Carbonyl Compounds
重氮化合物的亲核环化反应-北京大学工学院

2. 重氮化合物的亲核缩合反应
2.1 重氮化合物的Aldol缩合反应
Cu(II)-catalyzed synthesis of indoles and pyrroles from α-diazoketones
Reddy, B. V. S.; Reddy, M. R.; Rao, Y. G.; Yadav, J. S.; Sridhar, B. Org. Lett. 2013, 15, 464.
Likhar, P. R.; Roy, S.; Roy, M.; Subhas, M. S.; Kantam, M. L. Synlett 2008, 1283.
2. 重氮化合物的亲核缩合反应
2.1 重氮化合物的Aldol缩合反应
Xiao, F.; Liu, Y.; Wang, J. Tetrahedron Lett. 2007, 48, 1147
2. 重氮化合物的亲核缩合反应
2.2 串联1,2-迁移和括环
Allwood, D. M.; Blakemore, D. C.; Ley, S. V. Org. Lett. 2014, 16, 3064
2. 重氮化合物的亲核缩合反应
2.2 串联1,2-迁移和括环 R1基团的迁移和芳基的迁移
Hashimoto, T.; Naganawa, Y.; Maruoka, K. J. Am. Chem. Soc. 2008, 130, 2434. Kang, B. C.; Shim, S. Y.; Ryu, D. H. Org. Lett. 2014, 16, 2077.
2. 重氮化合物的亲核缩合反应
2.1 重氮化合物的Aldol缩合反应
Trost, B. M.; Malhotra, S.; Fried, B. A. J. Am. Chem. Soc. 2009, 131, 1674. Cui, H.-F.; Wang, L.; Yang, L.-J.; Nie, J.; Zheng, Y.; Ma, J.-A. Tetrahedron 2011, 67, 8470.
有机化学专业英语词汇常用前后缀

deca- 十 deci 10-1 -dine 啶 dodeca- 十二
-ene 烯 epi- 表 epoxy- 环氧 -ester 酯 -ether 醚 ethoxy- 乙氧基 ethyl 乙基
fluoro- 氟代 form 仿
-glycol 二醇
hemi- 半 hendeca- 十一 hepta- 七 heptadeca- 十七 hexa- 六 hexadeca- 十六 -hydrin 醇 hydro- 氢或水 hydroxyl 羟基 hypo- 低级的,次 hyper- 高级的,高
1有机化学专业英语词汇常用前后缀acetal醛缩醇acetal乙酰acid酸al醛alcohol醇aldehyde醛alkali碱allyl烯丙基propenyl丙烯基alkoxy烷氧基amide酰胺amino氨基的amidine脒amine胺ane烷anhydride酐anilino苯胺基aquo含水的ase酶ate含氧酸的盐酯atriyne三炔azo偶氮benzene苯bi在盐类前表示酸式盐bis双borane硼烷bromo溴butyl丁基carbinol甲醇carbonyl羰基carboxylicacid羧酸centi10chloro氯代cis顺式condensed缩合的冷凝的cyclo环deca十deci10dine啶dodeca十二21ene烯epi表epoxy环氧ester酯ether醚ethoxy乙氧基ethyl乙基fluoro氟代form仿glycol二醇hemi半hendeca十一hepta七heptadeca十七hexa六hexadeca十六hydrin醇hydro氢或水hydroxyl羟基hypo低级的次hyper高级的高ic酸的高价金属ide无氧酸的盐酰替胺酐il偶酰imine亚胺iodine碘iodo碘代iso异等同ite亚酸盐keto酮ketone酮lactone内酯mega10meta间偏methoxy甲氧基methyl甲基62micro10milli10monomon一单nano10nitro硝基nitroso亚硝基nona九nonadeca十九octa八octadeca十八oic酸的ol醇one酮ortho邻正原ous亚酸的低价金属oxa氧杂oxide氧化合物oxime肟oxo酮oxy氧化oyl酰para对位仲penta五pentadeca十五per高过petro石油phenol苯酚phenyl苯基pico10poly聚多quadri四quinque五semi半septi七sesqui一个半sulfa磺胺sym对称syn顺式同共63912ter三tetra四tetradeca十四tetrakis四个thio硫代trans反式超跨thio硫代tri三trideca十三tris三个undeca十一uni单一unsym不对称的偏位yl基ylene撑二价基价在不同原子上yne炔organiccompounds有机化合物compoundsofcarbon碳化合物hydrocarbonsan
羰基化合物的红外光谱

I. O-H:~3000 cm-1(b); II. C=O:1695 cm-1;III. O-H(oop):935 cm-1; III. CH2(oop):1350~1180 cm-1 IV. 2960,2850;V. 1450;VI. 715
(νC-N):只限伯酰胺,伸缩振动吸收峰出现在1420~ 1400 cm-1区间。
( C-N (oop ) ):伯酰胺在850~750;仲酰胺在 750~650。都是中等强度的吸收。
2020/5/16
32
丙酰胺的红外光谱图
I. N-H:3360,3200 cm-1(双峰)~伯酰胺; II. =C-H:2960,2850; III. C=O和 N-H (ip) :1635; IV. C-N:1430(仅限伯酰胺)
羰基化合物的红外光谱
Carbonyl Compounds
O C RX
酸酐
酰卤 酯 醛 酮 羧酸 酰胺
O OR
1810,1760
X 1760
OR’ 1735
H R’ OH 1725 1715 1710
NH2 1690
吸收波数降低
2020/5/16
2
样品的物态、邻近功能团的电子效应和空间效应、 共轭、是否形成氢键以及环等环境的影响。
1760(s)
1170~1050(s)
Cyclo anhydrides OOO
OOO
O O O
2020/5/16
1870~1820(s) 1800~1750(vs)
1800(s)1750(vs)
1805~1780(s) 1785~1755(vs)
1310~1210 952~909
硼氢化钠乙酸体系还原羰基机理

硼氢化钠乙酸体系还原羰基机理The reduction of carbonyl compounds using sodium borohydride (NaBH4) in acetic acid (CH3COOH) is a commonly employed reaction in organic chemistry. This process involves the transfer of hydride ions from NaBH4 to the carbonyl carbon, resulting in the formation of the corresponding alcohol. Understanding the mechanism of this reaction is crucial for optimizing reaction conditions and designing new synthetic routes. In this response, we will explore the various perspectives surrounding the reduction mechanism of carbonyl compounds using the NaBH4-acetic acid system.From a mechanistic point of view, the reduction of carbonyl compounds with NaBH4 in acetic acid proceeds through a nucleophilic addition-elimination pathway. Initially, the hydride ion (H-) from NaBH4 attacks the electrophilic carbonyl carbon, forming a tetrahedral intermediate. The addition of acetic acid to the reaction mixture serves to protonate the oxygen atom of thetetrahedral intermediate, facilitating the elimination of the leaving group. This elimination step results in the formation of the alcohol product and regenerates the borate ion (BH4-) as a byproduct.The role of acetic acid in this reaction is multifaceted. Firstly, acetic acid acts as a solvent, providing a suitable medium for the reaction to occur. Additionally, acetic acid serves as a proton source, protonating the oxygen atom of the tetrahedral intermediate and promoting the elimination of the leaving group. Moreover, acetic acid can also function as a catalyst, enhancing the rate of the reaction by facilitating the formation of the tetrahedral intermediate. The presence of acetic acid in the reaction mixture helps to maintain a favorable reaction environment and ensures the smooth progress of the reduction process.The choice of NaBH4 as the reducing agent in this system is based on its unique properties. NaBH4 is a mild and selective reducing agent, capable of reducing a wide range of carbonyl compounds without affecting otherfunctional groups present in the molecule. This selectivity is due to the relatively low reactivity of NaBH4 towards other functional groups, making it an ideal choice for carbonyl reduction reactions. Furthermore, NaBH4 is stablein acetic acid and can be easily handled, making it a convenient reagent for laboratory-scale reactions.The reduction mechanism of carbonyl compounds using NaBH4 in acetic acid has been extensively studied and supported by experimental evidence. Various spectroscopic techniques, such as nuclear magnetic resonance (NMR) and infrared (IR) spectroscopy, have been employed to monitorthe reaction progress and identify the intermediates involved. Additionally, computational studies using density functional theory (DFT) calculations have provided further insights into the reaction mechanism and energetics.In conclusion, the reduction of carbonyl compoundsusing NaBH4 in acetic acid proceeds through a nucleophilic addition-elimination pathway. Acetic acid plays multiple roles in this reaction, serving as a solvent, proton source, and even a catalyst. The choice of NaBH4 as the reducingagent is based on its mild and selective reactivity towards carbonyl compounds. Extensive experimental and computational studies have provided valuable insights into the mechanism of this reaction. Overall, understanding the reduction mechanism of carbonyl compounds using the NaBH4-acetic acid system is crucial for the development of efficient synthetic methodologies in organic chemistry.。
二苯基乙二酮的制备

安息香衍生物二苯乙二酮的合成及表征一、实验目的:1.学习安息香氧化制备α—二酮的原理与方法。
2.掌握薄层色谱的原理,薄层板的制作。
3.学习薄层色谱法跟踪反应进程。
二、实验原理:(一)薄层色谱的有关知识薄层色谱法是以薄层板作为载体,让样品溶液在薄层板上展开而达到分离的目的,故也称为薄层层析。
它是快速分离和定性分析少量物质的一种广泛使用的实验技术,可用于精制样品、化合物鉴定、跟踪反应进程和柱色谱的先导(即为柱色谱摸索最佳条件)等方面。
1.薄层色谱常用的吸附剂硅胶和氧化铝是薄层层析常用的固相吸附剂。
化合物极性越大,它在硅胶和氧化铝上的吸附力越强,所以吸附剂均制成活性精细粉末。
活化通常是加热粉末以脱去水分。
硅胶是酸性的,用来分离酸性或中性的化合物。
氧化铝有酸性、中性和碱性的,可用于分离极性或非极性的化合物。
商用的硅胶和氧化铝薄层板可以买到,这些薄板常用玻璃或塑料制成。
溶剂在薄层板上爬升的距离越长,化合物的分离效果越好。
宽的薄层板也可用于量较大的样品,具有1~2 mm厚的大板可用于50~1000 mg样品的分离制备。
2.样品的制备与点样样品必须溶解在挥发性的有机溶剂中,浓度最好是1~2 %。
溶剂应具有高的挥发性以便于立即蒸发。
丙酮、二氯甲烷和氯仿等是常用的有机溶剂。
分析固体样品时,可将20~40mg样品溶到2mL 的溶剂中。
在距薄层板底端约1cm处,用铅笔划一条线,作为起点线。
用毛细管(内径小于1mm)吸取样品溶液,垂直地轻轻接触到薄层板的起点线上。
样品量不能太多,否则易造成斑点过大,互相交叉或拖尾,不能得到很好的分离效果。
3.展开将选择好的展开剂放在层析缸中,使层析缸内空气饱和,再将点好样品的薄层板放入层析缸中进行展开。
使用足够的展开剂以使薄层板底部浸入溶剂3~5 mm,但溶剂不能太多,否则样点在液面以下,溶解到溶剂中,不能进行层析。
当展开剂上升到薄层板的前沿(离顶端5~10mm处)或各组分已明显分开时,取出薄层板放平晾干,用铅笔划出前沿的位置后即可显色。
碳陶复合材料英文专著

