激发三重态
激发三重态

发,跃迁回基态所发射的电磁辐射,称为荧光和磷光。现从分子结构理论来讨论荧光和磷光的产生机理。
每个分子中都具有一系列严格分立相隔的能级,称为电子能极,而每个电子能级中又包含有一系列的振动能级和转动能级。分子中电子的运动状态除了
电子所处的能级外,还包含有电子的多重态,用M=2S+1表示,S为各电子自旋量子数的代数和,其数值为0或1 。根据Pauli不相容原理,分子中同一轨道所
图14单重态系三重态激发示意图
处于分立轨道上的非成对电子,自旋平行要比自旋配对更稳定些(洪特规则),因此在同一激发态中,三重态能级总是比单重态能级略低。
图14.2为能级及跃迁示意图,其中S0、S1和S2分别表示分子的基态、第一和第二电子激发的单重态;T1和T2则分别表示分子的第一和第二电子激发的三重态
系间跨跃的速度较慢,经历的时间较长。
(5) 磷光发射(Phosphorescence emission,PE)——激发态的电子经系间跨跃后到达激发三重态,经过迅速的振动弛豫而跃迁至第一激发三重态
的最低振动能级,然后以辐射形式跃迁回基态的各振动能级,这个过程为磷光发射。磷光发射的跃迁仍然是自旋禁阻的,所以发光速度很慢。
以辐射的形式跃迁回基态(S0)的各振动能级,这个过程为荧光发射,发射的荧光波长为。由于经过振动弛豫和内部转移的能量损失,因此荧光发射的能量
比分子吸收的能量要小,荧光发射的波长比分子吸收的波长要长,即。第一激发单重态最低振动能级的平均寿命约为10-9~10-4s,因此荧光寿命也在
这一数量级。
程。图14.2表示分子激发和去活化的能量传递过程:
(1) 振动弛豫(Vibration relaxation,简写为VR)——当分子吸收光辐射(为图14.2中的λ1、λ2)后可能从基态的最低振动能级(V=0)跃迁到激发单重态
有机-过渡金属配合物的三重态发光问题

有机-过渡金属配合物的三重态发光问题吴世康;汪鹏飞【摘要】近年来,随着OLED器件的发展,有关有机-过渡金属配合物的三重态发光问题受到人们广泛的关注.这不仅是与电致发光器件效率的提高相关联,同时也是一个在光物理研究中值得深入研究的课题,文章对有机-金属化合物激发态的能级分裂,以及发光三重态亚态的零场-分裂(ZFS)等进行了讨论,对具不同电子构型中心金属离子,例如八面体结构与平面四方形构型结构等与它们光物理行为间的关系进行了讨论.特别对这类体系的发光三重态在自旋轨道偶合作用(SOC)和构型相互作用(CI)的影响下,导致单重态与三重态的混合和促进从最低三重态到基态的发光跃迁进行了讨论.%In recent years, the emission of triplet state of organic-transition metal complex at tracted extensive attentions with the development of OLED devices. It not only concerns with the increase the device efficiency, but also with some problems deeply in the field of photo physics. In this article, the energy level splitting of organic-metallic compounds, and the zero field splitting (ZFS) of emissive triplet state are discussed briefly. Simultaneously, the rela tionship of electronic configuration of centered metallic ions, such as the octahedral and square planar coordination and their photo-physical behaviors are also discussed. Especially, the emis sion state of these compounds, under the influence of spin-orbitalcoupling(SOC) and configu ration interaction (CI) which can induce the admixture of singlet and triplet state, then to im prove the emissive transition from the lowest triplet state to ground state, has also been dis cussed.【期刊名称】《影像科学与光化学》【年(卷),期】2011(029)002【总页数】18页(P81-98)【关键词】有机-过度金属配合物;零场-分裂(ZFS);自旋-轨道偶合;光物理行为;三重态的发光【作者】吴世康;汪鹏飞【作者单位】中国科学院,理化技术研究所,北京,100190;中国科学院,理化技术研究所,北京,100190【正文语种】中文【中图分类】O64自上世纪后期有机-电子学,特别是有机发光二极管(OL ED)得到迅速的发展以来,为提高器件的电-光转换效率和寻找新型三重态发光材料,人们对有机-过渡金属配合物,特别是它们的光物理性能进行了大量研究[1,2].众所周知,化合物的光致发光性质决定于分子轨道(MO)中那些被称为“前线轨道”的性质,分子最低激发态与基态间的跃迁对应其发光.同样,在电致发光过程中,器件的发光也同样取决于其分子轨道的性质,当然,二者激发态形成与“布居”过程有所不同.有机分子在受光激发下,其HOMO轨道上的电子跃迁到LUMO轨道形成激发态,导致其基态与激发态的电子构型不同.基态S0具有π2构型,而激发态π1π*1的构型则可有单重态(S1)和三重态(T1)之分,而其中的T1态又可分裂为3个亚态(substate).从上述两种构型的电子-电子相互作用考虑列出其能级图,通常可以看到存在两种分裂,一种是单重态-三重态间的能级分裂ΔE(S1-T1),另一种是T1态的分裂,亦即所谓的零场-分裂(ZFS).如下图所示[3].从上图左侧简单的HOMO-LUMO图中可以看到,所有来自π1π*1的4个态,具有相同的能量,它们是简并的(degenerate).然而从光谱研究知道,化合物分子被激发后,其激发单重态与三重态间的分裂甚大,例如,这里要讨论的配体化合物分子,其分裂能约为104cm-1(~1.24 eV).因此,在考虑与HOMO-LUMO激发相关联的电子-电子相互作用时,就可出现如上图右侧所示的能态图,可看到上述的两种分裂.对目前广泛应用于OL ED的三重态发光有机-过渡金属配合物,例如Ir(ppy)3和Pt(thpy)2等,考察它们的各种分子轨道,以及不同分子轨道间的跃迁,在配合物分子中可以有:以配体-中心(LC)的π-π*跃迁为特征的激发;以金属-中心(MC)的d-d*跃迁为特征的激发;金属-配体的电荷转移(MLCT)过程d-π*为特征的激发……图2列出的是有关金属配合物的HOMO-LUMO能级图,以及它们可能存在的能态模式.图2(a)中列出的为分子单个π、d和π*的轨道,以及其间可能存在的相应跃迁,如配体分子LC的π-π*跃迁,金属与配体间的电荷转移(MLCT)跃迁等,这里假设d*-轨道处于较高的能位,因此在一般情况下不存在d-d*的特征激发.右图(b)中列出的是与跃迁相关的能态,它包括由MLCT跃迁(1,3d-π*)形成的单重态与三重态能态,以及由LC(1ππ*)跃迁形成的单重态与三重态能态.由于三重态存在着3个亚态,因此总共可以得到8个能态,即单重态的1LC和1MLCT两个能态,以及3个3LC和3个3MLCT的亚态.值得注意的是,这些态都受构型的相互作用(CI),以及自旋-轨道偶合(SOC)的作用,即发生了实质上的量子力学混合,其中SOC的作用还引起了三重态的零场-分裂.因此,经过这种混合,列出的这些1,3LC以及1,3MLCT等能态不能再以一种“纯”的三重能态或单重能态来理解.上述的这种因激发态布居而引起的电子-电子相互作用(或在电激励的情况下,电子与空穴的逐步接近,电子与空穴波函数重叠所引起的相互作用)可导致单重态与三重态间的分裂ΔE(S-T)(~104cm-1),这是一种量子力学交换相互作用的结果(其值约为交换积分的2倍).交换积分有如下的表达形式[4]:式中的π和π*为HOMO与LUMO的波函数,r1与r2代表电子坐标,而r12则为两个电子间的距离.交换相互作用是一种量子力学效应,它与电子波函数的重叠大小相关,因此要考虑其中的自旋相关性.这意味着具有相反自旋取向的两个电子(单重态)有着比具有相同自旋取向的两个电子(三重态)有更多相互接近的机会,而三重态的两个电子则有相互避开的趋势,使三重态的能量较低.通过上面的讨论,可以定性地得到有关交换相互作用和波函数重叠的一些重要结论: 1.随着π和π*轨道共扼长度的增加,电子在(离域)分子内分布的区域变大,体系的交换积分K值变小.例如,对于有机分子从“苯”到“萘”、再到“蒽”,它们单重态-三重态间的分裂能ΔE(1π π*-3π π*)不断降低——从苯[5]的~18000 cm-1(约2.2 eV)降低到萘[6]的~12300 cm-1(~1.5 eV),再降到蒽[6]的10500 cm-1(~1.3eV).2.当分子轨道处于分子内不同空间区域时,分子轨道间的重叠变得很小,其交换积分也相应变小.例如:带有杂原子氧的有机分子——二苯酮的n-π*跃迁,是从二苯酮中氧原子上的n-电子向π*-LUMO轨道的跃迁,由于两者处于分子内的不同空间,其单重态与三重态间的分裂能ΔE(1nπ*-3nπ*)仅为1750 cm-1(0.22 eV)[7],如图3所示.类似于稠环芳香化合物ΔE值随共轭程度增大而不断减小的现象,也可以在具有不同共轭程度的高分子体系中观察到.如图4所示,随着齐聚物共轭程度的变大,ΔE(S1-T1)值逐步减小,说明其间的波函数重叠的程度也在不断减小.上述现象在有机-过渡金属配合物分子中也可作相应考虑.即:如有小量金属d-轨道或MLCT特征混入到配体中心(LC)态时,会因增大波函数的空间范围而使其交换积分减小,同时,也会引起被扰动的1LC(1π π*)和3LC(3π π*)间的单重态与三重态的分裂减小.例如:对于自由配体化合物2-噻吩吡啶(2-H(2-thpy))[9],其分裂能约在104cm-1(1.24 eV)左右,而其金属配合物二(2-噻吩吡啶)钯(Pd(2-thpy)2)[10]的ΔE(1LC-3LC)减小至5418 cm-1.在对有机-过渡金属配合物进行讨论之前,需要简单地对两种最常见的金属离子配位场作一介绍[11].下图左侧(A)为具有Oh对称的八面体配位场d-轨道的分裂,它使中心金属离子从原有能量简并的5个d轨道分为两组,即能量较高的eg和能量较低的t2g.右图(B)则为平面四方形配位,它可看作为八面体结构的上下伸长发生畸变的结果.图6中列出的是八面体与平面四方形两种构型简化了的能态模型,可以看出两者间存在着巨大的差异.在八面体的配位中,包括有配体的3π π*能级,以及在t2g中d1、d2轨道电子与π*间跃迁得到的能级1d2π*和3d1π*;平面四方形的配位中则包括处于高能位占有轨道与π*间的电子跃迁,得到单重态与三重态1π*和3π*能态,以及低能位占有轨道与π*间跃迁得到的1,3(dxzdyz)π*等.需要特别指出的是,不同轨道间的跃迁存在不同的自旋-轨道偶合(SOC)效应,以及不同的零场分裂(ZFS).从图6中可以看出,在八面体构型的条件下,无论是SOC或ZFS 都是较大的,而从最低三重态T1到基态S0的衰减时间也比平面四方形的短,充分说明这种具有八面体构型的发光配合物更适合在OL ED中应用.更为详细的情况,将于下文中做进一步说明.在图1和图2中还可看到,其中的三重态可分裂为3个亚态,这对将在OLED应用的有机-过渡金属化合物颇为重要.由于这一分裂系在零-磁场下发生的,因此被称为零场-分裂[12,13](ZFS).有机分子三重激发态3π π*的零场分裂,主要是经三重态中两个非成对电子的自旋-自旋相互作用而诱导发生的,由这种相互作用诱导而得的ZFS值约在0.1 cm-1(约1.2×10-5eV)的水平.而对有着较大配体中心3LC态的有机-过渡金属配合物,其3LC态的ZFS值也处于相同量级,例如[Rh(bpy)3]3+[14,15]所测得的ZFS值为0.125 cm-1(约1.55×10-5eV),而Pd(qol)2[16]的也约为0.25 cm-1(约3.1×10-5eV).这些有机-过渡金属化合物3LC(3π π*)态的ZFS值不易受外来微小扰动的影响,与“纯”的有机分子相类似.但要指出的是,由于金属的存在,它们仍可显著地增大其激发态的辐射和/或非辐射衰减的速度,即SOC的作用仍然有效.例如,它可使激发单重态向三重态的布居增大几个数量级,从而使ISC量子产率接近100%.此外,它也使一些以3LC态为发光机制的有机-金属配合物Pd(thpy)2和Pt(qol)2的ISC弛豫寿命分别快达τISC=800 fs[10]和500 fs[17].更为重要的是,它们T1→S0的辐射衰减速度也比“纯”有机分子快几个数量级,这意味着这类化合物的辐射衰减速度可超过非辐射失活过程,导致其高的发光量子效率.有机-过渡金属配合物的MLCT最低激发三重态同样也存在零场-分裂,其ZFS值的大小可作为这类配合物能否用作发光材料的一个重要量度.对于一种适用于OL ED的发光化合物,其ZFS值应处于10 cm-1(1.2×10-3eV)的水平,更大时更好.上文对图2的讨论中已经指出,存在有MLCT跃迁的体系情况比较复杂,体系中3LC、1LC、3MLCT以及1MLCT各态间的量子力学混合及相互影响,决定其最低三重态的性质,以及其在OLED中得到应用的可能.研究表明,一个化合物当它具有“纯”的3LC发光态时,并不适宜作为OL ED的发光物种,而具有3LC/3MLCT混合态的配合物则是一种很好的候选者.例如,一种主要为π-特征而有d-轨道混入的HOMO轨道,即可有πd→π*跃迁的混合体系出现,可因金属5d-轨道的存在引起自旋-轨道的偶合(SOC)作用.