HapMapTutorial
WorldHaplogroupsMaps
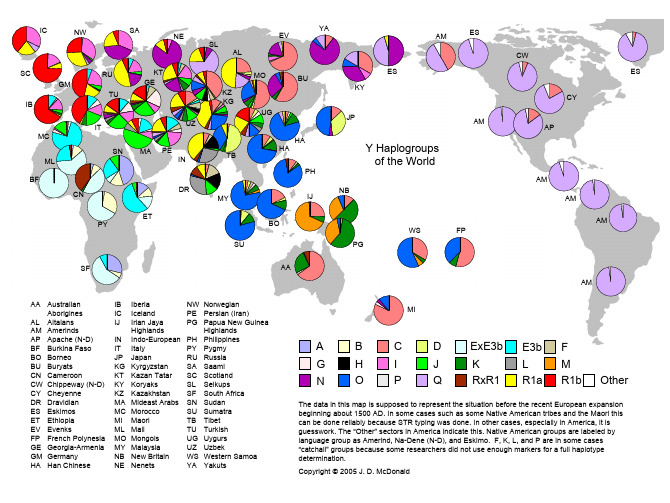
MI
A G N
B H O
C I P
D J Q
ExE3b K RxR1
E3b L R1a
F M R1b
Other
The data in this map is supposed to represent the situation before the recent European expansion beginning about 1500 AD. In some cases such as some Native American tribes and the Maori this can be done reliably because STR typing was done. In other cases, especially in America, it is guesswork. The “Other” sectors in America indicate this. Native American groups are labeled by language group as Amerind, Na-Dene (N-D), and Eskimo. F, K, L, and P are in some cases “catchall” groups because some researchers did not use enough markers for a full haplotype determination. Copyright © 2005 J. D. McDonald
Bortolini, Maria-Catira, et. al., Y-Chromosome Evidence for Differing Ancient Demographic Histories in the Americas, Am. J. Hum. Genet. 73:524–539, 2003 Bosch, Elena, et. al., High level of male-biased Scandinavian admixture in Greenlandic Inuit shown by Y-chromosomal analysis, Hum. Genet. 112 : 353–363, 2003 Capelli, Cristian, et. al., A Predominantly Indigenous Paternal Heritage for the Austronesian-Speaking Peoples of Insular Southeast Asia and Oceania, Am. J. Hum. Genet. 68:432–443, 2001 Cinnioglu, Cengiz, et. al., Excavating Y-chromosome haplotype strata in Anatolia, Human Genetics, 114: 127-148, 2004 Cruciani, Fulvio, et. al., A Back Migration from Asia to Sub-Saharan Africa Is Supported by High-Resolution Analysis of Human Y Chromosome Haplotypes, Am. J. Hum. Genet. 70:1197–1214, 2002 Faux, David, private communication Hammer, M. F., et. al., Jewish and Middle Eastern non-Jewish populations share a common pool of Y-chromosome biallelic haplotypes, Proc. Nat. Acad. Sci. USA, 97: 6769-6774, 2000 Jobling, Mark A.., and Tyler-Smith, Chris, The Human Y Chromosome: An Evolutionary Marker Comes of Age, Nature Rev. Genetics, 4: 598-612, 2003 Karafet, T. M., et. al., Ancestral Asian Source(s) of New World Y-Chromosome Founder Haplotypes, Am. J. Hum. Genet. 64:817–831, 1999 Karafet, Tatiana, et. al., Paternal Population History of East Asia: Sources, Patterns, and Microevolutionary Processes, Am. J. Hum. Genet. 69:615–628, 2001 Kayser, Manfred, et. al., Reduced Y-Chromosome, but Not Mitochondrial DNA, Diversity in Human Populations from West New Guinea, Am. J. Hum. Genet. 72:281–302, 2003 King, Roy, and Underhill, Peter. A., Congruent distribution of Neolithic painted potter and ceramic figurines with Y-chromosome lineages, Antiquity 76: 707-714, 2002 Lell, Jeffrey T., et. al., The Dual Origin and Siberian Affinities of Native American Y Chromosomes, Am. J. Hum. Genet. 70:192–206, 2002 Nasidze, I., et. al., Mitochondrial DNA and Y Chromosome Variation in the Caucasus, Ann. Hum. Genetics 68:205-221, 2004 Rosser, Zoë H., et. al., Y-Chromosomal Diversity in Europe Is Clinal and Influenced Primarily by Geography, Rather than by Language, Am. J. Hum. Genet. 67:1526–1543, 2000 Tambets, Kristiina, et. al., The Western and Eastern Roots of the Saami—the Story of Genetic “Outliers” Told by Mitochondrial DNA and Y Chromosomes, Am. J. Hum. Genet. 74:661–682, 2004 Underhill, Peter A., et. al., Y chromosome sequence variation and the history of human populations, Nature Genetics, 26: 358-361, 2000 Underhill, Peter A. , et., al., Maori Origins, Y-Chromosome Haplotypes and Implications for Human History in the Pacific, Human Mutation 17: 271-280, 2001 Wells, R. Spencer, et. al., The Eurasian Heartland: A continental perspective on Y-chromosome diversity, Proc. Nat. Acad. Sci. USA, 98: 10244–10249, 2001 Zegura, Stephen L., et. al., High-Resolution SNPs and Microsatellite Haplotypes Point to a Singlem Recent Entry of Native Americans Y Chrmosomes into teh Americas, Mol. Biol. Evol., 21: 164-175, 2004
HAPMAP