碳陶复合材料英文专著Carbon Ceramic Composite Materials: An English MonographAbstract:Carbon ceramic composite materials have attracted significant attention in various industries due to their unique properties and potential applications. This monograph provides a comprehensive overview of carbon ceramic composite materials in terms of their structure, properties, synthesis methods, and applications. The aim is to provide readers with insights into the advancements and future prospects of these materials.1. IntroductionCarbon ceramic composite materials, also known as carbon-carbon composites or C/C composites, are a class of materials that combine carbon fibers with a ceramic matrix. These composites exhibit exceptional mechanical, thermal, and chemical properties, making them suitable for a wide range of applications. This chapter introduces the background and significance of carbon ceramic composites, outlining their unique properties and potential applications.2. Structure of Carbon Ceramic CompositesThis chapter discusses the structure of carbon ceramic composites, focusing on the arrangement and orientation of carbon fibers within the ceramic matrix. The microstructure and macrostructure of these composites are explored, highlighting the role of fiber architecture in determining their mechanical properties and performance.3. Properties of Carbon Ceramic CompositesIn this section, the mechanical, thermal, and electrical properties of carbon ceramic composites are discussed in detail. The exceptional strength, stiffness, and wear resistance of these materials, along with their high thermal stability and low thermal expansion, make them ideal for applications in aerospace, automotive, and energy industries. The electrical conductivity and electromagnetic shielding properties of carbon ceramic composites are also addressed.4. Synthesis Methods of Carbon Ceramic CompositesVarious synthesis methods for carbon ceramic composites are presented, including chemical vapor infiltration (CVI), liquid silicon infiltration (LSI), and pyrolysis. Each method is described, highlighting the advantages, limitations, and challenges associated with their implementation. The effect of processing parameters on the microstructure and properties of carbon ceramic composites is also discussed.5. Applications of Carbon Ceramic CompositesThis chapter reviews the applications of carbon ceramic composites in different industries. Aerospace applications, such as aircraft brakes and thermal protection systems, are discussed, along with automotive applications in brake discs and engine components. The use of carbon ceramic composites in the energy sector, including nuclear fusion reactors and fuel cells, is also explored. Furthermore, potential future applications and emerging trends in the field are presented.6. Challenges and Future PerspectivesThe final chapter addresses the challenges and future perspectives of carbon ceramic composites. The limitations of current synthesis methods, such as high costs and complex processing requirements, are identified. The need for further research in areas such as interfacial bonding improvement, scalability, and recycling strategies is emphasized. Lastly, the future prospects of carbon ceramic composites in terms of advanced applications and market growth are discussed.Conclusion:Carbon ceramic composite materials exhibit exceptional properties and have diverse applications in various industries. This monograph provides a comprehensive overview of these materials, including their structure, properties, synthesis methods, and applications. Through understanding the advancements and challenges, it is evident that carbon ceramic composites have great potential for future development and innovation in materials science and engineering.Acknowledgements:The author would like to acknowledge the contributions of researchers and scientists in the field of carbon ceramic composites. Their valuable work and insights have greatly enriched the content of this monograph.。
monocarbonate 的化学式

monocarbonate 的化学式英文版Monocarbonate is a chemical compound that is composed of one carbonate ion and one metal ion. Its chemical formula is MCO3, where M represents the metal ion. Monocarbonates are commonly found in nature, such as sodium bicarbonate (NaHCO3) and potassium carbonate (K2CO3).Monocarbonates are important in various industries, such as the food industry where they are used as leavening agents in baking. They are also used in the pharmaceutical industry for their antacid properties.In conclusion, monocarbonates are versatile compounds that have a wide range of applications in different industries.完整中文翻译:单碳酸盐是一种由一个碳酸盐离子和一个金属离子组成的化合物。
其化学式为MCO3,其中M代表金属离子。
单碳酸盐在自然界中很常见,如碳酸氢钠(NaHCO3)和碳酸钾(K2CO3)。
单碳酸盐在各种行业中都很重要,比如食品工业中它们被用作烘焙中的发酵剂。
它们还被用于制药行业,因为具有抗酸性能。
总之,单碳酸盐是多功能化合物,在不同行业中有着广泛的应用。
甲基丙烯酰氧乙基三甲基氯化铵

甲基丙烯酰氧乙基三甲基氯化铵项目简介一. 概述甲基丙烯酰氧乙基三甲基氯化铵是一种性能优异的阳离子试剂,可和多种乙烯基单体反应合成各种功能高分子材料,其系列产品用途极广(如用于污水处理、纸张增强或油田开采。
还可用作合成、涂料的防沉淀剂和塑料、纤维及橡胶的抗静电添加剂等)。
在我国,DMC的主要用途是作为阳离子型污水絮凝剂的生产原料,目前主要依赖进口,年需求量达1500t 左右。
随着我国环保工作的加强,污水处理日显重要,对阳离子絮凝剂的需求与日俱增。
但由于DMC的短缺及价格等因素制约了阳离子絮凝剂的生产及推广应用,在一定程度上影响了环保工作的深入。
进行DMC的工艺开发,实现国产化并降低产品成本,成为环保行业的一致要求。
二.产品理化性质及质量指标甲基丙烯酰氧乙基三甲基氯化铵(简称DMAEMA-Q,DMC),英文名为2-Trimethylammonium ethyl methacrylate chloride, 分子式C9H18O2NCl, 结构式, 分子量207.67, CAS登录号5039-78-1。
DMC溶于水,不溶于酯、酮及烃。
可用自由基引发剂进行溶液聚合,悬浮液聚合和乳液聚合,可与其它单体均聚或共聚,由此给聚合物引入季铵盐基团。
产品质量指标外观:无色至淡黄色透明液体含量(m/m,%):79-81pH:3.0-7.0色泽(Hazen):≤ 200阻聚剂(MEHQ):1000ppm±100三.生产工艺简介N,N.二甲氨基乙醇(DMAE),过量的甲基丙烯酸甲酯(MMA),适量的阻聚剂吩噻嗪及催化剂投料,加热,维持反应物的温度98~l1O℃,分馏顶端温度63~65℃,生成的甲醇和过量的MMA形成共沸物馏出,反应4~5h.降压馏出剩余的MMA,然后分馏出产物甲基丙烯酸二甲氨基乙酯(DMAEMA).甲基丙烯酸二甲氨基乙酯(DMAEMA)水液通入一氯甲烷气体进行季铵化反应,再经稳定化处理即得成品DMC。
sTILLE羰基化偶联

Stille-Carbonylative cross-couplingA common alteration to the Stille coupling is the incorporation of a carbonyl group between R1 and R2, serving as an efficient method to form ketones. This process is extremely similar to the initial exploration by Migita and Stille (see History) of coupling organostannane to acyl chlorides. However, these moieties are not always readily available and can be difficult to form, especially in the presence of sensitive functional groups. Furthermore, controlling their high reactivity can be challenging. The Stille-carbonylative cross-coupling employs the same conditions as the Stille coupling, except with an atmosphere of carbon monoxide (CO) being used. The CO can coordinate to the palladium catalyst (9) after initial oxidative addition, followed by CO insertion into the Pd-R1 bond (10), resulting in subsequent reductive elimination to the ketone (12). The transmetalation step is normally the rate-determining step.Intramolecular Heck reactionThe Heck reaction is the palladium-catalyzed coupling of an aryl or alkenyl halide with an alkene to form a substituted alkene.[2]Intramolecular variants of the reaction may be used to generate cyclic products containing endoor exo double bonds. Ring sizes produced by the intramolecular Heck reaction range from four to twenty-seven atoms. Additionally, in the presence of a chiral palladium catalyst, the intramolecular Heck reaction may be used to establish tertiary or quaternary stereocenters with high enantioselectivity.[3] A number of tandem reactions, in which the intermediate alkylpalladium complex is intercepted either intra- or intermolecul arly before β-hydride elimination, have also been developed.[4]Mechanism and StereochemistryThe Neutral PathwayAs shown in Eq. 2, the neutral pathway of the Heck reaction begins with the oxidative addition of the aryl or alkenyl halide into a coordinatively unsaturated palladium(0) complex (typically bound to two phosphine ligands) to give complex I. Dissociation of a phosphine ligand followed by association of the alkene yields complex II, and migratory insertion of the alkene into the carbon-palladium bond establishes the key carbon-carbon bond. Insertion takes place in a suprafacial fashion, but the dihedral angle between the alkene and palladium-carbon bond during insertion can vary from 0° to ~90°. After insertion, β-hydride elimination affords the product and a palladium(II)-hydrido complex IV, which is reduced by base back to palladium(0).[5]The Cationic PathwayMost asymmetric Heck reactions employing chiral phosphines proceed by the cationic pathway, which does not require the dissociation of a phosphine ligand. Oxidative addition of an aryl perfluorosulfonate generates a cationic palladium aryl complex V. The mechanism then proceeds as in the neutral case, with the difference that an extra site of coordinative unsaturation exists on palladium throughout the process. Thus, coordination of the alkene does not require ligand dissociation. Stoichiometric amounts of base are still required to reduce the palladium(II)-hydrido complex VIII back to palladium(0).[6] Silver salts may be used to initiate the cationic pathway in reactions of aryl halides.[7]The Anionic PathwayReactions involving palladium(II) acetate and phosphine ligands proceed by a third mechanism, the anionic pathway.[8]Base mediates the oxidation of a phosphine ligand by palladium(II) to a phosphine oxide. Oxidative addition then generates the anionic palladium complex IX. Loss of halide leads to neutral complex X, which undergoes steps analogous to the neutral pathway to regenerate anionic complex IX. A similar anionic pathway is also likely operative in reactions of bulky palladium tri(tert-butyl)phosphine complexes.[9]Establishing Tertiary or Quaternary StereocentersAsymmetric Heck reactions establish quaternary or tertiary stereocenters. If migratory insertion generates a quaternary center adjacent to the palladium-carbon bond (as in reactions of trisubstituted or 1,1-disubstituted alkenes), β-hydride elimination toward that center is not possible and it is retained in the product.[3]Similarly, β-hydride elimination is not possible if a hydrogen syn to the palladium-carbon bond is not available. Thus, tertiary stereocenters can be established in conformationally restricted systems.[10]。
配合物及超分子