从图2中就可看到,由前线轨道,如HOMO-1、HOMO、LUMO 等所形成的电子(跃迁)态,以及由存在的各种混合和相互作用所引起的这些电子态性质的变化,包括MLCT的特征、SOC的影响、三重态的分裂、以及辐射衰减的速度等.总的说来,图2中列出的3个轨道模型中存在着两种激发,即相应分子轨道间的MLCT跃迁和LC跃迁.而所构成的8个能态都是通过构型相互作用(CI)和自旋轨道偶合作用(SOC)经受了实质上的量子力学混合,因此,它们也就不能再作为各种“纯”的能级分类.应当指出,上面所讨论的是有机-过渡金属配合物的一种简化模型,但它仍有重要的指导意义,并得到了一些一般性结论:1.图2的HOMO/LUMO模型中,并无单重态与三重态之分,所列出的HOMO-1与HOMO的相对位置,也不能直接预示相应能态的能级次序.从HOMO和HOMO-1的次序似可以建议3MLCT为最低能态,但图中示出的3LC却为最低能态,这可能与HOMO与HOMO-1间的能量差比交换积分间的能差要小有关.2.有机-过渡金属配合物简化了的模型中出现的能态数目仍相当多,如图2中所示,该模型就有两个单重态(1LC、1MLCT)和2×3=6个三重态亚态(3LC、3MLCT)共8个能态之多.3.不同电子构型间通过电子-电子相互作用以及SOC的作用,可诱导8个能态间发生量子力学混合,特别是1MLCT与3MLCT间的SOC作用,以及3MLCT与3LC间的构型相互作用(CI),可大大改变最低三重态的性质.处于最低能态的3LC态零场-分裂为亚态,导致这些态与基态间的跃迁变为允许,从而使发光衰减时间减小,光致发光量子产率增大,并导致发生光谱位移等.从上面的讨论可以看出,要定量地描述这类有机-过渡金属配合物的特征,即使采用了比上述简化模型更为复杂、涉及更多能态的模型体系,也不能认为就完全可行.然而,上述方法仍不失为一种有效的手段,对这类化合物的特征做出某种判断,同时对涉及不同分子轨道的空间分布、最终能态的状况等提供有益的信息,并对它们所具有的某些趋势性变化提供可参考的内容,以此来预测化学结构的变化、电子给体与受体取代基团的引入、共扼长度的变化、配位场强度及氧化还原电位的变动等对其光物理性质的影响.有关这类体系光物理行为定量化的工作主要基于从头算起的方法,如用时间依赖的密度泛函数理论[19.20](TDDFT)或CASSCF/CASPT2程序[21]等.由此所得的有关跃迁能、单重态-三重态振子强度、低阶三重态的数目、以及对ZFS 值的预测等,虽说多数还不能十分信赖,甚至还不能全部测出,但这些计算结果仍对光物理性质变化趋势的了解有所帮助.如果将SOC效应也同时加以考虑,预测将变得更加可信.对这类有重要应用价值复杂分子的理论计算存在一定的困难,如果能找出该类分子的某种结构参数,据此方便地对化合物的物理特性,特别是光物理特性作出某种经验性的次序排列,将大大有利于这类化合物的实际应用.这里指出的ZFS值[ΔE(ZFS)]就是一种可预测有机金属化合物重要光物理行为有价值的参数,它可用以直接显示1,3MLCT(1,3dπ*)组分在相应三重态中的重要程度.具体地说,如果ΔE(ZFS)值大于1—2 cm-1者,表明该体系三重态亚态波函数与1,3dπ*组分间存在着一定的关联;特别大的ΔE(ZFS)值则代表在相应三重态中存在重要的1,3MLCT组分.这种不同的关联或混合程度将大大影响三重态的发光,以及其它的光物理行为.图7列出的是处于高阶能态的MLCT对三重态的影响.这里所讨论的有机-过渡金属化合物的发光都是从最低激发三重态T1到电子基态S0跃迁产生的发光,在无自旋-轨道偶合SOC存在时,该跃迁是高度禁阻的,由于第三周期过渡金属化合物的存在,通过SOC作用使最低三重态到基态的跃迁成为可能.SOC作用还能诱导一些其他的重大效应,如三重态分裂为三个亚态,以及提高辐射衰减的速度等,这些后继效应的出现使化合物的磷光量子产率可达100%,而且使化合物的某些其他光物理性质也产生明显的变化.有机-过渡金属配合物最低三重态的发光,取决于三重态T1亚态I、II、III与基态S0间跃迁的特征和允许程度,而这种允许度又受控于处在高阶的1MLCT单重态的参与与否.图7中列出的1MLCT与T1亚态II、III间的短虚线表示其间的关联,而正是这种关联使三重态的两个高位亚态II、III有着较高的单重态混合程度,而最低三重态亚态I 由于不含明显的单重态组分,通常表现为“纯”的三重态特征,于是也就有着较长的寿命[22-24].十分有趣的是,只要有少量的单重态混入3LC(3π π*)态,更准确地说是有单重态混入到三重态亚态,就可增大后者的辐射跃迁几率.例如配合物Pd(thpy)2的发光衰减时间就可缩短到235μs(平均寿命),其中的τI=1200μs,τII=235μs,和τIII=130μs[25].对于Pt(thpy)2,因它有较大的MLCT混入最低3LC态,致使其衰减时间相对于纯的有机化合物可小6到7个数量级,其发光寿命短到1μs[10].图8中列出了一系列过渡金属配合物的ZFS值,它们相应的化学结构也列出于图9中[19,26,27].从图8所列不同-过渡金属配合物的ZFS值可以看出,配合物ZFS值的变化范围从最小到最大可跨越3—4个数量级.应当指出,图中所列不同配合物的ZFS序列与发光三重态中MLCT组分的增大有着密切关系,亦即配合物的ΔE(ZFS)值小于1 cm-1时,体系主要是以配体为中心的3LC(3π π*)发光态;而ZFS值处于中间范围时的配合物,其三重态就不再看作为“纯”的3LC态,其原有三重态特征3LC(3π π*)已显著地被1,3dπ*的混入所扰动;至于那些ZFS值大于50 cm-1的配合物,通常可被称为3MLCT发光化合物.常见的MLCT发光化合物有在上面列出的诸多配合物中可发现一个有趣的事实,即在含Pt(II)的准-平面四方形构型的有机金属化合物中,未发现具有大的ΔE(ZFS)值的(如大于40 cm-1)配合物,而对于准-八面体,如Re(I)、Os(II)、Ir(III),甚至第二周期过渡金属Ru(II)的配合物,它们都有很大的ΔE(ZFS)值,这一现象可以用后者易于发生强的SOC效应加以解释.对于准-八面体的配合物,如图9中所列出的配合物[31],其最低三重态3MLCT(3dπ*)可以和t2g系列中不同d-轨道电子跃迁形成的单重态1MLCT(1dπ*)间经历强的SOC作用,同时也与那些具有扭曲Oh对称的化合物最终轨道能量相接近.但对于准-平面四方形构型的Pt(II)配合物,其最低1MLCT和3MLCT态均来自相同的dz2→π*激发,而这些有着相同起始态间的SOC作用通常可以忽略.这是由于发生这种有效SOC的一个重要条件是在作用的二态之间需有不同的起始d-轨道存在,对于平面四方型的配合物,如不考虑处于高能位的dz2为起始激发的能量轨道,其候选者应是dxz和dyz轨道,它们在能量上则与dz2相远离,如从轨道的能量上加以估计,其间的差可以达到4000 cm-1(0.5 eV).因此,这一用于支配SOC有效性的能量分母,就要比八面体化合物的大许多,致使SOC的作用效果变差.对于这些有关八面体以及平面四方形构型的1,3MLCT相互作用的内容,均列于前面的图5中.另一点值得注意的是:MLCT与LC的混合途径,除了SOC外,还有所谓的构型相互作用(CI)作用途径.这种CI也是一种电子-电子间的相互作用,它能诱导3MLCT(3dπ*)与3LC(3π π*)的混合,而这种混合还可修饰最低的LC三重态(3π π*),使它能与3dπ*间发生间接的混合.因此,准-八面体的SOC途径与准-四面体配合物间的差别不仅只是3MLCT(3dπ*)态所表现的,还在3LC(3π π*)态上有所反映,准-八面体的ΔE(ZFS)值和辐射衰减速度都比准-平面四方形大.这种特殊的偶合途径使准-八面体发光物种有着相对短的发光衰减时间,使它比平面四方形材料能更好地应用于OLED之中.从前面列出的大量不同有机-过渡金属配合物的ZFS值可以看出,ZFS参数的大小范围可有2000倍之差,应当说这是由于LC的最低三重态3π π*与MLCT单重态和三重态间的SOC相互作用而引起最低三重态亚态波函数发生了重要变化的结果.对于多数化合物来说,其最低的发光三重态都是以LC(π π*)为其“原始能态”,而其ZFS值的增大则是上述不同相互作用的影响使三重态不断增大了与MLCT组分间关联的结果.大的ZFS值还可使原来简并的三重态发生较大的分裂,使共振式的转移成为可能,进一步提高了跃迁的允许程度.从图8中还可看到,准-平面四方形构型的金属配合物(如含Pt(II)配合物)的ZFS值均小于40 cm-1,有关它们的发光问题,可结合下列Pd(thpy)2和Pt(thpy)2配合物的发光状况加以说明,二者显示出十分不同的T1态与MLCT态的特征结合.上面指出的所谓SOC有效性,对于这类化合物的光物理性质十分重要,有效的SOC对增大态与态间的扰动起到重要的作用.Pd(II)和Pt(II)化合物所涉及的4dπ*和5dπ*SOC常数不同,因此,两种化合物的光物理行为有明显差别[10].图11中列出了这些化合物的能态变化特征,并与典型的有机分子的低阶3π π*和1π π*态相比较. 从图11可以清楚地看出,与纯的有机化合物相比较,配合物因MLCT的混入使单重态-三重态的分裂变小.即从配体有机分子的ΔE(S1-T1)~104cm-1量级(约1.24 eV)减小到有机-金属化合物Pd(thpy)2的5418 cm-1,其主要原因可认为是有小量MLCT(4dπ*)混入到LC(π π*)所致.对于有重要MLCT(5dπ*)混入3LC(π π*)态的Pt(thpy)2配合物,其电子波函数可扩展到配合物所涉及的配体与金属两者较大的空间中,因此,其ΔE(S1-T1)值降低到更低的3278 cm-1水平.从图11还可以看到,随着SOC诱导所引起的MLCT扰动的增大,三重态的零场-分裂(ZFS)从有机配体分子的0.1 cm-1增大到Pd(thpy)2的0.3 cm-1,到Pt(thpy)2增大至16 cm-1;相应的磷光发射寿命也从有机化合物的10 s减小到Pd(thpy)2的200μs,再到Pt(thpy)2的1μs;而系间窜越(ISC)寿命也同样从100 ns降低到800 fs,再到50 fs.所有这些光物理行为的变化都与“旋-轨偶合”的诱导增大了最低三重态中MLCT的扰动有关,而其中ZFS的数值变化则为一最直观的指标,可用于表示有机-过渡金属配合物作为三重态发光材料的优劣程度.最后要提到的是有关ZFS值的测定,除ESR方法外,最常用的方法是采用低温下激光激发的光学测定法,采用低温与激光的目的是为了消除光谱的不均匀加宽.图12列出的是在不同温度下所测得的结果[32].在常温下测定时,会因声子效应而引起对光谱的掩盖.从图12可以看到,随着温度的下降,这种掩盖效应可不断减小,图左侧的ZPL为零声子线(Zero Phonon line,ZPL),因此,一般的测定均应在零声子线的温度以下进行.光学法测定的装置[33]如图13所示.图中1为样品,2为样品的氦冷冻装置,3为氩离子激光器,4为压力扫描Fabry-Perot干涉仪,5为DFS-12光谱仪,6为光电倍增管,7为光子计数装置,8和9均为电磁快门,10为电子快门的控制系统,11为频率选择干涉仪.有机分子三重态能级的三重简并,严格地讲,在无外磁场存在下因自旋-自旋及自旋-轨道相互作用以及周围晶体场的影响而消失.在零场下自旋亚层(sublevel)的分裂可通过ZFS参数的D和E加以表征,但分子三重态的主要参数ZFS值很小,通常在0.01—0.1 cm-1量级,它可通过ESR法予以测定.采用光学光谱法来测定ZFS时,会因这种光谱的带宽大于所测定的ZFS值约几个数量级而存在困难,因此用光谱法来观察ZFS,直到上一世纪的70年代才得以实现.从图14中可以看到,只有当温度下降到1.35 K时方能对三重态亚态I和II加以分辨,并测出其能差[34].在前言中已经指出,近年来随着电致发光器件的长足发展,有关三重态发光问题得到广泛关注,一系列高水平的综述性论文相继出现,如文献[3]和[35]等.本文就是在这些文章的基础上对有关有机-过渡金属配合物的三重态发光问题,特别是有关三重态的零场-分裂(ZFS)与化合物光物理性质的关系进行简要的介绍.采用这种以ZFS值的大小排列的方法(见图8),可对在不同场合下选择应用的有机-金属发光化合物带来很大的方便,而且这种似乎是经验性的方法事实上也存在着深刻的理论依据.文章还对有关ZFS值的测定方法做了简单的介绍.需要特别指出的是,在有机-金属配合物的三重态发光问题中,尚有许多不够清晰的问题,需要我们进一步的努力.【相关文献】[1] Baldo M A,Thompson M E,Forrest S R.Phosphorescent materials for application to organic light emitting devices[J].Pure A ppl.Chem.,1999,71:2095.[2] Adachi C,Baldo M A,Thompson M E,Forrest S R.Nearly 100%internal quantum efficiency in an organic light-emitting device[J].J.A ppl.Phys.2001,90:5048.[3] Yersin H,Finkenzeller W J.Triplet emitters for organic light-emitting diodes:basic properties,inHighly Ef f icient OL EDs with Phosphorescent Materials[M].Ed.Hartmut Yersin,WILEY-VCH Verlag GmbH&Co.Weinheim,2008.[4] Klessinger M.Elektronenstruktur Organischer Molekule[M].VerlagChemie,Weinheim,1982.[5] McGlynn S P,Azumi T,Kinoshita M.Molecular S pectroscopy of the TripletState[M].Prentice-Hall,Englewood Cliffs,New Jersey,1969.[6] Turro N J.Modern Molecular Photochemistry[M].Benjamin-Cummings,MenloPark,Calif.1978.31.