-4 0 4 8 12 16 X
0.001 0.615 0.003 0.085 0.256 0.040 0.385
0.000 0.747 0.004 0.049 0.180 0.020 0.253
0.54 0.73 1.79 1.57 2.07 1.85
1.7×10-9 0.358 0.001 6.2×10-5 0.006 1.7× 10-9
Leukotriene A4 Hydrolase (LTA4H) gene Candidate Gene for Myocardial Infarction Resides in one LD block where there is no other gene
Variant of a gene located on chromosome 10q confers risk of type 2 diabetes mellitus Saturday Session #63
Struan F.A. Grant1, G. Thorleifsson1, I. Reynisdottir1, R. Benediktsson2,3, A. Manolescu1, J. Sainz1, H. Stefansson1, V. Emilsson1, A. Helgadottir1, U. Styrkarsdottir1, M.P. Reilly4, D.J. Rader4, Y. Bagger5, C. Christiansen5, V. Gudnason2, G. Sigurdsson2,3, U. Thorsteinsdottir1, J.R. Gulcher1, A. Kong1, K. Stefansson1
普开数据大数据培训课程讲解:Hadoop-MapReduce教程

JobClient.runJob(conf);
用法
假设环境变量 HADOOP_HOME 对应安装时的根目录, HADOOP_VERSION 对应 Hadoop 的当前安装版 本,编译 WordCount.java 来创建 jar 包,可如下操作: $ mkdir wordcount_classes $ javac -classpath ${HADOOP_HOME}/hadoop-${HADOOP_VERSION}-core.jar -d wordcount_classes WordCount.java $ jar -cvf /usr/joe/wordcount.jar -C wordcount_classes/ . 假设:
源代码
WordCount.java 1. 2. 3. 4. 5. 6. 7. 8. 9. import org.apache.hadoop.fs.Path; import org.apache.hadoop.conf.*; import org.apache.hadoop.io.*; import org.apache.hadoop.mapred.*; import java.io.IOException; import java.util.*; package org.myorg;
10. import org.apache.hadoop.util.*; 11. 12. public class WordCount { 13. 14. 15. 16. 17. 18. 19. 20. public void map(LongWritable key, Text value, OutputCollector<Text, IntWritable> output, Reporter reporter) throws IOException { String line = value.toString(); StringTokenizer tokenizer = new StringTokenizer(line); public static class Map extends MapReduceBase implements Mapper<LongWritable, Text, Text, IntWritable> { private final static IntWritable one = new IntWritable(1); private Text word = new Text();
Tassel 5.0关联分析软件 中文使用手册

3.1.5 投影校准(Projection Alignment)............................................................... 15 3.1.6 Phylip .............................................................................................................. 15 3.1.7 FASTA............................................................................................................. 16 3.1.8 Numerical Data(数值数据) ....................................................................... 16 3.1.9 Square Numerical Matrix(数值方阵) ........................................................ 17 3.1.10 Table Report(表格报告) .......................................................................... 18 3.1.11 TOPM(Tags on Physical Map,物理图谱上的标签)............................... 18 3.2 Export 导出 ....................................................................................................................... 18 3.3 转换(Transform) ......................................................................................................... 19 3.3.1 Genotype Numericalization(基因型数字化) .................................................... 19 3.3.2 Transform and/or Standardize Data 转换和/或标准化数据.................................. 20 3.3.3 Impute Phenotype 估算表现型.............................................................................. 21 3.3.4 PCA(主成分分析) ............................................................................................ 22 3.4 Synonymizer(举出分类单元名称的同义词)...................................................... 23 3.5 Intersect Join(交集合并) ............................................................................................. 25 3.6 Union Join(并集合并) ................................................................................................. 26 3.7 Merge Genotype Tables(合并基因型表格)................................................................. 26 3.8 Separate(分离) ............................................................................................................. 27 3.9 Homozygous Genotype(纯合的基因型) ..................................................................... 27 4 Impute(估算)菜单 .................................................................................................................. 27 5 Filter(过滤)菜单 ..................................................................................................................... 35 5.1 Sites(位点) ................................................................................................................... 35
MapX培训教程-(含多场景)

