Biographies_2013_Measuring-the-User-Experience-Second-Edition-
USANA的产品连续10年排名第一!

USANA的产品连续10年排名第⼀!〈营养补充品⽐较指南〉是加拿⼤卫⽣部对北美1500多种保健品进⾏严格科学的评测的刊物,具有很⾼的权威性和公信⼒,该书已经出版了3版,每年修订⼀次。
USANA的产品连续10年排名第⼀!下⾯是1-100名的公司和产品,安利、仙林蕾德等国⼈熟悉的国际品牌榜上⽆名。
排名公司产品分数注释1USANA Health Sciences Essentials (U.S.)96.1USANA2Douglas Laboratories Ultra Preventive X95.4 3Vitamin Research Products Extend Plus93.1 4Source Naturals Life Force Multiple 92.8 5Source Naturals Elan Vital91.8 6USANA Health Sciences Essentials (Canadian)90.2USANA7FreeLife Basic Mindell PlUS82.3 8Life Extension Foundation Life Extension Mix81.4 9Karuna Maxxum 479 10Ultimate Nutrition Super Complete75.8 11Douglas Laboratories Ultra Preventive Beta75.1 12SportPharma Multiguard74.9 13Dr. Julian Whitaker's Forward Multi-Nutrient74.7 14Douglas Laboratories Ultra Preventive III70 15Amni Added Protection III69.1 16Purify Products Perfect Mutti Focus Formula68.9 17DaVinci Laboratories Spectra Woman .66.8 18Doctor's Nutrition Mega Vites Woman66.8 19Mountain Naturals of Vermont Women's Superior66.8 20Douglas Laboratories Ultra Preventive IX 66.6 21Nutrition Dynamics Optimum Health Essentials66.6 22Karuna Maxxum 265.7 23DaVinci laboratories Spectra64.7 24FoodScience of Vermont Superior Care64.7 25Mountain Naturals of Vermont Superior Care64.7 26DaVinci Laboratories Spectra without Copper and Iron64.5 27Doctor's Nutrition Mega Vites without Copper & Iron64.2 28FoodScience of Vermont Women's Superior63.8 29DaVinci Laboratories Omni63.6 30Colgan Institute Formula MC863.3 31Rainbow Light Advanced Nutritional System63.2 32Nutrition Dynamics Day Star/Day End Essentials63 33DaVinci Laboratories Spectra Man62.5 34FoodScience of Vermont Men's Superior62.5 35Mountain Naturals of Vermont Men's Superior62.5 36Purity Products Purity's Perfect Multi62.2 37Rainbow Light Iron-Free Advanced Nutritional System62.2 38EAS Multi-Blend62.1 39American Longevity Ultimate Daily60.7 40Pharmacist's Ultimate Health Woman's Ultimate Formula60.7 41Amni Basic Preventive 160.2 42Amni Basic Preventive 360.2 43Amni Basic Preventive with Extra D60 44Nature's Way Alive! Whole rood Energizer (Iron-Free)60 45Amni Basic Preventive 559.9 46Amni Basic Preventive 2 59.6 47Amni Basic Preventive 459.6 48Dc Julian Whitaker's Forward Mufti-Nutrient58.5 49Jean Carper's Stop Aging Now!58.5 50Jean Carper's Stop Aging Now! Plus58.5 51MINES Pro Preventamins58.1 52Advanced Physician's Products Maximum Potency MuItiVitamin/Mineral with Iron57.4 53Advanced Physician's Products Maximum MuItiWtamin/Mineral without Iron57.2 54EcoNugenics Women's Longevity Rhythms Gold57 55Solaray Spectro Multi-Vita-Min57 56Solaray Vegetarian Spectra Multi-Vita-Min57 57Natural Factors MuItiStart Women's56.9 58Solaray Iron-Free Spectre Multi-Vita-Min56.7 59Solaray Spectro 3 Iron Free56.7 60Michael's Naturopathic Programs Active Senior Tabs56.6 61Lorna Vanderhaeghe FemmFssentials (Multiple only)56.5 62Solgar Omnium 56.5 63TwinLab Mega 6 Caps56.2 64Doctor's Nutrition UltraNutrient55.9 65Oregon Health Multi-Guard with CoQ1055 66Creative Nutrition Canada PLUS Formula54.8 67Doctor's Nutrition Mega Vites Senior 54.7 68Nature's Blend Vitaminix Ultimate54.7 69Nutrition for Life Grand Master Formula54.3 70Tyler Multiplex-2 without Iron54.2 71Bluebonnet Super Earth Formula54 72Pharmanex (Nu Skin)LifePak Prime52.7如新73Creative Nutrition Canada PRIME Formula52.4 74Body Wise International Right Choice AM/PM52.2 75Pilgrim's Sense of Energy51.7 76Amni Essential Basics51.5 77QCI Nutritionals Ultra Vitality #251.5 78Solgar Earth Source Multi-Nutrient51.4 79Essentials by Megafood Essentials for Life51.2 80KAL Multiple Energy51.2 81QCI Nutritionals Ultra Vitality #151.2 82genics Multigenics Intensive Care Formula51 83QCI Nutritionals Ultra Vitality #350.9 84genics Multigenics Intensive Care Formula without Iron50.5 85Options Androvite for Men50.1 86Solgar Female Multiple50.1 87All One Powder Multiple Vitamins and Minerals49.9 88KAL Enhanced Energy with Lutein, Iron-Free49.2 89Advanced Physician's Products Complete MuItiVttamin/Mineral with Iron49.1 90Advanced Physician's Products Complete MuItiVitamin/Mineral without Iron49.1 91SupraLife Maxum Essentials48.9 92Jarrow Mutti E-Z Powder48.7 93DaVinci Laboratories Spectra Vegetarian 48.6 94Great Earth TNT-Total Nutrition Tablet48.6 95QCI Nutritionals Daily Preventive #148.4 96QCI Nutritionals Daily Preventive #348.1 97Natural Factors MultiStart Men's 47.7 98Garry Null's Supreme Health Formula47.2 99Sisu Vegi-Mins 47.1 100GNC Ultra Mega Green46.9健安喜。
硫酸庆大霉素PLGA植入剂热融挤出

Fig.1:Effect of the admixture of PEG400on the glass transition temperature of PLGA and on the extrusion temperaturemization of the release profiles have been achieved inter alia by changes of the size of the dosage forms(Siepmann et al. 2004),by addition of monomers of the polymer(Yoo et al. 2004),by variation of the molecular weight(Kanellakopoulou et al.1999),by combining PLGA with other biodegradable polymers(Schmidt et al.1995)and of course by modify-ing the drug content(Cevher et al.2007).In this study,a plasticizer(PEG400)is added in different concentration to reduce the lag-phase and to gain a prolonged constant drug release.Gentamicin sulphate is an aminoglycoside broadspectrum antibiotic,which works by binding the50S subunit of the bacte-rial ribosome and hence interrupting the protein synthesis.The antibiotic is very potent and has a high temperature stability (Wang,Liu et al.2004).It is therefore often incorporated in polymer matrices via a melting process for extended drug release (Neut et al.2001).The production of the implants was performed by hot melt extru-sion(HME),an old manufacturing processfirst used in the plastics industry.Pharmacists have increasing interests in the HME technique as an alternative production method(Crowley et al.2007).The major advantages of this technique are that no solvents are involved in the process,only a few processing steps are needed,it is a continuous and well reproducible process, there are no requirements on the compressibility of the drug and the bioavaibility of the active ingredient can be increased enor-mously if it is molecularly dispersed in the polymer(McGinity et al.2007).Furthermore,hot melt extrusion allows produc-ing the favoured release behaviour by varying the composition of the formulations or by different geometry of the formed dosage form.The main disadvantage of HME is the thermal stress the active ingredient and the polymer are exposed to dur-ing processing(Breitenbach2002).In this study,biodegradable gentamicin sulphate loaded PLGA implants were produced via HME and PEG400was admixed as plasticizer to decrease the processing temperature and to affect the drug release behaviour beneficially.2.Investigations and results2.1.Plasticizer influence on glass transitionand extrusion temperatureGentamicin sulphate has no decreasing effect on the glass tran-sition temperature of PLGA(Fig.1).Therefore PEG400was added as plasticizer to produce the implants at lower extru-sion temperatures.With increasing PEG400-concentration up Fig.2:Drug content of the batches of all formulationsFig.3:Cumulative drug release of the20%GS batches with increasing PEG400 concentration compared to Septopal®to10%the glass transition temperature decreases linearly from 43◦C to about12◦C.Accordingly,it was possible to reduce the extrusion temperature from110◦C to85◦C and consequently minimize the temperature impact on the active ingredient.Nev-ertheless,the intense decrease of the glass transition temperature up to12◦C can eventually lead to stability problems during storage.2.2.Drug content of the extrudatesAs shown in Fig.2the measured drug contents reach well the specified contents with95%confidence intervals below 1.3%and each single extrudate exhibited an encapsulation effi-ciency of85%to115%.The p-values between the batches of the formulations exceeded0.1which indicates that there were no significant differences between the batches.Two exceptions were between0.05(>0.05indicates a significant difference)and 0.1,due to small confidence intervals.Thus,it can be stated that the manufacturing process led to reproducible implants.2.3.Release of gentamicin sulphate from melt extruded implantsFig.3shows the cumulative drug release of the20%gentam-icin sulphate batches with increasing concentration of PEG400 versus one Septopal®-sphere.After a burst effect,where about 20%of the drug amount is released due to the dissolving ofFig.4:Cumulative drug release of the25%GS batches with increasing PEG400 concentration compared to Septopal®gentamicin sulphate from the outer surface,water penetrates slowly into the extrudates and lets them swell.The drug that is further dissolved by intruding water diffuses into the release medium and thus generates pores which in turn accelerate the process of water penetration and drug liberation.In addition, the steady degradation of the polymer enhances drug release. PLGA hydrolytically decomposed into different products with chain lengths with free carboxyl groups at the end,which can-not penetrate out of the extrudate due to their size.Therefore the mass changes are only attributed to the loss in drug mass. The oligomers create an acidic microclimate inside the extru-date and hence accelerate the autocatalytic degradation process of PLGA(Shenderova et al.1999).In contrary the emerged frag-ments can diffuse away from the surface,so that the acidity here is neutralized by the buffer and the degradation of the surface is not autocatalysed.So,the outer layer is stable for a longer time.When the pores on the surface reach a size,where they allow an evacuation of the polymer fragments from the inside or when the oligomers degrade to dimers and monomers so that they can penetrate outside,a critical threshold is passed and the mass loss and the erosion of the extrudates starts(McGinity and O’Donnell1997).At this point the surface of the extru-dates can collapse and release an enlarged drug amount as can be seen at the20%gentamicin sulphate batch without plasti-cizer after approximately20days.By the end of the gentamicin sulphate release the liberation slows down until the drug is released almost completely after four tofive weeks.The increas-ing polyethylene glycol concentration does not significantly affect the release behaviour of the implants.In comparison to the release profile of the Septopal®-sphere,which releases gen-tamicin sulphate by diffusion from a non-biodegradable matrix system(polymethylmethacrylate)with release kinetics accord-ing to Higuchi the20%drug batches show more sigmoidal release curves.Although these batches release well above the minimal inhibitory concentration of Staphylococcus aureus,the main pathogen of osteomyelitis(Brady et al.2006),even over thefirst two weeks(data not shown),a higher drug release especially at the beginning of the antibiotic therapy would be preferable.The batches with25%gentamicin sulphate and increasing polyethylene glycol concentration,shown in Fig.4,exhibit a higher initial rate of release.After seven days about50%of the theoretical drug amount is liberated caused by the higher drug concentration yielding more pores.However,it is followed by a constant release over approximately four weeks.During this steady drug release the swelling of the extrudate and the liber-ation of the active ingredient equilibrate resulting in zero order Fig.5:Cumulative drug release of the30%GS batches with increasing PEG400 concentration compared to Septopal®kinetics with a coefficient of determination of0.99(formulation 4and6).The25%gentamicin sulphate batches,which again do not differ significantly with increasing plasticizer content, have release profiles equivalent to the Septopal®-sphere.Three extrudates with25%gentamicin sulphate are pharmaceutically equivalent to one Septopal®-sphere(containing7.5mg GS)and bioequivalence is anticipated by these release studies although this has to be verified in vivo.The release behaviour from30%drug content batches is shown in Fig.5.Basically,gentamicin sulphate is liberated quite fast, about55%,70%and80%drug respectively is set free depending on the plasticizer content after four days.In consideration of the fact that polyethylene glycol had no impact on the release rates of the batches with lower drug content,it can be assumed that the ebbing retarding effect of the batches is caused by the decreased PLGA-concentration.Although the30%gentamicin sulphate formulations combined with0%and5%plasticizer have the same PLGA-concentration as the25%drug batches combined with5%and10%PEG400,they show a less retarded drug release rate.This can be explained by the higher content of gentamicin sulphate,generating more pores and therefore triggering faster drug liberation.2.4.Water uptake and mass lossThe water uptake of all batches,ranked according to the PLGA-concentration,is illustrated in Fig.6.It becomes apparent that the water uptake corresponds mainly to the PLGA-content indepen-dent of further formulation ingredients.With a lower polymer Fig.6:Effect of the PLGA concentration on the water uptake of the extrudatesFig.7:Influence of the drug concentration and PEG 400concentration on the massloss of the extrudatescontent