质粒都有一段DNA复制起始点的序列,它帮助质粒DNA在宿主细胞中复制。
对细菌的某些代谢活动和耐药性表型具有一定的作用。
目前,已发现有质粒的细菌有几百种,已知的绝大多数的细菌质粒都是闭合环状DNA分子(简称cccDNA)。
细菌质粒的相对分子质量一般较小,约为细菌染色体的0.5%~3%。
根据相对分子质量的大小,大致上可以把质粒分成大小两类:较大一类的相对分子质量是40×106以上,较小一类的相对分子质量是10×106以下(少数质粒的相对分子质量介于两者之间)。
每个细胞中的质粒数主要决定于质粒本身的复制特性。
按照复制性质,可以把质粒分为两类:一类是严紧型质粒,当细胞染色体复制一次时,质粒也复制一次,每个细胞内只有1~2个质粒;另一类是松弛型质粒,当染色体复制停止后仍然能继续复制,每一个细胞内一般有20个左右质粒。
一般分子量较大的质粒属严紧型。
分子量较小的质粒属松弛型。
质粒的复制有时和它们的宿主细胞有关,某些质粒在大肠杆菌内的复制属严紧型,而在变形杆菌内则属松弛型。
在基因工程中,常用人工构建的质粒作为载体。
人工构建的质粒可以集多种有用的特征于一体,如含多种单一酶切位点、抗生素耐药性等。
质粒DNA酶切及琼脂糖电泳分析鉴定
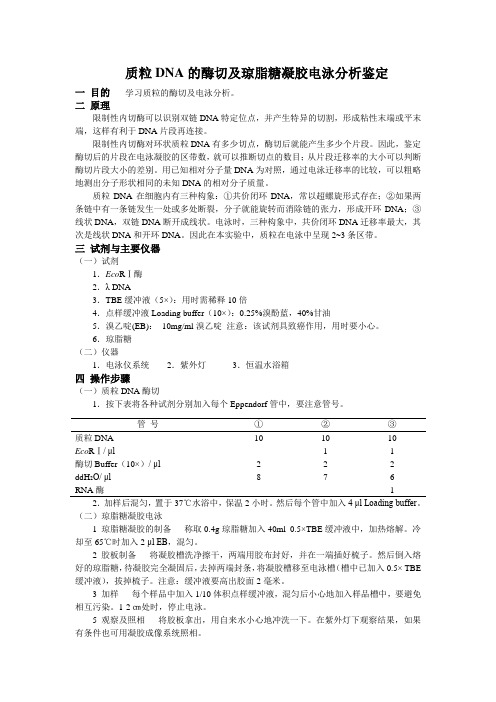
质粒DNA的酶切及琼脂糖凝胶电泳分析鉴定一目的学习质粒的酶切及电泳分析。
二原理限制性内切酶可以识别双链DNA特定位点,并产生特异的切割,形成粘性末端或平末端,这样有利于DNA片段再连接。
限制性内切酶对环状质粒DNA有多少切点,酶切后就能产生多少个片段。
因此,鉴定酶切后的片段在电泳凝胶的区带数,就可以推断切点的数目;从片段迁移率的大小可以判断酶切片段大小的差别。
用已知相对分子量DNA为对照,通过电泳迁移率的比较,可以粗略地测出分子形状相同的未知DNA的相对分子质量。
质粒DNA在细胞内有三种构象:①共价闭环DNA,常以超螺旋形式存在;②如果两条链中有一条链发生一处或多处断裂,分子就能旋转而消除链的张力,形成开环DNA;③线状DNA,双链DNA断开成线状。
电泳时,三种构象中,共价闭环DNA迁移率最大,其次是线状DNA和开环DNA。
因此在本实验中,质粒在电泳中呈现2~3条区带。
三试剂与主要仪器(一)试剂1.Eco RⅠ酶2.λ DNA3.TBE缓冲液(5×):用时需稀释10倍4.点样缓冲液Loading buffer(10×):0.25%溴酚蓝,40%甘油5.溴乙啶(EB):10mg/ml溴乙啶注意:该试剂具致癌作用,用时要小心。
6.琼脂糖(二)仪器1.电泳仪系统2.紫外灯3.恒温水浴箱四操作步骤(一)质粒DNA酶切1.按下表将各种试剂分别加入每个Eppendorf管中,要注意管号。
管号①②③质粒DNA 10 10 10Eco RⅠ/ μl 1 1酶切Buffer(10×)/ μl 2 2 2ddH2O/ μl8 7 6RNA酶 1 2.加样后混匀,置于37℃水浴中,保温2小时。
然后每个管中加入4 μl Loading buffer。
(二)琼脂糖凝胶电泳1 琼脂糖凝胶的制备称取0.4g琼脂糖加入40ml 0.5×TBE缓冲液中,加热熔解。
冷却至65℃时加入2μl EB,混匀。
质粒DNA的酶切和琼脂糖凝胶电泳鉴定

质粒DNA的酶切和琼脂糖凝胶电泳鉴定[实验原理]限制性内切酶识别短的DNA序列并在识别序列内或旁侧特异性切割双链DNA。