MapX培训教程引言MapX是一款强大的地图制作和地理信息系统(GIS)软件,广泛应用于地图制作、空间数据分析、地图发布等领域。
为了帮助用户更好地了解和掌握MapX的使用方法,本教程将详细介绍MapX的基本操作、功能模块和实际应用案例。
通过本教程的学习,用户将能够熟练使用MapX进行地图制作和空间数据分析,为工作和研究提供有力的支持。
第一章:MapX概述1.1MapX简介MapX是一款基于Windows操作系统的地图制作和地理信息系统软件,由美国Intergraph公司开发。
MapX提供了丰富的地图制作和空间数据分析功能,支持多种地图投影和坐标系,可以处理各种类型的地理数据。
1.2MapX的特点(1)强大的地图制作功能:MapX提供了丰富的地图制作工具和符号库,可以制作高质量的地图。
(2)灵活的空间数据分析:MapX支持多种空间分析功能,如缓冲区分析、叠加分析、网络分析等。
(3)易于使用的界面:MapX的界面直观易用,用户可以快速上手。
(4)与其他软件的兼容性:MapX可以与其他GIS软件和办公软件无缝集成,方便数据交换和共享。
第二章:MapX基本操作2.1安装和启动用户需要从官方网站MapX安装包,并按照提示完成安装。
安装完成后,双击桌面上的MapX图标即可启动软件。
2.2地图制作(1)打开地图文件:“文件”菜单,选择“打开”,在弹出的对话框中选择地图文件(.mxd)。
(2)添加图层:“图层”菜单,选择“添加图层”,在弹出的对话框中选择需要添加的图层。
(3)调整图层顺序:在“图层”面板中,拖动图层上下移动,以调整图层顺序。
(4)设置图层样式:在“样式”面板中,选择合适的符号和颜色,为图层设置样式。
(5)添加标注和图例:“标注”菜单,选择“添加标注”,在地图上添加标注。
“图例”菜单,选择“添加图例”,在地图上添加图例。
(6)保存和输出地图:“文件”菜单,选择“保存”,将地图保存为.mxd文件。
“文件”菜单,选择“输出”,将地图输出为图片或PDF 文件。
Python课程第四阶段第3课:海龟作图(三)——Python+课件(共18张PPT)

02 课堂知识
02 课堂知识
实战练习:画等腰直角三角形
首先我们来一起想一想怎样画出一个三角形 1.划出一条直线 2.旋转60度
3.重复上面的操作3次 那堂知识
实战练习:画五角星
首先我们来一起想一想怎样画出一个五角星 1.划出一条直线 2.旋转36度
使用Python的turtle模块
Turtle motion (运动控制) 本节中包含了 运动控制 中常用的一些函数
turtle.goto(x,y) 画笔定位到坐标(x,y) turtle.forward(distance) 向正方向运动 distance 长的距离 turtle.backward(distance) 向负方向运动 distance 长的距离 turtle.right(angle)
Python第四阶段第3课
海龟作图-其三
课程目标
课程内容 Python中的海龟作图复杂图形实战。 课程时间 60分钟 教学目标 1、海龟作图的综合运用。 教学难点 海龟作图 设备要求 音响、A4纸、笔
• 课前回顾 • 课堂知识 • 基础任务 • 升级任务 • 创意练习
01 课前回顾
01 课前回顾
向右偏 angle 度 turtle.left(angle)
向左偏 angle 度 turtle.home()
回到原点 turtle.circle(radius, extent=None, steps=None)
画圆形 radius 为半径,extent 为圆的角度
turtle.speed(speed) 以 speed 速度运动
3.重复上面的操作5次 那么这样的话,我们来试试循环吧
02 课堂知识
02 课堂知识
大挑战:五彩图片
Hapmap用户指南