a less tighter matrix system can be build up and water can more easily infiltrate the extrudate,whereby it is unimpor-tant if gentamicin sulphate or polyethylene glycol are embedded between the polymer chains and spacing them apart.The smaller the PLGA-concentration is,the faster and the more water is taken up.While the water uptake was observed to be dependent on the polymer weight fraction,the mass loss is dependent on the drug amount (Fig.7).The 20%gentamicin sulphate batches initiallyshow a mass increase,probably generated by not completely freeze dried samples.Apart from that the other formulations exhibit a mass loss according to the released amount of drug.For example the 25%drug batches have a mass loss of approx-imately 16.5%after 16days which correlates properly to the drug amount released (approx.62%of 25%drug amount equals 15.5%mass loss).The mass loss data display the dependency of the 30%batches on the plasticizer content or rather on the remaining PLGA-concentration as well as the release curves.According to the faster drug liberation a higher mass loss is vis-ible with decreasing PLGA-concentration.The erosion of the polymer starts slowly after 16days and intensifies after approx.20days,which can also be observed in the increasing drug release of the 20%gentamicin sulphate batches.2.5.SEM-pictures of the extrudatesThe morphological changes during drug release are illustrated in the scanning electron pictures.The cutting zones of the extrudates,shown in Fig.8,reveal the included spherical gen-tamicin sulphate particles in a nonporous and homogeneous matrix leaving little holes after exposure to the release medium (Figs.9and 10).In Fig.10the more structural roughness of the surface compared to Fig.9denote the incorporated polyethylene glycol in the polymer.The surface of the extrudates changes over time to less smooth and more porous.This results from the degradation and after 21days erosion of the polymer becomesvisible.Fig.8:SEM-pictures of the cutting zones of a pure PLGA-extrudate (left)and a gentamicin sulphate loaded extrudate(right)Fig.9:SEM-pictures of the surfaces of extrudates with 25%gentamicin sulphate and 0%PEG 400after 1,16and 21days (left to right)exposure to phosphate buffer pH 7.4Fig.10:SEM-pictures of the surfaces of extrudates with25%gentamicin sulphate and10%PEG400after1,16and21days(left to right)exposure to phosphate buffer pH7.4Table:Composition of the formulationsBatch no.GS(%)PEG400(%)PLGA(%) 120080 220575 3201070 425075 525570 6251065 730070 830070 9301060 3.DiscussionAlthough the admixture of plasticizer decreases the glass tran-sition temperature of PLGA with increasing concentration and therefore the extrusion temperature of the blends,drug liberation is not affected up to a specific remaining PLGA-content,which is necessary to provoke a prolonged drug release.The release pro-files of the extrudates differ depending on the drug amount from sigmoidal to exponential release curves,whereby25%gentam-icin sulphate amount lead to an initial high drug release followed by zero order kinetics over about four weeks.These formula-tions are consequently a good alternative to Septopal®without the disadvantage of a second surgery,although further in vivo investigations have to be done.4.Experimental4.1.MaterialsAs biodegradable polymer Resomer®RG503H(Boehringer Ingelheim, Ingelheim,Germany)was used.Resomer®RG503H is a poly(D,L-lactic-co-glycolic acid)with a ratio of50:50,an intrinsic viscosity of 0.32–0.44dL/g and a free carboxylic acid as end group.The antibiotic gentamicin sulphate purchased from Caelo(Hilden,Germany)was used as active ingredient.The plasticizer polyethylene glycol400was obtained by BASF(Ludwigshafen,Germany).O-Phthaldialdehyde thioglycolic acid (Merck,Darmstadt,Germany)and sodium1-heptane-sulfonate(Sigma Aldrich,Taufkirchen,Germany)were used for the analytical methods. 4.2.Methods4.2.1.Manufacture of the implantsThe manufacture of the antibiotic loaded implants was performed by hot melt extrusion.Nine formulations(Table)with a drug content of20,25and30% gentamicin sulphate and plasticizer content of0,5and10%were produced. The polymer and the active ingredient were mixed with a turbula-blender (Willy A.Bachofen AG,Basel,Switzerland)for15min and subsequently manually granulated with PEG400for at least10min.These blends were hot melt extruded by a twin screw extruder(Minilab II,Thermo Fisher Sci-entific,Karlsruhe,Germany)with a die diameter of2mm.The temperature was set as low as possible between85◦C and110◦C depending on the plasticizer content.After cooling down,extrudates of9.8to10.2mg weight were produced by cutting.4.2.2.Drug contentTo determine the encapsulation efficiency,the extrudates were individually dissolved in dichloromethane.After addition of4mL water the samples were mixed to let the active ingredient migrate to the aqueous phase for at least one hour.The gentamicin sulphate content of the aqueous phase was analysed by HPLC.For determination of gentamicin sulphate by HPLC the drug has to be derivatisated as it does not possess UV absorbing chromophores.As derivatisation agent for pre-column derivatisation with UV detection o-phthaldialdehyde(OPA)was used.The o-phthaldialdehyd reagent was prepared as described in the USP XXXI.For derivatisation1.0mL of the sample was mixed with0.4mL of o-phthaldialdehyd solution and1.1mL of isopropanol,incubated at60◦C for15min and in afinal step cooled down to room temperature.The analysis of the drug content of the prepared samples was carried out using reversed phase HPLC with a HPLC system by the Agilent Series1100(Agilent Technologies,Santa Clara,USA).As station-ary phase a Merck LiChroCart125–4mm column,filled with LiChrospher 100RP18,5m and a pre column LiChroChart4–4,LiChroSpher100, 5mm(all Merck KGaA,Darmstadt,Germany)were used.The mobile phase was composed of700mL methanol,25mL water,50mL glacial acetic acid (all obtained from Merck KGaA,Darmstadt,Germany)and5g sodium 1-heptane-sulfonate.Theflow rate wasfixed at1.0mL per min,the column temperature was25◦C,the injection volume was50L and the detection wavelength was set to330nm.4.2.3.Differential scanning calorimetryThe thermal properties of the loaded implants were analysed with a differen-tial scanning calorimeter from Perkin Elmer,Waltham,Massachusetts,USA (DSC7).Samples of approximately7mg were weighed into aluminium pans,sealed hermetically and analysed under a nitrogen atmosphere with a heating procedure from10◦C to80◦C at a scan rate of10◦C/min related to an empty reference pan.4.2.4.Scanning electron microscopyTo analyse surface morphology SEM pictures were made with a Philips XL20(Philips B.V.,Eindhoven,The Netherlands)scanning electron micro-scope.The samples werefixed on a carbonfibrefilm and sputtered with gold in an argon atmosphere at a sputter current of50mA for180s using a SCD005Sputter coater(BalTec,Balzers,Liechtenstein).4.2.5.In vitro releaseFive gentamicin sulphate loaded extrudates of approximately10mg weight of each batch or one Septopal®-sphere(containing7.5mg(w/w)genta-micin sulphate)were placed into3mL phosphate buffer pH7.4(containing 250.0mL0.2M monobasic potassium phosphate solution with393.4mL 0.1M sodium hydroxide solution)as dissolution medium.The extrudates were incubated at37◦C and horizontally shaken.At a time interval of72h starting at day one the whole dissolution medium was collected for analyzing via HPLC and replaced by fresh buffer.This procedure was repeated until no more gentamicin sulphate could be detected.4.2.6.Water uptake and mass lossTo determine the swelling and the erosion of the extrudates water uptake und mass loss were analysed.Therefore implants were weighed exactly and placed in release medium over specific time periods.The implants were weighed wet after removing the water from the surface using soft wipes.After freeze drying(freeze dryer Alpha1-4LDC-1M,Martin Christ Gefriertrocknungsanlagen GmbH,Osterode,Germany)over two days the extrudates were weighed again to investigate the mass loss.The measure-ment of the water uptake und mass loss was only possible as long as the extrudates stayed in shape.ReferencesAmbrose CG,Clyburn TA,Louden K,Joseph J,Wright J,Gulati P, Gogola GR,Mikos AG(2004)Effective treatment of osteomyelitis with biodegradable microspheres in a rabbit model.Clin Orthop Relat Res421: 293–299.Ambrose CG,Gogola GR,Clyburn TA,Raymond AK,Peng AS,Mikos AG(2003)Antibiotic microspheres:preliminary testing for potential treatment of osteomyelitis.Clin Orthop Relat Res415:279–285.Böstman O,Pihlajamäki H(2000)Clinical biocompatibility of biodegrad-able orthopaedic implants for internalfixation:a review.Biomaterials21: 2615–2621.Brady RA,Leid JG,Costerton JW,Shirtliss ME(2006)Osteomyelitis:Clin-ical overview and mechanisms of infection persistence.Clin Microbiol Newsl28:65–71.Breitenbach J(2002)Melt extrusion:from process to drug delivery technol-ogy.Eur J Pharm Biopharm54:107–117.Castro C,Evora C,Baro M,Soriano I,Sanchez E(2005)Two-month ciprofloxacin implants for multibacterial bone infections.Eur J Pharm Biopharm60:401–406.Cevher E,Orhan Z,Sensoy D,Ahiskali A,Kann PL,Sagirli O,Mulazimoglu L(2007)Sodium fusidate-poly(d,l-lactide-co-glycolide)microspheres: preparation,characterisation and in vivo evaluation of their effectiveness in the treatment of chronic osteomyelitis.J Microencapsul24:577–595. Crowley MM,Zhang F,Repka MA,Thumma S,Upadhye SB,Battu SK, McGinity JW,Martin C(2007)Pharmaceutical applications of hot-melt extrusion:part I.Drug Dev Ind Pharm33:909–926.Flick AB,Herbert JC,Goodell J,Kristiansen T(1987)Noncommer-cial fabrication of antibiotic-impregnated polymethylmethacrylate beads. Technical note.Clin Orthop Relat Res223:282–286.Friess W,Schlapp M(2002)Release mechanisms from gentamicin loaded poly(lactic-co-glycolic acid)(PLGA)microparticles.J Pharm Sci91: 845–855.Garvin K,Feschuk C(2005)Polylactide-polyglycolide antibiotic implants. Clin Orthop Relat Res437:105–110.Jain RA(2000)The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide)(PLGA)devices.Biomaterials 21:2475–2490.Kanellakopoulou K,Kolia M,Anastassiadis A,Korakis T,Giamarellos-Bourboulis EJ,Andreopoulos A,Dounis E,Giamarellou H(1999)Lactic acid polymers as biodegradable carriers offluoroquinolones:an in vitro study.Antimicrob Agents Chemother43:714–716.Lazzarini L,Mader JT,Calhoun JH(2004)Osteomyelitis in long bones.J Bone Joint Surg Am86-A:2305–2318.Lew DP,Waldvogel FA(2004)ncet364:369–379. McGinity JW,O’Donnell PB(1997)Preparation of microspheres by the solvent evaporation technique.Adv.Drug Deliv Rev28:25–42. McGinity JW,Repka MA,Koleng JJ,Zhang F(2007)Hot-Melt Extrusion Technology.In:Swarbrick J(ed)Encyclopedia of phar-maceutical technology.New York,Informa Healthcare USA.3: 2004–2020.Miller RA,Brady JM,Cutright DE(1977)Degradation rates of oral resorbable implants(polylactates and polyglycolates):rate modification with changes in PLA/PGA copolymer ratios.J Biomed Mater Res11: 711–719.Neut D,van de Belt H,Stokroos I,van Horn JR,van der Mei HC,Busscher JH(2001)Biomaterial-associated infection of gentamicin-loaded PMMA beads in orthopaedic revision surgery.J Antimicrob Chemother47:885–891.Schmidt C,Wenz R,Nies B,Moll F(1995)Antibiotic in vivo/in vitro release,histocompatibility and biodegradation of gentamicin implants based on lactic acid polymers and copolymers.J Control Release37: 83–94.Shenderova A,Burke TG,Schwendeman SP(1999)The acidic microclimate in poly(lactide-co-glycolide)microspheres stabilizes camptothecins. Pharm Res16:241–248.Shive MS,Anderson JM(1997)Biodegradation and biocompatibil-ity of PLA and PLGA microspheres.Adv Drug Deliv Rev28: 5–24.Siepmann J,Faisant N,Akiki J,Richard J,Benoit JP(2004)Effect of the size of biodegradable microparticles on drug release:experiment and theory. J Control Release96:123–134.Soundrapandian C,Datta S,Sa B(2007)Drug-eluting implants for osteomyelitis.Crit Rev Ther Drug Carrier Syst24:493–545.Wahlig H,Dingeldein E,Bergmann R,Reuss K(1978)The release of gentamicin from polymethylmethacrylate beads.An experimental and pharmacokinetic study.J.Bone Joint Surg.Br.60-B:270–275.Wang G,Liu SJ,Ueng SW,Chan EC(2004)The release of cefazolin and gentamicin from biodegradable PLA/PGA beads.Int J Pharm273:203–212.Wei G,Kotoura Y,Oka M,Yamamuro T,Wada R,Hyon SH,Ikada Y(1991) A bioabsorbable delivery system for antibiotic treatment of osteomyelitis. The use of lactic acid oligomer as a carrier.J Bone Joint Surg Br73: 246–252.Yoo JY,Kim JM,Khang G,Kim MS,Cho SH,Lee HB,Kim YS(2004) Effect of lactide/glycolide monomers on release behaviors of gentamicin sulfate-loaded PLGA discs.Int J Pharm276:1–9.。
CiteSpace中文手册

干燥综合症评分最终版

ESSPRI的实验结果
1.多变量回归分析证明,干燥症状、肢体痛、疲劳与PGA符合度好 ,但是精神疲倦与PGA符合度较差。 2.患者认为最需要改善的症状:干燥症状( 38.7%)、疲劳( 32.2% )、肢体痛(19.6%),最不重要的为精神疲倦
/
3.在干燥症状中,口干、眼干被认为最重要
Glandular domain [2] Please be careful of not rating glandular swelling not related to the disease (such as stone or infection) No activity Low activity Absence of glandular swelling Small glandular swelling with: 1
PROFAD评分
Measurement of fatigue and discomfort in primary Sjogren's syndrome using a new questionnaire tool. Rheumatology (Oxford). 2004 Jun;43(6):758-64. Epub 2004 Mar 23.