[7] Mataga N,Kubota T.Molecular Interactions and Electronic S pectra[M].Marcel Dekker,NewYork,1970.187.[8] Kohler A,Bassler H.Triplet states in organic semiconductors[J].Materials Science and Engineering R,2009,66:71-109.[9] Maestri M,Sandrini D,Balzani V,Chassot L,Jolliet P,A.von Zelewsky.Luminescence of ortho-metallated platinum(ii)complexes[J].Chem.Phys.Lett.,1985,122,375.[10] Yersin H,Donges D.Low-lying electronic states and photophysical properties of organometallic Pd(II)and Pt(II)compounds.Modern research trends presented in detailed case studies in:Transition Metal and Rare Earth Compounds.Excited States,Transitions and Interactions,Vol.II S pringer-Verlag Berlin,ed.Yersin H.,Top.Curr.Chem.2001,214:81.[11]Jolly W L.Modern Inorganic Chemistry[M].McGraw-Hill,New York,1984.[12]Yersin H,Humbs W,Strasser J.Characterization of excited electronic and vibronic states of platinum metal compounds with chelate ligands by highly frequency-resolved and time-resolved spectra.Electronic and Vibronic S pectra ofTransition Metal Complexes,Vol.II S pringer-Verlag Berlin,ed.Yersin H.,Top.Curr.Chem.1997,191:153.[13] Yersin H,Humbs W.Spatial extensions of excited states of metal-organicPt(II)compound in a Shpolskii matrix[J].Inorg.Chem.,1999,38:5820.[14]Glasbeek M.Excited state spectroscopy and excited state dynamics of Rh(III)andPd(II)chelates as studied by optically detected magnetic resonancetechniques[J].Top.Curr.Chem.,2001,213:96.[15] Komada Y,Yamauchi S,Hirota N.Phosphorescence and zero-field optically detected magnetic resonance studies of the lowest excited triplet states of organometallic diimine complexes.1.Rhodium bipyridine and rhodium phenanthrolinecomplexes[Rh(bpy)3]3+and[Rh(phen)3]3+[J].J.Phys.Chem.,1986,90:6425.。
三重态激子的演化机制与应用探索

三重态激子的演化机制与应用探索1.引言1.1 概述概述三重态激子是指在固体材料中形成的一种电子-空穴对和激子之间的复合物。
这种激子具有特殊的电子结构和能量级别,使其在光电子学、能源和材料科学领域中具有广泛的应用前景。
在过去的几十年里,科学家们对三重态激子的性质和演化机制进行了广泛的研究。
通过实验和理论模拟,我们可以了解到三重态激子的形成和衰变过程,以及其在光学和电学性质上的独特特性。
本文将首先介绍三重态激子的定义和特性,包括其形成机制和能量特征。
接着,我们将重点探讨三重态激子的演化机制,包括电子-空穴对的形成、激子的激发和衰变过程等。
这些研究在进一步理解三重态激子的行为和性质方面具有重要意义。
在结论部分,我们将对三重态激子演化机制的理解与应用进行总结和讨论。
我们将重点介绍三重态激子在光电子学和能源领域的潜在应用,如光电转换、光催化和能量传输等。
此外,我们还将探讨未来研究的方向,包括材料设计和制备、激子动力学的研究等,以期进一步推动三重态激子相关领域的发展。
通过对三重态激子的演化机制和应用进行全面的探索,我们可以更好地理解和利用这一复合物的特性,为光电子学和材料科学领域的发展做出贡献。
希望本文能够为关于三重态激子的研究和应用提供一定的参考和启示。
文章结构是一篇文章的骨架,它需要清晰地展示整篇文章的逻辑顺序和章节安排。
对于本文《三重态激子的演化机制与应用探索》,文章结构如下:1. 引言1.1 概述1.2 文章结构1.3 目的2. 正文2.1 三重态激子的定义和特性2.2 三重态激子的演化机制3. 结论3.1 对三重态激子演化机制的理解与应用3.2 未来的研究方向在引言部分之后,我们进入正文,其中包含了两个主要部分。
第一部分是对三重态激子的定义和特性的介绍,这部分将对三重态激子进行全面而准确的阐述,包括其定义、形成条件、稳定性等方面的内容。
第二部分是对三重态激子演化机制的探究,这部分将重点讨论三重态激子在化学反应中的形成、变化和衰变机制,以及与其他物质相互作用的过程。
氮氧自由基对苯甲酮三重激发态猝灭反应的TR_EPR研究

飞, 卞伟伟, 朱光来, 许新胜郑 ( 安徽师范大学 原子与分子物理研究所,安徽 芜湖 241000)摘 要: 利用 T R-EP R 技术并结合瞬态吸收光谱方法研究了乙二醇( EG ) 均相溶液中稳态自由基TEMP O 对光诱导苯甲酮三重激发态 3 BP * 的猝灭反应,分析了其中的光化学反应中间体,讨论光解自由基的 C I DEP 机理,测量了 TE MP O 对3 BP *的猝灭速率常数. 结果表明,EG 均相溶液中 BP * 与 溶剂分子 E G 之间存在氢原子转移反应,生成苯甲酮中性自由基 BP H ·和乙二醇羰自由基 E g l [-H ]· . E + E / A 的 C I DEP 信号说明 TM 机理是体系 C I DEP 的主要机理. 另外,T E MP O 对3 BP * 也有明 显的猝灭作用,猝灭速率常数为 1. 40 × 107 L . m o l - 1 ·s - 1 . 关键词: 苯甲酮; TE MPO ; T R-EP R; 猝灭动力3 + 2; O643. 12中图分类号: O562. 文献标志码: A文章编号: 1001 - 2443( 2014) 05 - 0449 - 05 苯甲酮 ( b e n z o ph e n o n e ,BP ) 是一种常用的药物、香料、杀虫剂中间体,广泛使用于香水和皂用香料 中[1 - 3]. 苯甲酮也是一种典型的紫外线吸收剂,能够有效吸收 290 - 360nm 的紫外光,因此化妆品行业中常 把它用于生产防晒霜和防晒膏. 然而,进入人体的苯甲酮在紫外光的照射下( 皮肤中就有这样的过程) 会形成高氧化性的激发三重态 3 BP * ,高氧化性的激发三重态 3 BP *有对人体造成伤害的风险. 一方面,高氧化性的激发三重态3 BP *可能会通过夺电子或质子直接攻击蛋白质、DN A 造成生物损伤,另一方面,它们也可作为基 态氧3 O 的敏化剂形成1O 间接氧化蛋白质、DN A 而造成生物损伤[4 - 8]. 正是由于激发三重态3 BP * 有对生 2 2 组织造成损伤的可能,使得研究激发三重态3 BP * 的猝灭反应成为一项有意义的课题.很多研究表明,氮氧自由基( 2,2,6,6-四甲基-1-哌啶酮,TE MPO ) 是光敏分子激发态自旋极化的有效猝 灭剂,TE MP O 与光敏分子激发单重态或激发三重态之间通过磁相互作用使前者产生自旋极化的机理分别称 之为二重态母体的自由基 - 三重态对机理( DP -RTPM ) 和四重态母体的自由基 - 三重态对机理 ( QP - RTP M ) [9]. 本实验室曾系统地开展过 DP -RTPM 及 QP -RTP M 的理论计算以及 TEMPO 对蒽醌、对苯醌、丙酮以及杜醌等三重激发态猝灭反应的实验研究[10 - 14]. 但有关 TE MP O 对 BP 激发三重态的猝灭机理和猝灭动 力学研究在文献调研范围内还未见开展.T R-EP R 技术是探测和鉴定光化学反应过程中顺磁性自由基或三重态等反应中间体的强有力手段. 本 文采用 T R-EP R 技术并结合瞬态吸收光谱的方法,在乙二醇( EG ) 均相溶液中开展 TEMPO 对光诱导苯甲酮 激发三重态的猝灭反应研究,分析其中的光化学反应中间体,讨论稳态自由基 TE MPO 对 BP 激发三重态的 猝灭机理,并测量稳态自由基 TE MPO 对苯甲酮三重态3 BP *的猝灭动力学.实验试验中所用 T R-EP R 波谱仪已在相关文献[15]中做详细介绍,这里简单介绍如下: 微波频率约为9. 4G H z , 功率约为 15mW ; 微波系统采用平衡反射桥式电路和零差拍平衡混频方式,无高频磁场调制; 用 TE 102 矩形腔 作样品腔,样品放置的位置是微波磁场的波腹处; 平衡混频器的输出信号送入宽带差动放大器,其响应时间 为 50ns . 放大器的输出或送至数字存储示波器记录固定磁场下 C I DEP 的时间演化信号,或送至1 收稿日期: 2013 - 12 - 20基金项目: 国家自然科学基金 ( 20903004、21173002 ) ; 安 徽 省 自 然 科 学 基 金 ( 1308085MB20 ) ; 安 徽 高 校 省 级 自 然 科 学 研 究 重 点 项 目 ( K J2010A145) .作者简介: 郑飞( 1989 - ) ,男,安徽六安人,硕士研究生; 许新胜( 1975 - ) ,安徽芜湖市人,博士、教授、博士生导师.引用格式: 郑飞,卞伟伟,朱光来,许新胜. 氮氧自由基对苯甲酮三重激发态猝灭反应的 T R-EP R 研究[J ]. 安徽师范大学学报: 自然科学版,2014,37( 5) : 449 - 453450安 徽 师 范 大 学 学 报 ( 自 然 科 学 版 ) 2014 年Boxcar 平均器取样平均后,通过磁场的线性扫描,在计算机上记录 C I DEP 磁场谱.激光光源为 Nd : YAG 激光器三倍频输出的 355nm 激光,单脉冲能量为 8m J ,重复频率为 20H z . 样品管为 厚度约 0. 5mm 的扁平石英管. 为了防止激光照射下样品的局部过热,样品经蠕动泵循环流过谐振腔. Boxcar 平均器取样门宽为 0. 3μs ,B o xc a r 取样门相对激光激发的延时为 0. 6μs . 实验中所用试剂均为分析纯. 光解物质苯甲酮 ( 99. 5% ) 未经进一步提纯直接使用,溶剂为乙二醇( EG ) . 苯甲酮在溶液中的浓度均为 0. 05m o l ·L - 1 . 在 C I DEP 测量前用氮气对配制好的样品鼓泡除氧.瞬态吸收光谱的测量在中国科学技术大学微尺度物质科学实验室完成.结果与讨论3 光解 BP / E G 体系的 C I D E P图1 为 BP / EG 体系在 N2 饱和条件下经过 355nm 激光照射后记录到的 C I DEP 信号. 图 1 中的 CIDEP 谱 线可以分为两组,一组是低场侧和高场侧都有 的、磁场范围较宽的超精细双线,这部分谱线在光解对苯醌 / EG [16]、蒽醌 / EG [17]等体系时也观 察到过,可将其归属为乙二醇羰基自由基 E g l3. 1 [- H ]·( CH ( OH ) C HO H ,g = 2. 0041,a = H ( α) 2 7mT ,a 2H ( β,CH ) 93mT ,a H ( β,OH )1. = 0. = 0. 2 95mT ) [18]. E g l [- H ]· 自由基的超精细线应该 是六条超精细双线,图 1 中明显分辨的是低场 两条和高场侧的两条,另外的两条位于中心磁 场附近,因与谱线叠加而未能明显分辨. 另一组 强度较大,位于中心磁场处的较宽的超精细线,可归属为苯甲酮中性自由基( BP H · ) [1]. 由此可见,光解过程中存在着3 BP *从 EG 上夺氢的氢原子转移反应,生成苯甲酮中性自由基 BP H ·和乙二醇羰基自由基 E g l [- H ]·. 另外,图 1 中的 C I DEP 信号整体是发射的,而且呈现出明显的图 1 室温下光解 BP / EG 体系的 C I DEP 谱, [BP ]= 50 × 10 - 3 mo l ·L - 1 ,延时 0. 6μs低场侧信号强、高场侧信号弱即发射 + 发射 / 吸收( E + E / A ) 的特点,可见氢原子转移过程中自由基 CIDEP的形成机理主要是三重态机理( TM ) ,同时 RP M 机理也有相当份额的贡献[19].为了进一步确认上述氢原子转移反应,我们测量了光解 BP / EG 体系的瞬态吸收光谱,如图 2 所示. 图 2中 541nm 附近的吸收峰可归属于3 BP * ,632nm 附近的吸收峰属于 BP H · [20]. 而且随着激光激发后延时的增 加,541nm 处3 BP * 吸收峰强度减弱的同时 632nm 处 BP H · 的吸收峰在增强. 可见,光解 BP / EG 体系过程中的确存在着3 BP *从 EG 上的夺氢的反应过程.