欧盟医疗器械CE认证技术 编写Technical file editting
CE认证TCF技术文件编写技术服务

CE认证TCF技术文件编写技术服务
keywords:TCF技术文件编写,CE认证TCF文件编写
CE认证TCF技术文件
TCF文件是申请CE认证的制造商向CE认证机构提度交的一份重要文件,它是认证机构审核发证的重要依据。
编制TCF文件必须全部使知用英文。
欧盟法律要求,加贴了CE标签的产品投放到欧洲市场后,其技术文件(TechnicalFiles)必须存放于欧盟境内供监督机构随时检查。
技术文件中所包涵的内容若有变化,技术文件也应及时地更新。
技术文件通常应包括下列内容:
a.制造商(欧盟授权代表(欧盟授权代理)AR)名称,商号,地址
b.产品的型号,编号
c.产品使用说明书
d.安全设计文件(关键结构图,即能反映爬申距离、间隙、绝缘层数和厚度的设计图)
e.产品技术条件(或企业标准)
f.产品电原理图
g.产品线路图
h.关键元部件或原材料清单
i.测试报告((TestingReport)
j.欧盟授权认证机构NB出具的相关证书((2010年后欧盟规定CE认证证书必须是指定机构或者是能力机构出具)
k.产品在欧盟境内的注册证书(对于某些产品比如:ClassI医疗器械,普通IVD体外诊断医疗器械)等等......。
医疗器械CE认证的基本介绍

医疗器械CE认证的基本介绍医疗器械CE认证是指医疗器械制造商根据欧盟医疗器械指令(Medical Device Directive,简称MDD)或欧盟医疗器械规例(Medical Device Regulations,简称MDR)的要求,申请并获得CE认证的过程。
CE认证是产品在欧洲市场上销售和使用的必要条件之一,它代表着制造商对产品的合规性和质量的承诺。
CE认证的过程包括以下几个关键步骤:1.审查文件:制造商需要准备和提交一系列的技术文档,包括产品设计和制造过程的说明、使用说明书、性能测试报告、质量管理体系等。
这些文件将被认证机构用于审核和评估产品的合规性。
2.评估产品:认证机构将对产品进行技术评估,以确认其是否符合欧盟医疗器械指令或规例的要求。
评估的范围包括产品的设计、性能、安全性等方面。
3.审核质量管理体系:制造商需要建立并实施符合相关标准要求的质量管理体系,并通过认证机构的审核确认其有效性。
4.风险评估:制造商需要对产品进行风险评估,以确定可能存在的风险并采取相应的控制措施,确保产品安全可靠。
5.进行现场检查:认证机构可能进行现场检查,以确保制造商的生产设施和质量管理体系符合要求,并能够保证产品的一致性和可追溯性。
6.发放证书:如果产品经过评估和审核符合要求,认证机构将向制造商发放CE认证证书,授权其在欧洲市场上销售和使用该产品。
CE认证的优势:1.欧洲市场准入:CE认证是进入欧洲市场的重要条件之一,对于想要将产品销售到欧洲国家的制造商而言,CE认证是不可或缺的。
2.提升产品竞争力:CE认证代表着产品的合规性和质量保证,可以提升产品的竞争力,增加消费者对产品的信任度。
3.强调品牌价值:CE认证标志能够使产品具备更高的品牌价值,有助于建立品牌形象和口碑。
4.符合法律法规:欧盟对医疗器械的安全和质量要求非常严格,CE认证可以确保产品符合相关法律法规的要求,降低企业的法律风险。
5.提高产品质量:通过认证过程,制造商需要建立符合标准要求的质量管理体系,这有助于提高产品的质量和可靠性。
(完整word版)无源医疗器械CE技术文档和CE设计档案材料指南(中英文 也是极好的)