2
Limited cutaneous vasculitis involve <18% body surface area; Diffuse Cutaneous vasculitis involve >18% body
surface area
Body surface area (BSA) is defined using the rules of nines (used to assess extent of burns) as follows: Palm (excluding fingers) = 1% BSA; each lower limb = 18% BSA; each upper limb = 9% BSA; torso (front) = 18% BSA; torso (back) = 18% BSA
欧洲标准变应原联合化妆品筛选变应原对女性面部皮炎患者的斑贴试验

圭堡医堂羞堂麓嶷盘查垫塑生!!魍整!§盎筮!塑£b虹LM趟△!!!b坠鱼§璎£!!Q£!Q鲢12塑2:YQl:;§:艘2:§欧洲标准变应原联合化妆品筛选变应原对女性面部皮炎患者的斑贴试验周成霞李械李利【摘要l蹦的采矮蚊溯标准嶷辙鳆联合化妆赫筛遴燮应原对女性llli鄙发炎患者进行斑贴试验,薅壹主要致竣藤;方法霹弱诊女瞧露帮安炎患者果弼讫绶曩蘩选变绽琢联合黢鞘标灌变应蘸送行褒雅试验,按嚣繇接魅建皮炎瓣究组捶荐标准巅潦缝祭。
结果4i灏焱褴患者逢幸亍了38静德妆酷筛选变虑原和26种欧洲标准变腹原的斑贴试验。
熊巾阳性率最高ff缸他妆鼎筛选变应原计裔鸟洛托品(12.20%)、硫柳汞(9.76%)、双咪唑烷基脲(7.32%)及DMDM海因(7.32%),阳性率躐高的欧洲标准嶷成原汁有硫酸镍(22.20%)、甲醛(14.63%)、对苯二胺(9.76%)及香料混骨芍势f9。
弱%)。
绥谂辘臻耱,争鏊、岛渗托瑟、骧秘汞、对答:藏、蚕辫浸合耢,双曝迹靛基骣,DMDM海毽等是女瞧嚣零瘦灸患毒主要簸被骧。
【关键词l欧渊标准变琏原;化妆品筛选变赢愿#巅部皮炎;疆髂试簸PatchtestingforselectionofcosmeticalllergensinfemalefaeialdermatitisbyEuropeanstandardofc08mmeticallergenszH(彤Cheng-xia,L{w武,乙iLi.DepartmentofDermatology,We啦ChinaHos“pitat,Si商#口镕汝iverMtY,酝ongdu§10041,China[Abstract]ObjectiveToidentifytheeomnqonallergensofthefemalepatientswkhfaeiaidef—matitiswithEuropeanstandardofcosmeticallergens.MethodsFemalepatientswithfaciaidermatitisweretestedwithEuropeanstandardofcosmeticallergens.ThereactionstoallergensweredocumentedhyfollowingtheInternationalContaetDermatitisResearchGrouprecommendations.ResultsTotal4lfemalepatientswithfacialdermatitisweretestedwithEuropeanstandardofcosmeticallergens,themaincosmeticallergensvcerehexamine(12,20),thimerosal(9。
《2024年丹皮酚(Pae)对高脂血清损伤人内皮细胞的保护作用及其分子机制》范文

《丹皮酚(Pae)对高脂血清损伤人内皮细胞的保护作用及其分子机制》篇一一、引言心血管疾病是全球范围内威胁人类健康的主要疾病之一,其中高脂血症是导致动脉粥样硬化和心血管疾病的重要因素。
在血管损伤中,内皮细胞起着至关重要的作用。
因此,研究高脂血清损伤人内皮细胞的保护机制及潜在的药物或治疗方法具有十分重要的意义。
丹皮酚(Pae)是从丹参等植物中提取出的活性成分,被认为具有抗氧化、抗炎等多种药理作用。
本研究旨在探讨丹皮酚对高脂血清损伤人内皮细胞的保护作用及其可能的分子机制。
二、研究方法本研究使用体外细胞实验,选用人内皮细胞作为研究对象,通过高脂血清诱导细胞损伤,并观察丹皮酚对细胞的保护作用。
采用荧光显微镜、流式细胞仪等手段进行细胞活性、凋亡、氧化应激等指标的检测。
同时,通过分子生物学技术如PCR、Western Blot等手段检测相关基因和蛋白的表达变化。
三、实验结果1. 丹皮酚对高脂血清损伤的人内皮细胞的保护作用实验结果显示,在受到高脂血清损伤后,人内皮细胞的活性明显降低,细胞凋亡率显著增加。
然而,在加入丹皮酚后,细胞的活性得到显著提高,细胞凋亡率明显降低。
荧光显微镜观察结果显示,丹皮酚能够显著减轻细胞内的氧化应激水平。
2. 丹皮酚对相关基因和蛋白表达的影响通过PCR和Western Blot等分子生物学技术检测发现,丹皮酚能够显著上调内皮细胞中抗氧化酶基因和蛋白的表达水平,如超氧化物歧化酶(SOD)和过氧化氢酶(CAT)。
同时,丹皮酚还能够抑制炎症相关基因和蛋白的表达,如NF-κB和COX-2等。
3. 丹皮酚的作用机制探讨根据实验结果,我们推测丹皮酚可能通过以下机制发挥对高脂血清损伤人内皮细胞的保护作用:首先,丹皮酚能够提高内皮细胞的抗氧化能力,减轻氧化应激对细胞的损伤;其次,丹皮酚能够抑制炎症反应,减少炎症介质对细胞的损害;最后,丹皮酚可能通过调节相关基因和蛋白的表达,从而发挥其保护作用。
四、讨论本研究表明,丹皮酚对高脂血清损伤人内皮细胞具有显著的保护作用。
USP 1132 HCP

Summary, Conclusions, and References (Guidances)
8
HCP Critical Reagents: the Antigen/Standard Design, Preparation, Characterization
Immunogen = (reformulated) standard
2
HCP Expert Panel Charter and Members
Provide stakeholders with best practices (USP chapter <1132>) for preparation and characterization of assay reagents, and development and validation of Host Cell Protein (HCP) measurement procedures (focus on immunoassays)
5
Draft Chapter Terminology, cont’d.
Process-Specific: A proprietary set of standards and antibodies used for a single product that fall in 2 major classes:
– Upstream process-specific: an assay designed from material where the upstream culture process deviates significantly from the platform. This is prior to any purification and may be applied to more than one product if these parameters are similar.
Interplay between the Cancer Genome and Epigenome

Leading EdgeReviewInterplay between the CancerGenome and EpigenomeHui Shen1and Peter ird1,*1USC Epigenome Center,University of Southern California,Room G511B,1450Biggy Street,Los Angeles,CA90089-9061,USA*Correspondence:plaird@/10.1016/j.cell.2013.03.008Cancer arises as a consequence of cumulative disruptions to cellular growth control with Darwinian selection for those heritable changes that provide the greatest clonal advantage.These traits can be acquired and stably maintained by either genetic or epigenetic means.Here,we explore the ways in which alterations in the genome and epigenome influence each other and cooperate to promote oncogenic transformation.Disruption of epigenomic control is pervasive in malignancy and can be classified as an enabling characteristic of cancer cells,akin to genome instability and mutation.IntroductionCancer develops through successive disruptions to the controls of cellular proliferation,immortality,angiogenesis,cell death, invasion,and metastasis.This evolutionary process requires new malignant traits to be stably encoded so that oncogenic events can accumulate in clonal lineages.Genetic mechanisms of mutation,copy number alteration,insertion,deletion,and recombination are particularly well suited as vehicles of persis-tent phenotypic change.For this reason,cancer has long been viewed as a disease based principally on genetics.Nevertheless, genetic events occur at low frequency and are thus not a partic-ularly efficient means for malignant transformation.Some cancer cells overcome this bottleneck by acquiring DNA repair defects, thus boosting the mutation rate.Mechanisms of epigenetic control offer an alternative path to acquiring stable oncogenic traits.Epigenetic states areflexible yet persist through multiple cell divisions and exert powerful effects on cellular phenotype. Although cancer cells have long been known to undergo epige-netic changes,genome-scale genomic and epigenomic anal-yses have only recently revealed the widespread occurrence of mutations in epigenetic regulators and the breadth of alterations to the epigenome in cancer cells(You and Jones,2012).It is now clear that genetic and epigenetic mechanisms influence each other and work cooperatively to enable the acquisition of the hallmarks of cancer(Hanahan and Weinberg,2011).Shaping the EpigenomeEpigenetic mechanisms allow genetically identical cells to achieve diverse stable phenotypes by controlling the transcrip-tional availability of various parts of the genome through differen-tial chromatin marking and packaging.These embellishments include direct DNA modifications,primarily CpG cytosine-5 methylation(Jones,2012),but also hydroxylation,formylation, and carboxylation(Ito et al.,2011),as well as nucleosome occu-pancy and positioning(Gaffney et al.,2012;Valouev et al.,2011), histone variants,and dozens of different histone modifications (Tan et al.,2011b),interacting proteins(Ram et al.,2011),and noncoding RNAs(Fabbri and Calin,2010;Lee,2012).These epigenetic marks do not act in isolation but form a network of mutually reinforcing or counteracting signals.Genome-scale projects charting the human epigenome are rapidly extending our understanding of epigenetic marks and how they interact (Adams et al.,2012;Dunham et al.,2012;Ernst et al.,2011).A key facet of epigenetics is that these marks can be stably maintained yet adapt to changing developmental or environ-mental needs.This delicate task is accomplished by initiators, such as long noncoding RNAs,writers,which establish the epigenetic marks,readers,which interpret the epigenetic marks, erasers,which remove the epigenetic marks,remodelers,which can reposition nucleosomes,and insulators,which form bound-aries between epigenetic domains.Epigenetic writers are directed to their target locations by sequence context,existing chromatin marks and bound proteins,noncoding RNAs,and/or nuclear architecture.Those marks are then recognized by reader proteins to convey information for various cellular functions.The establishment,maintenance,and change of epigenetic marks are intricately regulated,with crosstalk among the marks and writers to help guide changes to the epigenetic landscape. DNA MethylationDe novo methylation of DNA is catalyzed by the enzymes DNMT3A and DNMT3B and is then maintained by the major DNA methyltransferase DNMT1,with participation from DNMT3A and DNMT3B(Jones and Liang,2009).DNA methylation patterns are guided in part by primary DNA sequence context(Cedar and Bergman,2012;Lienert et al.,2011)and are influenced by germ-line variation(Gertz et al.,2011;Kerkel et al.,2008).Much of the mammalian genome consists of vast oceans of DNA sequence containing sparsely distributed but heavily methylated CpG dinucleotides,punctuated by short regions with unmethylated CpGs occurring at higher density,forming distinct islands in the genome(Bird et al.,1985).These CpG islands(CGIs)are pro-tected from DNA methylation in part by guanine-cytosine(GC) strand asymmetry and accompanying R loop formation(Ginno et al.,2012)and possibly also by active demethylation mediated by the TET family members(Williams et al.,2012).The unmethy-lated state of CpG islands in the germline,along with biasedgene38Cell153,March28,2013ª2013Elsevier Inc.conversion,helps to preserve CpG islands despite ongoing attri-tion of methylated CpG dinucleotides by cytosine deamination throughout most of the genome (Cohen et al.,2011).Transition zones between CpG islands and CpG oceans are called CpG shores and display more tissue-specific variation in DNA methyl-ation (Irizarry et al.,2009).CpG islands span the transcription start sites of about half of the genes in the human genome,largely representing genes that are either actively expressed or poised for transcription (Figure 1).Methylated DNA is recognized by methyl-CpG binding domains (MBD)or C2H2zinc fingers.The MBD-containing DNA methylation readers include MBD1,MBD2,MBD4,and MeCP2,whereas Kaiso (ZBTB33),ZBTB4,and ZBTB38proteins use zinc fingers to bind methylated DNA.MBDs and Kaiso are believed to participate in DNA methylation-mediated transcrip-tional repression of tumor suppressor genes with promoter DNA methylation.Histone ModificationsPosttranslational modifications of histones are coordinated by counteracting histone methyltransferases (HMTs)and demethy-lases (e.g.,KDMs),histone acetyltransferases (HATs)and de-acetylases (HDACs),and writers and erasers of phosphorylation,as well as many other modifications (Chi et al.,2010;Tan et al.,2011b ).These histone modifiers generally act in complexes,such as the repressive Polycomb (PcG)and activating Trithorax (TrxG)group complexes,which counterbalance each other in the regulation of genes important for development but which have also been implicated in cancer (Mills,2010).Polycomb repres-sive complexes (PRCs)are guided to their targets in part by intrinsic signals in the genome sequence (Ku et al.,2008;TanayFigure 1.Representative Epigenetic StatesExamples of representative epigenetic states are shown for several typical categories of genes and in different cellular contexts.(A)CpG-poor promoters are often tissue specific and/or reside in inducible genes that can be readily turned on or off.Transcription factor (TF)binding initiates nucleosome-depleted regions (NDR)at regulatory elements and at the promoter.(B)Many genes with CpG island promoters are constitutively expressed housekeeping genes.(C)Some genes with CpG island promoters,such as TF master regulators of differentiation and development,are repressed by the Polycomb complexes in stem cells and are kept in a bivalent state with both active and repressive marks.(D)Polycomb targets in stem cells are predisposed to cancer-associated promoter hypermethylation.Repressive marks are shown in red,and active marks are shown in blue.Cell 153,March 28,2013ª2013Elsevier Inc.39et al.,2007).The histone H3K27me3mark deposited by Poly-comb repressive complex 2(PRC2)provides docking sites for PRC1,whose enzymatic core unit RING1B monoubiquitinylates histone H2A at lysine 119(H2AK119ub1),thereby blocking RNA polymerase II elongation.The Trithorax group complex,contain-ing MLL,which lays down the H3K4methylation mark,counter-acts Polycomb function.The transcription factors encoding master regulators of differentiation and development are tar-geted by PRC2in embryonic stem cells and are held in a bivalent chromatin state poised for transcription,with both the activating H3K4me3and the repressive H3K27me3(Bernstein et al.,2006)(Figure 1).During differentiation,the Trithorax demethylase KDM6A/UTX removes the repressive H3K27me3mark,allowing transcription elongation to proceed for genes required in that particular lineage,whereas genes not required in that cell type lose the H3K4me3active mark and undergo spreading of theH3K27me3repressive mark (Hawkins et al.,2010).Other histone marks have various readers with binding motifs,including bro-modomain,PHD domain,chromodomain,and tudor domain (Musselman et al.,2012)(Figure 2).Trithorax and Polycomb complexes recruit HATs and HDACs,respectively,to counteract each other,and the establishment of histone acetylation can block Polycomb binding (Mills,2010).Histone VariantsHistone variants provide an additional layer of regulation.The main histone genes have multiple copies in the genome and are expressed during S phase.Single-copy variants are also ex-pressed at other phases of the cell cycle and have distinct func-tions and/or locations.H2A has the largest number of variants,including H2A.Z,MacroH2A,H2A-Bbd,H2AvD,and H2A.X (Ka-makaka and Biggins,2005).The H3variants include H3.3and centromeric H3(CenH3or CENP-A),as well as amammalianFigure 2.Histone H3Lysine Writers,Erasers,and ReadersAlthough many other important histone modifications also occur,only major histone H3lysine modifications (Ac:Acetylation;me1:monomethylation;me3:trimethylation)with well-defined functions are shown above a representative gene.The distribution of the marks is shown as colored bars and wedges to indicate approximate abundance.Repressive marks are shown in red,and active marks are shown in blue.Epigenetic regulators are listed to the right of each mark.Acetylation across different lysines shares