于是,光解 BP / EG 体系过程中可能存在的光物理与光化学过程如下:hv I SC31 * *( I ) BP → BP → BP 3 T - 113BP *→BP( II )( III )k sq+ BP →BP + BP3BP * kr+ CH ( OH ) CH OH →+ CH ( OH ) C HOH3BP * ( I V ) 2 2 2OH( I ) 式表示基态苯甲酮分子吸收 355nm 光子跃迁到激发单重态1 BP * ,然后再经各向异性的系间窜越( I S C )产生自旋极化的激发三重态3 BP * . ( II ) 表示激发三重态3 BP * 的自旋晶格弛豫过程,3 T自旋晶格弛豫时间. 1 ( III ) 式表示的是3 BP * 的自猝灭过程,自猝灭速率常数是 k . ( IV ) 表示3 BP * 从乙二醇分子上夺氢生成极化sq郑 飞,卞伟伟 ,朱光来,等:氮氧自由基对苯甲酮三重激发态猝灭反应的 T R-EP R 研究45137 卷第 5 期的苯甲酮中性自由基 BP H ·和乙二醇羰基自由基 E g l [- H ]· ,k 表示3 BP * 与 EG 之间的氢原 r子转移速率常数. 光解 BP / E G / TE MP O 体系的 C I D E P图3( a ) 是未经高频调制的 TE MPO 的稳态 ES R 吸收 信 号,超 精 细 常 数 为 1. 6mT [9]. 图 3( b ) 是 光 解 BP / EG / TEMPO 体系测量得到的C I DEP 谱. 图 3 ( b ) 中 BP H · 和 E g l [- H ]· 的 C I DEP 信号变弱,强度较大的三条超精细线,其 超精细常数也是 1. 6mT ,而且是发射的,属于TE MP O . 可见,光解过程中母体三重态3 BP * 的自 旋极化转移给了 TE MP O . 另外,图 3 ( b ) TE MP O 的 C I DEP 信号强度还表现出较强的 E + E / A 的 超精细相关性,这说明 TE MPO 的极化形成机理主要是 QP -RTPM [19]. 于是,我们认为光解 BP /3. 2 图 2 光解 BP / EG 均相体系( Ar 2 饱和) 的瞬态吸收光谱EG / TEMP O 体系过程中除了反应( I ) - ( IV ) 外,还存在如下的反应图 3 TEMP O 的稳态 ES R 谱( a ) 及光解 BP / EG / TEMP O 体系的C I DEP 谱( b ) ,[TE MP O ]= 10 × 10 - 3 mo l ·L - 1 ,延时 0. 6μs图 4 光解 BP / EG / TEMP O 体系 I [Eg [l - H ]·]0 / I [Eg l [ - H ]·]随[TEMPO ]的变化曲线TE MP O 对 3 BP * 的猝灭速率常数基于反应( I ) - ( V ) ,可得如下的 S t e r n -V o l m e r 方程3. 3 I [E g l [ - H ·]]= 1 + k [TEMPO ]( 1) ( 2) ( 3)s I [E g l [-H ]·]k s = k q τ其中1 τ = 3 T - 1+ k sq + k r1 ( 1) 式中 I [E g l [ - H ]·] 和 I E g l[ - H ]· 分别表示未加 TE MP O 和加入 TE MPO 后 E g l [- H ] 的 C I DEP 信号强度,k s 是 ·452安 徽 师 范 大 学 学 报 ( 自 然 科 学 版 ) 2014 年I [E g l [ - H ]·]0 / I [E g l [ - H ]·]随 TE MPO 浓度[TE MP O ]变化的斜率,( 2 ) 式中 τ 表示 BP 的寿命. 由( 1 ) - ( 3 ) 式可 3 *见,要测量 TE MP O 对3 BP *的猝灭速率常数 k ,首先要测量 I 随 [TEMPO ]变化的斜率/ I [E g l [ - H ]·]0 [E g l [ - H ]·]q k s ,然后再测量 BP 的寿命 τ.3 *表1 光解 BP / EG / T EM PO 体系的动力学参数表 1 列出了 不 同 TEMPO 浓 度 下 I [E g l [ - H ]·] / 0I [E g l [ - H ]·]值,将其对 TEMPO 浓度[TEMPO ]作图可 得如图 4 所示的 S t e r n -v o l m e r 曲线,线性拟合可得其I / TE M P O ( 10 - 3mo l / L ) [Eg l [ - H ] ]0 k ( L ·mo l - 1) τ( μs ) k ( L ·mo l - 1·s - 1) · s q I [Eg l [ - H]·]2 46 810 1. 235 1. 364 1. 712-1 斜率为 k s = 165. 85L m o l . 图 5 是光解 BP / EG 体系 541nm 处3 BP * 的瞬态吸收衰减曲线,单指数拟合得 到 3 BP *的寿命 τ 为 11. 80μs . 于是根据( 2) 式 1. 4 ×107165. 85 11. 80 可计算得到光解 BP / EG 体系 TE MP O 对3 BP * 的猝灭速率常数为 1. 40 × 107 L ·m o l - 1·s - 1.从上面的结果可以看出,稳自由基 TEMPO对 3 BP * 的猝灭速率常数在 107 L ·m o l - 1 ·s - 1量 级. 我们知道,溶液中进行的光化学反应一般都 是近似受扩散控制的,而且乙醇、乙腈溶液中, 扩散控制的反应速率常数一般为 109 L m o l - 1 s- 1 量级[21],可见我们这里的 TE MPO 对3 BP *的猝 灭速率常数与其相差 2 个数量级. 事实上,溶液 中受扩散控制的反应,其反应速率常数与反应 物扩散系数成正 比,而 扩 散 系 数 可 由 S t o k es - E i n s t e i n 关系式给出,即k B T ( 4)D =6πηr式中 η 是溶剂的粘度,r 是反应物分子半径,k B是玻尔兹曼常数,T 是绝对温度. 由( 4 ) 式可知, 图5 光解 BP / EG 体系 541nm 处3BP * 的瞬态吸收衰减曲线扩散系数 D 与溶剂粘度成反比. 由于乙二醇的粘度是 1. 16 × 10 - 2 P as [22],而乙醇和乙腈粘度分别是 9. 97 × 10 - 4 Pas 和 3. 45 × 10 - 4 P as [23],它们相差数十倍,因此,TE MPO 对3 BP * 的猝灭速率常数在 107 L ·m o l - 1 ·s - 1 量级是可以理解的,这也说明 TE MP O 对3 BP *的猝灭反应也是近似受扩散控制的.结论4 本文利用 T R-EP R 技术并结合瞬态吸收光谱的方法研究了乙二醇均相溶液中激光诱导的稳态自由基TE MPO 对 3 BP * 的猝灭反应. 光解 BP / EG 体系,3BP * 从溶剂分子 EG 上夺氢形成苯甲酮中性自由基 BP H · 和 乙二醇羰自由基 E g l [- H ]· ,光解自由基 C I DEP 形成的主要机理是 TM 机理,同时 RPM 机理也有相当的贡 献. 当体系中加入 TE MPO 时,观察到发射的 TE MPO 的 C I DEP 信号,T E MP O 对 3 BP * 具有明显的猝灭作用,猝 灭反应过程伴随着自旋极化的转移,猝灭速率常数为 1. 40 × 107 L ·m o l - 1 ·s - 1,反应近似受扩散控制.参考文献:[1] Turr N i cho l as J ,Khudya ko v I go r V . An e l ec tr on sp i n po l a r i za t i on study of the i n t e r ac t i on of pho t oexc i t ed tr i p l e t mo l ecu l es w i t h mono andpo l yn i tr oxy l s t ab l e fr ee Rad i ca l s [J ]. J Phys Chem ,1993,( 97) : 1138 - 1144. INBA R S ,LI NSC HI TZ H ,COHEN S G . P r i ma r y quantum y i e l ds of k e t y l r ad i ca l s i n pho t o r educ t i on by am i nes . A bs tr ac t i on of hyd r ogen from n i tr ogen [J ]. J Am Chem Soc ,1980,( 102) : 1419 - 1421.S I MON J D ,PET E RS K S . So l ven t effects on the p i cosecond dynam i cs of the pho t o r educ t i on of benzophenone by a r oma t i c am i nes [J ]. J Am Chem Soc ,1981,( 103) : 6403 - 6406.B RUNMA RK A ,C A DEN A S E . Redox and add i t i on chem i s t r y of qu i no i d compounds and i t s b i o l og i ca l i mp li ca t i ons [J ]. Free Rad B i o l Med ,1989,( 7) : 435 - 477.KUMAGAI Y ,KOIDE S ,T A GUC HI K ,et a l . Ox i da t i on of p r ox i ma l p r o t e i n su l f hyd r y l s by phenan t h r aqu i none ,a component of d i ese l exhaus t pa rt i c l es [J ]. Chem Res Tox i co l ,2002,( 15) : 483 - 488.S HI M A H ,KOIKE E ,S HI N O HA RA R,et a l . Ox i da t i ve ab ili t y and t ox i c i t y of n -H exane i nso l ub l e f r ac t i on of d i ese l exhaust pa rt i c l es [J ]. Tox i co l[2] [3][4] [5] [6]郑 飞,卞伟伟 ,朱光来,等:氮氧自由基对苯甲酮三重激发态猝灭反应的 T R-EP R 研究45337 卷第 5 期Sc i ,2006,( 91) : 218 - 221.CHUNG S W ,CHUNG H Y ,TO RIBA A ,et a l . An env i r onmen t a l qu i no i d po l ycy c li c a r oma t i c hyd r oca r bon ,acenaph t henequ i none ,modu l a t es cyc l ooxygenase -2 exp r ess i on through r eac t i ve oxygen spec i es gene r a t i on and nuc l ea r factor kappa B ac t i va t i on i n A549 ce ll s [J ]. Tox i co l Sc i ,2007,( 95) : 348 - 355.SUG I M OT O R,KUMAGAI Y ,NAKAI Y ,et a l . 9,10-Phenan t h r aqu i none i n d i ese l exhaust pa rt i c l es down r egu l a t es Cu ,Zn – SOD and H O -1 i n human pu l mona r y ep i t he li a l ce ll s : I n t r ace ll u l a r i r on scavenger 1,10-phenan t h r o li ne affords r o t ec t i on aga i ns t apop t os i s [J ]. Free Rad i c B i o l Med , 2005,( 38) : 388 - 395.KAWAI A ,OBI K . A new mechan i sm of e l ec tr on sp i n po l a r i za t i on gene r a t i on through r ad i ca l -exc i t ed mo l ecu l e i n t e r ac t i ons [J ]. Res Chem I n t e r med ,1993,( 19) : 865 - 894.XU X i nsheng ,J IA L i x i a ,S HI L e i ,CU I Zh i f eng . Quench i ng dynam i cs study on pho t o i nduced exc i t ed tr i p l e t du r oqu i none by TEMP O i n 1,2- p r opand i o l [J ]. Res Chem I n t e r med ,2010,( 36) : 259 - 267.XU X i nsheng ,J IA L i x i a ,S HI L e i ,CU I Zh i f eng . V i scos i t y effect study on quench i ng of pho t o i nduced exc i t ed tr i p l e t du r oqu i none by TEMPO [J ]. Spectrosc Lett ,2010,( 43) : 310 - 316. 许新胜,朱光来,陆同兴,崔执凤. 自由基 - 三重态对化学诱导动态电子极化强度的全哈密顿计算[J ]. 