对环状DNA有多少切口,就能产生多少个酶解片段,因此鉴定酶切后的片段在电泳凝胶中的区带数,就可以推断酶切口的数目,从片段的迁移率可以大致判断酶切片段大小的差别。
DNA分子在琼脂糖凝胶中泳动时有电荷效应和分子筛效应。
DNA分子在高于等电点的pH溶液中带负电荷,在电场中向正极移动。
由于糖—磷酸骨架在结构上的重复性质,相同数量的双链DNA几乎具有等量的净电荷,因此它们能以同样的速度向正极方向移动。
在一定的电场强度下,DNA分子的迁移速度取决于分子筛效应,即DNA分子本身的大小和构型。
具有不同的相对分子质量的DNA片段泳动速度不一样,可进行分离。
DNA分子的迁移速度与相对分子质量的对数值成反比关系。
凝胶电泳不仅可分离不同相对分子质量的DNA,也可以分离相对分子质量相同,但构型不同的DNA分子。
如上次实验提取的质粒,有3种构型:超螺旋的共价闭合环状质粒DNA(covalently closed circular DNA,简称CCCDNA),开环质粒DNA,即共价闭合环状质粒DNA 1条链断裂,(open circular DNA,简称OCDNA),线状质粒DNA,即共价闭合环状质粒DNA 2条链发生断裂(linear DNA,简称L DNA)。
这3种构型的质粒DNA分子在凝胶电泳中的迁移率不同。
因此电泳后呈3条带,超螺旋质粒DNA泳动最快,其次为线状DNA,最慢的为开环质粒DNA。
[仪器、材料与试剂](一)仪器与材料恒温水浴槽、电泳仪、电泳槽、紫外线透射仪、移液枪、质粒、HindI II酶、EcoRI酶(二) 试剂1 000 mL 5xTBE:Tris 54 g硼酸27.5 g 0.5 mol/L EDTA 20 mL(pH 8.0)凝胶加样缓冲液(6x):溴酚蓝0.25%蔗糖40%琼脂糖溴化乙锭溶液(EB) 0.5ug/mL[实验步骤](一) 酶切取5uLDNA溶液,加1uL酶切缓冲液,EcoRI酶1uL(2U),无菌水补至总体积10uL,37保温3h,加凝胶上样缓冲液(6X)2uL,准备下个实验进行电泳,分析质粒DNA的限制性酶切图谱。
质粒DNA的提取、酶切及其琼脂糖凝胶电泳实验报告

(2) TAE和TBE均为常用的缓冲液。
TBE比TAE有相对高的缓冲能力。
(3)加样染料溴酚蓝可与长度约为0.5 kb的DNA一起迁移,可用于指示迁移率最高的片段。
(4) DNA的迁移速率取决于以下因素:①DNA的分子大小—分子量越小,迁移越快。
②琼脂糖浓度—浓度越低,迁移越快。
③DNA的构象—环状的或带切口环状的DNA通常比线状的DNA迁移要快。
④两个电极之间单位厘米的电压——电压越高,迁移越快。
(5)如果DNA条带不够窄且不够均匀,可能是内以下原因所引起:①DNA过载②电压过高③加样孔破损④凝胶中有气泡(6)在紫外灯下观察凝胶电泳所得结果应该戴上防护眼镜,因为紫外线对眼睛有伤害作用;二.实验内容1.实验现象与结果:将电泳后的凝胶放在紫外灯的照射下观察到的和用凝胶电泳成像系统进行拍照得到的DNA电泳条带图如下所示意:图注:x’:代表经过酶切的质粒DNA样品的电泳条带;x :代表未经过酶切的质粒DNA样品的电泳条带(x相同的互为对照组);marker:DNA相对分子质量标准物。
pUC19质粒DNA标准参照条带图像:2.对实验现象、实验结果的分析及其结论:(1)对实验现象的分析及其结论:从上述所示的DNA电泳条带图可以看出:不管在DNA Sample中还是在经过酶切处理后的DNA样品中均具有电泳迁移速率处于中间的线性质粒DNA。