国际人类基因组单倍体图计划网站用户指南Gudmundur A. Thorisson1*, Albert V. Smith*, Lalitha Krishnan, and Lincoln D. Stein Cold Spring Harbor Laboratory, 1 Bungtown Road, Cold Spring Harbor, NY 11724翻译:Hejidong国际人类基因组单体型计划网()是获取国际人类单体型图计划部分基因分型数据的主要门户网站(Gibbs et al. 2003)。
在计划的第一阶段,来自全世界4个人群的270个样本共检测出了110多万个SNP位点(Consortium 2005)。
该网站向研究者提供用于数据分析的工具以及允许其下载数据以便进行本地分析。
本文提供使用这些工具的详细指南,包括:检索基因分型和基因频率数据,关联性研究中标签SNP的选择,单体型作图,相互关系的排列测验。
国际人类单体型图计划的目标是对人类基因组中常见遗传多态性作图和推断,以推动针对人类疾病遗传因素的研究。
该计划第一个重要的里程碑是在2005年春天,完成了对4个人群超过110万个SNP的基因分型检测,第二阶段准备完成另外460万个SNP的检测,并计划于2005年秋完成。
该计划的数据可在HapMap()网站无限制获取。
该网站提供数据集的批量下载,以及独有的交互数据浏览与分析工具。
自2003年9月向公众开放以来,来自100多个国家的研究者已经下载了HapMap数据集50万余次。
该网站目前每月处理超过3万余次的静态页面请求,其中1万4千次是批量下载请求,以及每月处理10万余次HapMap交互式浏览器登录。
本文介绍Hapmap网站及其用于查看、检索和分析数据的工具。
我们将展示如何进行一些有用和常见的任务,以及概述正在完善和开发中的新工具。
Hapmap网概述Hapmap网()主要由三个部分组成,均可从网页顶部的横标题进入。
Hapmap用户指南
国际人类基因组单倍体图计划网站用户指南Gudmundur A. Thorisson1*, Albert V. Smith*, Lalitha Krishnan, and Lincoln D. Stein Cold Spring Harbor Laboratory, 1 Bungtown Road, Cold Spring Harbor, NY 11724翻译:Hejidong国际人类基因组单体型计划网()是获取国际人类单体型图计划部分基因分型数据的主要门户网站(Gibbs et al. 2003)。
在计划的第一阶段,来自全世界4个人群的270个样本共检测出了110多万个SNP位点(Consortium 2005)。
该网站向研究者提供用于数据分析的工具以及允许其下载数据以便进行本地分析。
本文提供使用这些工具的详细指南,包括:检索基因分型和基因频率数据,关联性研究中标签SNP的选择,单体型作图,相互关系的排列测验。
国际人类单体型图计划的目标是对人类基因组中常见遗传多态性作图和推断,以推动针对人类疾病遗传因素的研究。
该计划第一个重要的里程碑是在2005年春天,完成了对4个人群超过110万个SNP的基因分型检测,第二阶段准备完成另外460万个SNP的检测,并计划于2005年秋完成。
该计划的数据可在HapMap()网站无限制获取。
该网站提供数据集的批量下载,以及独有的交互数据浏览与分析工具。
自2003年9月向公众开放以来,来自100多个国家的研究者已经下载了HapMap数据集50万余次。
该网站目前每月处理超过3万余次的静态页面请求,其中1万4千次是批量下载请求,以及每月处理10万余次HapMap交互式浏览器登录。
本文介绍Hapmap网站及其用于查看、检索和分析数据的工具。
我们将展示如何进行一些有用和常见的任务,以及概述正在完善和开发中的新工具。
Hapmap网概述Hapmap网()主要由三个部分组成,均可从网页顶部的横标题进入。
tutorial 中文手册2.2

感 谢本tutorial Manual 2.2翻译文档在许多网友的关心和支持下,得以翻译成功,在此对他们表示热烈地感谢:蝈蝈、fiona、kailinziv、Yan、杀毒软件、tiny0o0、timothy、prolee等关心TG手册翻译的热心朋友。
关于本文档内容说明:由于本手册由不同网友翻译,可能对某些概念有不同的理解,翻译可能不大一样,但决不影响理解,欢迎大家探讨。
再一次对各位网友的努力和汗水表示感谢!如有什么问题可以联系我:Mail:tiny0o0@注:本文档内容版权归X Y Z scientific company 所有,谢绝任何意图商用。
I、TrueGrid介绍True Grid是一套优秀的、功能强大的通用网格生成前处理软件。
它可以方便快速生成优化的、高质量的、多块结构的六面体网格模型。
作为一套简单易用,交互式、批处理前处理器,True Grid支持三十多款当今主流的分析软件。
True Grid是基于多块体结构(multiple-block-structured)的网格划分工具,尽管这个指南手册开始会提供一些介绍信息,新手还是强烈要求阅读用户手册(True Grid® User’s Manual)的前2章,用户指南和参考手册。
True Grid是几何和网格形成过程是分开进行。
曲面和曲线形成的方式有以下几种:内部产生,从CAD/CAE系统导出IGES格式导入TG,或用vpsd命令导入多边曲面。
块网格(block mesh)初始化然后通过各种变换与几何模型匹配形成最后的有限元模型。
True Grid网格划分过程:运用block命令初始化块网格;块网格部分会被删掉以使拓扑结构与划分目标对应;块网格部分通过移动,曲线定位,曲面投影等方法进行变换;网格插值、光滑和Zoning(控制边界节点分布)等技术可以用来形成更好的网格;块网格之间独立形成,然后通过块边界面(BB)和普通节点合并命令(指定容差范围内合并)将各块网格合并成完整的有限元模型。
GWAS分析详解