3.4---配合物与超分子Coordination compound & supramolecule一、配位键coordination bond1.concept :配位键,又称配位共价键,简称配键,是一种特殊的共价键;当共价键中共用的电子 对是由其中一原子独自供应,另一原子提供空轨道时,就形成配位键(coordination bond ),配位 键形成后就与一般共价键无异;【配位键算σ键】2.配位体(ligand ):应含有孤电子对,如N .H 3、H 2O .、F .-、O .H -、C .O 等; 3.成键的性质:共用电子对对两原子的电性作用;4.成键条件:一方是能够提供孤电子对,另一方具有能够接受孤电子对的空轨道;5.配位键的表示方法:6.提供孤电子对的原子其电负性越大,形成的配位键越不牢固;二、配合物coordination compound1.concept :一般指由金属的原子或离子(价电子层的部分d 轨道和s 、p 轨道是空轨道)与含有孤对 电子的分子(如CO 、NH 3、H 2O)或离子(如---2NO CN Cl 、、等)通过配位键结合形成复杂的离子 或分子,称为配位单元,凡是含有配位单元的化合物叫做配位化合物,简称配合物(complex ); 显然:配合物中一定含有配位键;含配位键的化合物不一定是配合物;NH 4Cl 、FeCl 3、AlCl 3、CO 不是配合物;2.组成配合物由中心原子(或离子,提供空轨道)和配体(提供孤电子对)组成,分成内界和外界;如Cu(NH 3)4]Cl 2、[Cu(NH 3)4]SO 4可表示为:①配合物的中心一般都是带正电的阳离子,但也有带电中性的原子;如Ni(CO)4,Fe(CO)4,Cr(CO)6中Ni 、Fe 、Cr 都是电中性的;②配合物的中心原子绝大多数是金属离子,而过渡金属离子最常见;③配体(ligand ):必须是含有孤电子对的原子,常是V A 、VIA 、VIIA 族元素的原子;配体既可以是阴离子,如X -、OH -、SCN -、CN -、RCOO -、C 2O 42-、PO 43-等,也可以是中 性分子,如H 2O 、CO 、NH 3、醇、胺、醚等;④配合物在电离时,内界不电离,只电离出外界部分;⑤配位数(coordination number ):直接同中心原子配位的原子或离子数目;3.按照配位原子种类的不同,配体分为以下几种:①含氮配体--NH3、NO(亚硝基)、NO2(硝基)等;②含氧配体--H2O、OH-、CO32-、ONO-(亚硝酸根)、R-OH、R-O-R、R-COO(羧基);③含碳配体--CN、CO(羰基)等;④含硫配体--S2-、SCN-(硫氰酸根)、RSH(硫醇)、R-S-R(硫醚);⑤含磷配体--PH3(膦)、PR3、PF3、PCl3、PBr3等;⑥卤素配体--F-、Cl-、Br-、I-等;4.命名:关键在于配合物内界(即配离子)的命名①配离子其命名顺序应“先无机后有机”“先阴离子、后中性分子”,再写“合”字(表示配位结合),然后说明形成体(中心离子)的名称,如:[Cu(NH3)4]2+四氨合铜(II)离子;[Co(NH3)4Cl2]+二氯四氨合钴(III)离子;[Pt(NH3)2Cl2]二氯二氨合铂(Ⅱ);②配合物的命名,按照一般无机物命名原则,如:K2[PtCl6]六氯合铂(IV)酸钾;K[Pt(NH3)Cl5]五氯一氨合铂(IV)酸钾;[Co(NH3)4Cl2]Cl氯化二氯四氨合钴(III);Na3[AlF6]六氟合铝(III)酸钠(冰晶石);[Cr(H2O)5Cl]Cl2·H2O一水二氯化一氯五水合铬(III);K[Pt(C2H4)Cl3〕三氯乙烯合铂(Ⅱ)酸钾;③金属羰基化合物Metal carbonyl compoundsFe(CO)5五羰基铁;Ni(CO)4四羰基镍;Cr(CO)6六羰基铬;Mo(CO)6六羰基钼;5.配合物的结构①配合物中的配位键配离子[Ag(NH3)2]+是由中心原子Ag+与配位体氨分子通过配位键结合而成的,这种配位键的本质是中心原子提供空轨道来接受配位体上的孤电子对而形成配位键;在[Ag(NH3)2]+配离子中,中心原子Ag+采用sp杂化轨道接受配位体NH3中配位原子N的孤电子对形成配位键;②配离子的空间构型配位数杂化轨道类型空间构型实例2sp直线形[Ag(NH3)2]+,[Ag(CN)2]+4sp3四面体形[ZnCl4]2-,[Cd(CN)4]2-4sp2d平面四方形[Pt(NH3)4]2+,[PtCl4]2-,[Ni(CN)4]2-5sp3d三角双锥Fe(CO)5,PF55d3sp正方锥SbF52-6sp3d2正八面体形[AlF6]3-,[Co(NH3)6]3+6.几种常见的配合物①[Cu(H2O)4]SO4将白色的硫酸铜固体放入水中后,溶液变为天蓝色,这是因为生成了[Cu(H2O)4]2+;在该配离子中,H2O提供孤电子对(配体),Cu2+提供空轨道(中心原子),实际上变成了H2O 与Cu2+共用电子对,形成了一种特殊的共价键---配位键,将H2O与Cu2+紧紧连接在一起;②铜氨配合物向盛有硫酸铜溶液的试管里逐滴加入氨水,形成的氢氧化铜难溶于水,反应的离子方程式为:Cu2++2NH3·H2O=Cu(OH)2↓+2NH4+;向上述溶液中继续滴加氨水,难溶物溶解,得到深蓝色的透明溶液,反应的离子方程式为:Cu(OH)2+4NH3=[Cu(NH3)4]2++2OH-;在四氨合铜离子中,NH3分子的氮原子提供孤电子对(配位体),Cu2+提供空轨道(中心原子),形成了配位键;7.配合物的稳定性及应用①配合物的稳定性配合物具有一定的稳定性.配合物中的配位键越强,配合物越稳定.当作为中心原子的金属离子相同时,配合物的稳定性与配位体的性质有关;②配合物的应用※可利用配离子的特殊颜色鉴定离子的存在;※在催化剂的研制、燃料工业等方面用途很广;※生物体内许多酶与金属离子的配合物有关;【例题】硼砂是含结晶水的四硼酸钠,其阴离子X m-(含B、O、H三种元素)的球棍模型如图所示,则配位键存在于________原子间;卤素12nd二、超分子supra-molecule1.concept超分子通常是指由两种或两种以上分子依靠分子间相互作用结合在一起,组成复杂的、有组织的聚集体,并保持一定的完整性使其具有明确的微观结构和宏观特性;【超分子定义中的分子是广义的,包括离子】【超分子大环主体有DNA、冠醚、环糊精、杯芳烃、杯吡咯、杯咔唑、瓜环葫芦脲、柱芳烃等】2.超分子基本功能:分子识别、自组装;3.分子识别molecular recognition①concept:是两个或以上的分子之间通过非共价键结合相互作用;②实现分子识别条件※要求两个分子的结合部位是结构互补的;※要求两个结合部位有相应的基团,相互之间能够产生足够的作用力,使两个分子能够结合在一起;③存在:分子识别是一种普遍的生物学现象,糖链、蛋白质、核酸和脂质各自间以及他们相互之间都存在分子识别;④例:抗体与抗原之间,酶与底物之间,激素与受体之间的专一结合;4.自组装self-assembly①concept:自组装过程并不是大量原子、离子、分子之间弱作用力的简单叠加,而是若干个体之间同时自发地发生关联并集合在一起形成一个紧密而又有序的整体,是一种整体的复杂的协同作用;②图例5.冠醚crown ether①concept :是分子中含有多个-氧-亚甲基-[--22CH OCH ]结构单元的大环多醚;②冠醚最大的特点就是能与正离子,尤其是与碱金属离子络合,并且随环的大小不同而与不同③冠醚有一定的毒性,必须避免吸入其蒸气或与皮肤接触;对眼睛具有一定的刺激性如果接触眼睛,立即使用大量清水冲洗并送医诊治④冠醚有其独特的命名方式,命名时把环上所含原子的总数标注在“冠”字之前,把其中所含氧原子数标注在名称之后,如15-冠(醚)-5、18-冠(醚)-6、12-冠(醚)-4、二苯并-18-冠(醚)-6;⑤化学性质※与碱金属离子络合由于冠醚是一种大分子环状化合物,其内部有很大的空间,因此它能与正电离子特别是碱金属离子发生络合反应,把无机物带入有机物中,相转移催化剂就是基于这个原理;※醚的性质和氧化剂、还原剂、活泼金属、碱、稀酸等不起反应,与强酸性物质可发生某些化学反应;※氢键与客体分子生成配合物冠醚作为主体分子,可通过氢键与客体分子形成配合物,多发生在冠醚与铵离子之间;。
脯氨酸催化的不对称Aldol反应的研究进展