而在此次试验中出现线性质粒DNA是因为pUc质粒DNA在提取的过程中DNA双链在相对应的两条链上同时产生切口。
这说明质粒制备过程个出现线性DNA说明存在核酸酶污染或实验操作有问题。
可能在其中混有少量的蛋白质(图中,位置在marker组最后一电泳条带后方的很可能就是蛋白质组分),或者在实验的提取过程中加入溶液Ⅱ所经历的时间过长,在碱性条件下基因组DNA片断会慢慢断裂,从而使提取的质粒DNA样品混入了基因组 DNA。
但是其中各组也存在超螺旋DNA(与marker组对照,电泳速率最快,跑在最前面的)。
实验一 质粒提取、酶切及琼脂糖电泳检测

实验一质粒提取、酶切及琼脂糖电泳检测一、实验内容1.采用碱裂解法提取质粒DNA2.采用限制性内切酶对质粒进行酶切3.采用琼脂糖电泳对所提取的质粒DNA及其酶切产物进行鉴定二、实验目的和要求1.掌握碱裂解法提取质粒DNA的原理和方法2.掌握限制性内切酶酶切DNA的操作过程3.掌握琼脂糖电泳的原理和方法三、实验仪器、材料和试剂1.仪器:摇床、无菌操作台、离心机、电泳仪、凝胶成像仪等2. 材料:移液枪、枪头、离心管、三角烧瓶等3. 试剂:LB培养基,卡那霉素,质粒提取试剂盒,琼脂糖,电泳缓冲液TAE,核酸染料,DNA Marker(DL2000,λ-Hind III digest DNAMarker), Eco R I内切酶等。
四、实验操作1. 质粒提取(1)挑取单菌落至3 mL LB液体培养基,37℃摇动(200 rpm)培养过夜。
(2)转移菌液至1.5 mL Eppendorf管中,12000 rpm离心45 sec,尽量弃尽上清。
(3)加入250 l 预冷的PI(请先检查是否加入RNaseA),使用移液器彻底混匀细菌沉淀,充分悬浮,静置2 min。
注意:如果有未彻底悬浮的菌块,会影响裂解,导致提取量和纯度的偏低。
(4)向离心管中加入250ul溶液P2,温和地上下翻转6-8次使菌体充分裂解。
注意:温和地混合,不要剧烈振荡,以免打断基因组DNA,造成提取的质粒中混有基因组DNA片段。
此时菌液应变得清亮粘稠,所用时间不应超过5min,以免质粒收到破坏。
如果未变得清亮,可能菌体过多,裂解得不彻底,应减少菌体量。
(5)向离心管中加入350ul 溶液P3,立即温和地上下翻转6-8次,充分混匀,此时将出现白色絮状沉淀。
12,000rpm离心1min,此时在离心管底部形成沉淀。
注意:P3加入后应立即混合,避免产生局部沉淀。
如果上清中还有微小的白色沉淀,可再次离心后取上清。
(6)向吸附柱CP3中(吸附柱放入收集管中)加入500ul的平衡液BL,12,000rpm 离心1min,倒掉收集管中废液,将吸附管重新放入收集管中。
综合性实验报告 质粒的提取及酶切

当迁移足够距离后,就可以通过Gelview染色来观察DNA片断
Gelview是一种荧光染料。它可以在做胶时混入其中在电泳时进行染色,也可以待电泳完成后将凝胶浸泡在稀释的Gelview溶液中进行染色。但必须将凝胶置于紫外透射仪中才可以对凝胶中的DNA或RNA进行观察。
To visualize DNA or RNA, the gel is placed on a ultraviolet transilluminator.