writers and erasers,whereas methylation usually has dedicated enzymes.Readers (which can also be writers and erasers themselves)recognize different chromatin states and propagate the signal in various ways,including self-reinforcement or crosstalk,transcriptional activation or repression,or DNA repair.Crosstalk can also occur between histone modification and DNA methylation because DNMT3A ,DNMT3L ,and UHRF1all contain reader domains for chromatin states.40Cell 153,March 28,2013ª2013Elsevier Inc.testis-specific histone H3variant called H3.4.Nucleosomes con-taining H3.3and H2A.Z are located at dynamic regions requiring nucleosome mobility and exchange,such as at actively ex-pressed gene promoters(Jin et al.,2009).Wide presence of H2A.Z in embryonic stem cells(Zhu et al.,2013)suggests prev-alent chromatin exchange,which is consistent with the emerging idea that the genome of ESC is generally kept highly accessible. During differentiation,H2A.Z quickly redistributes.The mecha-nisms of recruitment have not been fully delineated,but various chromatin remodeler complexes and/or chaperones have been shown to be involved.For example,SRCAP is involved in H2A.Z loading into promoter/TSS,whereas H3.3is loaded to telomeric/pericentric regions by the ATRX/DAXX complex and promoter/TSS by HIRA(Boyarchuk et al.,2011).Nucleosome Positioning and RemodelingThe positioning of nucleosomes displays a weak10bp period-icity associated with minor sequence compositionfluctuations in phase with the DNA helical repeat.Some nucleosomes are more consistently positioned in phased arrays anchored by sequence-specific binding of proteins such as CTCF or adjacent to nucleosome-free regions at transcription start sites(Gaffney et al.,2012;Valouev et al.,2011).CpG islands have been asso-ciated with transcription-independent nucleosome depletion at mammalian promoters(Fenouil et al.,2012).ATP-dependent chromatin-remodeling complexes are responsible for sliding of the nucleosomes,as well as insertion and ejection of histone oc-tamers,processes that are important for transcriptional repres-sion and activation,and other important cellular functions such as DNA replication and repair.The remodeling complexes can be divided into four families:SWI/SNF,CHD(chromodomain and helicase-like domain),ISWI,and INO80(including SWR1, or SRCAP in mammals).InsulatorsThe CCCTC-binding factor CTCF and its paralog CTCFL/BORIS (expressed in the germline)are the only insulator proteins that have been identified so far in vertebrates.CTCF has a strong binding motif,and there is extensive overlap of the occupied CTCF-binding sites among different cell types(Kim et al., 2007).CTCF binds to enhancer blocking elements to prevent enhancer interactions with unintended promoters(‘‘enhancer blocking insulator’’)and also demarcates active and repressive chromatin domains(‘‘barrier insulator’’).Nuclear ArchitectureThe genome can be compartmentalized based on nuclear archi-tecture and associated genomic features into mostly heterochro-matic late-replicating regions attached to the nuclear lamina at the nuclear periphery and more gene-rich early-replicating regions closer to the nuclear interior(Dunham et al.,2012;Meule-man et al.,2013).Lamina-associated sequences(LASs)enriched for a GAGA motif are bound by transcriptional repressors and appear to contribute to the establishment of lamina-associated domains(LADs)in the mammalian genome(Zullo et al.,2012). Maintaining the Epigenetic StateThe persistence of epigenetic traits in a growing tumor requires that the epigenome be faithfully copied during cell division.The chromatin structure is dismantled for passage of the replication fork(RF).Newly synthesized DNA and histone octamers are then assembled at the RF by chromatin assembly factor I(CAF1),which is tethered to the RF by PCNA.Similarly,the dedicated maintenance DNA methyltransferase DNMT1and the euchro-matic H3K9methyltransferase G9a,among other epigenetic maintainers,are loaded to RFs and copy the epigenetic marks. The Trithorax and Polycomb complexes are recruited prior to replication and are distributed evenly to the mother and daughter strands at the RF,and they restore the correct marks on the daughter molecules during G1(Petruk et al.,2012).The his-tone marks are self-reinforcing and self-propagating,as PcG, SUV39H1/2,SETDB1,and TrxG all bind to the marks that they are responsible for catalyzing via an intrinsic reader domain or by interacting with a reader protein,thus helping to maintain the epigenetic state.Nucleosomes containing methylated DNA also stabilize DNMT3A/3B,which is a self-reinforcing mecha-nism for DNA methylation maintenance(Sharma et al.,2011). Disruption of Epigenetic Control in CancerMost studies of cancer epigenetics have focused on DNA meth-ylation as the epigenetic mark that most easily survives various forms of sample processing,including DNA extraction,and even formalinfixation and paraffin embedding(Laird,2010). However,other epigenetic marks also undergo broad changes, including long noncoding RNAs and miRNAs(Baer et al.,2013; Baylin and Jones,2011;Dawson and Kouzarides,2012;Sandoval and Esteller,2012)and histones,including loss of K16acetyla-tion and K20trimethylation at histone H4(Fraga et al.,2005; Hon et al.,2012;Kondo et al.,2008;Seligson et al.,2005;Yama-zaki et al.,2013).Loss of5-methylcytosine in cancer cells was dis-cussed more than three decades ago(Ehrlich and Wang,1981), with global DNA hypomethylation reported in cancer cell lines(Di-ala and Hoffman,1982;Ehrlich et al.,1982)and reduced levels of DNA methylation found at selected genes in primary human tumors compared to normal tissues(Feinberg and Vogelstein, 1983).The widespread loss of DNA methylation contrasted starkly with the subsequentfinding of hypermethylation of CpG islands in cancer(Baylin et al.,1986),including of promoter CpG islands of tumor-suppressor genes(Jones and Baylin, 2002).These seemingly contradictoryfindings have been widely reported for many types of cancer(Baylin and Jones,2011). The causal relevance of epigenetic changes in cancer was initially questioned,but this concern has now largely been laid to rest.First,many known tumor-suppressor genes have been shown to be silenced by promoter CpG island hypermethylation (Jones and Baylin,2002).Importantly,thefinding that these silencing events are mutually exclusive with structural or muta-tional inactivation of the same gene,such as the case for BRCA1in ovarian cancer(Cancer Genome Atlas Research Network,2011)and for CDKN2A in squamous cell lung cancer (Cancer Genome Atlas Research Network,2012a),reinforces the concept that epigenetic silencing can serve as an alternative mechanism in Knudson’s two-hit hypothesis(Jones and Laird, 1999).Second,mouse models of cancer have been shown to require epigenetic writers and readers for tumor development (Laird et al.,1995;Prokhortchouk et al.,2006;Sansom et al., 2003).Third,some DNA methylation changes appear to be essential for cancer cell survival,suggesting an acquired addic-tion to epigenetic alterations(De Carvalho et al.,2012).Finally, a plethora of significantly mutated epigenetic regulators haveCell153,March28,2013ª2013Elsevier Inc.41now been reported for many types of human cancer,as dis-cussed further below.Long-Range Coordinated Disruptions and Nuclear ArchitectureThe genome of undifferentiated embryonic stem cells is uniformly heavily methylated across CpG oceans,punctuated by unmethy-lated CpG islands.As stem cells differentiate and proliferate, the late-replicating lamin-associated domains(LADs)undergo progressive loss of DNA methylation within CpG oceans,and the LADs become recognizable as long partially methylated domains(PMDs),which become even more strikingly demar-cated as hypomethylated domains in cancer cells(Berman et al.,2012;Hansen et al.,2011;Hon et al.,2012;Lister et al., 2009).This loss of DNA methylation is associated with an increase of repressive chromatin with large organized chro-matin-lysine-(K)modification regions(LOCKs)(Hansen et al., 2011;Hon et al.,2012;Lister et al.,2009).CpG island hyperme-thylation is enriched in the hypomethylated domains,suggesting that these two events may be mechanistically linked but confined to distinct areas of the genome near the nuclear periphery(Ber-man et al.,2012).These long regions of DNA hypomethylation and repressive chromatin are consistent with prior reports of coordinated epigenetic silencing events located across mega-base distances,a phenomenon termed long-range epigenetic silencing(LRES)(Clark,2007;Coolen et al.,2010).It is noteworthy that the euchromatic part of the genome asso-ciated with the interior of the nucleus is generally much more epigenetically stable during cell differentiation,aging,and malig-nant transformation.However,loss of the DNA methyltrans-ferase Dnmt3a can promote tumor progression with uniform hypomethylation across the genome and moderate deregulation of genes in euchromatic regions(Raddatz et al.,2012). Disruption of Differentiation and Development Differences between cell types are guided by the expression of tissue-specific transcription factors and consolidation of associ-ated epigenetic states.Therefore,the epigenome of a cancer cell is determined in part by the cell of origin for that cancer and in-cludes passenger hypermethylation events at genes not required in that particular lineage(Sproul et al.,2012).Epithelial to mesen-chymal transition(EMT)of cancer cells is partly under reversible epigenetic control(De Craene and Berx,2013).For example, primary breast tumors display heterogeneous and unstable silencing of the CDH1(E-cadherin)gene,which facilitates the plasticity required during extravasation,metastasis,and estab-lishment of a solid tumor at the metastatic site(Graff et al.,2000). It has long been debated whether cancer cells arise by dedif-ferentiation or instead originate from stem cells or early progen-itors by a differentiation block.Polycomb repressors mark genes in stem cells encoding master regulators of differentiation and development,poised to either be turned on to coordinate differ-entiation of a lineage or to be fully repressed if it is not needed in that particular lineage(Bernstein et al.,2006).These genes occu-pied by Polycomb repressors in stem cells are particularly prone to acquiring CpG island hypermethylation during cell prolifera-tion,aging,and particularly malignant transformation(Ohm et al.,2007;Schlesinger et al.,2007;Teschendorff et al.,2010; Widschwendter et al.,2007)(Figure1).Although the genes affected by this process are primarily those not required or expressed in that particular cell lineage,cancer cells do also show evidence of silencing of genes essential for differentiation of their cell of origin(Berman et al.,2012;Easwaran et al., 2012;Gal-Yam et al.,2008;Mohn et al.,2008;Teschendorff et al.,2010).This predisposition of Polycomb target genes to aberrant permanent epigenetic silencing is consistent with a model in which stem cells slowly acquire irreversible silencing of poised master regulators required for successful differentia-tion.As a consequence,some stem cells lose their ability to properly differentiate while retaining their self-renewal capabil-ities and become attractive candidates for malignant transfor-mation by subsequent genetic and epigenetic events.One provocative implication of this model is that thefirst steps of oncogenesis may in some cases be an epigenetic defect affecting the differentiation capabilities of stem cells,as opposed to a gatekeeper mutation.Hematopoietic cell lineages and their corresponding malig-nancies also offer insights into the role of epigenetics in differen-tiation and transformation.For example,the DNMT3A gene is commonly mutated in human cases of acute myeloid leukemia (AML)(Ley et al.,2010;Yan et al.,2011),whereas loss of Dnmt3a in mice progressively impairs hematopoietic stem cell differenti-ation(Challen et al.,2012),suggesting that epigenetic perturba-tion can lead to differentiation block and subsequent malignant transformation.CpG Island Methylator PhenotypesAberrant DNA methylation of promoter CpG islands in cancer was initially viewed as a spontaneous or stochastic event with selection for functionally relevant silencing events.However, the discovery of cases of colorectal cancer with an exceptionally high frequency of CpG island hypermethylation suggested a coordinated event,possibly attributable to an epigenetic control defect.This phenomenon was referred to as a‘‘CpG island methylator phenotype’’(CIMP)(Toyota et al.,1999), analogous to the mutator phenotypes observed in mismatch repair-deficient cancers.Although the existence of CIMP sub-sets of cancer was initially disputed(Yamashita et al.,2003), more recent genome-scale analyses have unambiguously docu-mented distinct epigenetic subtypes for some types of cancer, such as colorectal cancer(Hinoue et al.,2012;Cancer Genome Atlas Network,2012b)and glioblastoma(Noushmehr et al., 2010),and not for others,such as serous ovarian cancer(Cancer Genome Atlas Research Network,2011).The most distinct examples of CIMP show exceptionally strong associations with other molecular or pathological features of the tumors,lending further validity to the biological relevance to this classification. For example,colorectal CIMP is very tightly associated with the V600E mutation of the BRAF oncogene(Weisenberger et al.,2006),whereas glioma CIMP(G-CIMP)is exceptionally tightly associated with mutation of the IDH1gene(Noushmehr et al.,2010).In the case of G-CIMP,IDH1mutation appears to be a causal contributor