无源医疗器械技术文件和设计文档指南Whereas the term “Technical File“ is used for Medical Devices of class I, class IIa and class IIb, the term “Design Dossier“ is used for the class III products.标题中的“技术文件”适用于I类,IIa类,IIb类医疗器械,“设计文档”适用于III类医疗器械。
Technical Files are retained in the premises of the manufacturer or the Authorized Representative for potential review of Competent Authorities and Notified Body.Part B of the Technical File may be available at the manufacturer only.技术文件是保留在制造商或授权代表单位的主管部门和认证机构。
部分技术文件B部分只保留在制造商处。
Whereas Design Dossiers have to be submitted to the Notified Body for review prior to CE-Marking of the product (use form Application for CE Conformity Assessment (Product)MED_F_03.03). We will assign a project manager who will entrust one or more further experts with the review of particular modules. All experts are at your disposal directly or indirectly through the project manager. After successful review, the Notified Body issues a design examination certificate according to Annex II.4 of the Council Directive certifying compliance with the relevant provisions of Annex I of the MDD.设计档案材料已被提交到公告机构用于需要CE认证前的产品审查(用CE合格评定(产品)MED_F_03.03规定的格式)。
欧盟CE认证详细内容简介

2019/5/30
Introduction of CE
23
2.CE相关指令介绍
2.CE相关指令介绍
2019/5/30
Introduction of CE
24
2.CE相关指令介绍
2.1 目前欧盟制定并颁布的指令
欧盟CE认证指令名称
指令编号
机械(MD)
2006/42/EC
承压设备(PED)
97/23/EC
2019/5/30
Introduction of CE
16
1.CE的概述
技术文件(TCF)
定义:技术文件是合格评定活动的文件化表述形式,证明产品对指令 的符合性。该文件由生产厂家负责制定,旨在提供产品有关的设计、 生产和使用的信息。 文件的内容包括证实产品对于适用要求的符合性证据。 技术文件应涉及产品设计阶段、生产阶段和产品运行阶段。 技术文件应证明产品符合相关指令基本要求。 技术文件需以欧盟成员国语言(英语)。 随时提供(48小时),技术文件必须至少保留10年以上。
Introduction of CE
14
1.CE的流程 1.4 CE认证流程
总流程: 合同订立—项目开案—工程师介入—项目通知企业开案(认证资料清单+ 申请表等)--企业按照资料清单提供认证所需文件—资料审核—按照指令 确认是否需要审厂—安排工厂审核(如需要)--测试及技术文件(TCF) 建立—签署符合性声明—签发证书—产品标贴CE标志
2006/42/EC
低压电气安全(LVD)
2006/95/EC
电磁兼容(EMC)
2004/108/EC
承压设备(PED)
97/23/EC
建筑用品(CPD)
2011/305/reg
欧盟医疗器械CE认证技术文档编写Technicalfileeditting

Module 3 – CE技术文档编写
技术文档的语言
? 如果欧盟指令未包括对文档语言的特 别规定时,生产者必须对成员国的需 求进行评估。
? 可能不会出现更多状况来影响到相关 的翻译件。 (例如经过验证的)
13
Module 3 – CE技术文档编写
技术文档-第一部分
包括与符合性评价程序有关的,最重要 技术数据: ? 制造商信息 ? 欧洲代表信息 ? 质量系统所涉及的全部制造场所的清
19
示例
6.设计验证
如果生产者能证明这些年来市场验证后是一 种安全设计且其性能按设计要求,则不需要。
否则,按如下步骤提供文件:
-设计合格检验
-关于预期用途的设计预测(如果有关,参 考其他产品)
7.风险分析
-器械的风险评估
20
示例
8.符合基本要求和调查标准
-列出所有使用的调整标准或,如果使用不 完整,详细叙述如何满足基本要求。
物理性能报告
? 不同的产品有不同的物理性能
? 例如:带针注射器
尺寸
针管韧性和刚性
推拉力
器身密合性
残留容量
52
化学性能报告
? 不同的产品有不同的化学性能
? 例如:带针注射器
酸碱度Βιβλιοθήκη 可萃取金属含量易氧化物环氧乙烷残留(生物性能)
53
可用性报告
? EN 62366:2008
54
EMC测试报告 电气安全测试报告
綠色能源 利用自然,開發綠色能源
綠色生活 讓環保融入您生活的各個層面
綠色產品 更低的環境影響, 帶來更高的市占率
綠色循環 負責的回收再利用
始於產品設計
德國萊因 綠色解決方案
医疗产品TCF技术文件编写技术服务