质粒是存在于几乎所有细菌中染色体之外的外状DNA分子。质粒通常携带有染色体上所不存在的基因,并表现出一些有用的性状,如抗生素抗性,耐受重金属等。质粒拥有自己的复制原点,因此可以不依赖于染色体而进行独立复制。一些小的质粒利用宿主细胞的酶进行复制,而较大的质粒则自身携有编码与复制有关的酶。
Agarose is typically used at concentrations of 0.5% to 2%.
The distance DNA has migrated in the gel can be judged by visually monitoring migration of the tracking dyes.
质粒分为:严紧型(低拷贝数)和松弛型(高拷贝数)。
质粒通常用作克隆载体,而碱裂解法是制备质粒DNA最为常用的方法之一。
本方法是依据共价闭合环状质粒DNA与染色体DNA在变性和复性之间存在差异进行的。
当细胞悬浮于NaOH和十二烷基酸钠(SDS)溶液中时,在高pH(碱)的作用下细胞发生裂解,此外,蛋白质和染色体DNA发生变性与细胞碎片一起沉淀下来。
The higher the agarose concentration, the "stiffer" the gel. Higher concentrations of agarose help in the separation of small DNAs, while low agarose concentrations allow resolution of larger DNAs.
质粒DNA的琼脂糖凝胶电泳

(3)胶浓度 (4)电场强度:根据需要选择合适电压。
为了尽快得到实验结果,所用电场强度约为5V/cm, 但分辨率不高。 精确测定DNA分子大小时,电压1V/cm。 (5)溴化乙锭 简称EB,电泳中的染色剂。
具有扁平 结构,能嵌入到DNA碱基对间,对线状分子 与开环分子影响较小而对超螺旋态的分子影响较大。
(6)电泳缓冲液 常用DNA电泳缓冲液(1000ml) TAE (50×) 242gTris 57.1ml 冰乙酸 100ml 0.5mol/L EDTA (pH8.0)
TBE(5×) 54gTris 27.5硼酸 20ml 0.5mol/L EDTA (pH8.0)
TPE (10×) 108g Tris 15.5ml 磷酸 (85%,1.679g/ml) 40ml 0.5mol/L EDTA (pH8.0)
(2)荧光染料 GoldViewTM核酸染料
三、实验操作步骤
• 琼脂糖凝胶的制备(浓度依照质粒大小确定). • 凝胶板的制备 • 加样(加样体积在10~30ul) • 电泳 • 结果观察
1234
1:DNA Marker2; 2~3:Hind Ⅲ/EcoR Ⅰ双酶切的pET/dhaT质粒; 4:pET-28a-c(+)空质粒