to the phenotype(Turcan et al.,2012), whereas BRAF mutation does not appear to be directly impli-cated in colorectal CIMP(Hinoue et al.,2009).The affected gene subsets differ between colorectal CIMP and glioblastoma G-CIMP,and their predisposition to aberrant methylation appears to be distinct from the susceptibility of stem cell poly-comb targets in lamin-attachment domains(Hinoue et al.,42Cell153,March28,2013ª2013Elsevier Inc.2012),which is generally not restricted to cancer subtypes. Despite a clear rationale for the association of IDH1mutation with G-CIMP,the mechanistic basis for the coordinated hyper-methylation events in most cases of CIMP is unknown and will remain an active area of investigation.Epigenetic Influences on Genomic IntegrityMutation rates vary strikingly across the genome,with strong local influences of base composition on single nucleotide varia-tion(SNV)and regional effects of sequence composition,chro-matin structure,replication timing,transcription,and nuclear architecture,among others,on both SNVs and structural alter-ations(Hodgkinson and Eyre-Walker,2011).Despite widespread misuse of the term in the literature,it should be recognized that mutation rates of a tumor cannot be inferred directly from observed mutation numbers or frequencies in a tumor without consideration of the number of cell divisions that have occurred since a shared reference genome,although comparisons across the genome obviate the need for Luria-Delbru¨ckfluctuation modeling and analysis.Epigenetic mechanisms can influence both the rates at which lesions arise and the rates at which they are repaired.For example,the epigenetic mark5-methylcytosine undergoes spontaneous deamination at higher rates than do unmethylated cytosines(Wang et al.,1982),whereas epigenetic silencing of the MLH1mismatch repair gene increases mutation frequencies by several orders of magnitude,providing an adap-tive advantage to mismatch repair-deficient cancer cells. Unmethylated and methylated cytosine residues both under-go spontaneous hydrolytic deamination but yield uracil and thymine,respectively.Uracil is not a normal constituent base in DNA and is repaired much more efficiently than thymine in a mismatch with guanine.As a consequence,the rate of C-to-T mutations in the context of CpG dinucleotides,most of which contain methylated cytosines,is about10-fold higher than any other SNV in the human genome(Hodgkinson and Eyre-Walker, 2011).This effect is particularly pronounced in highly proliferative tissues because deamination of5-methylcytosine in the parent strand just prior to DNA replication results in a full T:A base substitution that is not recognizable as a lesion for repair. Approximately a quarter of all TP53mutations in human cancer are thus attributable to this epigenetic mark(Olivier et al.,2010). Regional Effects of Chromatin OrganizationChromatin regulators play a role in maintaining genomic integrity (Papamichos-Chronakis and Peterson,2013),and regional chro-matin structure has a major impact on mutation frequencies. Megabase regions of repressive chromatin,represented by the H3K9me3mark,are positively correlated with single-nucleotide variations in cancer(Schuster-Bo¨ckler and Lehner,2012), whereas open chromatin associated with DNase I hypersensitive sites(DHS)have a lower inferred mutation rate,but this is partly due to evolutionary constraints on this compartment(Hodgkin-son and Eyre-Walker,2011).Transcription-coupled repair may also play a role in suppressing observed mutation frequencies in gene-rich euchromatic regions.Other types of mutation and structural change also appear to be associated with chromatin states.For example,retrotranspo-sition occurs more frequently in hypomethylated regions(Lee et al.,2012).Genes resistant to cancer-associated hypermethy-lation are more likely to have SINE and LINE retrotransposons near their transcription start sites than methylation-prone genes (Este´cio et al.,2010).Severe hypomethylation appears to be associated with genomic instability.Mouse models of DNA methyltransferase deficiency display chromosomal instability (Eden et al.,2003),and germline mutations of the DNMT3B gene cause ICF syndrome,characterized by centromeric insta-bility(Okano et al.,1999).Indeed,areas of hypomethylation in the human germline showed higher frequencies of structural mutability(Li et al.,2012).DNA breakpoints associated with somatic copy number alterations are also enriched in hypome-thylated domains(De and Michor,2011).Epigenetic Influences on DNA RepairDepletion of DNA methyltransferases causes increased micro-satellite instability(Guo et al.,2004;Kim et al.,2004),destabiliza-tion of repeats(Dion et al.,2008),and dramatically increased telomere length,telomeric recombination,and alternative telo-mere lengthening(Gonzalo et al.,2006).These effects of DNA methyltransferase depletion appear to be mediated in part by a drop in DNA repair proteins as part of DNA damage response (Loughery et al.,2011).The Dnmt1protein has also been shown to be recruited to areas of irradiation-induced DNA damage, possibly to facilitate repair of epigenetic information following DNA repair(Mortusewicz et al.,2005).It is increasingly appreci-ated that chromatin can serve as a cellular sensor for DNA damage and other genomic events(Johnson and Dent,2013). Epigenetic silencing of DNA repair genes such as MLH1, MGMT,BRCA1,WRN,FANCF,and CHFR can boost mutation rates and promote genomic instability in cancer cells(Toyota and Suzuki,2010).Familial cases of tumors with microsatellite instability(MSI)in Lynch syndrome result from germline muta-tions in mismatch repair genes,primarily MSH2and MLH1. However,most MSI-high tumors arise from an epigenetic defect in sporadic cases of cancer.Approximately15%of sporadic cases of colorectal cancer display MSI as a consequence of epigenetic silencing of the MLH1mismatch repair gene by promoter CpG island hypermethylation(Herman et al.,1998)in the context of CIMP(Toyota et al.,1999;Weisenberger et al., 2006).MSI caused by epigenetic silencing of MLH1has also been reported in other types of cancer,including about a quarter of sporadic endometrial cancers(Simpkins et al.,1999).Germline variants of MLH1and MSH2can predispose to extensive somatic epigenetic silencing of these genes and thereby increase cancer risk(Hitchins et al.,2011;Ligtenberg et al.,2009).Such familial cases of systemic epigenetic abnormalities can mas-querade as germline transmission of epigenetic defects.True transgenerational epigenetic inheritance is evident in genomic imprinting and in mouse models but has been difficult to demon-strate directly in human populations,although there is indirect evidence for its existence(Daxinger and Whitelaw,2012).The O6-Methylguanine DNA methyltransferase(MGMT)en-zyme repairs O6-alkylated guanine residues in genomic DNA. O6-methylguanine pairs with thymine and would lead to a G-to-A transition during DNA replication if left unrepaired. MGMT promoter methylation in colorectal cancer is associated with G-to-A mutations in KRAS(Esteller et al.,2000b)and in TP53(Esteller et al.,2001).Alkylating agents such as temozolo-mide are the current standard of care for malignant glioblastomaCell153,March28,2013ª2013Elsevier Inc.43。
RSC-2013soft matt-酰阱衍生物有机凝胶-支持数据
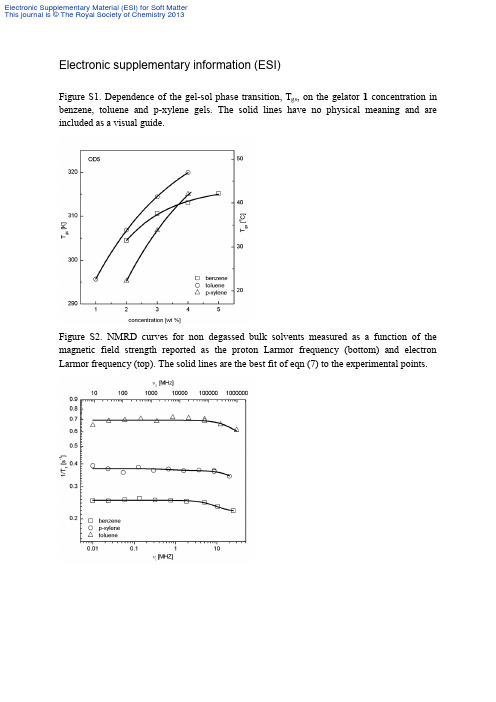
Electronic supplementary information (ESI)Figure S1. Dependence of the gel-sol phase transition, T gs, on the gelator 1 concentration in benzene, toluene and p-xylene gels. The solid lines have no physical meaning and are included as a visual guide.Figure S2. NMRD curves for non degassed bulk solvents measured as a function of the magnetic field strength reported as the proton Larmor frequency (bottom) and electron Larmor frequency (top). The solid lines are the best fit of eqn (7) to the experimental points.Table S1. Experimental and calculated vibrational transitions for gelator 1molecule; ν -wavenumbers (cm-1), I -IR intensity (km mol-1), A – Raman scattering activity (Å4 amu).Experiment(a) Calculation(b)Approximate description(d) νIR νRaman ννnor(c) I A2175w 164 158 5.4769 1.3468 def oop209vw 215 207 0.7117 5.7643 def ip245w 238 229 4.0521 2.3991 def oop279w 285 274 10.0547 1.7881 ring def oop318m 310 298 6.8261 7.2669 ring def oop334w 334 321 31.5746 2.2067 N-N-C b ip359w 359 345 4.1742 1.6383 benzene def oop376m 378 363 12.9336 6.3084 benzene def oop 426vw 426w 420 404 5.3618 5.724 benzene def oop466vw 462w 484 465 3.0401 5.3972 ring def ip488vw 487vw 514 494 116.4822 6.5129 N-H b oop519vw 522w 564 542 13.4105 6.9406 C-C-C b oop567w 617 593 2.0383 6.0833 C-C-N b ip 646w 650s 656 631 20.6637 14.7442 C-C-C b ip677vw 681w 681 655 10.4994 2.8726 C-C-C b oop713vw 714vw 733 705 10.0646 3.6574 C-C-C b oop756w 753s 770 740 13.5028 7.0123 C-N b oop+C-H b oop769sh 769w 774 744 9.9448 4.9048 C-N b oop+C-H b oop833w 852 819 1.6359 2.044 C-O str+CH2 rck846w 851m 860 827 20.7645 11.8539 NO2 scis+N-N-N b ip859vw 862m 872 838 5.6494 10.8539 CH2 rck917w 920w 952 915 16.1423 7.1553 C-C-C b ip+NH2 wag942w 943w 972 934 90.6577 11.413 C-H b oop+CH2 rck977w 980vw 1029 989 3.7884 10.325 C-N str1016vw 1060 1019 34.1106 18.4477 C-N-C b ip 1044w 1045w 1084 1042 12.9285 2.4798 CH2 rck1067w 1068vw 1098 1056 22.2418 4.8487 C-N str1081vw 1080w 1106 1063 2.3469 6.8098 C-N str+NH2 wag1113m 1112w 1131 1087 90.8016 13.079 C-O str+C-C str1118sh 1122vw 1168 1123 20.8797 7.7378 C-N str+C-H b ip1150vw 1150vw 1196 1150 54.4868 20.0334 C-N str+C-H b ip1176vw 1178sh 1216 1169 11.9833 73.1072 N-N str+C-C str1191w 1191m 1238 1190 18.2149 16.6461 CH2 tw1215w 1218m 1260 1211 171.0506 26.8502 C-N str+CH2 tw1236m 1241m 1281 1231 50.7907 5.9455 C-H b ip+CH2 tw1257w 1261w 1302 1252 228.5949 139.6885 C-C str+N-N str+C-H b ip1276w 1281s 1314 1263 28.9892 39.1447 C-C str+C-H b ip1293w 1290sh 1327 1276 57.652 24.3401 CH2 tw1302w 1299s 1333 1281 33.8362 24.4048 CH2 tw1333m 1351 1299 8.0097 6.8558 NH2 tw1345m 1348vs 1361 1308 205.3756 108.2845 C-N str1353sh 1413 1358 93.8967 4.1007 C-N str+CH2 wag1380w 1375sh 1436 1380 21.9244 28.5344 C=C str+C-N str1450w 1449vw 1490 1432 187.7616 29.9071 N-H b ip+CH2 scis1457sh 1464vw 1502 1444 94.2035 4.4488 N-H b ip+CH2 scis1496m 1496vw 1539 1479 29.2301 31.0322 C-N str1521vs 1521vw 1566 1505 69.9167 76.6854 C=C str+N=O asym str1555sh 1555m 1584 1523 210.2549 21.457 C=C str+N=O asym str1616vs 1619m 1652 1588 323.6646 264.2258 C=C str1636m 1705 1639 122.068 3.9605 NH2 scis+C=O str1657m 1650m 1724 1657 191.4801 170.8803 C=O str+NH2 scis2832vw 2961 2846 45.8613 137.7087 CH2 sym str2860w 2863vw 2972 2857 61.2436 235.341 CH2 sym str2898vw 2896vw 2992 2876 48.4888 65.512 CH2 sym str2923vw 2924vw 3017 2900 36.0166 68.4949 CH2 sym str2963vw 2968vw 3093 2973 13.3159 45.4619 CH2 asym str3043vw 3157 3035 6.9933 29.2384 CH2 asym str 3077vw 3080vw 3196 3072 0.1211 52.6515 C-H str in benzene3102vw 3100vw 3215 3091 5.4163 115.6617 C-H str in benzene3310m 3311vw 3461 3327 2.2865 197.7792 NH2 sym str3343m 3340vw 3527 3391 8.5385 75.331 NH2 asym str3480sh 3621 3481 43.4442 41.3151 N-H str(a) vs=very strong, s=strong, m=medium, w=weak, vw=very weak, sh=shoulder.(b) Calculations have been performed for OD5 at BLYP/6-311++G(d,p) theory level.(c) Calculated frequencies were scaled by the factor of 0.9613 [Wong1996].(d) Approximate mode description: str=stretching, def=deformation, b=bending, wag=wagging, tw=twisting, rck=rocking,scis=scissoring, ip=in-plane, oop=out of plane, sym=symmetrical, asym=antisymmetrical.Table S2. Excitation energies (∆E in eV), wavelength (λ in nm), oscillator strengths f, and characterization of the excited singlet states for the gelator molecule.Experiment Theory(a)Charakter(b)∆Eλ∆Eλf3.12 398 3.02 411 0.0411 HOMO→LUMO (91%) π→π*4.28 290 4.28 290 0.1914H-7→LUMO (10%), H-3→LUMO(30%), HOMO→L+1 (46%)π→π*+n→π* 4.75 261 0.1328H-7→LUMO (21%), H-6→LUMO(15%),H-4→LUMO (26%)5.17 240 5.17 240 0.0476 H-10→LUMO (20%), H-9→LUMO(20%),H-7→LUMO (26%), H-6→LUMO(12%)π→π*+n→π*(b)H means the highest occupied molecular orbital (HOMO), L means the lowest unoccupied molecular orbital (LUMO)。
体外重建人体皮肤模型刺激试验的验证

23 重 建皮 肤模型 特 征 的分析 . 重 建 皮 肤 模 型 的特 征 分析 重 点应 放 在 组 织 形
到 的 目标 值 ,也 即 当评 估 使 用 参 考 物质 最 小 清 单
时 , 实 验 方 法 应 满足 的标 准 。
态 、脂 质 组 成 和 屏 障 功 能 等 方 面 。 231 形 态特征 分析 .. 除常 规组 织学 技术 外 ,还可 以采 用分 子生物 学技 术或 免疫组化 技 术分析 细胞 分
31 实验 方 法 的科 学 原理和 局 限性 . 皮 肤刺激 的发生 是 由于刺 激物质 破坏皮 肤角质 J 拜障进入 表 皮层 ,并 且对 底 层细 胞 产 生细 胞 毒性 ,
而细胞 培养 与组织 重建 技术 的发展 使得 体外 制备 的 皮肤替 代模 型具有 与体 内相 似 的组织 结构和 屏 障功 能【。这样 就可 以在 体外 进 行皮 肤刺 激 性 的测试 和 其它 相 关研 究 。而 且与 单 层细 胞 不 同 ,3 皮 肤模 D
脂 类 进行 半 定 量分 析 是 一项 有 效 简 便 的方 法 。
围 为 48h 87h。E I KI M模 型 用 S S作 用 . ~. P S N丁 L
1 , 均 I 5为 23 / , 受范围 为 1 / ~ 8 平 h C 0 . mgml接 2 .mg 0 ml
性 ,关 键 步 骤 描述 和 质 量 控 制 ,这 些 内容 可 以确
保新 方法 所基于 的原 理和 预期 目的相 一致 。 要素 二 是参考 化 学物清 单 ,用于 评价 实验 方法 的准确 性和
可 靠性 。要 素三 是 可靠 性和 预测 能 力( 确性 ) 准 应达
特 定 毒 理 学 终 点 的测 试 , 如 应 用 于 皮 肤 光 毒 性 、 皮肤 刺 激性或 皮肤 吸收 时应满 足对 己知化 学物质 正
重组猪源抗菌肽Cecropin P1在毕赤酵母中的分泌表达与活性研究

抗 菌肽 ( er pn 是 一类 被 称 为“ 二 防御 体 系 ” C co i) 第 的生 物 活性 小 肽 Ⅲ , 有 广 谱 、 】具 ] 高效 的 抑 菌 活性 , 能攻
ATC 5 2 标 准 株 , C2 9 2 自行保 存 ; ihap s rsX 3 、 P c i a t ‘ 一3 oi
0 5 甲醇 ,. Ag r d H2 . 15 a ,d O。 1 2 试 剂 . 限 制 性 内切 酶 Hi m、 o 、 a 、 v 、 分 子 na Nc S c A r1低 I I I
确 翻译 和 翻译 后加 工 , 而且 能分 泌许 多 蛋 白质产 物 , 使
生 物 素,
内二硫 键 , 有强 碱 性 的 N一 和 强 疏 水 性 的 C 端 , 一 端 _ c 端
酰胺 化 。其 分 子 内含有 两 亲性 一 旋 , 间 是形 成 柔 螺 中
tat 1 3 YNB, ×1 rc, . 4 4 0
生 物素 ,. 甘 油 , 0 05 10
mmo ・ 磷 酸钾缓 冲溶 液 ( H 值 6 0 。 l L p . ) B MMY 培 养 基 : P lp po e 1 Yes x 2 oy e tn , ate ~
收 集 菌体 , 上 清 , 菌 体 重 悬 于 B 弃 将 MMY 培养 基 中, 使 其 oD 。为 1 然后继 续在 3 。 , O℃摇 床 培养 , 每隔 2 4h
向B MMY培 养基 中加 入 甲醇至 终浓 度为 0 5 , 按 . 并
照 2 、6h 4 、 0h 7 、4 h 9 、 2 4h 3 、8h 6 、 2h 8 、 6h 1 0h时 间