第17卷第4期化学研究Vol .17No .42006年12月CHE M I CAL RESE ARCH Dec .2006脯氨酸催化的不对称Aldol 反应的研究进展柯桢,马楠,王筱平3,韩超(同济大学化学系,上海200092收稿日期:2006-06-23.摘要:不对称合成是手性药物制备的核心环节,A ldol 反应是重要的形成C —C 键的反应之一,在全合成中有广泛应用.脯氨酸的两个异构体均价廉易得,作为一个非金属不对称催化试剂,它催化的不对称A ldol 反应立体选择性高,有很好的应用前景.本文就近二十年来脯氨酸催化A ldol 反应的机理,溶剂效应,最新进展三方面进行了介绍.关键词:脯氨酸;不对称催化剂;A ldol 反应中图分类号:O 621.3文献标识码:A 文章编号:1008-1011(200604-0096-06Advance of Asy mmetri c Aldol Reacti ons Cat alyzed by Proli n eKE Zhen,MA Nan,WANG Xiao 2p ing 3,HAN Chao(D epart m ent of Che m istry,Tongji U niversity,Shanghai 200092,China Abstract:The A ldol reacti on is an excep ti onally useful strategic C —C bond 2f or m ing reacti on f or thestereoselective constructi on of cyclic and acyclic molecules .The synthetic value of the A ldol reacti onshas been p r oven by their app licati on in the t otal synthesis of natural p roducts .The advantages of p r o 2line based aldolisati on reacti on are that the methodol ogy is metal free and that both enanti omers of thecatalyst are cheap and easily available .Pr oline catalysed aldolisati on reacti on shows both high yieldsand stereoselectivity .The catalytic enanti oselective versi on of this reacti on has received considerableattenti on in recent years .Keywords:p r oline;unsy mmetric catalysts;A ldol reacti on近年来,脯氨酸(p r oline 催化的不对称A ldol 反应在不对称合成中应用广泛,不仅在于它是廉价易得的手性原料,而且与其结构也有很大关系.首先,它含有羧基、氨基双官能团,既能起酸催化剂又能起碱催化剂的作用,或者起协同作用,在这一点上类似于酶的作用.另外,作为一个双齿配体,它可与金属形成金属配合物.与其它氨基酸不同的是,脯氨酸中的氨基为吡咯环二级胺,可与金属形成双环[3,3,0]辛烷类物质,它的氨基易于形成亚胺,烯胺中间体.1分子内不对称A ldol 反应20世纪70年代,Haj os 和Parrish 首次报道了脯氨酸催化的分子内不对称A ldol 反应[1].随后Eder,Sau 2er 和W iecher 等人报道了在脯氨酸和HCl O 4共同催化下,高选择性地得到A ldol 缩和产物(Sche me 1.因此,脯氨酸催化的分子内不对称A ldol 环化反应被命名为Haj os 2Parrish ΟEder ΟSauer 2W iechert 反应.继而,该反应被人们用来合成许多有用的化合物,如类固醇和许多天然产物.第4期柯桢等:脯氨酸催化的不对称A ldol反应的研究进展97Sche me12003年,Chandrakala等人首次发现了不对称enolex o2A ldol反应[2].作者认为Haj os2Parrish2Eder2Sauer2 W iechert反应的过渡态对应于本文机理部分的过渡态E,属于enolendo类型的分子内A ldol反应.作者发现反应(Sche me2,属于enolex o类型,对应于本文机理部分的过渡态F.同时,Douglas M将p r oline催化的enolexo2A ldol反应应用到(+ΟCocaine的合成中,用以形成基本骨架[3](Sche me3.Sche me2 Sche me32分子间不对称A ldol反应2.1醛酮间的A ldol反应2000年,L ist小组[4]报道了脯氨酸催化的分子间不对称A ldol反应.文中提到,用大大过量的丙酮与醛反应,才能实现分子间的不对称A ldol反应(Sche me4.Figure1中列出的是在相同条件下,醛与酮反应的A ldol产物(结构、产率和ee值.从Figure1中可以看出,反应的对映选择性与醛的结构密切相关,当丙酮与芳香醛反应,ee值在70%左右,产率在54%~94%之间;丙酮与α位有支链的脂肪醛反应,选择性和产率普遍较高;而丙酮与三级醛反应,ee值甚至超过99%.2001年,L ist小组[5]又报道了丙酮与α位没有支链的脂肪醛反应的研究.结果发现,当用丙酮或者氯仿代替DMS O时,减少脯氨酸用量(10%~20%,则以30%的收率和70%左右的ee值得到交叉Adl ol反应产物.同时可以看出,当醛的β位较大时,如叔丁基,选择性和产率将明显下降.Sche me4由于酮要过量,使得脯氨酸催化的不对称A ldol反应只适合使用廉价的小分子酮,如丙酮、丁酮、环戊酮和羟基丙酮等.在报道了脯氨酸催化下的丙酮与醛的不对称A ldol反应后不久,L ist小组[6]又考察了羟基丙化学研究2006年98酮与醛的不对称A ldol反应,以较高的收率和选择性得到了anti二醇.Figure1从Figure2中(dr:非对映异构体比例,本文特指anti:syn;ee值:主要产物的对映体过量可以看出,用邻氯苯甲醛时,反应的dr和ee值均比较低;用α位有支链的环己醛和异丁醛时,反应产率中等,但反应的dr 和ee值较高;用α位有支链的(R2甘油醛时,反应的对映选择性比较高(97%ee,但非对映选择性和反应收率不是很好.Figure2Thomas B[7]利用羟胺类物质和p r oline催化的A ldol产物进行原位反应,以高产率和高ee值得到N2alkyl2 C2hydr oxy2nitr ones(Sche me5.Jesús Casas将此反应发展了醛酮的α2羟甲基化反应[8],其它研究小组相继报道了脯氨酸作为催化剂在不对称A ldol反应中的应用,该反应被广泛的应用到合成中.Sche me52.2醛醛间不对称A ldol反应2002年,Mac M illan小组[9]报道了醛与醛在脯氨酸催化下发生交叉A ldol反应,以较好的收率和较高的选择性得到了交叉A ldol反应产物(Sche me6.Figure3中列出的是醛与醛在脯氨酸催化下发生交叉A ldol 反应得到的产物(结构、产率、dr和ee 值.可以看出,产物的ee值普遍较高,但反应的非对映选择性受作为电子受体的醛的影响较大,用异丁醛作为电子受体时,反应的非对映选择性明显要比其它醛作为电子受体高.Sche me6A lan首次报道了α2氧代醛之间的A ldol反应[10],且产物可作为合成糖类化合物的前体(Sche me7. Rajes wari[11]等人用α2氨基醛与其它醛反应制得β2羟基氨基醛,可方便地制备β羟基氨基酸类衍生物.第4期柯桢等:脯氨酸催化的不对称A ldol反应的研究进展99Figure3 Sche me73溶剂效应Pr oline催化的A ldol反应常采用干燥的DMS O作为溶剂,不利于后处理.近年来,逐渐考察了其它溶剂. Peter Kotrusz[12],Teck2Peng[13]等考察了质子性溶剂[bm i m]PF6(12n2butyl23Οmethy2l2i m idaz olium hexafluor o phos phate对A ldol反应的影响,[b m i m]PF6可减少p r oline的用量.此外,可以将p r oline负载在其它载体上进行非均相催化,催化剂可重复利用,简化后处理.A r mando Córdova[14]首次报道了在此溶剂中进行的两分子醛间的A ldol 缩合反应(Sche me8.T omoya Kitazume[15]讨论了在[e m i m][OTf]类质子性溶剂中进行的α2卤代酮和醛的A ldol反应,并进一步得到光学活性的环氧丙烷类单元.Sche me8Annika等人发现加入适量水可提高反应速度和对映选择性[16].A r mandoCórdova[17]报道了在BPS(0.01 mol・L-1磷酸盐缓冲液,2.7mmol・L-1KCl,137mmol ・L-1NaCl,pH=7.4缓冲溶液中进行的p r oline 催化的A ldol反应,并发现加入十二烷基硫酸钠(S DS有利于反应进行.Maurizi o[18]将(2S,4R242羟基脯氨酸负载在poly(ethyleneglycol(M5000上实现非均相A ldol反应.w4机理研究目前对脯氨酸催化的A ldol反应机理共提出了如下几种过渡态(Figure4.A,B,C,D,E为分子内A ldol反应的过渡态,F,G为分子间反应的过渡态.A由Haj os[1]提出,在这个过渡态中,环上羰基被活化为Carbinola m ine,进而发生亲核取代反应生成C—C键,这个机理很快遭到Jung[19]的反对,因为机理中涉及到一个S2反应,但是构象仍然保留.随后Jung和Eschenmoser讨论了侧链烯胺的N单分子p r oline催化的机理[20].Aga m i提出侧链烯胺的双分子p r oline催化机理(B,其中一个p r oline参与侧链形成烯胺,另一个作为质子转移的中介[21].动力学研究和所观察到的非线性效应支持了双分子p r oline参与的非对映选择的决定性步骤[22].Benja m in L ist研究认为,非线性效应是由测量手段的不精确导致,采用较精确的HP LC得到很好的线性效应[23].由于p r oline在很多有机溶剂中的溶解度不是很好,S wa m inathan提化学研究2006年100出在晶体表面进行协同催化的过渡态(C,然而很多p r oline催化的A ldol反应是在均相条件下进行的.通过密度泛函分析计算,Houk[24]提出了D模型.E模型是Chandrakala Pidathala等人根据D模型提出的enolexoΟA ldol反应模型.F模型跟D 有相似之处,是根据金属烯醇化物反应Zi m mer mannΟTraxler模型提出的.然而根据量子化学计算表明N—H键并不能降低过渡态能量[25],进而提出G模型,G模型由密度泛函分析计算得出,并得到实验证实[26].L inh Hoang通过对逆向A ldol反应的动力学测定间接证实了一分子p r oline参与过渡态的机理.Figure45结束语脯氨酸的两个异构体来源广泛、廉价、稳定,结构简单,由它催化的A ldol反应立体选择性高,无须金属参与,有酶催化的特点,显示了优良的催化性能,有很好的应用前景,同时其催化机理也有重要的理论研究价值.总之,脯氨酸作为天然手性分子中的一员,必将为不对称催化开拓一个新的领域.参考文献:[1](aHaj os Z G,Parrish D R.A sy mmetric synthesis of op tically active polycyclic organic compounds[J].Ger O ffen,1971,DE2102623.(bHaj os Z G,Parrish D R.A sy mmetric synthesis of bicyclic inter mediates of natural p r oduct chem istry[J].J O rg Che m,1974,39(12:1615-1621.[2]Chandrakala P,L inh H,Benja m in L.D irect catalytic asy mmetric enolexo aldolizati ons[J].A nge w Che m Int Ed,2003,42(24:2785-2788.[3]DouglasM M,W illiam H P.T otal synthesis of(+Οcocaine via desy mmetrizati on of a mes oΟdialdehyde[J].O rg L ett,2004,6(19:3305-3308.[4]L ist B,Lerner R A,Barbas C F.Pr olineΟcatalyzed direct asy mmetric aldol reacti ons[J].J Am Che m Soc,2000,122(10:2395-2396.[5]L ist B,Pojarliev P,Castell o C.Pr olineΟcatalyzed asy mmetri c aldol reacti ons bet w een ket ones and unsubstituted aldehydes[J].O rg L ett,2001,3(4:573-575.[6]L ist B.Pr olineΟcatalyzed asy mmetric reactions[J].Tetrahedron,2002,58(28:5573-5590.[7]Anders B,Thomas P,W ei Z,et al.For mati on of op tically active functi onalizedΟhydr oxy nitr ones using a p r oline catalyzed A ldolreacti on of aldehydes withcarbonyl compounds and hydr oxyla mines[J].Synlett,2003,12:1915-1918.[8]Jesús C,Henrik S.D irect organocatalytic asy mmetricΟhydr oxy methylati on of ket ones and aldehydes[J].Tetrahedron L ett,2004,45(32:6117-6119.[9]Northrup A B,Mac M illan D W C.The first direct and enanti oselective crossΟaldol reacti on of aldehydes[J].J Am Che m Soc,第 4期 2002, 124 ( 24 : 6798 - 6799. 柯桢等 : 脯氨酸催化的不对称 A ldol反应的研究进展101 [ 10 ] A lan B , Ian K M. Enantioselective organo Ο catalytic direct aldol reactions of oxy aldehydes: Step one in a two Ο step synthesis of carbohydrates [ J ].A ngew Chem In t Ed, 2004, 43 ( 16 : 2152 - 2154. [ 11 ] Rajeswari T, Fujie T, Carlos FB. D irect organocatalytic asymmetric aldol reactions of am ino aldehydes: expedient syntheses of highly enantiomerically enriched antihydroxyl am ino acids [ J ]. O rg L ett, 2004, 6 ( 20 : 3541 - 3544. m un, 2002, 21: 2510 - 2511. hed ron L ett, 2002, 43 ( 48 : 8741 - 8743. [ 12 ] Peter K, Iveta K ProlineΟ . catalyzed asymmetric aldol reaction in the room temperature ionic liquid [ bm im ] PF6 [ J ]. Chem Com 2 [ 13 ] TeckΟ Peng Loh. L Ο Proline in an ionic liquid as an efficient and reusable catalyst for d irect asymmetric aldol reactions [ J ]. Tetra 2 [ 14 ] A r mandoC. D irect catalytic asymmetric crossΟ aldol reactions in ionic liquid media [ J ]. Tetrahed ron L ett, 2004, 45 (20 : 3949 - 3952. [ 15 ] Tomoya K Synthesis of fluorinated materials catalyzed by p roline or antibody 38C2 in ionic liquid [ J ]. J F luor Chem , 2003, . 121 (2 : 205 - 212. [ 16 ] Annika I N , Annina U , Petri M P. Proline Ο catalyzed ketone Ο aldehyde A ldol reactions are accelerated by water [ J ]. S yn lett, 2004, 11: 1891 - 1896. 2002, 24: 3024 - 3025. [ 17 ] A rmando C, W olfgang N , Carlos F B. D irect organocatalytic aldol reactions in buffered aqueous media [ J ]. Chem Comm un , [ J ]. A dv S yn th Ca ta l, 2002, 344 ( 5 : 533 - 542. [ 19 ] Jung M E. A review of annulation [ J ]. Tetrahed ron , 1976 32: 3 - 31. 3108 - 3135. [ 20 ] B rown K L , Dunitz J D , Eschenmoser A , et a l Structural studies on crystalline enam ines [ J ]. Helv Ch im A cta , 1978, 61 (8 : . ric Robinson cyclization; structure of two inter mediates A symmetric dehydration [ J ]. Tetrahed ron, 1984, 40: 1031 - 1038. . A symm etry , 1999, 10: 1631 - 1634 [ 18 ] M aurizio B. Poly ( ethylene glycol Ο supported p roline: A versatile catalyst for the enantioselective aldol and im inoaldol reactions [ 22 ] Puchot S O , Dunach Z S, Swam inathan S Proline mediated asymmetric ketol cyclization: a temp late reaction [ J ]. Tetrahed ron: . [ 23 ] Benjam in L , L inh H , Harry J M. Newmechanistic studies on the p roline Ο catalyzed aldol reaction [ J ]. PNAS, 2004, 101 (16 : 5841. [ 25 ] L ist B. A symmetric am inocatalysis [ J ]. S yn lett, 2001, 11: 1675 - 1686. [ 24 ] ( a Bahmanyar S, Houk K N. The origin of stereoselectivity in p roline Ο catalyzed intramolecular aldol reactions [ J ]. J Am Chem S oc , 2001, 123: 12911 - 12912. ( b Bahmanyar S, L ist B. Quantum mechanical p redictions of the stereoselectivities of p roline [ 21 ] Agam i C, M eynier F, Puchot C, et a l Stereochem istry Ο New insights into the mechanism of the p roline Ο . 59. catalyzed asymmet2 [ 26 ] Clemente F R , Houk K N. Density functional calculations: Computational evidence for the enam ine mechanis of intramolecular m aldol reactions catalyzed by p roline [ J ]. A ngew Chem In t Ed, 2004, 43 ( 43 : 5766 - 5768. Ο catalyzed asymmetric intermo lecular aldol reactions [ J ]. J Am Chem S oc, 2003, 125 ( 9 : 2475 - 2479.。
常见有机物pKa表