3、DNA电泳上样缓冲液 主要作用如下: (1) 螯合Mg2+,防止电泳过程中DNA被降解 (2) 增加样品密度以保证DNA沉入加样孔内,一 般上样缓冲液中加入一定浓度的甘油或蔗糖,这样 可以增加样品的比重。大片段电泳中采用Ficoll(聚 蔗糖),可减少DNA条带的弯曲和托尾现象。 (3)指示剂监测电泳的行进过程,一般加入泳动速 率较快的溴酚蓝指示电泳的前沿,它的速率约与 300bp的线状双链DNA相同。
DNA的琼脂糖凝胶电泳
实验一、质粒DNA的提取及琼脂糖凝胶电泳

因此电泳后呈3条带,超螺旋质粒DNA泳动最快,其 次为线状DNA,最慢的为开环质粒DNA。
三、仪器、材料与试剂
(一)仪器 1.微波炉 2.琼脂糖凝胶电泳系统 3.移液器 4.凝胶成像系统
(二)试剂
1.(1)5×TBE (50倍体积的TBE贮存液)配1000 mL 5×TBE Tris 54 g 硼酸 27.5 g 0.5mol/L EDTA 20 mL pH 8.0 或(2) 50×TAE(50倍体积的TAE贮存液) Tris 242 g 乙酸 57 mL 0.5mol/L EDTA 100 mL pH 8.0 2.凝胶加样缓冲液(6x) 3.琼脂糖 4.溴化乙锭溶液(EB) 0.5ug/mL
溶液II:由SDS(十二烷基硫酸钠)DS的作用是解聚核蛋白并与蛋白质分子结合使之变
性(裂解细胞和蛋白质变性作用)
② NaOH(PH>12)的作用是破坏氢键,使DNA分子变性
(DNA变性作用)
Solution II
0.4mol/LNaOH,2%SDS, 用前等体积混合
2.当溴酚蓝染料移动到距凝胶前沿1~2cm处, 停止电泳。
五、实验结果
在紫外灯(360nm或 254nm)下观察染色后的电泳 凝胶。DNA存在处应显出桔 红色荧光条带(在紫外灯下观 察时应戴上防护眼镜,紫外 线对眼睛有伤害作用)。
M 1
五.问题与讨论:
1.简要叙述溶液Ⅰ、溶液Ⅱ和溶液Ⅲ的作用,以及 实验中分别加入上述溶液后,反应体系出现的现 象及其成因? 2.简要叙述酚-氯仿抽提DNA体系后出现的现象及其 成因? 3.沉淀DNA时为什么要用无水乙醇及在高盐、低温条 件下进行? 4.若提取的质粒电泳为单一一条条带,如何判断其 构型?
上层水相转移到新EP管,加入2倍体积无水乙醇,颠倒混匀, -20 ℃ 冰箱中放置15min,12000g10min
质粒DNA提取原理、步骤、凝胶电泳分析及其应用

分混匀(涡旋振荡),室温放置10min
•
加入200μL溶液Ⅱ(新鲜配制),盖紧
盖子,快速温和颠倒离心管数次,以混匀内
容物 (千万不要振荡),冰浴3分钟
•
加入150μL溶液Ⅲ(冰上预冷),盖紧
盖子,颠倒数次使混匀。冰上放置3~5min
• 12000r/min离心15min,将上清转至另一 离心管中
• 向上清中加入等体积酚:氯仿:异戊醇 (25:24:1),反复混匀,12000r/min离 心2min,将上清转至另一离心管中
• 向上清中加入2倍体积异丙醇,混匀后, 室温放置2min, 12000r/min 离心1min.