example.fam pop.phe qt.phe
PLINK tutorial, December 2006; Shaun Purcell, shaun@
The Truth…
Chinese Japanese Case Control 34 11 7 38 Case Control
gPLINK / PLINK in “remote mode”
Secure Shell networking
Server, or cluster head node
gPLINK & Haploview: initiating and viewing jobs
WWW
PLINK, WGAS data & computation
Load and filter binary PED file
Basic association analysis
~11 minutes
~5 minutes
PLINK tutorial, December 2006; Shaun Purcell, shaun@
/purcell/plink/ /mpg/haploview/
PLINK tutorial, December 2006; Shaun Purcell, shaun@
PLINK tutorial, December 2006; Shaun Purcell, shaun@
A simulated WGAS dataset Summary statistics and quality control Whole genome SNP-based association Whole genome haplotype-based association Assessment of population stratification
map的操作方法

map的操作方法一、map的基本概念。
1.1 map就像是一个超级收纳盒。
它可以把各种东西按照一定的规则放在不同的小格子里。
在编程的世界里呢,map是一种数据结构,它主要的功能就是存储键值对。
就好比你有很多双鞋子,每双鞋子都有一个对应的鞋盒,这个鞋盒就像是键,鞋子就是值。
1.2 它特别的灵活。
可以用来处理各种各样的数据关系。
比如说,你要管理一个班级学生的成绩,每个学生的名字就是键,他对应的成绩就是值。
这就像每个学生都有自己专属的“成绩小格子”,方便你随时找到想要的信息。
2.1 创建map。
创建map就像搭建一个新的收纳架。
在很多编程语言里,都有专门创建map的语法。
比如说在Java里,你可以用HashMap或者TreeMap。
这就好比你选择不同类型的收纳架,有的收纳架可能是按照顺序摆放东西(TreeMap类似这种有序的),有的就比较随意(HashMap就无序一些)。
创建的时候就像是你先把这个收纳架的框架搭好,准备往里面放东西。
2.2 添加元素。
往map里添加元素就像往收纳盒里放东西。
还是以学生成绩为例,你想把小明的成绩90分放进map里,你就把“小明”这个键和90这个值对应起来放进map。
这就好比你把写着“小明”的纸条贴在装着90分试卷的小盒子上,然后把这个小盒子放在收纳架上。
操作起来很简单,只要按照编程语言规定的语法来就行,就像按照收纳架的使用说明来放东西一样。
2.3 查找元素。
查找元素的时候呢,map就像一个贴心的小助手。
你只要告诉它你要找的键,它就能快速地把对应的值找出来给你。
比如说你想知道小明的成绩,你只要在map里查找“小明”这个键,它就能把90分这个值给你。
这就像你在收纳架上找贴着“小明”纸条的小盒子一样,只要纸条标记清楚了,就能很快找到。
三、map的优势。
3.1 高效性。
map查找元素的速度那是相当快的。
就像一个经验丰富的快递员,能够在众多包裹中迅速找到你要的那个。
不管你map里存储了多少个键值对,它都能快速定位。
hapmap 样本标号规则

hapmap 样本标号规则(实用版)目录1.HapMap 计划简介2.HapMap 样本标号规则3.样本标号的意义4.HapMap 计划对我国遗传学研究的贡献正文【1.HapMap 计划简介】HapMap(Haplotype Map)计划是一项旨在解析人类基因组多态性的国际合作研究计划。
该计划的主要目标是绘制人类基因组的单倍型图谱,以揭示不同人群、不同地域的遗传变异规律。
HapMap 计划的实施对于遗传病的研究、药物研发及基因治疗等方面具有重要的指导意义。
【2.HapMap 样本标号规则】在 HapMap 计划中,为了确保样本的唯一性和准确性,采用了一套严格的样本标号规则。
样本标号由 9 位数字组成,其中前 5 位数字表示个体的 ID 号,后 4 位数字表示该个体在该研究计划中的样本编号。
例如,个体 ID 号为 12345 的样本,其标号为 12345-0001。
【3.样本标号的意义】样本标号的设立有助于研究人员在海量的基因组数据中快速准确地找到所需样本。
同时,样本标号也便于研究人员对不同个体、不同地域、不同种族的遗传信息进行比较和分析。
【4.HapMap 计划对我国遗传学研究的贡献】HapMap 计划为我国遗传学研究提供了宝贵的资源。
通过参与 HapMap计划,我国科研人员获得了大量关于我国人群遗传特征的数据,为疾病基因的发现、药物研发及基因治疗等领域的研究提供了重要依据。
此外,HapMap 计划的成功实施也提升了我国在国际遗传学研究领域的地位和影响力。
综上所述,HapMap 计划中严格的样本标号规则有助于研究人员在遗传学研究中准确查找和分析数据。
map函数 python用法