(24.7) (24.4) (17.7) (12.7) (13.3) (22.7) (10.2) (21.6) (22.85) (21.1) (16.9) (18.6) (20.3) (14.6) (7.7) (11.4)
O
(28.1)
O
(29.0)
O
(25.5)
O
(32.4)
Me Me
*Values <0 for H2O and DMSO, and values >14 for water and >35 for DMSO were extrapolated using various methods.
有机物pka表 values>14 dmsowere extrapolated using various methods. 0.79 sulfinic sulfonicacids peroxides pka (dmso) (dmso) pka pka (dmso) pka's oxo-acids8.2 11.5 12.5 15.5 15.7 (11.1) (12.3) (1.6) (0.3) -14 (7.9) (12.9) (15) (1.8) (0.9) (32) (dmso) cis-co 3.6,10.3 3.77 -0.25 0.65 1.29 -8.0 11.6 -3.0, 1.99 1.9, 7.21 4.00 2.12, 7.21, 12.32 7.00 -1.7 15.7 -1.3 3.29 4.72 9.24 3.17 -0.98, 6.50 9.4 -2.6 -10 7.5 -9.00 9.23 inorganic acids chem 206 substrate substrate 1.92,6.23 -12.4 -7.8 -6.2 -3.8 -2.05 -2.2 -2.6 2.1 -1.8-6.5 1.682.66 2.86 2.86 3.12 4.76 4.2 o-o 2.172.45 3.44 2.94 3.83 3.99 1.37 p-(ch 3.434.47 4.25 3.02, 4.38 substrate substrate (13.7) (18.5) protonated species carboxylic acids (31.2) (27.9) (29.3) (23.5) (18.2) (29.4) 10.2 7.1 8.4 p-omec ohp-o ohm-o oh9.95 (18.0) 2-napthol (17.1) (10.8) (19.1) alcohols 16.5 17.0 c-hex coh24.0 oximes hydroxamicacids 8.88 11.3 (20.1) pka phph phoh ohph ohph oh oh ph ph ch ohme me meph comprehensivecompilation bordwellpka data see: /areas/reich/pka
浙工大论文封面模板

11.Mukherjee S, ListB. Radical catalysis [J]. Nature, 2007, 447:1522153.
12.聂尧,徐岩,生物催化立体选择性氧化还原中存在问题及其发展策略,生物加工过程,2008,6(2):1-9.
14.杨忠华,曾嵘,颜晓潮等,酵母细胞不对称还原4-氯苯乙酮合成相应手性醇[J].精细化工,2007,24(1):63-66.
15.Yang Z H, Zeng R,Wang Y, et al.Isolation of microbe for asymmetric reduction ofprochiral aromatic ketone and its reaction characters[J].Front Chem Eng China,2007,1(4):416-420.
生物体由于其自身的不对称性可以有效识别不同立体构型的对映异构体,进而导致反应及产物构型的不对称性[10-11]。利用生物体组织、细胞或酶催化手性化合物转化已逐渐成为获得光学纯手性产品的一种重要途径[12]。
活性生物细胞可以催化前手性羰基的不对称还原,使氧化还原酶催化的不对称还原和辅酶再生在胞内耦合,是一种很有学术价值和应用前景的方法[8]。目前已有多种活性生物细胞成功地用于催化多种前手性羰基不对称还原合成相应的手性醇[12-17]。但研究主要集中在酵母不对称还原上,利用植物组织细胞进行不对称还原的研究很少。
值得指出的是,在不对称催化合成研究依然处在方兴未艾的发展阶段的今天,许多与手性相关的科学问题仍有待解决。如:手性催化剂大部分只对特定的反应、甚至特定的底物有效,却没有广泛适用的万能手性催化剂,而且大多数手性催化剂存在转化数较低,稳定性不高,难以回收和重复使用等等亟待解决的问题[3],有待于我们进一步研究解决。
3-甲基-1-苯基-2-磷-1-氧化物