• 倒去上清液,将离心管倒扣在吸水纸 上,吸干液体
• 用1mL70%乙醇洗涤质粒DNA沉淀, 振荡,12000r/min离心30s,倒去上清 液,空气干燥10min
☆原理示意图
操作步骤
• 培养细菌:将带有质粒的大肠杆 菌接种到5~10ml液体培养基中, 37 ̊ C震荡培养12~16h,到细菌浓 度远超过对数生长期时收获菌种 。
• 吸取1.5mL培养物加入1.5mL 离心管中,4 ̊ C 12000r/min室 温离心1min
• 小心吸去培养液,将管倒置于 卫生纸上数分钟,使液体流尽( 细菌沉淀尽可能干燥)
M: DL5000 DNA Marker 1, 4,7,10 为质粒DNA 2, 5,8,11 为PCR产物 3, 6,9,12 为酶切片段
酶切长片段
含目的DNA片段 的酶切短片段
琼脂糖凝胶电泳的应用
• 1、DNA的制备 • ⑴ 适合分离大片段DNA。
琼脂糖凝胶约可区分相差100bp的DNA片段,其分辨 率虽比聚丙烯酰胺凝胶低,但它制备容易,分离范 围广,尤其适于分离大片段DNA。普通琼脂糖凝胶 分离DNA的范围为0.2-20kb,利用脉冲电泳,可分离 高达10^7bp的DNA片段。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Plasmid DNA Preparation for Recombination Lin Chengyu Bio 04 2010030007 ; Cooperator: Yuan Xiaowen Experiment Date: 2012-03-15; Submit Date: 2012-03-181Introduction1.1Background InformationPlasmid is a small, independently replicating and cytoplasmic DNA molecule that canbe transferred from one organism to another. It is a kind of double-stranded and usuallycircular DNA, whose size range from 1 kb to 200 kb.Figure 1 Sketch of plasmid nucleoid and cellBecause of its property, plasmid is often used as cloning vector carrying foreign genesinto the host cells. In order to make plasmid ready for recombination, severalpreparative processes shall be done, including plasmid DNA extraction, restrictionenzyme digestion and agarose gel electrophoresis.1.2Objectives(1)Learn the characteristics of plasmid.(2)Purify the plasmid DNA by alkaline lysis method.(3)Measure the DNA concentration by spectrophotometer.(4)Learn the characteristics of restriction endonuclease, and use Eco R I and Xho I toperform restriction enzyme digestion.(5)Perform agarose gel electrophoresis to separate DNAs, and compare the differenceamong original plasmids, single digestion, and double digestion.1.3Major principles1.3.1Plasmid DNA extraction(1)Some special elements, including heat, extreme pH, organic solution candenature DNA and will bring DNA into sediment;(2)When denaturing conditions are eliminated, DNA will be renatured, and itsspeed will depend on the size and ratio over protein;(3)The isolation kit has special material on the column and can specificallybind with DNA molecules. Thus, plasmids can be purified;(4)Double-strand DNA has optical density at wavelength 260nm. Itsconcentration can be measured by spectrophotometer.1.3.2Restriction enzyme digestion(1)Type II restriction endonuclease can recognize specific nucleic acidsequence and incise double strain DNA in specific sites in this sequence.Figure 2 illustrate the specific sequence Eco R I recognize and its cuttingsite;Figure 2 Cutting site of Eco R I(2)Most plasmids have multiple cloning sites (MSC) which is artificiallydesign for genetic engineering (See Figure 3). Restriction endonuclease candigest in the specific site, produce sticky ends for recombination.Figure 3 Plasmid profile (pCMV - Myc)1.3.3Agarose gel electrophoresisIn order to confirm that the preparation above is successful, agarose gel electrophoresis is essential.Agarose gel has small pores in it, whose size depends on agarose concentration. In the electric field and buffer in neutral pH, negatively charged nucleic acid will migrate toward the positive pole. Molecular weight of DNAand DNA conformation will be the major factors that determine the mobilityspeed of DNA. Thus, DNA of different sizes and conformations can beseparated. If a DNA maker exists, the size of sample can be known as well. (SeeFigure 4)Figure 4 Agarose gel electrophoresis:(1) plasmids (2) Eco R I digestion (3) double digestion12Experiment Operation2.1MaterialE.coli DH5 harboring pCMV-Myc-T10 (SIPAR),Eco R I and Xho I restriction endonuclease (in glycerol, for details see Table 1),Table 1 Recognition Sequence and cutting site of Eco R I and Xho I2NEB 1kb DNA Ladder (for details see Figure 5);Figure 5 NEB 1kb DNA Ladder32.2Chemicals and apparatus2.2.1SolutionsTable 2 Solutions2.2.2Apparatus(1)Pipettes;(2)Eppendorf tube;(3)Biodev plasmid isolation kit;(4)Centrifuge;(5)Spectrophotometer;(6)Electrophoresis apparatus;(7)UV gel imaging system.2.3Procedure2.3.1Plasmid extraction and purification(1)Pipette 1.4 ml LB medium containing E.coli into one Eppendorf tube,centrifuge at 12,000 rpm for 2 min, and discard the supernatant. Thenpipette another 1.4 ml, and repeat;(2)Add 100 µl Solution I (contains DNase-free RNase), resuspend pelletthoroughly. V ortex might be necessary. Incubate for 5 min at R.T.;Notes: Solution I provides a buffer system for materials in E.coli,avoiding harsh environment.