一、什么是map函数在学习Python编程语言时,经常会遇到map函数的使用场景。
map 函数是Python内置的高阶函数,它接收一个函数和一个可迭代对象作为参数,然后返回一个新的可迭代对象,其中的每个元素都是将传入函数作用于原可迭代对象中对应元素的结果。
二、map函数的基本语法在Python中,map函数的基本语法如下所示:```map(function, iterable, ...)```其中,function是一个函数,iterable是一个或多个可迭代对象。
三、map函数的使用示例我们来看一个简单的示例,假设我们有一个列表,里面存储了一些数字,我们想要对这些数字进行平方运算,可以使用map函数来实现:```pythondef square(x):return x * xnumbers = [1, 2, 3, 4, 5]squared_numbers = map(square, numbers)```这里,我们定义了一个square函数,然后将它作为参数传入map函数,同时将numbers作为可迭代对象传入。
squared_numbers中存储了数字1至5的平方值。
四、map函数的灵活性除了可以使用普通函数作为map函数的参数外,还可以使用匿名函数(lambda表达式)来简化代码。
下面是一个使用lambda表达式的示例:```pythonnumbers = [1, 2, 3, 4, 5]squared_numbers = map(lambda x: x * x, numbers)```这里,我们直接使用lambda表达式来定义函数,避免了额外的函数定义,使代码更加简洁。
五、map函数的返回值map函数返回的是一个map对象,它是一个迭代器,可以使用list 函数将其转换为列表,或者直接用for循环遍历其中的元素。
下面是两种方式的示例:```pythonsquared_numbers_list = list(squared_numbers)for num in squared_numbers:print(num)```六、map函数的应用场景map函数在实际开发中有着广泛的应用场景,特别是在对数据进行批量处理时,可以大大简化代码的编写。
paintmap包用户指南说明书

Package‘paintmap’October14,2022Type PackageTitle Plotting PaintmapsVersion1.0Date2016-08-31Author Daniel GreeneMaintainer Daniel Greene<************.uk>Description Plots matrices of colours as grids of coloured squares-aka heatmaps,guaranteeing legible row and column names,without transformation of values,without re-ordering rows or columns,and without dendrograms.License GPL(>=2)RoxygenNote5.0.1NeedsCompilation noRepository CRANDate/Publication2016-08-3120:47:04R topics documented:paintmap-package (2)color_matrix (2)colour_matrix (3)inches_tall (3)inches_wide (4)lines_between_hm_and_labels (4)margin_lines (5)paintmap (5)Index61paintmap-package Plotting paintmapsDescriptionPlots matrices of colours as grids of coloured squares-aka heatmaps,guaranteeing legible row and column names,without transformation of values,without re-ordering rows or columns,and without dendrograms.DetailsThe function‘bhm’takes a matrix of colours(i.e.a character matrix of descriptions like red or hex-codes),and creates a plot using‘grid’graphics.Author(s)Daniel Greene Maintainer:Daniel Greene<************.uk>Examplespaintmap(matrix(heat.colors(9),3,3,dimnames=list(letters[1:3],letters[4:6]))) color_matrix Convert numeric matrix to color(character)matrixDescriptionGiven a numeric matrix,assign to each cell a color(character)value based on linearly interpolatinga given vector of colors.Usagecolor_matrix(x,colors=heat.colors(10))Argumentsx Numeric or logical matrix.colors Character vector of colors.ValueCharacter matrix.colour_matrix Convert numeric matrix to colour(character)matrixDescriptionGiven a numeric matrix,assign to each cell a colour(character)value based on linearly interpolatinga given vector of colours.Usagecolour_matrix(x,colours=heat.colors(10))Argumentsx Numeric or logical matrix.colours Character vector of colours.ValueCharacter matrix.inches_tall Get number of inches high a putative heatmap will beDescriptionGet number of inches high a putative heatmap will beUsageinches_tall(x,row_lines=1)Argumentsx Character matrix of coloursrow_lines Numeric value determining number of lines width each row of the heatmap should occupy.ValueNumeric value.4lines_between_hm_and_labels inches_wide Get number of inches across a putative heatmap will beDescriptionGet number of inches across a putative heatmap will beUsageinches_wide(x,col_lines=1)Argumentsx Character matrix of colourscol_lines Numeric value determining number of lines width each column of the heatmap should occupy.ValueNumeric value.lines_between_hm_and_labelsLines of space between the heatmap and row/column labelsDescriptionLines of space between the heatmap and row/column labelsUsagelines_between_hm_and_labelsFormatAn object of class numeric of length1.margin_lines5 margin_lines Lines of space at margins of paintmapDescriptionLines of space at margins of paintmapUsagemargin_linesFormatAn object of class numeric of length1.paintmap Plot paintmapDescriptionPlot paintmapUsagepaintmap(x,add=FALSE,...)Argumentsx Character matrix of coloursadd Add ink to current viewport....Other graphical parameters for the rectangles of the grid to pass to grid function gpar,in turn passed to grid function grid.rect.ValuePlots heatmap.Examplespaintmap(matrix(heat.colors(9),3,3,dimnames=list(letters[1:3],letters[4:6])))Index∗datasetslines_between_hm_and_labels,4margin_lines,5∗heatmappaintmap-package,2color_matrix,2colour_matrix,3inches_tall,3inches_wide,4lines_between_hm_and_labels,4margin_lines,5paintmap,5paintmap-package,26。
hapmap如何寻找SNP位点