原子与分子物理学报,2008, ( 25) : 93 - 98.XU X i nsheng ,Z HA N G X i any i ,C U I Zh i f eng ,e t a l . Theo r e t i ca l study of hype rf i ne r e l a t ed C I DEP of Rad i ca l -tr i p l e t Pa i r s [J ]. Chem Phys Lett , 2003,( 369) : 579 - 583.ZHANG X i any i ,X U X i nsheng ,L U Tongx i ng . CIDEP i nves t i ga t i on of the T r i p l e t -Rad i ca l System i n the So l u t i on of the T r i p l e t Quenche r[J ]. Ch i n J Chem ,2000,( 18) : 683 - 670.何光龙,危启正,喻其山,季学韩,陆同兴. 高时间分辨电子自旋共振波谱仪设计中的几个问题[J ]. 安徽师范大学学报: 自然科学版, 1994,17( 2) : 59 - 63.许新胜,张为俊,张先邁,季学韩. 光解对苯醌 / 甲酰胺体系的 C I DEP 实验研究[J ]. 原子与分子物理学报,2004,( 4) : 351 - 354. 陆同兴,许新胜,洪新,李干佐. 蒽醌在乙二醇 / T r i t on X 胶束与乙二醇 / 三乙胺溶液中光还原的时间分辨电子顺磁共振研究[J ]. ACTA CHIMICA S I N I C A ,2006,( 17) : 1824 - 1830. KONKIN A L ,ROTH H K ,SC H ROE D NE R M ,et a l . T i me -r eso l ved ES R s t ud i es on tr ans i en t r ad i ca l s photogenerated i n so l u t i ons of me l am i ne i n e t hy l ene g l yco l [J ]. Chem Phys ,2006,( 324) : 563 - 572.史蕾,许新胜,崔执凤. 光解自由基化学诱导动态电子极化( C I DEP ) 的四种机理[J]. 安徽师范大学学报: 自然科学版,2010,33 ( 2 ) : 125 - 130.HAYON E ,IBATA T ,LICH t i n N N ,et a l . E l ec t r on and hydrogen atom attachment to a r oma t i c ca r bony l compounds i n aqueousso l u t i on . A bso r p t i on spectra and d i ssoc i a t i on constants of k e t y l r ad i ca l s [J ]. J Phys Chem ,1972,( 76) : 2072 - 2078.B I SB Y R H ,PA RKE R A W . Reac t i ons of exc i t ed tr i p l e t du r oqu i none w i t h α-t ocophe r o l and asco r ba t e : a nanosecond l ase r f l ash pho t o l ys i s and t i me -r eso l ved resonance raman i nves t i ga t i on [J ]. J Am Chem Soc ,1995,( 117) : 5664 - 5670.EVESON R W ,M CL A U C HLA N K A . E l ec tr on sp i n po l a r i za t i on ( CIDEP ) s t ud i es of the dynam i cs of gem i na t e free r ad i ca l r eac t i ons [J ]. Mo l Phys ,1999,( 96: 133 - 142.DYMON D J H ,AWAN M A ,GLEM N F ,et a l . Transport p r ope rt i es of none l ec tr o l y t e m i x t u r es . IX . V i scos i t y coe ff i c i en t s for ace t on i tr il e and for three m i x t u r es of t o l uene + ace t on i tr il e from 25 to 100° c at pressures up to 500 Mpa ,I n t . J . Thermo phy s [J ]. 1991,( 12) : 433 - 447.[7][8][9] [10] [11] [12] [13][14] [15] [16] [17] [18][19] [20] [21] [22] [23]A T R-EP R I nv e s t i ga t i o n on the Qu e n c h i ng Re a ct i o n of Ph oto i n d u ce dE x c i te d T ri p l et Benzopheonoe by TE M POZHENG F e i , BIAN W e i -w e i , ZHU G u a n g -l a i , XU X i n -s h e n g( I ns t i t u t e of At om i c and Mo l ecu l a r Phys i cs ,A nhu i No r ma l Un i ve r s i t y ,Wuhu 241000,C h i na )Ab st r a c t : The qu e nc h i n g r ea c t i o n of ph o t o -i nduc e d e xc i t e d tr i p l e t of benzophenone ( 3 BP * ) by s t a b l e r a d i c a l TEMPO i n e t hy l e n e g l yc o l ( EG ) homogeneous s o l u t i o n have been s t ud i e d by u s i n g t i m e -r es o l e d e l e c tr o n p a r a m ag n a n e t i c resonance ( T R-EP R) t e chn i qu e and tr a n s i e n t a b s o r p t i v e spec trum method . The tr a n s i e n t r ea c t i o n i n t e r m e d i a t es and c h e m i c a l i nduc e d dy n a m i c e l e c tr o n p o l a r i z a t i o n ( CIDEP ) m e c h a n i s m have b ee n a n a l y z e d . The qu e nc h i n g rate constant of 3BP*by TEMPO has been measured . The e x p e r i m e n t a l r es u l t si nd i c a t e that there i s hydrogen atom transfer r ea c t i o n from EG to 3BP *,and the n e u tr a l s e m i qu i n o n e r a d i c a l and k e t y l r a d i c a l are generated . The E + E / A pattern C I DEP means that tr i p l e t m e ch a n i s m ( TM ) i s the m a i n m e c h a n i s m . TEMPO i s an e ff e c t i v e quencher to 3BP*and the qu e nc h i n g rate constant i s 1. 40 × 107 L ·m o l- 1·s- 1.K e y wo r d s : b e n z o ph e n o n e ; TEMPO ; T R-EP R; qu e nc h i n g。
ADF教程:如何计算单重态到三重态的激发(相对论、许可跃迁)
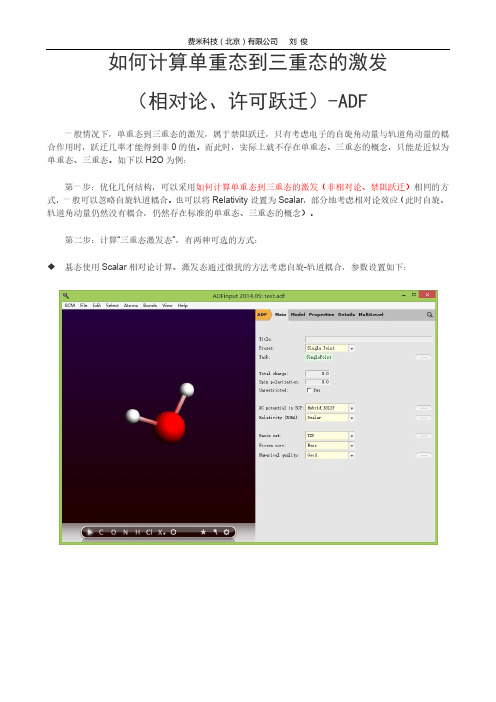
如何计算单重态到三重态的激发
(相对论、许可跃迁)-ADF
一般情况下,单重态到三重态的激发,属于禁阻跃迁,只有考虑电子的自旋角动量与轨道角动量的耦合作用时,跃迁几率才能得到非0的值。
而此时,实际上就不存在单重态、三重态的概念,只能是近似为单重态、三重态。
如下以H2O为例:
第一步:优化几何结构,可以采用如何计算单重态到三重态的激发(非相对论、禁阻跃迁)相同的方式,一般可以忽略自旋轨道耦合。
也可以将Relativity设置为Scalar,部分地考虑相对论效应(此时自旋、轨道角动量仍然没有耦合,仍然存在标准的单重态、三重态的概念)。
第二步:计算“三重态激发态”,有两种可选的方式:
基态使用Scalar相对论计算、激发态通过微扰的方法考虑自旋-轨道耦合,参数设置如下:
基态计算考虑自旋-轨道耦合、激发态精确地考虑自旋-轨道耦合,参数设置如下:
这两种方式,后者精确度更高,但前者可以计算自旋轨道耦合矩阵元。
后者计算得到的跃迁几率非常小(例如0.0000001以下)的激发态,并且三个激发态能量很接近,这就是对应的T1态劈裂开的3个激发态,具体可以参考三重态的劈裂。
前者得到的三个态能量完全一样,不能处理三重态的劈裂问题。
荧光知识补充-激发三重态

在一般温度下,大多数分子处在基态的最低振动能级。
处于基态的分子吸收能量(电能、热能、化学能或光能等)后被激发为激发态。
激发态是很不稳定的,它将很快地释放出能量又重新跃迁回基态。
若分子返回基态时以发射电磁辐射(即光)的形式释放能量,就称为“发光”。
如果物质的分子吸收了光能而被激发,跃迁回基态所发射的电磁辐射,称为荧光和磷光。
现从分子结构理论来讨论荧光和磷光的产生机理。
每个分子中都具有一系列严格分立相隔的能级,称为电子能极,而每个电子能级中又包含有一系列的振动能级和转动能级。
分子中电子的运动状态除了电子所处的能级外,还包含有电子的多重态,用M=2S+1表示,S为各电子自旋量子数的代数和,其数值为0或1 。
根据Pauli不相容原理,分子中同一轨道所占据的两个电子必须具有相反的自旋方向,即自旋配对。
若分子中所有电子都是自旋配对的,则S=0,M=1,该分子便处于单重态(或叫单重线),用符号S表示。
大多数有机化合物分子的基态都处于单重态。
基态分子吸收能量后,若电子在跃迁过程中,不发生自旋方向的变化,这时仍然是M=1,分子处于激发的单重态;如果电子在跃迁过程中伴随着自旋方向的变化,这时分子中便具有两个自旋不配对的电子,即S=1,M=3,分子处于激发的三重态,用符号T表示。
处于分立轨道上的非成对电子,自旋平行要比自旋配对更稳定些(洪特规则),因此在同一激发态中,三重态能级总是比单重态能级略低。
处于激发态的分子是很不稳定的,它可能通过辐射跃迁和非辐射跃迁的形式去活化(去激发)释放出多余的能量而返回基态。
辐射跃迁主要涉及到荧光,延迟荧光或磷光的发射;无辐射跃迁是指以热的形式释放多余的能量,包括振动弛豫、内部转移、系间跨越及外部转移等过程。
(3) 荧光发射(Fluorescence emission,FE)——处于激发单重态的电子经振动弛豫及内部转移后到达第一激发单重态(S1)的最低振动能级(V=0)后,以辐射的形式跃迁回基态(S0)的各振动能级,这个过程为荧光发射,发射的荧光波长为。
仪器分析名词解释

仪器分析名词解释1指示电极:在电化学电池中借以反映待测离子活度,发生所需电化学反应或激发信号的电极。
2参比电极:在恒温恒压条件下,电极电位不随溶液中待测离子活度的变化而变化,具有基本恒定电位值的电极。
3钠差(碱差):当电极测定PH>9.5或钠离子浓度较高的溶液时,ph值的测定值低于真实值,偏小而产生负误差。
4酸差:测定pH<1的强酸溶液时,PH的测定值高于真实值,产生正误差。
5原子光谱法:以测量气态原子离子外层或内层电子能级跃迁所产生的光谱为基础的成分分析方法。
6分子光谱法:以测量分子转动能级,分子中原子的振动能级(包括分子转动能级),分子的电子能级(包括振-转能级)等的能级跃迁而产生的分子光谱为基础的定性定量和物质结构分析的分析方法。
7生色团(Chromphre)含有π键的不饱和基团,能产生n-π某,π-π某跃迁。
8助色团(Au某ochrome)含有非键电子对的饱和基团,本身没有生色功能:与生色团相连时,发生n-π共轭作用,增强生色团的生色能力。
9红移(Redhift)某化合物的最大吸收波长向长波方向移动。
10蓝移(Bluehift)某化合物的最大吸收波长向短波方向移动。
11增色/减色效应(Hyperchromic/Hypochromiceffect):吸收强度(摩尔吸光系数)增大/减小的现象。
12荧光发射(Fluorecenceemiion)当激发态分子经过内转换或振动弛豫到达第一电子激发态的最低振动能级后,以辐射形式发射光量子,回到基态的过程。
13磷光发射(Phophorecenceemiion)经过体系间跨越的分子再通过振动弛豫降至激发三重态的最低振动能级,跃迁回基态的各个能级并辐射发光的过程。
14振动弛豫(Vibrationalrela某ation)激发态分子与溶剂分子碰撞,以热能形式损失部分能量,以极快速度降至同一电子激发态的最低振动能级上。
15内转换(Internalconverion)当2个电子能级靠近或有重叠时,发生电子由高能级以无辐射跃迁的方式返回低能级,将激发能转变成热能。