(3)Add 150 µl Solution II, mix by inverting the tube 4-6 times gently. Solutionin the Eppendorf tube becomes clear. Incubate the tube on ice for 2 min;Notes: Solution II contains NaOH and SDS, which provide a stronglydenaturing environment. It will lyse the cell and release genomic DNA,plasmid DNA and protein into the solution. DNA is denatured and itssolubility in water is lowered.(4)Add 150 µl Solution III, mix by inverting the tube gently several times.White pellet is formed as Solution III is added. Incubate the tube at R.T. for5 min to get genomic DNA precipitate completely, and centrifuge at 12,000rpm for 8 min;Notes: Solution III can neutralize the denaturing environment caused bySolution II, so that plasmid DNA molecules renature. Sodium ion in SDSwill be replaced with potassium ion in Solution III, and becomes PDS(Potassium Dodecyl Sulfate), which is insoluble in water. PDS,combined with protein and genomic DNA, will coprecipitate into pellet.(For details, see discussion session.) Using centrifuge can removegenomic DNA;(5)Add 420 µl Binding buffer to the mini-spin column to activate its ability tobind DNA molecules;(6)Transfer the supernatant from Step 4 to the mini-spin column, mix withbinding buffer by pipette, place the mini-spin column in the collection tube,and centrifuge at 12,000 rpm for 30 sec. Discard the waste solution in thecollection tube;Notes: At this time, plasmids specifically bind to the material in themini-spin column;(7)Add 700 µl Wash buffer (contains 3 folds volume of absolute ethanol) tothe mini-spin column, and centrifuge at 12,000 rpm for 1min. Discard thewaste solution in the collection tube. Then repeat with another 500 µl;Notes: Wash buffer can wash off the non-DNA substance thatnon-specifically binds to the mini-spin column. Ethanol is essentialbecause plasmids will be washed off with its absence.(8)Centrifuge the mini-spin column as well as collection tube at 12,000 rpmfor 2 min to get rid of ethanol;Notes: Ethanol is harmful for most enzymes. Its existence will greatlyinfluence the following digesting step. Step 8 is designed for eliminateethanol from the column.(9)Replace the collection tube with a new Eppendorf tube, add 50 µl ddH2O tothe mini-spin column, incubate 5 min at R.T., and centrifuge at 12,000 rpmfor 1 min;(10)L abel the Eppendorf tube containing plasmids at initial concentration withclass, name, group on it;(11)P ipette 96 µl ddH2O into a new Eppendorf tube, and add 4 µl of plasmidsolution. Measure its concentration by spectrophotometer.Notes: [dsDNA] = 50 * (OD260– OD310) * dilution factor.2.3.2Restriction endonuclease digestion(1)Label 3 Eppendorf tube as No.1, 2, 3;(2) Add substrate, solutions, and enzymes as shown in Table 3;Table 3 Scheme of restriction endonuclease digestion(3) Incubate 3 tubes at 37 ℃ for 30 min; 2.3.3Agarose gel electrophoresis(1) During enzyme digestion, place a clean electrophoresis mold on ahorizontal surface, carefully insert a comb (15 µl slots) into the mold;(2) Pour the agarose gel solution (contains 0.8 g agarose in 100 ml TAE) intothe mold gently to avoid bubbles. The height of gel shall be slightly higherthan the black line on the side face;(3) Wait for approximately 30 min for solidification, when enzymatic reactionscomplete as well;(4) Add 10 µl 3×Loading buffer to each Eppendorf tube, and mix with pipette;(5) Carefully remove the comb, place the mold in the electrophoresis chamber,and add 1×TAE buffer in order to cover the gel to a depth ofapproximately 1 mm;(6) Load 15 µl from each Eppendorf tube into the slots of the gel: one slot forone tube. Load 4 µl of 1kb DNA ladder into the slot beside the sample;Notes : this step shall be done as quickly as possible. If electrophoresisstarts late after samples loading, samples might diffuse in the gel and causethe band get in the end not parallel with the slot.