3-甲基-1-苯基-2-磷-1-氧化物3-甲基-1-苯基-2-磷-1-氧化物是一种有机磷化合物,化学式为C7H9PO。
它是一种无色液体,具有特殊的气味。
本文将从结构、性质、合成方法以及应用等方面介绍3-甲基-1-苯基-2-磷-1-氧化物。
一、结构3-甲基-1-苯基-2-磷-1-氧化物的结构中,一个磷原子连接着两个氧原子,同时与一个甲基基团和一个苯基基团相连。
这种结构使得它具有特殊的化学性质和广泛的应用价值。
二、性质3-甲基-1-苯基-2-磷-1-氧化物是一种可燃液体,能溶于有机溶剂如醇、醚等,但不溶于水。
它具有较低的沸点和高度不稳定,容易发生自燃、爆炸等危险。
三、合成方法3-甲基-1-苯基-2-磷-1-氧化物的合成方法有多种,下面介绍其中一种常用的方法。
首先,将亚甲基二苯基膦和过氧化氢反应,生成3-甲基-1-苯基-2-磷-1-氧化物。
这个反应一般在低温下进行,以避免危险的自燃和爆炸。
四、应用由于3-甲基-1-苯基-2-磷-1-氧化物具有特殊的化学性质,因此在不同领域有着广泛的应用。
1. 医药领域:3-甲基-1-苯基-2-磷-1-氧化物是一种重要的有机磷化合物中间体,可用于合成抗癌药物、抗病毒药物等。
它在药物研究中扮演着重要的角色。
2. 农药领域:3-甲基-1-苯基-2-磷-1-氧化物具有一定的杀虫作用,可用于合成农药。
它能有效地控制害虫的繁殖,提高农作物的产量和质量。
3. 助剂领域:3-甲基-1-苯基-2-磷-1-氧化物可作为阻燃剂、润滑剂、增塑剂等助剂的原料。
它可以提高材料的阻燃性能,减少摩擦系数,改善材料的加工性能。
4. 电子材料领域:3-甲基-1-苯基-2-磷-1-氧化物可用于制备光学薄膜、半导体材料等。
它在电子器件的制造过程中具有重要的应用价值。
总结:本文对3-甲基-1-苯基-2-磷-1-氧化物的结构、性质、合成方法和应用进行了详细的介绍。
作为一种重要的有机磷化合物,它在医药、农药、助剂和电子材料等领域都有着广泛的应用。
用人单位劳动防护用品管理规范

用人单位劳动防护用品管理规范第一章总则第一条为规范用人单位劳动防护用品的使用和管理,保障劳动者安全健康及相关权益,根据《中华人民共和国安全生产法》、《中华人民共和国职业病防治法》等法律、行政法规和规章,制定本规范。
第二条本规范适用于中华人民共和国境内企业、事业单位和个体经济组织等用人单位的劳动防护用品管理工作。
第三条本规范所称的劳动防护用品,是指由用人单位为劳动者配备的,使其在劳动过程中免遭或者减轻事故伤害及职业病危害的个体防护装备。
第四条劳动防护用品是由用人单位提供的,保障劳动者安全与健康的辅助性、预防性措施,不得以劳动防护用品替代工程防护设施和其他技术、管理措施。
第五条用人单位应当健全管理制度,加强劳动防护用品配备、发放、使用等管理工作。
第六条用人单位应当安排专项经费用于配备劳动防护用品,不得以货币或者其他物品替代。
该项经费计入生产成本,据实列支。
第七条用人单位应当为劳动者提供符合国家标准或者行业标准的劳动防护用品。
使用进口的劳动防护用品,其防护性能不得低于我国相关标准。
鼓励用人单位购买、使用获得安全标志的劳动防护用品。
第八条劳动者在作业过程中,应当按照规章制度和劳动防护用品使用规则,正确佩戴和使用劳动防护用品。
第九条用人单位使用的劳务派遣工、接纳的实习学生应当纳入本单位人员统一管理,并配备相应的劳动防护用品。
对处于作业地点的其他外来人员,必须按照与进行作业的劳动者相同的标准,正确佩戴和使用劳动防护用品。
第二章劳动防护用品选择第十条劳动防护用品分为以下十大类:(一)防御物理、化学和生物危险、有害因素对头部伤害的头部防护用品。
(二)防御缺氧空气和空气污染物进入呼吸道的呼吸防护用品。
(三)防御物理和化学危险、有害因素对眼面部伤害的眼面部防护用品。
(四)防噪声危害及防水、防寒等的听力防护用品。
(五)防御物理、化学和生物危险、有害因素对手部伤害的手部防护用品。
(六)防御物理和化学危险、有害因素对足部伤害的足部防护用品。
化合物英文命名规则

II. Nomenclature of organic compounds (有机化合物的命
名)
Nonfunctional Compounds (非功能性化合物)
A.
1. Alkane(烷烃) Alkene (olefin)(烯 烃) Alkyne (炔烃)
IUPAC names (systematic names) trivial names (popular names)
I. Nomenclature of inorganic compounds
When there are more than two oxidation states of the electronegative element, prefix used. The prefix “hypo-” meaning “below”, is used in the name of the lowest oxidation state. The prefix “per-” meaning “highest”, is used when it is in the highest oxidation state. 当电负性较低的元素有超过两种价态时,就使用前缀。 前缀“hypo-” 表示“低于”,用于低价态之前;前 缀“per-” 表示“高于”,用于高价态之前
Supplementary Information of Specialty English(专业英语补充 信息)
——Nomenclature of compounds(化合物 构词法)
Zhang Baohua College of Environmental & Chemical Engineering zhangbh@
I. Nomenclature of inorganic compounds
伯胺单甲基化反应小结
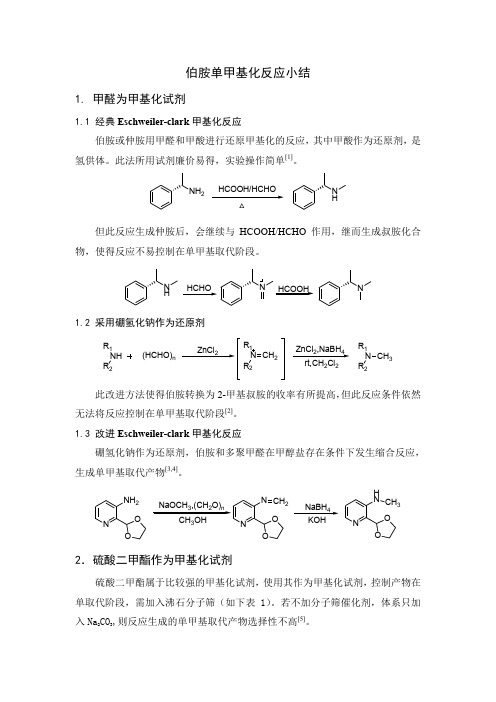
伯胺单甲基化反应小结1. 甲醛为甲基化试剂1.1 经典Eschweiler-clark 甲基化反应伯胺或仲胺用甲醛和甲酸进行还原甲基化的反应,其中甲酸作为还原剂,是氢供体。
此法所用试剂廉价易得,实验操作简单[1]。
NH 2NH但此反应生成仲胺后,会继续与HCOOH/HCHO 作用,继而生成叔胺化合物,使得反应不易控制在单甲基取代阶段。
N HNHCOOHHCHO N1.2 采用硼氢化钠作为还原剂NH R 2R 1(HCHO)nZnCl 2NR2R 1CH 2ZnCl 2,NaBH 422N R 2R 1CH 3此改进方法使得伯胺转换为2-甲基叔胺的收率有所提高,但此反应条件依然无法将反应控制在单甲基取代阶段[2]。
1.3 改进Eschweiler-clark 甲基化反应硼氢化钠作为还原剂,伯胺和多聚甲醛在甲醇盐存在条件下发生缩合反应,生成单甲基取代产物[3,4]。
NOO NH 22NaBH 4NOO HN CH32.硫酸二甲酯作为甲基化试剂硫酸二甲酯属于比较强的甲基化试剂,使用其作为甲基化试剂,控制产物在单取代阶段,需加入沸石分子筛(如下表1)。
若不加分子筛催化剂,体系只加入Na 2CO 3,则反应生成的单甲基取代产物选择性不高[5]。
表1. 胺与硫酸二甲基的甲基化反应碱转化率(%)PhMeNH:PhMe2N- 34 2.2:1 KX(0.25g)59 4.0:1KX(0.5g)63 12:1KX(1.0g)57 32:1NaX(0.5g)52 3.0:1KY(0.5g)71 8.9:1KY(1.0g)58 21:1NaY(0.5g)53 2.6:1Na2CO3(0.5g)66 3.0:1DBU(0.5mmol) 2.8 -a) 反应条件:PhNH2(0.5mmol),Me2SO4(0.25mmol),溶剂:己烷(3ml),反应时间:9h 3.碳酸二甲酯作为甲基化试剂有文献显示,碳酸二甲酯作为甲基化试剂,加入Y-或者X-型沸石,均能将反应控制在单甲基化取代阶段:ArNH2CH3OCOOCH31ArNHCH3CH3OH CO21) Y-或者X-型沸石,120-180℃;Ar = Ph, p-O2NC6H4, p-NCC6H4, o-MeO2CC6H4,2,6-Me2 C6H3其中:转化率:93%,N-单甲基化产物选择率:92%-98%[6]4.甲醇作为甲基化试剂有文献指出,使用超临界甲醇(Tc=239℃, Pc=8.1MPa,and Pc=0.272 g/cm3)作为甲基化试剂,可以将反应控制在单甲基取代阶段[7]。
在α-位的羰基化合物的取代反应