激发三重态(Excitedthreestates)

激发三重态(Excited three states)At most temperatures, most molecules are at the lowest vibrational level of the ground state. Molecules absorbed in the ground state absorb energy (electric energy, thermal energy, chemical energy, or light energy, etc.) and are excited to be excited. The excited state is very unstable,It will release energy quickly and re jump back to the ground state. When the molecules return to the ground state, the energy is emitted in the form of emission of electromagnetic radiation (light), known as luminescence". If the molecules of matter absorb light energy, they are stimulatedThe electromagnetic radiation emitted by the transition back to the ground state, known as fluorescence and phosphorescence. The mechanism of fluorescence and phosphorescence is discussed in terms of molecular structure theory.Each molecule has a series of strictly separated energy levels, called electron energy poles, and each electron energy level contains a series of vibrational energy levels and rotational energy levels. The state of motion of electrons in moleculesIn addition to the energy levels, the electrons contain multiple states of electrons. In M=2S+1, S is the sum of the quantum numbers of each electron spin quantum, with a value of 0 or 1. According to the principle of Pauli incompatibility, the same orbital in the moleculeThe two electrons occupied must have opposite spin directions, namely spin pairing. If all electrons in the molecule are spinpaired, then S=0, M=1, the molecule is in a singlet state (or a single line), expressed in symbolic S.The ground states of most organic compounds are in the singlet state. When the ground state molecules absorb energy, if the electron does not change in the direction of spin during the transition, it is still M=1, and the molecules are excited at a single weightIf the electron is accompanied by a change in the spin direction during the transition, then the molecule has two spin unpaired electrons, S=1, M=3, and the molecule is in the excited three state, expressed in symbolic T.Fig. 14.1 is a schematic diagram of electronic states.Fig. 14.1 sketch of excitation of three heavy states in a singlet stateThe unpaired electrons in discrete orbits are more stable than spin pairs (especially the rules), so in the same excited state, the energy levels of the three states are always slightly lower than those of the singlet state.Fig. 14.2 is a diagram of energy levels and transitions, in which S0, S1 and S2 represent the ground states of the molecules, the first and second electron excited singlet states, respectively, and T1 and T2 represent the three and second electron excited states of the molecule respectively. V=0, 1, 2, 3,... Represents the vibrational level of theground state and excited state.Fig. 14.2 energy level diagram of fluorescence and phosphorescence systemThe molecules in the excited state are very unstable, which may be activated by means of radiative transitions and nonradiative transitions (de excitation), releasing excess energy and returning to the ground state.Radiation transitions are mainly related to fluorescence, delayed fluorescence or phosphorescence emission; nonradiative transition is the release of excess energy in the form of heat, including vibrational relaxation, internal transfer, intersystem crossing and external transferCheng. Fig. 14.2 represents the energy transfer process of molecular excitation and deactivation:(1) vibrational relaxation (Vibration, relaxation, abbreviated as VR) - the transition from the lowest vibrational energy level (V=0) of the ground state to the excited singlet state may be possible when the molecules absorb the radiation of light (as shown in lambda 1, lambda 2 in Fig. 14.2)Higher vibrational levels of Sn (as shown in S1 and S2). Then,In the gas phase where the liquid phase or the pressure is high enough, the collision probability between molecules is large, and the molecules may pass excess vibrational energy to the surrounding region in the form of heatEnvironment, and its transition from the high vibrational energy level of the excited state to the lowest vibrational level of the electron energy level, this process is called vibrational relaxation. The vibrational relaxation time is 10 - 12s orders of magnitude.(2) internal transfer (Internal, conversion, abbreviated as IC) - when the low vibrational energy levels in the high electron levels overlap with the high vibrational energy levels in the lower electron levels, electrons frequently occur from the high electron energy levelsThe transition from nonradiative to low electron levels. As shown in Fig. 14. and 2, the low vibrational kinetic energy levels in S2 and T2 overlap with the high vibrational kinetic energy levels in S1 and T1, and electrons can transition from S2 to S1 through the superposition of vibrational levels, or fromT2 transition to T1. This process is called internal transfer. Time transfer for 1011s ~ 1013s magnitude. The rate of vibrational relaxation and internal transfer is much faster than the direct emission of photons by a highly excited state,Therefore, no matter which excited singlet state excited by the radiation energy, the molecules can jump to the lowest vibrational level via vibrational relaxation and internal transfer to the lowest (first) excited singlet state.(3) fluorescence emission (Fluorescence, emission, FE) - afterthe vibrational relaxation and internal transfer of electrons in the excited singlet state, reach the lowest vibrational level (V=0) of the first excited singlet (S1),The vibrational levels of the ground state (S0) transition in the form of radiation. The process is fluorescence emission with a fluorescence wavelength of. The energy loss due to vibrational relaxation and internal transfer, and hence the fluorescence emission energyThe energy is smaller than the molecular absorption, and the wavelength of fluorescence emission is longer than the wavelength of molecular absorption. The average lifetime of the lowest vibrational level in the first excited singlet state is about 10-9 - 10 - 4S, so the fluorescence lifetime is also in the rangeThis order of magnitude.