(7) Start electrophoresis immediately samples loading at 100 V, and stop whenBromophenol blue is 2/3 gel length from the starting line;(8) Place the gel into EB working solution for 20 min, and observe under UVat 310 nm. The red fluorescent bands show where DNA is and theirdarkness show the quantity. Then take photograph for records;3 Result & Discussion3.1 Plasmids extractionTable 4 Plasmid concentration and purityTable 4 shows that the plasmid extracted was 188.4 ng / µl, but not pure enough. OD 260 / OD 280 shall be 1.8~2.2. The major reason might be that during transferring supernatant from the Eppendorf tube to the mini-spin column, the pellet was loosely attached, causing some protein solute, and making the extraction not pure enough.Q1: During plasmid extraction, when Solution II was added, why can’t incubate longerthan 2 min?A1: NaOH can produce a strong alkaline environment to lyse cells, but genomic DNA will also degraded slowly under alkaline situation.Q2: When Solution III is added, white pellet can be see immediately. What is it?A2: In order to study the nature of the pellet, I designed an experiment and perform it in the lab.Table 5 Experiment designed to study the pellet after Solution III addedFrom the result, we can know that NaOH is mainly used for lyse, and denatured DNA is still soluble! Pellet produced in experiment No.2 is mostly protein that precipitate under acidic and high salt concentration environment.The pellet we see in experiment No.1 is actually mainly caused by PDS, as demonstrated among experiment 4, 5, and 6. As a result, PDS precipitate in a large amount, and its hydrophobic group binds to protein and makes it coprecipitate. Genomic DNA molecules are bind with proteins, and are brought into pellet in the same time. 3.2Gel electrophoresisThe result is shown in Figure 6.Figure 6 Plasmid DNA gel electrophoresis:Lane I – Plasmid without treatment;Lane II –Eco R I single digestion;Lane III –Eco R I and Xho I double digestion;Lane M – 1 kb DNA LadderAs shown in Figure 6, Lane III, which contains plasmids treated by double digestion,has bands at approx. 1.8 kb and 4.0 kb, and add up to approx. 5.8 kb; Lane II, whichcontains plasmids treated by Eco R I single digestion, has band at approx. 6.0 kb, whichis about the same length as the sum of two composition from Lane III.This phenomenon suggests that in pCMV-Myc-T10, the number of Eco R I recognitionsites and Xho I are the same. According to the plasmid profile, both enzyme have andonly have one recognition site, which confirm our conjecture.As for Lane I, there is no apparent band as shown in other groups. It is mainly becauseof a loading mistake. When we were loading the first sample, we failed to put the pipettenear the bottom of the slot, but the middle of it, causing a lot of DNA moleculesoverflow from the slot.4ConclusionThe plasmid sample extracted from E.coli is 188.4 ng / µl. And it is a circular DNA molecule composed of approx. 6 kilobasepairs, has 1 Eco R I and 1 Xho I restriction enzyme recognition site.5Reference【1】Results of student experiment, 2004-03-15【2】/rebase/rebase.html【3】/nebecomm/products/productN3232.asp6Appendix6.1Difference between plasmid DNA and genomic DNA extractionPlasmid DNA extraction procedure includes denaturing and renaturing, in order toremove genomic DNA; genomic DNA extraction tends to focus more on proteinremoval, and will inevitably mix with plasmid DNA if without further purification.6.2Restriction MapThe gene sequence comes from NCBI, which coding for PDCD11 (Programmed CellDeath), also named as ALG-4 (Apoptosis-Linked Gene). It is composed of 6000 bp.Using Bioedit to analyze:Use “Sequence -> Select positions” to select the whole sequence;Use “Sequence -> Nucleic acid -> Restriction map”:Figure 7-1Click “Generate Map”, here shows analysis from 881 to 960 bp:Figure 7-2And those enzymes that have no recognition sites are listed as follows:Figure 7-3。