Substitution reactions of carbonyl compounds at the α-position Carbonyl compounds are acidic at α-C (e.g. CHR 2COR ); this is because of the electrophilic nature of carbonyl C=O bond. The pKa values of simple carbonyl compounds are generally in between 15-20; loss of an α hydrogen is stabilized by the C=O dipole.Thus under basic conditions, bases immediately deprotonate carbonyl compounds to give an enolate.-resonance-stabilized enolateHow important is the electronegative oxygen atom to the stabilization of the enolate?pKa of an α hydrogen of CH 3CH=CH 2 = 43 pKa of an α hydrogen of CH 3CH=O = 16.7Under acidic conditions, carbonyl compounds are protonated on O first, thenweak base deprotonates at the α-C to give enol.OHresonance-stabilized enolH 3O+/H 2OIn base or in acid, carbonyl compounds that have α-hydrogens are in equilibrium with their enolate or enol forms.So, where does the equilibrium lie between the carbonyl compound and its enol form, and how does it depend on structure?acetaldehyde and acetone have less than 1% of the enol tautomer present at equilibrium – the C=O bond is stronger than the C=C bond and dominates (see section 23.2 in your textbook).More complex carbonyl compounds can be much more enolized; for example, 1,3-dicarbonyls (β-dicarbonyls) exist primarily in their enol formsOHO76% enol tautomerspKa = 9See Table 23.2 for typical pKa values for carbonyl compounds and nitriles. Reactions of Enols and EnolatesBoth enolate and enol are nucleophilic at C (and O).For example, enolates react in the presence of good electrophiles by undergoing substitution of the alpha carbon.The addition of electrophiles to the alpha carbon via the acid- or base-induced formation of enols or enolates is quite common.1. Reaction under acidic conditions (i.e. reaction of enols):1a. Acid-Catalyzed Halogenation of Aldehydes and KetonesAldehydes can easily be mono-halogenated at the alpha position by simply mixing the aldehyde with a halogen (usually Br2 or I2) and a trace of acid. IMPORTANT NOTE - if there are no alpha hydrogens, this reaction will not take place!when various possible enols are formed under equilibrating conditions, usually the MOST STABLE enol is the one formed in the highest concentration and thus, is the one that reacts with the halide. For example, methyl cyclohexanone canform two different enols - the more highly substituted one is the most stable,and thus predominates:There are two major uses for these bromo-ketones and aldehydes. The first is elimination to form conjugated carbonyl compounds. This is a classic (andsimple) method for preparing such compounds:1b. The Hell-Volhard-Zelinskii ReactionThe acid-promoted alpha halogenation only works with aldehydes and ketones. What if you need to brominate a carboxylic acid? That’s where the HVZ reaction comes in. The reaction basically takes a difficult-to-enolize carboxylic acid, and first turns it into a much more enolizable acid bromide. Aqueous workup returns the acid bromide to the carboxylic acid state. Workup with an alcohol would, ofcourse, produce the ester.the first step is formation of the acid bromide, which is in equilibrium with its enol form. Bromination of the enol form gives an isolable α-bromo acid bromide.PBr3Br2acid bromideenol formThe latter reacts like an acid chloride and can be used to generate α-bromo acid, ester, and amide.HOR 2NH 3H 2OPlease remember that while this reaction can be used to form an ester, it cannot be used with an ester as starting material – you must start with the carboxylic acid!2. Reaction under basic conditions (i.e. Reaction of enolates)Under sufficiently basic conditions, an enolate ion can be formed:However, we must generally be careful with our choice of bases – if the base is also a good nucleophile, then attack at the carbonyl carbon becomes a more likely pathway. The most common bases used to form enolate ions are sodium hydride (very basic, non-nucleophilic) and Lithium Diisopropylamide (LDA, very bulky, thus non nucleophilic). Occasionally, hydroxide ion is used, but this is usually for very specific reactions (e.g. the haloform reaction).Discuss Table 23.3– HO - and RO - bases are more useful for forming enolates from 1,3-dicarbonyl compounds or in reactions that involve both the enolate and carbonyl starting material.The enolate ion has two nonequivalent resonance forms (compare this with both the allyl anion and the carboxylate anion). The form where the charge is localized on the oxygen predominates (because of the electronegativity of oxygen), but the form with the charge localized on carbon is more nucleophilic, and is thus the form that typically reacts with electrophiles:There are a few exceptions to this rule; generally acyl and silyl halides (TMS-Cl or CH 3COCl) will react with the oxygen anion, to give silyl enol ethers and enolacetates, respectively. As you might expect, alkylation of an enolate is a powerful tool for the formation of carbon-carbon bonds under relatively mild conditions.The Haloform Reaction:While enolates are great for forming new carbon-carbon bonds, the first reaction we’ll look at is a method for the destruction of a carbon-carbon bond. Essentially, the haloform reaction takes a methyl ketone (or a molecule which can be oxidized to a methyl ketone), and turns it into a carboxylic acid with one less carbon:How does it work? As you probably expect, the enolate is formed, and is then halogenated. The protons on the halogenated compound are even more acidic, thus facilitating further enolization and halogenation. When the compound is fully halogenated (in this case, forming a CI3 group), there are no more acidic protons, so we look for the next possible mechanistic route: nucleophilic attack. Addition-elimination (as shown) leads to the carboxylate anion and a haloform (in this case, iodoform):2b. Alkylation of EnolatesNormal enolates formed by the action of LDA or NaH can generally be alkylated with alkyl iodides, bromides or tosylates, or benzylic or allylic halides. However, these reactions can sometimes be difficult to perform in high yield. A few methods do exist which allow enolate alkylations in high yield, and both take advantage of highly stabilized enolate anions: The Malonic Ester synthesis, and the Acetoacetic Ester synthesis. We’ll look at each of these in detail.The Malonic Ester Synthesis.Esters of malonic acid (in this case, diethyl malonate) are easily deprotonated to form the highly stabilized enolate (note sodium ethoxide is used as base – why not NaOMe?):As you can see, the negative charge is delocalized over one carbon and two oxygens – a very stable anion! The only significantly nucleophilic form is the one with the negative charge on the carbon, and this is the form that gets alkylated. You should note that there are two acidic protons on a malonic ester - thus it can be alkylated twice if desired. If an alkyl compound with two halogens is added, cyclic compounds can be formed.But the really cool thing about malonic esters (and acetoacetic esters, which we’ll see in a minute) is a reaction called decarboxylation. Once a malonic ester is hydrolyzed, then treated with acid, the two carbonyl groups interact, and through a 6-membered ring transition state (remember how organic molecules love to react through 6-membered ring - e.g. think of the Diels Alder reaction!), one of the carbonyl groups is blown off as CO2:Note that this is not a common reaction, and only happens with 1,3-dicarbonyl groups!!!!! Thus, malonic ester chemistry is an ideal way to make substituted acetic acids (which can then be converted to acid chlorides). Let’s look at an example:Acetoacetic Ester SynthesisThe acetoacetic ester synthesis is almost exactly like the malonic ester synthesis – invloves a 1,3-dicarbonyl compound that is easily alkylated, and which also decarboxylates. But instead of making substituted acetic acid derivatives, this reaction makes substituted acetones.Just like in the malonic ester synthesis, the protons between the carbonyl groups are particularly acidic. They can be deprotonated with alkoxide (e.g.NaOEt), or sodium hydride, to yield the stabilized anion. Alkylation is also straightforward. The only difference comes in the decarboxylation step, where the only carboxyl group is lost to leave a methyl ketoneSome things to note for both the acetoacetic ester and malonic ester synthesis (all of which are noted for the scheme below):1) These are highly stabilized anions, and will thus add to conjugated carbonyl compounds in a 1,4 fashion. Decarboxylation leads to the vinyl-substituted compound.2) Saponiofication and decarboxylation can occur in one step - provided the temperature is high enough (usually, 110 - 120°C will do it).3) Note that all esters in the molecule are saponified, but only the 1,3-dicarbonyl compounds can undergo decarboxylation.4) As you’ll see in lecture, many different electron withdrawing groups can stabilize enolate charges, and lead to a large variety of different compounds. However, you’ll only get decarboxylation if you have at least one group that can be hydrolyzed to an acid, which is separated from another carbonyl group by one carbon. For example:Of course, this same ketone could be alkylated directly:The product is the same, and there are fewer steps, but the reaction conditions are much more harsh. Furthermore, if we were not working with a symmetrical ketone, a mixture of products would result. Sometimes taking a few extra steps is worth it...With simple esters and ketones, however, direct alkylation of the enolate is usually the way to go:Notes:1) These non-stabilized enolated are very basic - alkylation generally won’t happen with anything other than primary, allylic or benzylic halides. Secondary and tertiary halides will give elimination products...2) Again, if there is more than one way to form the enolate, you’ll get a mixture of products...。
F.A.Carey-最全最完备的高等有机化学习题 详细解答与分析 完美版