游 引 物进行 鉴定 。 1 3 4 阳性 菌 株 的诱 导 表达 . .
定量药理学名词中英对照表

English中文absolute prediction error(s) (APE)绝对预测误差absorption, distribution, metabolism, elimination (ADME)吸收、分布、代谢、消除active transport主动转运adaptive design自适应性设计additive error加和性误差adherence依从性administration给药affinity亲和力agonist激动剂allometric scaling异速生长antagonist拮抗剂area under curve (AUC)曲线下面积assumptions假设auto-induction自诱导backward elimination逆向剔除法base model基础模型baseline基线below the limit of quantification (BLQ)低于定量下限between-subject variability (BSV)个体间变异bias偏差biliary clearance胆汁清除率bioavailability生物利用度bioequivalence生物等效性biomarker生物标志物biopharmaceutics classification system (BCS)生物药剂学分类系统blood血body mass index (BMI)体质指数body surface area (BSA)体表面积bolus推注bootstrap自举法bottom-up appraoch自下而上的模式capacity-limited metabolism能力限制型代谢categorical data分类数据catenary compartment model链式模型causality因果chi-square test卡方检验clearance清除率Clinical trial simulation临床试验模拟clinical utility index临床效用指数Cmax峰浓度coefficient of variation (CV)变异系数Compartmental analysis房室模型分析competitive inhibition竞争性抑制compliance依从性concomitant medication effect联合用药效应condition number条件数conditional probability条件概率conditional weighted residuals (CWRES)条件加权残差confidence interval置信区间constitutive model本构模型continuous data连续数据convergence收敛correlation相关correlation coefficient相关系数correlation matrix相关矩阵count data计数数据covariacne matrix协方差矩阵covariance协方差covariate evaluation协变量评价covariate model协变量模型creatinine clearance肌酐清除率cross-over design交叉设计data analysis plan数据分析计划dataset assembly/construction数据集建立dataset specification file数据库规范文件degrees of freedom自由度dependent variable (DV)因变量determinant行列式deterministic identifiability确定性可识别性deterministic simulation确定性模拟diagonal matrix对角矩阵dichotomous二分类direct-effect model直接效应模型discrete离散disease progression疾病进程disease-modifying effect疾病缓解效应dose dependence剂量依赖性dose-normalized concentrations剂量归一化浓度double-blind双盲drug accumulation药物蓄积drug-drug interaction药物-药物相互作用duration of infusion输注持续时间efficacy功效eigenvalues特征值empirical Bayesian estimates (EBEs)经验贝叶斯估计endogenous内源性enterhepatic circulation肝肠循环estimate估计值estimation求参exogenous外源性exploratory data analysis (EDA)探索性数据分析exponential指数型external validation外部验证extrapolation外推extravascular administration血管外给药fasted禁食fed进食first in human (FIH) trial首次人体试验first-order absorption一级吸收first-order conditional estimation method (FOCE)一阶条件估计法first-order method (FO)一阶评估法first-pass effect首过效应Fisher information matrix Fisher信息矩阵fixed effect固定效应flip-flop翻转forward selection前向选择fraction of unbound (fu)游离分数full agonist完全激动剂gastric emptying胃排空generic products仿制药genetic polymorphism遗传多态性genome-wide association study (GWAS)全基因组关联研究genotype基因型global minimum全局最小值global sensitivity analysis全局敏感性分析glomerular filtration rate (GFR)肾小球滤过率goodness of fit拟合优度gradient梯度half maximal inhibitory concentration (IC50)半数抑制浓度half-life半衰期hepatic clearance肝清除率hierarchical层级homeostasis稳态homoscedasticity方差齐性hysteresis滞后identity matrix单位矩阵ill-conditioned matrix病态矩阵immunogenicity免疫原性in silico经由电脑模拟in situ原位in vitro体外in vivo体内independent variable自变量indirect response model间接反应模型individual parameter estimates个体参数估计individual prediction (IPRED)个体预测值individual residuals (IRES)个体残差individual weighted residuals (IWRES)个体加权残差infusion输注initial estimate起始参数估计inter-individual variability (IIV)个体间变异internal validation内部验证inter-occasion variability场合间变异interpolation插值intestinal absorption肠道吸收intra-individual variability个体内变异intramuscular administration (i.m.)肌肉注射intravenous administration (i.v.)静脉给药intrinsic clearance内在清除率inverse agonist反向激动剂inverse of matrix逆矩阵isobologram等效线图Jacobian matrix雅可比矩阵lag time滞后时间large-scale systems model大型系统模型lean body weight瘦体重level 1 random effect (L1)一级随机效应level 2 random effect (L2)二级随机效应ligand-receptor binding配体-受体结合likelihood ratio test似然比检验linear models线性模型linear pharmacokinetics线性药物动力学local minimum局部最小值local sensitivity analysis局部敏感性分析locally weighted scatterplot smoothing (LOWESS)局部加权散点平滑法logistic regression Logistic回归logit transform Logit变换log-normal distribution对数正态分布log-transformation对数变换maintenance dose维持剂量marginal probability边际概率mean均值mean absolute prediction error percent (MAPE)平均绝对预测误差百分比mean prediction error (MPE)平均预测误差mean residence time (MRT)平均滞留时间mean squared error (MSE)均方误差mechanism-based inhibition基于机制的抑制median中位数Michaelis-Menten constant米氏常数Michaelis-Menten kinetics米氏动力学missing dependent variable (MDV)缺失应变量mixed effect混合效应mixture models混合模型model diagnostic plots模型诊断图model evaluation模型评价model misspecification模型错配model specification file (MSF)模型规范文件model validation模型验证Model-based drug development基于模型的药物研发moment矩Monte Carlo simulation蒙特卡洛模拟multivariate linear regression多元线性回归negative feedback负反馈nested嵌套Non-compartmental analysis非房室模型分析noncompetitive inhibition非竞争性抑制nonlinear mixed effect models (NONMEM)非线性混合效应模型nonlinear pharmacokinetics非线性药物动力学normal distribution正态分布normalized prediction distribution errors (NPDE)归一化预测分布误差numerical predictive check (NPC)数值预测性能检查objective function value (OFV)目标函数值observation观测occupancy占有occupational model受体占有模型one-/two-compartment model一/二室模型onset of effect起效operational model操作模型optimal sampling最优采样Optimal study design优化试验设计oral口服ordered data有序数据outlier离群值parallel design 平行设计partial agonist部分激动剂peak concentration峰浓度perfusion灌注permeability渗透性Pharmacodynamics药效动力学Pharmacogenomics药物基因组学Pharmacokinetics 药物动力学Pharmacometrics定量药理学phase I reaction第一相反应phase II reaction第二相反应phenotype表型Physiologically based pharmacokinetics (PBPK)生理药物动力学piecewise linear models分段线性模型placebo安慰剂plasma血浆Poisson distribution泊松分布Poisson regression泊松回归population pharmacokinetics群体药物动力学positive feedback正反馈post hoc事后posterior distribution后验分布posterior predictive check (PPC)后验预测性能检查posterior probability后验概率potency效价强度power function幂函数precision精密度pre-clinical study临床前研究prediction (PRED)群体预测值prediction error (PE)预测误差prior distribution先验分布prodrug前药proof of concept study概念验证研究proportional error比例型误差Q-Q plot分位图quality assurance (QA)质量保证quality control (QC)质量控制random effect随机效应randomisation 随机化rate constant速率常数rate-limiting step限速步骤reference group对照组relative bioavailability相对生物利用度relative standard deviation (RSD)相对标准偏差relative standard error (RSE)相对标准误renal clearance肾清除率reparameterization重新参数化repeat dose重复剂量resampling重采样residual (RES)残差residual unexplained variability (RUV)残留不明原因的变异·rich sampling密集采样robust鲁棒性root mean square error (RMSE)均方根误差rounding errors舍入误差saturable可饱和的semi-logarithmic plot半对数图shirinkage收缩signalling transduction信号转导simulation模拟single dose单剂量singular奇异sparse sampling稀疏采样standard error (SE)标准误steady state (SS)稳态stochastic simulation随机模拟stratification分层structural identifiability结构可识别性subcutaneous administration (s.c.)皮下注射superposition叠加surrogate endpoint替代终点survival analysis生存分析symptomatic effect对症疗效synergism协同作用Systems pharmacology系统药理学target-mediated drug disposition靶点介导的药物处置therapeutic drug monitoring (TDM)治疗药物监测therapeutic index治疗指数time after dose (TAD)给药后时间time varying时间变化time-to-event analysis事件史分析tissue组织titration design滴定式设计tmax达峰时间tolerance耐受性top-down approach自上而下的模式total body weight总体重transit compartment model中转室模型transporter转运体transpose转置trough concentration谷浓度tubular reabsorption肾小管重吸收tubular secretion肾小管分泌turnover置换typical value paramters参数的群体典型值uncompetitive inhibition反竞争性抑制variance-covariance matrix方差协方差矩阵visual predictive check (VPC)可视化预测性能检查volume of distribution表观分布容积weighted residuals (WRES)加权残差well-stirred model充分搅拌模型within-subject variability个体内变异zero-order absorption零级吸收。
An Estimation Model for Test Execution Effort

An Estimation Model for Test Execution EffortEduardo Aranha1,2ehsa@cin.ufpe.br1Informatics Center Federal University of Pernambuco PO Box7851,Recife,PE,Brazil+55812126-8430Paulo Borba1phmb@cin.ufpe.br2Mobile Devices R&DMotorola Industrial LtdaRod SP340-Km128,7A-13820000 Jaguariuna/SP-BrazilAbstractTesting is an important activity to ensure software qual-ity.Big organizations can have several development teams with their products being tested by overloaded test teams.In such situations,test team managers must be able to properly plan their schedules and resources.Also,estimates for the required test execution effort can be an additional criterion for test selection,since effort may be restrictive in practice. Nevertheless,this information is usually not available for test cases never executed before.This paper proposes an estimation model for test execu-tion effort based on the test specifications.For that,we de-fine and validate a measure of size and execution complexity of test cases.This measure is obtained from test specifica-tions written in a controlled natural language.We evalu-ated the model through an empirical study on the mobile application domain,which results suggested an accuracy improvement when compared with estimations based only on historical test productivity.1.IntroductionIn competitive markets,such as the mobile phone mar-ket,companies that release products with poor quality may quickly lose their clients.Consequently,companies should ensure that their products conform to the clients’expecta-tions.A usual activity performed to ensure software quality is testing.Software testing is being considered so important that organizations can assign teams only for test activities.For example,a big organization may have several development teams with their products being tested by few test teams.In such situations,test managers must be able to properly plan their schedules and resources.They must should be able to estimate the required effort to execute a given set of tests (test suite)and to justify requests for more resources or for extending deadlines.In addition,Model-based testing(MBT)has become popular in recent years.MBT is a technique for generat-ing test cases from system specifications[15].Using MBT, a high number of test cases can be automatically generated. However,it may not be possible to execute all generated test cases,since test resources are limited.For this reason,test cases are usually selected using some criteria such as the coverage metrics[19][16].An additional criterion that can be useful for test selec-tion is the execution effort,since effort may be restrictive in practice.However,in this case,we need a model that esti-mates the effort to execute each test case individually,even for test cases never executed before.Several software development estimation models have been proposed over the years.However,these models are not appropriate to estimate the effort for executing a given set of test cases,since their estimations are based on soft-ware size and development complexity,instead of test size and execution complexity.In this work,we address the problem of supporting test managers to plan their schedules and resources.We propose a test execution effort estimation model that is based on test specifications.We define and validate a measure of test size and execution complexity.This measure is obtained from test specifications written in a controlled natural language. Also,we want to provide execution effort estimates about generated test cases as an additional criterion for test selec-tion.The rest of this paper is organized as follows.In Sec-tion2,we discuss about existent software estimation mod-els.After,Section3introduces a controlled natural lan-guage used for specifying tests.In Section4,we propose a measure of test size and execution complexity and present a model for estimating test execution effort based on thisFirst International Symposium on Empirical Software Engineering and Measurementmeasure.After that,Section5presents the results of an empirical study on the mobile application domain.Then, we discuss about the cost of using our model in Section6. Finally,our conclusions are presented in Section7.2.Existing SW effort estimation modelsDuring the last few decades,several models and tech-niques were created for estimating size,complexity and ef-fort on software development.The surveys presented in[2], [12]and[7]summarize the software estimation evolution so far.Some of the related and renowned software estimation models are discussed here.Thefirst model discussed here is Function Points Analy-sis(FPA)[6].FPA gives a measure of the size of a system by measuring the complexity of system functionalities offered to the user.The size of system is determined in function points(FP),a unit-of-work measure,and this count is used for estimating the effort to develop it.The Use Case Point Analysis(UCP)[11]is an extension of FPA and estimates the size of a system based on use case specifications.Both UCP and FPA regard the development complexity of a system,while our proposed model regards the size and execution complexity of test cases.The Constructive Cost Model(COCOMO)[3]converts size measures such as FP and SLOC(source lines of code) into effort estimation for developing systems.Its formula uses effort multipliers and scale factors,and their values are defined according to the characteristics of the development environment,teams and processes used in the project.Similar to UCP,Test Point Analysis[13]is a method for estimating the effort required to perform all functional test activities based on use case points.This model estimates the effort required for all test activities together,such as defin-ing,implementing and executing all the tests.For example, it is not possible to estimate only the effort to execute test cases that were automatically generated.3.Test specification languageTests are usually specified in terms of precondition,pro-cedure(list of test steps with inputs and expected outputs) and post-condition[8].These specifications are commonly written in natural language,often leading to problems such as ambiguity,redundancy