(4) department (Intersystem Crossing, ISC Kuayue) - between leap refers to the non radiative transition process between different multiplets, it relates to the electronic excited spin state change.Such as the transition from the first excited singlet state S1 to the first excited three heavy state T1, so that the two spin pairs of electrons are no longer paired. This transition is prohibited (not in conformity with the spectral selection rule),But if the two energy layers have a large overlap, the minimumvibrational level of S1 in Figure 14.2 overlaps with the higher vibrational levels of T1, and it is possible to achieve this transition by spin orbit coupling.Between leap slower and experience a long time.(5) (Phosphorescence emission, PE luminescence emission) - the electronic excited state by the Department after three leap reach the excited state, after the rapid relaxation of vibrational relaxation and the transition to the first excited state threeThe lowest vibrational energy levels are then converted in the form of radiation back to the vibrational levels of the ground state, which are emitted by phosphorescence. The transition of the phosphorescence emission is still the spin forbidden, so the light speed is very slow.The life of 10-4 ~ 100s phosphor. Therefore, after the external light source is stopped, phosphorescence remains a short time. After the system vibration leap and T1 lost a part of the energy relaxation,So the phosphorescence wavelength is longer than the fluorescence wavelength, that is, the wavelength is longer than the wavelength of the phosphor.It must be pointed out that T1 may also be re excited by thermal excitation back to S1, i.e., T1S1, and then converted back to S0 by radiation from S1, S1S0, which emits fluorescence, which is called delayed fluorescence,Its lifetime is similar to that of phosphorescence, but its wavelength is shorter than phosphorescence.(6) external transfer (External, convertion, EC) - excited state molecules collide with solvent molecules or other solute molecules, and the process of energy transfer is called external transfer.External transfer can weaken or even weaken the intensity of fluorescence or phosphorescence. This phenomenon is called quenching or quenching.。
分子发光

( 3)基态单重态到激发单重态的激发为允许跃迁,基 态单重态到激发三重态的激发为禁阻跃迁。
(4)激发三重态的能量较激发单重态的能量低。
2、分子能级结构与分子发射光谱
处于激发态的电子,通常以辐射跃迁方式或无辐射
跃迁方式再回到基态。
辐射跃迁:荧光、磷光的发射。 无辐射跃迁:振动弛豫(VR)、内转化(ic)、体系间 窜跃(isc)等。
A
(2.3 A) 2 (2.3 A)3 I f I o [2.3 A ] 2 3
如果吸光度A<0.05, 方括号中其他各项与第一项相比 均可忽略:
I f 2.3 I o A
由于A=bc,故在实验条件固定时,荧光强度与浓
度成正比,即:
I f 2.3I 0 A
抗体、抗原
酶联免疫吸附分析示意 Enzyme-linked immunosorbent assay (ELISA)
2. 荧光与有机化合物结构的关系
(1)跃迁类型 对于大多数荧光物质,首先经历激发,然后经过
振动弛豫或其他无辐射跃迁,再发生 跃迁而得到荧光。
(2)共轭效应 容易实现激发 的芳香族化合物容易发生荧光。 增加体系的共轭度荧光效率将增大,主要是由于增大荧 光物质的摩尔吸光系数,有利于产生更多的激发态分子。
类型 转入三重态猝灭: 溶解氧与荧光物质。 发生电子转移反应猝灭: 猝灭剂与荧光物质。 浓度较高单重激发态的分子在 荧光物质的自猝灭: 发生荧光之前和未激发的荧光 物质分子碰撞而引起的自猝灭。 29
二、荧光分析仪
Cary Eclipse 荧光分光光度计 荧光、磷光化学/生物发光 美国瓦里安技术中国有限公司
抗磁性。
当分子吸收能量后,在跃迁过程中不发生电子自旋方
单线态和三线态

单线态和三线态是指分子的激发态大多数分子含有偶数电子,在基态时,这些电子成对地存在于各个原子或分子轨道中,成对自旋,方向相反,电子净自旋等于零:S= 12 +(-12 )=0,其多重性M=2S+仁1 (M为磁量子数),因此,分子是抗(反)磁性的,其能级不受外界磁场影响而分裂,称单线态”当基态分子的一个成对电子吸收光辐射后,被激发跃迁到能量较高的轨道上,通常它的自旋方向不改变,即S=0,则激发态仍是单线态,即单线(重)激发态”如果电子在跃迁过程中,还伴随着自旋方向的改变,这时便具有两个自旋不配对的电子,电子净自旋不等于零,而等于S=12+1/2=1其多重性:M=2S+仁3即分子在磁场中受到影响而产生能级分裂,这种受激态称为三线(重)激发态”三线激发态”比单线激发态”能量稍低。
但由于电子自旋方向的改变在光谱学上一般是禁阻的,即跃迁几率非常小,只相当于单线态-单线态过程的10-6~10-7。
当激发态的分子通过振动驰豫--内转换--振动驰豫到达第一单线激发态的最低振动能级时,第一单线激发态最低振动能级的电子可通过发射辐射(光子)跃回到基态的不同振动能级,此过程称荧光发射”如果荧光几率较高,则发射过程较快,需10-8秒。
第一电子三线激发态最低振动能级的分子以发射辐射(光子)的形式回到基态的不同振动能级,此过程称为磷光发射”发生过程较慢约10-4~10秒。
第二章分子发光分析分子一吸收能量一激发为激发态一释放出能量亠基态称为“发光光致发光分子发光化学发光荧光磷光电能化学能光能辐射跃迁I光的形式释放非辐射跃迁以热的形'释放分子荧光分析法一.基本原理(一)荧光和磷光的产生 从分子结构理论来讨论-振动能级Y・转动能级'S=0, J=1单重态s 表示(所有电子都是自旋配对的)大多数基态分子都处于单重态 「S 二J=3 三重态T 表不 电子在跃迁过程中伴随着 自旋方向的变化(自旋平行)基态单亟态S 激发态单重态S 激发态三垂态T激发单重态S 与激发三重态T 的不同点: ⑴S 是抗磁分子,T 是顺磁分子⑵t s = 108s, tp =丄〜Is ;(发光速度很慢) ⑶基态单重态到激发单重态的激发为允许跃迁, 基态单重态到激发三重态的激发为禁阻跃迁;⑷激发三重态的能量较激发单重态的能量低r 电子所处的能级分子中电子 的能量状态Y亠电子的多重态2J=2S+1S :为各电子自旋量子 数的2.分子内的光物理过程其中S。
单重态激子和三重态激子

单重态激子和三重态激子单重态激子和三重态激子是材料科学中经常被提及的概念。
它们是指在固体中由电子和空穴形成的激发态。
这些激子在半导体、电池、有机太阳能电池等领域的应用中具有重要的作用。
单重态激子,也称为S1激子,是由电子和空穴的自旋方向相反组成的。
当一个电子和一个空穴在材料中相遇时,它们会形成一个S1激子。
这种激子具有很长的寿命和高的光量子效率,因此在光伏材料中应用广泛。
S1激子的能量通常在1.8-2.0 eV之间,这是太阳辐射的主要部分之一。
因此,S1激子在太阳能电池中的应用非常重要。
三重态激子,也称为T1激子,是由电子和空穴的自旋方向相同组成的。
当一个电子和一个空穴在材料中相遇时,它们会形成一个T1激子。
与S1激子相比,T1激子的能量较低,寿命较短。
T1激子的能量通常在0.5-1.0 eV之间,因此它们在太阳能电池中的应用相对较少,但在有机发光二极管(OLED)等领域中具有重要作用。
在有机太阳能电池(Organic solar cells)中,单重态激子和三重态激子的作用是相反的。
光子被吸收后,它会产生一个激子,这个激子可以进一步分裂成一个S1激子和一个T1激子。
S1激子会进一步分裂成电子和空穴,这些载流子可以被收集并转化为电流。
而T1激子则会通过非辐射复合的方式衰减成热能。
因此,在有机太阳能电池中,提高S1激子的数量和稳定性是非常重要的。
同时,通过控制T1激子的数量和寿命,可以提高有机太阳能电池的效率。
因此,对于有机太阳能电池的研究,单重态激子和三重态激子的研究非常重要。
在半导体材料中,单重态激子和三重态激子的作用也非常重要。
在光电器件中,S1激子可以通过光电效应产生电子和空穴,从而产生电流。
而T1激子则可以通过热激发的方式产生电子和空穴,也可以通过热发光的方式产生光子。
单重态激子和三重态激子是材料科学中非常重要的概念。
它们在太阳能电池、有机发光二极管、半导体器件等领域都有广泛的应用。
对它们的研究和控制,可以帮助我们设计和制造更高效、更稳定的光电器件。
仪器分析名词解释

仪器分析名词解释:1指示电极:在电化学电池中借以反映待测离子活度,发生所需电化学反应或激发信号的电极。
2参比电极:在恒温恒压条件下,电极电位不随溶液中待测离子活度的变化而变化,具有基本恒定电位值的电极。
3钠差(碱差):当电极测定PH>9.5或钠离子浓度较高的溶液时,ph值的测定值低于真实值,偏小而产生负误差。
4酸差:测定pH<1的强酸溶液时, PH的测定值高于真实值,产生正误差。
5原子光谱法:以测量气态原子离子外层或内层电子能级跃迁所产生的光谱为基础的成分分析方法。
6分子光谱法:以测量分子转动能级,分子中原子的振动能级(包括分子转动能级),分子的电子能级(包括振-转能级)等的能级跃迁而产生的分子光谱为基础的定性定量和物质结构分析的分析方法。
7生色团(Chromphre)含有π键的不饱和基团,能产生n-π*,π-π*跃迁。
8助色团(Auxochrome)含有非键电子对的饱和基团,本身没有生色功能:与生色团相连时,发生n-π共轭作用,增强生色团的生色能力。
9红移(Red shift)某化合物的最大吸收波长向长波方向移动。
10蓝移(Blue shift)某化合物的最大吸收波长向短波方向移动。
11增色/减色效应(Hyperchromic/Hypochromic effect):吸收强度(摩尔吸光系数)增大/减小的现象。
12荧光发射(Fluorescence emission)当激发态分子经过内转换或振动弛豫到达第一电子激发态的最低振动能级后,以辐射形式发射光量子,回到基态的过程。
13磷光发射(Phosphorescence emission)经过体系间跨越的分子再通过振动弛豫降至激发三重态的最低振动能级,跃迁回基态的各个能级并辐射发光的过程。
14振动弛豫(Vibrational relaxation)激发态分子与溶剂分子碰撞,以热能形式损失部分能量,以极快速度降至同一电子激发态的最低振动能级上。
激发三重态名词解释

激发三重态名词解释
嘿,你知道啥是激发三重态不?这可真是个超级有趣的东西呢!咱
就拿白天和黑夜来类比一下哈。
白天阳光灿烂,那就是基态,一切都
正常有序;而激发三重态呢,就像是黑夜里突然出现的神秘光芒。
比如说,在一个分子世界里,分子们本来好好地待在基态,就像我
们平常过着普通的日子。
但突然,有了某种能量的输入,就好比一道
闪电划过夜空,分子就被激发啦,进入了激发态。
这激发态又分为单
重态和三重态呢!
单重态就好像是个急性子,活跃但不持久;而三重态呢,就像个沉
稳的家伙,虽然行动慢点,但耐力超强。
你想想,这多有意思呀!