医疗产品TCF技术文件编写技术服务
TCF文件是医疗器械向欧盟市场做CE认证,需要提供的一套技术文件。
是欧盟医疗器械指令中很重要的一个事项,它的目的是要求企业准备充份的技术资料和证明,供主管机关抽查,或发生诉讼纠纷时使用。
各欧盟指令对于" 技术文件" 的要求有所差别,在这里谨以中国出口企业最常用的“医疗器械”的要求为例,加以说明。
主要内容如下:
1 、产品名称、分类及引用标准条款的简要描述
2 、产品概述(包括类型和预期用途)
3 、使用该产品的调和标准/ 或其它标准
4 、风险分析评估结论和预防措施(EN1441 产品服务危险分析报告)
5 、生产质量控制
产品资料和控制文档(包括产品生产工艺流程图)
产品的灭菌方法和确认的描述
灭菌验证
产品质量控制措施
产品稳定性和效期的描述
6 、包装和标识
7 、技术评价
8 、潜在风险评价
产品潜在风险测试报告及相关文献
潜在风险的概要及权威观点
9 、临床评价
产品临床测试报告及相关文献
临床使用概述及权威观点
附1 、产品出厂检测报告
附2 、产品型式检测报告
附3 、基本要求检查表。
(完整word版)无源医疗器械CE技术文档和CE设计档案材料指南(中英文也是极好的)

无源医疗器械技术文件和设计文档指南Whereas the term “Technical File“ is used for Medical Devices of class I,class IIaand class IIb,the term “Design Dossier“ is used for the class III products.标题中的“技术文件”适用于I类,IIa类,IIb类医疗器械,“设计文档”适用于III类医疗器械. Technical Files are retained in the premises of the manufacturer or the Authorized Representative for potential review of Competent Authorities and Notified Body.Part B of the Technical File may be available at the manufacturer only.技术文件是保留在制造商或授权代表单位的主管部门和认证机构。
部分技术文件B部分只保留在制造商处。
Whereas Design Dossiers have to be submitted to the Notified Body for review prior to CE—Marking of the product (use form Application for CE Conformity Assessment (Product)MED_F_03。
03). We will assign a project manager who will entrust one or more further experts with the review of particular modules. All experts are at your disposal directly or indirectly through the project manager. After successful review,the Notified Body issues a design examination certificate according to Annex II。
医疗器械 欧盟技术文档清单 MDD CE Checklist
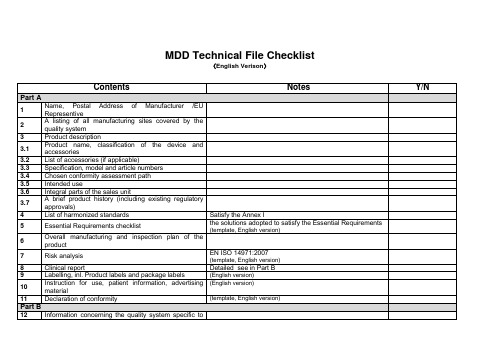
3.7
简要的产品历史(包括现有的管理审批)
4
适用的标准清单
符合医疗器械指令附录I
5
基本要求检查表
符合医疗器械指令附录I的方案
(有固定模板,需提交英文文件)
正在填写
6
产品的总体生产或质量控制方案
7
风险分析
EN ISO 14971:2007
有固定模板,需提交英文文件
正在索要
8
临床报告
详细的临床数据见Part B
9
Labelling, inl.Product labels and package labels
(English version)
10
Instruction for use, patient information,advertising material
(English version)
11
Declaration of conformity
MDDTechnical FileChecklist
(English Verison)
Contents
Notes
Y/N
Part A
1
Name, Postal Address of Manufacturer /EU Representive
2
A listing of all manufacturing sites covered by the quality system
13.2
Packaging and specification
(English version)
13.3
Description of the manufacturing processes
CE认证自我声明