and lack of writing standard.All these problems make difficult test understanding and exe-cution complexity estimation.Nevertheless,they can be avoided using controlled natural languages.A controlled natural language(CNL)[17]is a subset of natural language with restricted grammar and lexicon in or-der to have sentences written in a more concise and standard way.This restriction reduces the number of possible ways to describe an event,action or object.The test specifications considered by this work are writ-ten using a CNL described here.In a simplified way,each sentence(test step)in the specification conforms to the fol-lowing structure:a main verb and zero or more arguments. Table1shows an example of test procedure written in a con-trolled natural language defined for the mobile application domain.Table1.Example of a test procedure writtenin a controlled natural language.Step Description Expected Results1Start the message cen-ter.The phone is in mes-sage center.2Select the new mes-sage option.The phone is in mes-sage composer.3Insert a recipient ad-dress into the recipi-entsfield.The recipientsfield isfilled.4Insert a SMS contentinto the message body.The message body ispopulated.5Send the message.The send messagetransient is displayed.The message is sent.The verb identifies the action of the test step to be per-formed during the test.The arguments provide additional information about the action represented by the verb.For instance,the sentence Start the message center has the verb start(action of starting an application)and the required ar-gument the message center(application to be started).The CNL can have its lexicon and grammar extended for specific application domains.For example,the list of possible verbs and arguments may be different between the mobile and the Web application domains.The context of this work is related to testing mobile ap-plications for Motorola Brazil Test Center site at the Infor-matics Center/UFPE.Hence,in this work,the considered controlled natural language reflects this domain[18][9].4Test execution effort estimation modelIn this section,we present a new test effort estimation model developed during our research.As illustrated by Fig-ure1,the input of our estimation model is a test suite and the output is the estimated effort in man-hours required to execute all tests in the suite.Our test execution effort estimation model works as fol-lows.First of all,(1)we analyze each test case in the suite. During this analysis,(2)we assign to each test case a num-ber of execution points,a unit of measure defined in thisFigure1.Estimating the effort to execute atest suite.work for describing the size and execution complexity of test cases.After that,(3)we sum all the execution points measured from the analyzed test cases.This total describes the size and execution complexity for the whole test suite.Finally, (4)we estimate the required effort in man-hours to execute all tests in the test suite based on the total number of execu-tion points.Next,we present the details about this estimation model.4.1Test size and execution complexityOur estimation model is based on the size and execution complexity of test cases in a test suite.Test size means the amount of steps required to execute the test.Test execution complexity is related to the relationship(complexity of in-teraction)between the tester and the tested product required during the test.These definitions are adaptations of the idea of size and development complexity for software products [14][4][5].Since we are proposing a measure of size and execution complexity of a test case,it is important to have an intuitive understanding of this test case attribute.This leads us to the identification of empirical relations between test cases with respect to their size and execution complexity:•The relation bigger than indicates that one test has a bigger size and execution complexity than another.•The relation similar to indicates that one test has a sim-ilar size and execution complexity when compared to another.These relations were defined intuitively by analysing how experts create associations between test cases with re-spect to their size and execution complexity.We consider that a test t1can be bigger than a test t2only if t1is not similar to t2.This assumption reflects the difficult to intu-itively compare test cases considered similar with respect to their size and execution complexity.Here,we call T as the set of all existing test cases.The set of all identified empirical relations is called R.Then,we call(T,R)as the empirical relation system for the attribute test size and execution complexity[5].To measure test size and execution complexity that is characterised by(T,R),we must define a mapping M of(T, R)into(E,P),in which test cases in T are mapped into num-bers(called execution points)in E and empirical relations in R are mapped to numerical relations in P.In this way,we can validate our measure demonstrating empirically that the mapping is valid for the attribute size and execution com-plexity.The set E of all possible numbers of execution points consists of the nonnegative integers and the set of numerical relations P consists of the relations>ep and≈ep,defined as follows.a>ep b=false if a≈ep ba>b otherwisea≈ep b=true if|a−b|a≤p100and|a−b|b≤p100false otherwiseAs we can see,the expression a>ep b is equivalent to the expression a>b,except in the case of similar numbers (≈ep)of execution points a and b.The definition of≈ep shows that numbers a and b are considered similar if the absolute difference between them is less than or equal to p percent of a and of b.The value of p is discovered empir-ically,as discussed later in Section5.3.The relations of R and P are mapped following the order of their presentations in this section.In practice,the execution point count of a test case gives us a quantitative reference about its size and execution com-plexity.For instance,a test case rated with700execution points is bigger than others rated with590and350.In ad-dition,it allows us to better compare test productivity or ca-pacity.For example,a tester that executed5tests rated with 500execution points each one is faster than another that ex-ecuted15tests rated with100execution points during the same amount of time.4.2The measurement methodThis section presents how we measure the size and exe-cution complexity of a test case.All required information is extracted from the test specification.Although not essential, we consider in this paper that test specifications are written in the CNL discussed in Section3.The CNL simplifies theFigure2.Assigning execution points to a testcase.use of our model and also efficiently supports a high level of automation of our measurement method.Figure2illustrates how our measurement method works. First,(a)we individually analyze each test step of the test specification.This step by step analysis was defined with the objective to support the method automation.We analyze each test step according to a list of characteristics(C1to C n).These characteristics represent some general functional and non-functional requirements exercised when the test step is executed.Examples of possible characteristics are number of navigations between screens,number of pressed keys and use of network.The list of characteristics may not be the same for different application domains,as discussed later in Section4.4.Each characteristic considered by the model has an im-pact in the size and execution complexity of the test and(b) this impact is rated using an ordinal scale(Low,Average and High).Later,Section4.4presents how to create guide-lines to help us to objectively choose the more appropriate impact level for each characteristic.After that,(c)we assign execution points for each char-acteristic according to its impact level.The objective here is to transform the qualitative rate(impact level)into a quan-titative value.For instance,a characteristic C1rated with the Low value could be assigned to30execution points.However,a more relevant characteristic rated with the same Low value may be assigned to a higher number of execution points.Section 4.4also discusses about guidelines provided for assigning the correct value for each possible characteristic value.To calculate the total number of execution points of atesting execution points and a con-version factor to calculate test execution ef-fort.step,(d)we sum the points assigned for each characteristic. Then,(e)we measure the size and execution complexity of a test case by summing the execution points of each one of its test steps.4.3.Test effort estimationThe execution point count of a test suite gives us a refer-ence about its tests size and execution complexity.The es-timated effort is calculated based on this information and a conversion factor(CF).The conversion factor represents the relation between test execution effort and execution points, which varies according to the productivity of the test team.The conversion factor is given in seconds per execution point,indicating the number of seconds required to exe-cute each execution point of a test case.For calculating the conversion factor,testers can measure the test size and execution complexity of several test cases.Then,the exe-cution time of the tests should be collected from a historical database(if available)or by executing them.As illustrated by Figure3,(f)the conversion factor is calculated by dividing the total effort by the total number of execution points.This information is then used for estimat-ing the execution effort of new test suites,we just need to (g)multiply its number of execution points by the conver-sion factor.For instance,a test manager may verify a conversion fac-tor of3.5seconds per execution ing this value,a new test case with120execution points is estimated to be executed in7minutes.Similar approach is used by other existing estimation models[6][11][13][14].In this approach,we assume that the test productivity and environment conditions are stable over time.For example, improvements in the test team,tools or environment may change the test productivity and consequently the conver-sion factor.In this case,the conversion factor should be recalculated using data collected after the improvements.In summary,the conversion factor used in the estima-tions should properly represent the current situation.In ad-dition,the conversion factor should be calculated for each different test team,test type or tested product,since they may have significant differences in productivity.4.4Model configurationOur proposed test execution effort model should be con-figured according to the target application domain in order to maximize the estimation accuracy.This section presents what,why and how to configure our estimation model.Controlled natural languageTest specifications written in CNL are the input of our model.As shown in Section3,the CNL grammar and lexicon are defined according to the target application do-main.For example,on the mobile application domain you have the verb take that accepts the term picture as argument. Hence,a possible test step is Take a picture.The list of verbs and possible arguments can be con-structed by analysing requirement documents and existing test specifications.Besides,new verbs and terms may arises over the time due to the specification of new requirements, technology changes,etc.The CNL grammar and lexicon is stored in a database that can be updated whenever neces-sary.System characteristicsDuring the test execution effort estimation,all test steps are analyzed according to a list of characteristics.These characteristics represent some general functional and non-functional requirements exercised when the test step is exe-cuted.They may depend on the target application domain.We use the Delphi method[10]for obtaining a consensus from a group of experts about the list of relevant character-istics.Examples of possible characteristics are the number of navigations between screens,the number of pressed keys and the use of network.The Delphi panel consists of3to7experienced testers invited from different teams for attending two or more rounds.In each round,they have the opportunity to add or remove characteristics from the list.Examples of real test cases are provided as a source for identifying types of test actions,software configurations,use of tools or specific hardwares and other characteristics that may impact the size or the execution complexity of a test case.All this process is anonymous and,in each round, a moderator provides the participants with a summary of the experts’decisions and their reasons for that.An alternative technique that can be used to identify rel-evant characteristics is the survey.When we have several testers in the organization,we can survey them about the relevant characteristics using questionnaires or other survey instrument.GuidelinesOnce the experts have defined the list of characteristics to be considered by the estimation model,the experts continue attending the Delphi panels,but with different objectives.First,they have to define the possible values that each identified characteristic may have.For example,if the type of camera is selected as a relevant characteristic for test size and execution complexity,its possible values would be au-tomatic shooting,required manual zoom,required use of flash,etc.After identifying the possible values of each selected characteristic,the experts group these values into three im-pact levels(low,average and high).The choices are made based on the impact of each value in the test size and ex-ecution