entire data set. – Download the entire data set in bulk.
Finding HapMap SNPs in a Region of Interest
2c. Press “Next”
2b. Select “SNPs found in Exons – synonymous coding
SNPs”
4. Select output fields
2c. Press “Export”
4a. Choose among several pages of fields.
included, excluded, or design scores.
9: Generate Reports
9. Select the desired “Download” option and press
“Go” or “Configure”
Available Downloads: •Pairwise LD values •Allele & Genotype frequencies •Raw genotypes •Tag SNPs
6: Adjust Track Settings (cont)
6c. Adjust populations and display settings.
6d. PTurn on Tag SNP Track
7a. Activate the “tag SNP Picker” and press
3. Select “latest”
4. Choose the Dataset
haploview安装使用说明

Haploview 安装使用说明
131017
• Haploview是一个进行单倍型分析的一个软 件,该软件具有如下功能: • 1.连锁不平衡与单倍型分析 • 2.单倍型人群频率估算 • 3.SNP与单倍型关系分析 • 4.相互关系的排列测验 • 5.可以从HapMap上直接下载基因型信息
• Data file需要输入的数据格式要求6列
• 1列:家系的ID,如果你分析的是无关个体,建议 用自然序号1,2,3….来替代。 • 2列:个体的ID,做无关个体的研究则每个个体的 编号不能重复 • 3列,4列:分别表示父亲的ID 、母亲的ID,做无 关个体的研究,则第三列,第四列都赋值为0。 • 5列:个体的性别信息。1代表男性,2代表女性。 • 6列:个体的患病状态。0表示疾病状态未知;1 表示、软件: • 1) Haploview必须在JAVA环境下才能运行, 因此在安装该软件之前,必须先安装一个 “JAVA”
• 2)java安装完之后需要改变环境变量
Java_home变量:C:\Program Files (x86)\Java\jre6
Classpath变量:C:\Program Files (x86)\Java\jre6\lib\dt.jar; C:\Program Files (x86)\Java\jre6\lib\tools.jar; C:\Program Files (x86)\Java\jre6\bin
整理myd88有关snp位点,以及snp位点的position 将其copy至txt文档,命名为
HapMap使用手册