Chapter 1 Effect of Substituted in Organic molecule1. 试判断下列各对基团,那一个具有强的-I 效应(即强的吸电子诱导效应):(1) -COOH , -COO -(2) C HN OCH 3 , C H N N(CH 3)2CH 3+(3) C OCH 3 , C CH 2CH 3, (4) SO 2H ,SO 3H(5) OCH 3 , SCH 3 (6) C H C H CH 3 ,C C CH 3(7) N (CH 3)2 , P(CH 3)2 (8) Si(CH 3)3 ,Si(CH 3)2CH 3(9)N(CH 3)3+,NH 2 (10) CN ,CH 2NH 2 (11) SiCH 3 , Cl (12) C C CH 3 ,C H C H CH 3(13),(14)NO 2,NO 2(15) O 2SCH 3, O 2SBr2. 指出下列各对酸中哪一个酸性强(1) H 3NCH 2CH 2COOH , HOCH 2CH 2COOH (2) HC C COOH ,H 2C C H COOH(3) C 6H 5COCH 2COOH , C 6H 5CHOHCH 2COOH(4) C 4H 9CHCOOn,C C CH 3CH 3CH 3CH 3COOHOOC(5) COOH OH, COOH OH(6) BrCH 2CH 2COOH , CH 3CHBrCOOH(7)(H 3C)2CH 2CHCOOH,H 2CC HCH 2COOH(8) HC C COOH , SCOOH(9) CH 2(COOH)2 ,HOOCH CCOOCl(10) CH 3OCH 2CH 2COOH , CH 3SCH 2CH 2COOH (11) CH 3SCH 2COOH ,CH 3SO 2COOH(12)OHCCOOH,COOHCHO(13)OHC(CH 3)3(H 3C)3C,OH3)3CH 3(H 3C)3C(14) H 3COCOOH ,OCH 3COOH(15) C 6H 5CH 2SeH ,H 3CSeH3. 预料以下各对化合物,何者具有更强的酸性? (1) CH 3NO 2 ,(CH 3)2CHNO 2(2) CH 2(SO 2C 6H 5)2 ,CH 2(SOC 6H 5)2(3) H 3CCH(C 6H 5)2,(C 6H 5)2CHCH 2C 6H 5 (4) CH 3COCH 2COOCH 3 ,CH 3COCH 2CONH 2 (5) CH 3COCH 2COCH 2F ,CH 3COCHFCOCH 3(6)NOCH 3,NO H 3C(7)SO 2O 2S ,OO(8) NO 2CH 3CH 3H 3C ,NO 2CH 3H 3CCH 3(9)CH3,CH3(10) CH(C 6H 5)2,CH(C 6H 5)2(11) (CH)3Se , (CH 3)2O(12) ,4. 解释以下现象:(1). 杯烯 (Calcene) 的偶极距很大,μ= 5.6 D.(2). 吡咯 μ = 1.80 D ,吡啶 μ = 2.25 D ,且极性相反,如图:NN H5. 比较下列化合物的碱性的强弱:NN(CH 3)2N(C 2H 5)2NH 2N6. 9,10-二氢蒽-1-羧酸(A )和9,10-乙撑蒽-1-羧酸(B )的酸性取决于8-位上取代基X的性质。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
CARBONYL COMPOUNDS - Aldehydes and KetonesStructure•carbonyl groups consists of a carbon-oxygen double bond •the bond is polar due to the difference in electronegativity •aldehydes / ketones differ in what is attached to the carbon ALDEHYDES- at least one H attached to the carbonyl groupHCHO orCH 3CHO C 6H 5CHOKETONES - two carbons attached to the carbonyl groupCH 3COCH 3C 2H 5COCH 3C 6H 5COCH 3Bonding •the carbonyl carbon is sp 2 hybridised and three sigma (σ) bonds are planar •the unhybridised 2p orbital of carbon is at 90° to these •it overlaps with a 2p orbital of oxygen to form a pi (π) bond •as oxygen is more electronegative than carbon the bond is polarNaming •aldehydes end in... AL •ketones end in...ONE•pick the longest chain of carbon atoms which includes the C=O •substituent positions are based on the carbon with the O attached CH 3CH 2CH 2CH 2CH 2CHOCH 3COCH 2CH 2CH 2CH 3hexanalhexan-2-oneC OO C OC Q.1 Draw structures for, and name, all the carbonyl compounds with molecular formulae;a) C 4H 8Ob) C 5H 10Oc) C 6H 12OC=O CCC=O HCC=O HHFormation of carbonyl compounds from alcoholsAldehydes • Oxidation of primary (1°) alcohols - risk of oxidation to acidsegCH 3CH 2OH (l) + [O] —> CH 3CHO (l) + H 2O (l) ethanol ethanalhowever, this can happenCH 3CHO (l) + [O] —> CH 3COOH (l)ethanal ethanoic acid• it is essential to distil off the aldehyde before it gets oxidised to the acid • the alcohol is dripped into a warm solution of acidified K 2Cr 2O 7• the aldehyde has a low boiling point - no hydrogen bonding - it distils off • if it didn’t distil off it would be oxidised to the equivalent carboxylic acid • to oxidise an alcohol straight to the acid you would reflux the mixtureKetones • Oxidation of secondary (2°) alcohols.egCH 3CHOHCH 3(l) + [O] —> CH 3COCH 3(l) + H 2O (l) propan-2-ol propanoneDISTILLATIONgives an ALDEHYDE REFLUXING gives aCARBOXYLIC ACIDQ.2Which alcohol would you use to make the following?• C 2H 5CHO• C 2H 5COCH 3• hexanal• 3-methylhexan-2-one• 3-methylpentanalCHEMICAL PROPERTIES OF CARBONYL COMPOUNDSOXIDATION• provides a way of differentiating between aldehydes and ketones• mild oxidising agents are best• aldehydes are easier to oxidise• powerful oxidising agents oxidise ketones to carboxylic acid mixturesALDEHYDES easily oxidised to acids e.g. RCHO(l) + [O] ——> RCOOH(l)CH3CHO(l) + [O] ——> CH3COOH(l) KETONES only oxidised under vigorous conditions to acids with fewer carbons.e.g. C2H5COCH2CH3(l) + 3 [O] ——> C2H5COOH(l)+ CH3COOH(l)Q.3What product (if any) is formed when the following undergo mild oxidation?• C2H5CHO• C2H5COCH3• hexanal• 3-methylhexan-2-one• 3-methylpentanal• cyclohexanoneIDENTIFYING A CARBONYL COMPOUNDMethods• characteristically strong peak at 1400-1600 cm-1in the infra red spectrum or • formation of orange crystalline precipitate with 2,4-dinitrophenylhydrazineBUT to narrow it down to an aldehyde or ketone you must do a second test Differentiation• to distinguish an aldehyde from a ketone you need a mild oxidising agent ...Tollens’ Reagent• ammoniacal silver nitrate• contains the diammine silver(I) ion - [Ag(NH3)2 ]+• acts as a mild oxidising agent and will oxidise aldehydes but not ketones• the silver(I) ion is reduced to silver Ag+(aq) + e¯ —> Ag(s)• the test is known as THE SILVER MIRROR TESTFehling’s Solution• contains copper(II) ions complexed with tartrate ions• on warming, it will oxidise aliphatic (but not aromatic) aldehydes• copper(II) is reduced to a red precipitate of copper(I) oxide, Cu2OThe silver mirror test is the better alternative as it works with all aldehydes.Ketones do not react with Tollens’ Reagent or Fehling’s Solution.Q.4Which of the following produce an orange precipitate with 2,4-dinitrophenylhydrazine?• C2H5OH• 3-methylhexan-2-one• C2H5COCH3• cyclohexanol• hexanal• 3-methylpentan-1-olQ.5Which of the following produce a silver mirror with Tollens’ reagent?• C2H5CHO• 3-methylhexan-2-one• C2H5COCH3• cyclohexanone• hexanal• 3-methylpentanalNUCLEOPHILIC ADDITION REACTIONSMechanism •occurs with both aldehydes and ketones•involves addition to the polar C=O double bond •attack is by nucleophiles at the positive carbon centre •alkenes are non-polar and are attacked by electrophilesREDUCTIONReagentsodium tetrahydridoborate(III) (sodium borohydride), NaBH 4Conditions aqueous or alcoholic solutionMechanism Nucleophilic addition (also reduction as it is addition of H¯)NucleophileH¯ (hydride ion)Product(s) Aldehydes REDUCED to primary (1°) alcohols KetonesREDUCED to secondary (2°) alcoholsEquation(s) CH 3CHO + 2[H] ——> CH 3CH 2OH CH 3COCH 3 + 2[H] ——> CH 3CHOHCH 3Step 1H¯ is a nucleophile and attacks the C δ+An electron pair from the C=C moves onto O making it -ive Step 2A lone pair on oxygen removes a proton from water Overall, there is addition of hydrogen (reduction)BondPolarity Attacked by Result Carbonyl C=O Polar Nucleophiles Addition AlkeneC=C Non-polarElectrophilesAdditionC HOCH 3OHH C CH 3H CCH 3HOH HOHO H+HQ.6Draw a diagram to indicate the bonding in NaBH 4.Alternative MethodReagenthydrogenConditions catalyst - nickel or platinumReaction type Hydrogenation, reduction Equation(s)CH 3CHO + H 2 ——> CH 3CH 2OH CH 3COCH 3 + H 2 ——> CH 3CHOHCH 3NoteHydrogen also reduces C=C bondse.g.CH 2 = CHCHO + 2H 2 ——> CH 3CH 2CH 2OHQ.8Draw structures of the organic products formed when the following are reduced using...NaBH 4H 2OOHOCH = CHCH CHO 22Q.7Why are C=C double bonds NOT reduced when NaBH 4 is used?HCN Reagent hydrogen cyanide - HCN (in the presence of KCN)Conditions reflux in alkaline solution Nucleophile cyanide ion CN¯Product(s)hydroxynitrile (cyanohydrin)EquationCH 3CHO + HCN ——> CH 3CH(OH)CN2-hydroxypropanenitrileMechanismNucleophilic additionStep 1CN¯ acts as a nucleophile and attacks the slightly positive COne of the C=O bonds breaks; a pair of electrons goes onto the O Step 2A pair of electrons is used to form a bond with H +Overall, there has been addition of HCNNotes •HCN is a weak acid ; HCN H + + CN¯ few CN¯ ions produced•the reaction is catalysed by alkali - produces more of the nucleophilic CN¯•watch out for the possibility of optical isomerism in hydroxynitrilesCN¯ attacks from aboveCN¯ attacks from belowCH OCH 3N C———_HC CH 3NC OH+HC CH 3NC OHCH OCH 3N C———_HCCH 3N COH+H CCH 3N C OHC HOCH 3N C———_OHH C CH 3NC HCCH 3NC OH +2,4-DINITROPHENYLHYDRAZINEC 6H 3(NO 2)2NHNH 2Theory •reacts with carbonyl compounds (aldehydes and ketones )•used as a simple test for aldehydes and ketones•makes orange crystalline derivatives - 2,4-dinitrophenylhydrazones •derivatives have sharp, well-defined melting points •also used to characterise (identify) carbonyl compounds .Identification A simple way of characterising a compound (finding out what it is) is to measure• the melting point of a solid • the boiling point of a liquid The following structural isomers have similar boiling points because of similar van der Waals forces and dipole-dipole interactions. They would be impossible to identify with any precision using boiling point determination.Boiling point of compound 213°C 214°C 214°C Melting point of 2,4-dnph derivative 209°C 248°C 265°CBy forming the 2,4-dinitrophenylhydrazone derivatives and taking the melting point of the purified , crystalline product, it is easy to identify the original compound.Typical equationMechanism ADDITION-ELIMINATION2NH NH2NO 2NO ClCHOClCHO ClCHOisomericchlorophenylmethanalsNO 2NO 22NH NH 3CH CHO+NO 2NO 2N N C 3CH H H2H O +Q.6Suggest a method for differentiating between the following. What would be seen?• C2H5CHO and C2H5COCH3• C3H7COCH3 and C2H5COC2H5• hexanal and hexanoneQ.5Write out equations for the reactions between HCN and...• C2H5CHO + HCN——>• CH3COCH3 + HCN ——>• hexanal + HCN ——>Indicate which reactions give rise to optically active organic compounds?Why is the addition of HCN such a useful reaction?。