complexity.This part of the guideline will help us to objectively choose the more appropriate impact level of a test step according to each characteristic.Finally,the experts must define for each characteristic the number of execution points to be assigned for each one of its impact levels.The experts proceed as follows.Each characteristic is weighted from1to10.These weights indi-cate the significance of each characteristic for the test size and execution complexity.Then,the experts give a weight from1to10for the lev-els Low,Average and High of each characteristic.These weights indicate the significance of each level for the char-acteristic.In general,values3,5and8are used to weight the levels Low,Average and High,respectively.After that, the number of execution points assigned for a level is cal-culated by multiplying its weight by the weight of its char-acteristic.After ending thefirst version of the model,one or two experts are enough for updating the guidelines when neces-sary.4.5Model automationOne of the objectives of this work was to develop an es-timation model that can be automated.This automation is important for supporting the development of new test gen-eration and test selection tools.In practice,companies may not be able to execute all test cases generated by such tools,since its resources are limited.For this reason,the test execution effort should be taken in consideration for test selection.A test generation tool,for instance,can consider a minimal requirement cov-erage and a maximum execution effort as its stop criteria.The use of CNL for specifying tests supports the devel-opment of an estimation tool that automatically reads and interprets these specifications.In addition,all the informa-tion required for using the model,such as the list of charac-teristics,guidelines and the conversion factor can be stored in a database.Actually,the CNL grammar and lexicon is also stored in the database[18].During the analysis of thefirst test cases,the estimation tool will ask the user to rate the characteristics of each test step.This information is stored in the database.Since the use of CNL reduces the number of possible ways to describe a test step,it is reasonable that the same test step(or very similar ones)occurs many times in the same test case and in different ones.For this reason,the necessity for manual assistance during the estimations tends to reduce as much as you use the tool.At the moment this paper was written,only an initial pro-totype was created for validating the model automation ca-pability.5.Empirical studyThis section presents the empirical study we run using our test execution effort estimation model on the mobile application domain.First,we configured our estimation model for the target domain.After that,we applied the es-timation model in a controlled experiment.Then,we vali-dated the test size and complexity measure we proposed.5.1Model configuration for the mobileapplication domainThe CNL used in this empirical study was defined in [18],we just needed to reuse its definition.To define the list of characteristics to use in our estimation model,we invited 6experienced testers.They identified the relevant charac-teristics and defined the guidelines in a Delphi panel that took four hours(two sessions of two hours).The results are presented in Table2.5.2ExperimentWe tested the presented model running an experiment. The main goal of this study was to analyze the accuracy of test execution effort estimates when using the estimation model proposed in this paper.In order to do that,we com-pared the estimates given by our model with the ones cal-culated using historical averages of test execution times,a simple and common estimation method used in practice.Following the goal-question-metric approach[1],we re-fined our goal for this empirical study to the questions pre-sented next:Q1:Is the average estimation error lower when using our model rather than using historical execution times?Q2:Is the average percentage of estimates within20%of the actual values higher when using our model rather than using historical execution times?The answer for Q1will indicate if the use of our estima-tion model results in a small error when regarding all esti-mates together.In its turn,the answer for Q2will indicate if the number of estimates within20%of the actual values increased when using our estimation model.For the study,we selected33test cases of a messaging application feature for mobile phone.These test cases were written in a controlled natural language and their size and complexity were measured using our method.We wanted to compare the precision of estimates made using historical information with estimates made using our model.As both approaches require information related to test productivity,we split the collected execution times into two sets of data,one for training and other for testing.The tests were randomly split,where the training set contained approximately65%of the tests.All test cases were then executed by a tester.The execution times were collected and stored in a spreadsheet for analysis.We used the test execution times of the training set to calculate:•The average test execution time(for the historical data approach).•The average time required to execute each execution point of a test case(the conversion factor for our pro-posed model).With this information,we estimated the test execution ef-fort of the testing set using both approaches.The following metrics were collected for answering Q1and Q2.•Mean magnitude of the relative estimation error.MMRE=Tt=1MRE tTwhere:MRE t=abs(estimated t−actual tactual t)T=number of tests•Average percentage of estimates that were within20% of the actual values.P RED(.20)=Tt=1(1,ifMRE t≤.20,0,otherwise)TIn order to avoid bias,we repeated the process two more times with different training and testing sets.Table3com-pare the metrics collected from the two analyzed estimation approaches.In all tests we achieved better or equivalent es-timation precision.In thefirst test,for example,the number of estimates within20%of the actual values increased by 100%.We also applied t-tests and confirmed the signifi-cance of the results.5.3Test size and complexity measure val-idationIn Section4.1,we proposed a measure of the size and execution complexity attribute of a test case.We can vali-date this measure demonstrating empirically that the map-ping between the empirical relation system(T,R)to the nu-merical relation system(E,P)is valid[5].During the experiment presented in the previous section, we mapped several test cases in T into execution points in E.It is necessary to verify that the mapped relations(in R and P)is valid considering the collected data.We used expert judgement and effort information to identify similar tests and tests bigger than others with re-spect to their size and execution complexity.We verified that tests intuitively identified as similar tests had different measured numbers of execution points.However,the differ-ences between these measures were within20%of their val-ues and this percentage value(p)can be used for identifying similar test cases from their number of execution points.We also verified that tests identified as bigger than others had bigger measures for their size and execution complexity attribute.In summary,we demonstrated empirically that, for all t a and t b in T:t a bigger than t b⇔ep(t a)>ep ep(t b)t a similar to t b⇔ep(t a)≈ep ep(t b)where ep(t)is the number of execution points measured from t.6.DiscussionIn our empirical study,we observed the cost to use our proposed model.This cost can be decomposed into the costs to:•Define a controlled natural language;•Identify relevant system characteristics;•Define guidelines;•Evaluate the size and execution complexity of test steps.The cost to define a controlled natural language depends on the way we define it.For instance,we can only define general rules,such as:each test step must be an imperative sentence giving a direct command to the tester,the main verb in infinitive form that defines the test action must starts the sentence,etc.In this case,we do not have a significant cost.However,we can also specify the list of all possible verbs that define test actions,their possible arguments,the vocab-ulary to be used,etc.In this case,we have to analyze exist-ing requirement and test documents,a process that can be done incrementally.We also need support tools to store this information and to check the conformity of the test cases.In this way,we maximize the benefits of the controlled natural language:reduced grammar and lexicon,writing standard, etc.In our study,the cost to identify the relevant system char-acteristics and to define the guidelines was low.We did a Delphi assessment that took four hours of six experienced testers.Nevertheless,this cost will increase when consider-ing a large scope.Finally,the cost to evaluate the size and execution com-plexity of test steps is the most significant one.Although it will usually take less than a minute to evaluate a test step, there may exist hundreds of test steps to be evaluated.How-ever,we did not necessarily need to evaluate all test steps, since it is common to have the same test step occurring sev-eral times in different test cases or even in the same test. After evaluating a test step,we just need to assign the same number of execution points to its other occurrences.In our experiment,we observed that most of times we can evaluate a test step based only on the main verb of its sentence,independently of the verb arguments.For in-stance,the act of launching an application has the same complexity for most applications and only the exceptions need to have a specific evaluation.Also,we use a controlled natural language that reduces the vocabulary and consequently increases the use of the same verb in different test steps that only change the verb。
谢勇博士和美国哈佛医学院专家研究获得β细胞活性肽茶

谢勇博士和美国哈佛医学院专家研究获得β细胞活性肽茶谢勇博士和美国哈佛医学院专家研究获得β细胞活性肽茶β-cell活性肽茶:1992年4月,一封来自大洋彼岸的信寄到了第二军医大学长海医院,信的落款是美国哈佛大学医学院,收到这封来自异国的信函,谢勇心里也充满了疑惑。
打开来信,信的内容让他激动中又有犹豫,信是美国哈佛医学院海外交流中心负责人梅里斯教授写的。
梅里斯教授对谢勇来说并不陌生,他是国际糖尿病联盟的学刊《世界糠尿病》的特约撰稿人,也是国际糖尿病领域著名的学者。
谢勇在参加《世界糖尿病》杂志年会时曾经和梅里斯教授有过交流,梅里斯教授对谢勇在糖尿病方面的一些独特见解十分欣赏。
尤其是谢勇在年会上提出糖尿病不能单独依靠药物治疗的理念更是推崇。
在信中,梅里斯教授代表哈佛医学院邀请他前往该院进行讲学和深入的合作,共同进行糖尿病医学的研究,美国哈佛医学院海外交流中心将让他担任项目负责人,并为他提供可观的研究实验经费。
年轻的谢勇彷徨了,以内心来说,他不愿意离开自己成长学习的地方,更希望能够在自己岗位上为患者治病救人,正如他所说,这是我的责任。
但另一方面,国内的医学科研,无论从硬件设施、政策规定以及其他客观方面都不完善,学术界的论资排辈,研究项目僧多粥少,很多研究资金被无谓的浪费,而一些关键急需的项目却又无从上马,人情、资历、关系都制约着发展。
而美国哈佛医学院海外中心的邀请和承诺,将会是他实现飞跃的一个良好平台。
取舍之间,情理之间,该何去何从?第三天晚上,谢勇来到了导师顾老的家里,他拿着邀请信,把情况告诉了顾老,想征求顾老的意见。
顾老看完信之后,沉默了许久,拿起一柄小锄头到后院的小花园里松土。
当焦急的谢勇快要憋不住时,顾老拿起一块土块放到谢勇的手里,问他:“你的心在那里,你的根在那里,你会丢掉自己的根吗?”谢勇站在那里深思了许久,终于他对顾老坚定地说:我的心是患者,要把他们的病治好,我的根在这里,生养我的土地,我不会改变,我相信自己会回来的!改变时代的发现1992年11月10日,谢勇携带简单的行李,走进了现代医学圣地美国哈佛医学院。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Biographies Thomas S. (Tom) Tullis is Vice President of User Experience Research at Fidelity Investments and Adjunct Professor in Human Factors in Information Design at
Bentley University. He joined Fidelity in 1993 and was instrumental in the devel-
opment of the company’s User Research department, whose facilities include
state-of-the-art Usability Labs. P rior to joining Fidelity, he held positions at
Canon Information Systems, McDonnell Douglas, Unisys Corporation, and Bell Laboratories. He and Fidelity’s usability team have been featured in a number
of publications, including Newsweek, Business 2.0, Money, The Boston Globe, The
Wall Street Journal, and The New Y ork Times. Tullis received his B.A. from Rice University, M.A. in Experimental Psychology from New Mexico State University,
and Ph.D. in Engineering Psychology from Rice University. With more than 35
years of experience in human–computer interface studies, Tullis has published
over 50 papers in numerous technical journals and has been an invited speaker
at national and international conferences. He also holds eight United States pat-
ents. He coauthored (with Bill Albert and Donna Tedesco) “Beyond the Usability
Lab: Conducting Large-Scale Online User Experience Studies,” published by
Elsevier/Morgan Kauffman in 2010. Tullis was the 2011 recipient of the Lifetime Achievement Award from the User Experience Professionals Association (UXPA)
and in 2013 was inducted into the CHI Academy by the ACM Special Interest
Group on Computer–Human Interaction (SIGCHI). Follow Tom as @TomTullis.
William (Bill) Albert is currently Executive Director of the Design and Usability
Center at Bentley University and Adjunct P rofessor in Human Factors in Information Design at Bentley University. Prior to joining Bentley University,
he was Director of User Experience at Fidelity Investments, Senior User Interface Researcher at Lycos, and Post-Doctoral Researcher at Nissan Cambridge Basic Research. Albert has published and presented his research at more than 30
national and international conferences. In 2010 he coauthored (with Tom
Tullis and Donna Tedesco), “Beyond the Usability Lab: Conducting Large-Scale
Online User Experience Studies,” published by Elsevier/Morgan Kauffman. He
is co-Editor in Chief of the Journal of Usability Studies. Bill has been awarded prestigious fellowships through the University of California Santa Barbara and
the Japanese government for his research in human factors and spatial cogni-
tion. He received his B.A. and M.A. degrees from the University of Washington (Geographic Information Systems) and his P h.D. from Boston University (Geography-Spatial Cognition). He completed a Post-Doc at Nissan Cambridge
Basic Research. Follow Bill as @UXMetrics.。