HapMap使用手册HapMap使用手册1、简介1.1 HapMap的定义和背景1.2 HapMap的目标和用途1.3 适用的研究领域和项目类型2、数据资源2.1 HapMap项目的数据库2.1.1 HapMap数据库的结构和组织 2.1.2 数据和访问方式2.2 数据文件的格式和内容2.2.1 SNP标记的命名和注释2.2.2 基因型数据的表示方法2.3 数据处理和分析工具2.3.1 常用的数据处理软件2.3.2 基因型数据的质量控制步骤2.3.3 数据分析的方法和策略3、数据挖掘与分析3.1 基本统计和可视化分析3.1.1 基因型频率和遗传多样性的计算3.1.2 主成分分析和群体结构检测3.1.3 基因型与表型关联分析3.2 基因组关联研究3.2.1 单个SNP分析和基因组关联图谱3.2.2 基因区间的关联分析和功能富集分析 3.3 跨种族比较和人种遗传学研究3.3.1 人种间的遗传差异和迁移历史分析3.3.2 超种系的遗传关系分析和进化研究4、数据管理和共享4.1 数据备份和存储4.1.1 数据备份策略和方法4.1.2 储存设备和系统要求4.2 数据共享和社群协作4.2.1 共享数据的许可和权益4.2.2 数据使用的合作和共同发表原则附件:1、数据文件样例2、数据处理工具的教程和示例代码法律名词及注释:1、数据共享许可:指在符合一定条件下可以共享数据的许可协议。
2、版权:指作品的创作权及其衍生权,受到法律保护。
3、个人隐私:指个人信息保护的法律原则,保护个人的隐私权利。
本文档涉及附件,详见正文中的附件部分。
本文所涉及的法律名词及注释已在正文中标注。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Release Notes
• Phase 1+2: Latest Release #24, October 2008 (NCBI build 36):
3.9 M unique QC+ SNPs -- > 1 SNP/700 bp /00README.releasenotes_rel24 – Added back chrX SNPs dropped in previous releases – Corrected allele flips from rel#23a
3: This exonic region has many typed SNPs. Click on ruler to re-center image.
3: Examine Region (cont)
Use the Scroll/Zoom buttons and menu to change position & magnification
3: Mouse over a SNP to see allele frequency table
As you zoom in Click to gothe SNP further, to details page display changes to include more detail
• PLINK format: /genotypes/2008-07_phaseIII/plink_format • HapMap3 sites:
Broad - /~debakker/p3.html Sanger - /humgen/hapmap3/ Baylor - /projects/human/
• Data merged with PLINK (concordance over 249,889 overlapping SNPs = 0.9931) • Alleles on the (+/fwd) strand of NCBI b36
Phase 3: Draft Release 1
samples 71 ASW 162 CEU 82 CHB 70 CHD 83 GIH 82 JPT 83 LWK 71 MEX 171 MKK 77 TSI 163 YRI QC+ SNPs 1,632,186 1,634,020 1,637,672 1,619,203 1,631,060 1,637,610 1,631,688 1,614,892 1,621,427 1,629,957 1,634,666 poly QC+ SNPs 1,536,247 1,403,896 1,311,113 1,270,600 1,391,578 1,272,736 1,507,520 1,430,334 1,525,239 1,393,925 1,484,416
alleles: C1 C2 POP allele freqs: A (80%) a (20%) B (60%) b (40%) SNP1 A/a SNP2 B/b
genotypes:
Person 1 Person 2 Person 3
AA BB phased haplotypes (C1/C2): A B A B
• EXCLUDED from QC+ data set:
– Samples with low completeness, and SNPs with low call rate in each pop (< 80%) and not in HWE (p < 0.001) – Overall false positive rate: ~3.2%
AA Bb
Aa Bb
A A
B b
A a OR A a
B b B
HapMap Glossary
• LD (linkage disequilibrium): For a pair of SNP alleles, it’s a measure of deviation from random association (i.e., no recombination). Measured by D’, r2, LOD • Phased haplotypes: Estimated distribution of SNP alleles. Alleles transmitted from Mom are in same chromosome haplotype, while Dad’s form the paternal haplotype. • Tag SNPs: Minimum SNP set to identify a haplotype. r2= 1 indicates two SNPs are redundant, so each one perfectly “tags” the other. • Questions? help@
* Population is made of family trios
Phase 3
• 11 panels & 1,115 samples
– 558/557 males/females – 924/191 founders/non-founders
• Platforms:
– Illumina Human 1M (Sanger) – Affymetrix SNP 6.0 (Broad)
3: Examine Region
Chromosome-wide summary data is shown in overview
Region view puts your ROI in genomic context Default tracks show HapMap genotyped SNPs, refGenes with exon/intron splicing patterns, etc.
1: Surf to the HapMap Browser
1a. Go to
1b. Select “HapMap phase 3”
2: Search for TCF7L2
2. Type search term – “TCF7L2”
Search for a gene name, a chromosome band, or a phrase like “insulin receptor”
Goals of This Tutorial
This tutorial will show you how to:
• Find HapMap3 SNPs near a gene or region of interest (ROI)
– – – – – – Visualize allele frequencies in HapMap3 populations Download SNP genotypes in ROI for use in Haploview 4.1 Identify GWA hits in the vicinity of ROI & visualize in the context of all chromosomes (karyogram) Add custom data onto the GWAs karyogram Add custom tracks of association data onto ROI Create publication-quality images
• Phase 3: Draft release #1 (NCBI build 36)
/genotypes/2008-07_phaseIII/00README.txt
– HapMap3 sites @ Broad Institute, Sanger Center and Baylor College
HapMap Project
Phase 1 Samples & POP panels Genotyping centers Unique QC+ SNPs Reference 269 samples (4 panels) HapMap International Consortium 1.1 M Nature (2005) 437:p1299 Phase 2 270 samples (4 panels) Perlegen 3.8 M (phase I+II) Nature (2007) 449:p851 Phase 3 1,115 samples (11 panels) Broad & Sanger 1.6 M (Affy 6.0 & Illumina 1M) Draft Rel. 1 (May 2008)
a b a b A B a b
A b
A B A B A b
OR
a B a B A B
A b etc…
High LD -> No Recombination (r2 = 1) SNP1 “tags” SNP2
Low LD -> Recombination Many possibilities
Basic Concepts
Resources at
HapMap3 Tutorial
Marcela K. Tello-Ruiz Cold Spring Harbor Laboratory
Basic Concepts
Parent 1 A a B A Parent 2 B b
X
b a
A B A B a b A B
Phase 3 Data
• HapMap format: /genotypes/2008-07_phaseIII/hapmap_format
* Excluded 1,527 SNPs with strandedness issues & 411 indels