再打个比方,激发三重态就像是一场冒险之旅。
分子们勇敢地踏上
这条不同寻常的道路,带着满满的新奇和未知。
在这个过程中,它们
会经历各种奇妙的事情,也许会遇到阻碍,也许会发现新的机会。
“哎呀,那这激发三重态到底有啥具体作用呀?”你可能会这么问。
嘿,它的作用可大啦!在化学、物理等好多领域都有着重要的地位呢。
比如在一些发光材料中,激发三重态的存在就决定了材料能不能发出
漂亮的光。
我觉得呀,激发三重态就像是一个隐藏在分子世界里的宝藏,等待
着我们去探索和发现。
它让我们看到了物质世界中更多的可能性和神
奇之处。
所以呀,可别小瞧了这激发三重态哟!。
三重激发态和紫外可见光谱

三重激发态和紫外可见光谱1.引言1.1 概述概述:三重激发态和紫外可见光谱是在化学和物理领域中广泛研究的重要课题。
在过去几十年里,人们对这两个领域进行了深入的研究,并取得了许多重要的成果。
三重激发态是指分子或原子在光照射下能够吸收能量并达到的第三个能级,其能级比起激发态更高。
相比于单一的激发态,三重激发态在化学反应和光电转换等方面具有独特的性质和应用。
紫外可见光谱是一种常用的分析技术,通过测量样品在紫外可见波长范围内吸收光的强度来研究其结构和性质。
紫外可见光谱能够提供关于物质吸收和能级跃迁等信息,从而帮助研究人员了解分子或原子的能级分布和电子结构。
在化学、生物学和材料科学等领域中,紫外可见光谱广泛应用于溶液浓度分析、颜色测量、药物研究和环境监测等方面。
本文将重点介绍三重激发态和紫外可见光谱的基本原理、实验方法以及在不同领域中的应用。
通过深入了解三重激发态和紫外可见光谱的研究现状和进展,可以更好地理解分子和原子的光电性质,进而为相关领域的研究和应用提供理论和实验依据。
此外,本文还将展望三重激发态和紫外可见光谱的未来发展方向和可能的研究热点,以期推动相关领域的进一步探索和创新。
文章结构部分的内容可以如下所示:1.2 文章结构本文将从三个方面来论述三重激发态和紫外可见光谱的相关内容。
首先,我们将在引言部分概述本文的主题,并介绍三重激发态和紫外可见光谱的基本概念和重要性。
其次,在正文部分2.1中,我们将详细探讨三重激发态的形成机制、特性和应用领域。
然后,在正文部分2.2中,我们将介绍紫外可见光谱的基本原理、测量方法和在化学、生物学、环境科学等领域的应用。
最后,在结论部分3.1中,我们将对本文进行总结,总结三重激发态和紫外可见光谱在科学研究中的重要性,并提出未来的研究展望。
通过对三重激发态和紫外可见光谱的全面分析和讨论,旨在提高读者对这两个领域的理解和应用能力,促进相关研究的进一步发展和应用。
1.3 目的本文的目的是探讨三重激发态和紫外可见光谱在材料科学领域的重要性和应用。
单线态和三线态讲解学习
2. 荧光与有机化合物结构的关系 (1)跃迁类型
实验证明,对于大多数荧光物质,首先经历 激发,然后经过振动弛豫或其他无辐射跃 迁,再发生 跃迁而得到荧光。 (2)共轭效应 实验证明,容易实现激发 的芳香族化合物 容易发生荧光,增加体系的共轭度荧光效率一般 也将增大,主要是由于增大荧光物质的摩尔吸光 系数,有利于产生更多的激发态分子
激发态三重态T
激发单重态S与激发三重态T的不同点: ⑴ S是抗磁分子,T是顺磁分子
⑵ tS = 10-8s, tT = 10-4~1s;(发光速度很慢) ⑶ 基态单重态到激发单重态的激发为允许跃迁,
基态单重态到激发三重态的激发为禁阻跃迁; ⑷ 激发三重态的能量较激发单重态的能量低
4
2.分子内的光物理过程
18
(3) 刚性平面结构 实验发现,多数具有刚性平面结构的 有机分子具有强烈的荧光。
因为这种结构可以减少分子的振动, 使分子与溶剂或其它溶质分子的相互作用 减少,也就减少了碰 撞去活的可能性。
19
(4)取代基效应
给电子基团,荧光增强(-OH、-OR、-CN、-NH2)
芳环上 取代基
产生了p-共轭作用,增强了电子共轭程度,使最低 激发单重态与基态之间的跃迁几率增大。
11
(二 )荧光的激发光谱和发射光谱
激发光谱:(ex)
以不同波长的入射光激发荧光物质,在荧 光最强的波长处测量荧光强度 即以激发光波长为横坐标,以荧光强度为 纵坐标绘制曲线即可得到激发光谱曲线。
发射光谱:(em)
固定激发光波长(最大) 然后测定不同的波长时所发射的荧光强度 即可绘制荧光发射光谱曲线
例如S1→T1就是一 种系间窜跃。
通常,发生系间 窜跃时,电子由S1 的较低振动能级转 移至T1的较高振动 能级处。
三重态湮 双光子吸收

三重态湮和双光子吸收三重态湮灭和双光子吸收都是光学领域的重要概念。
三重态湮灭三重态湮灭是一种光化学过程,其中两个处于激发三重态的物质分子相互碰撞,释放出一个光子。
三重态湮灭的能量等于两个激发三重态分子的能量之和,因此其波长比激发三重态分子的吸收波长短。
三重态湮灭的应用包括:1.用于光催化反应:三重态湮灭可以产生高能单线态,进而激活光催化剂,促进光催化反应的进行。
2.用于生物成像:三重态湮灭可以产生荧光,因此可用于生物成像。
3.用于光电探测:三重态湮灭可以产生光子,因此可用于光电探测。
双光子吸收双光子吸收是一种光学过程,其中一个物质分子同时吸收两个光子,进入激发态。
双光子吸收的能量等于两个光子的能量之和,因此其波长比单光子吸收的波长长。
双光子吸收的应用包括:●用于光学显微镜:双光子显微镜可以实现更高的光学分辨率。
●用于光谱学:双光子光谱可以对物质的能级结构进行更精细的分析。
●用于光催化反应:双光子吸收可以产生更高的能量单线态,进而提高光催化反应的效率。
三重态湮灭和双光子吸收的区别三重态湮灭和双光子吸收都是光学领域的重要概念,但它们之间存在一些区别。
1.激发态不同:三重态湮灭需要两个处于激发三重态的物质分子,而双光子吸收需要一个物质分子同时吸收两个光子。
2.能量不同:三重态湮灭的能量等于两个激发三重态分子的能量之和,而双光子吸收的能量等于两个光子的能量之和。
3.波长不同:三重态湮灭的波长比激发三重态分子的吸收波长短,而双光子吸收的波长比单光子吸收的波长长。
4.应用不同:三重态湮灭的应用包括光催化反应、生物成像、光电探测等,而双光子吸收的应用包括光学显微镜、光谱学、光催化反应等。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
它将很快地释放出能量又重新跃迁回基态。若分子返回基态时以发射电磁辐射(即光)的形式释放能量,就称为“发光”。如果物质的分子吸收了光能而被激
发,跃迁回基态所发射的电磁辐射,称为荧光和磷光。现从分子结构理论来讨论荧光和磷光的产生机理。
。V=0、1、2、3、…表示基态和激发态的振动能级。
图14.2 荧光和磷光体系能级图
处于激发态的分子是很不稳定的,它可能通过辐射跃迁和非辐射跃迁的形式去活化(去激发)释放出多余的能量而返回基态。
辐射跃迁主要涉及到荧光,延迟荧光或磷光的发射;无辐射跃迁是指以热的形式释放多余的能量,包括振动弛豫、内部转移、系间跨越及外部转移等过
占据的两个电子必须具有相反的自旋方向,即自旋配对。若分子中所有电子都是自旋配对的,则S=0,M=1,该分子便处于单重态(或叫单重线),用符号S表示。
大多数有机化合物分子的基态都处于单重态。基态分子吸收能量后,若电子在跃迁过程中,不发生自旋方向的变化,这时仍然是M=1,分子处于激发的单重
态;如果电子在跃迁过程中伴随着自旋方向的变化,这时分子中便具有两个自旋不配对的电子, 即S=1,M=3,分子处于激发的三重态,用符号T表示。
磷光的寿命为10-4~100s。因此,外光源照射停止后,磷光仍可持续一短时间。由于经过系间跨跃及T1 中振动弛豫丢失了一部分能量,
所以磷光波长比荧光波长要长,即。
必须指出的是 T1还可能通过热激发而重新跃回S1 即T1S1,然后再由S1经辐射跃迁回S0,即S1S0,发出荧光,这种荧光称为延迟荧光,
图14.1为电子重态示意图。
图14.1 单重态系三重态激发示意图
处于分立轨道上的非成对电子,自旋平行要比自旋配对更稳定些(洪特规则),因此在同一激发态中,三重态能级总是比单重态能级略低。
图14.2为能级及跃迁示意图,其中S0、S1和S2分别表示分子的基态、第一和第二电子激发的单重态;T1和T2则分别表示分子的第一和第二电子激发的三重态
每个分子中都具有一系列严格分立相隔的能级,称为电子能极,而每个电子能级中又包含有一系列的振动能级和转动能级。分子中电子的运动状态除了
电子所处的能级外,还包含有电子的多重态,用M=2S+1表示,S为各电子自旋量子数的代数和,其数值为0或1 。根据Pauli不相容原理,分子中同一轨道所
环境,而自身从激发态的高振动能级跃迁至该电子能级的最低振动能级上,这个过程称为振动弛豫。发生振动弛豫的时间为10-12s数量级。
(2) 内部转移(Internal conversion,简写为IC)——当高电子能级中的低振动能级与低电子能级中的高振动能级发生重叠时,常发生电子从高电子能级以
(4) 系间跨跃(Intersystem Crossing, ISC)——系间跨跃是指不同多重态之间的无辐射跃迁过程,它涉及到受激发电子自旋状态的改变。
如由第一激发单重态S1跃迁至第一激发三重态T1,使原来两个自旋配对的电子不再配对。这种跃迁是禁阻的(不符合光谱选律),
但如果两个能态的能层有较大重叠时,如图14.2中S1的最低振动能级与T1的较高振动能级重叠,就有可能通过自旋一轨道耦合等作用实现这一跃迁。
以辐射的形式跃迁回基态(S0)的各振动能级,这个过程为荧光发射,发射的荧光波长为。由于经过振动弛豫和内部转移的能量损失,因此荧光发射的能量
比分子吸收的能量要小,荧光发射的波长比分子吸收的波长要长,即。第一激发单重态最低振动能级的平均寿命约为10-9~10-4s,因此荧光寿命也在
这一数量级。
系间跨跃的速度较慢,经历的时间较长。
(5) 磷光发射(Phosphorescence emission,PE)——激发态的电子经系间跨跃后到达激发三重态,经过迅速的振动弛豫而跃迁至第一激发三重态
的最低振动能级,然后以辐射形式跃迁回基态的各振动能级,这个过程为磷光发射。磷光发射的跃迁仍然是自旋禁阻的,所以发光速度很慢。
所以,分子吸收辐射能后不管激发到哪一个激发单重态,都能通过振动弛豫及内部转移而跃迁到最低(第一)激发单重态的最低振动能。
(3) 荧光发射(Fluorescence emission,FE)——处于激发单重态的电子经振动弛豫及内部转移后到达第一激发单重态(S1)的最低振动能级(V=0)后,
程。图14.2表示分子激发和去活化的能量传递过程:
(1) 振动弛豫(Vibration relaxation,简写为VR)——当分子吸收光辐射(为图14.2中的λ1、λ2)后可能从基态的最低振动能级(V=0)跃迁到激发单重态
Sn(如图中S1、S2)的较高振动能级上。然后,在液相或压力足够高的气相中,分子间的碰撞几率很大,分子可能将过剩的振动能量以热的形式传递给周围
无辐射跃迁形式转移至低电子能级。如图14.2中,S2和T2中的低振动能级与S1和T1中的高振动能级重叠,电子可以通过振动能级的重叠从S2跃迁至S1,或从
T2跃迁至T1。这个过程称为内部转移。内部转移的时间为10-11s~10-13s数量级。振动弛豫及内部转移的速率比由高激发态直接发射光子的速率快得多,
其寿命与磷光相近,但波长比磷光短。
(6) 外部转移(External convertion,EC)——激发态分子与溶剂分子或其它溶质分子相互碰撞,并发生能量转移的过程称为外部转移。
外部转移能使荧光或磷光的强度减弱甚至消失,这种现象称为猝灭或熄灭。