CE认证,如何进行自我声明(DOC)?产品自我声明适用于:1.用于简单的、大批量的、无危害产品,仅适用应用欧洲标准生产的厂家。
2.工厂自我进行合格评审,自我声明。
3.技术文件提交国家机构保存十年,在此基础上,可用评审和检查来确定产品是否符合指令,生产者甚至要提供产品的设计、生产和组装过程供检查。
4.不需要声明其生产过程能始终保证产品符合要求。
CE认证对于大多数的产品都可以利用自我声明,此时制造方或其欧盟的授权代表机构要签署“符合性声明”(Self Declaration of Conformity)。
符合性声明的内容根据不同指令而有所不同。
需要注意的是,必须按指令准备符合性声明,如果一个产品有两个适用的指令,则应分别准备和签署两份符合性声明。
很多企业以为拿到某个检测机构的或认证机构的CE证书就可以使用CE标志了,这是一种错误理解。
既然是自我声明,企业自己就要对使用CE标志负责,第三方机构颁发的CE证书只是间接证明而已。
但是,如果CE证书是欧盟指定机构颁发的,就不必签发自我声明了。
仅签署符合性声明是不够的,制造方还必须提交技术机构档案TCF(Technical Construction File),包括检测报告,产品技术说明,线路图等技术文件或技术档案,以证明产品在技术上是符合指令要求的。
准备一份符合要求的技术档案也是很重要的,欧盟指定机构为客户准备TCF的所收取的费用和颁发证书的费用(不含检测费用)是一样的。
当制造方签署了符合性声明并附有符合要求的TCF后,制造方就可以在产品上使用CE标志了。
目前有很多CE证书不被欧洲的客户或管理机构承认,主要是对CE标志制度不了解所造成的。
要注意以下两点对点LVD、EMC和R&TTE三个指令在进行自我声明时必须注意:所声明的、或技术文件中提到的符合欧洲协调标准的有效期(终止期)(Date of Cessation ”DOC”),超过该终止后,按过期的标准生产的产品将不允许再在EU/EEA市场销售,也就是说,自我声明此时无效。
三类医疗器械ce认证流程

三类医疗器械ce认证流程Title: The Process of CE Certification for Class III Medical DevicesTitle: 三类医疗器械CE认证流程Medical devices fall into three classes, Class I, Class II, and Class III, with Class III devices posing the highest risk to patients.CE marking is a requirement for these devices to be sold in the European Union, and it indicates compliance with欧盟医疗器械指令.三类医疗器械根据风险程度分为三个等级,其中第三类器械风险最高。
CE标志是这些设备在欧盟销售的法律要求,它表示符合欧盟医疗器械指令。
The process of obtaining CE certification for Class III medical devices involves several stages, including the preparation of a technical file and a clinical evaluation report, and the completion of a comprehensive questionnaire.The technical file should contain all relevant documentation, such as design specifications, manufacturing processes, and quality control procedures.三类医疗器械获得CE认证的过程包括几个阶段,包括准备技术文件和临床评估报告,以及完成一份详尽的问卷。
医疗器械CE认证
医疗器械CE认证医疗器械CE认证简介:所有进入欧盟市场的产品,企业必须具有表示自我符合声明的CE标志,以说明产品符合欧盟制定的相关指令。
医疗器械需要满足的指令有《有源植入性医疗器械指令》(AIMDD, 90/385/EEC)、《医疗器械指令》(MDD,93/42/EEC)和体外诊断器械指令(IVDD, 98/79/EC)。
医疗器械指令(MDD),MDD指令适用于大多数进入欧盟销售的医疗设备。
它根据不同的要求共分为6个等级,供认证机构评估。
- 设计阶段生产阶段I级自我符合声明自我符合声明I级(级测量功能)自我符合声明申报机构I级(灭菌)自我符合声明申报机构IIa级自我符合声明申报机构IIb级申报机构申报机构III级申报机构申报机构认证机构的统一评估包括根据指令规定的基本要求评审技术文件、根据标准EN 46001 或 EN/ISO 13485评审质量体系。
医疗设备CE认证技术档案所需内容:生产商/或欧洲代表名址;产品及型号描述;EC符合声明书;风险评估;基本安全点检表;适用之调合标准/或其他标准;市场反馈及抱怨分析;使用说明及标签;授权代表;线路、图表(适用的话);计算书、测试报告或其它证明材料;检验过程及过程描述;灭菌或其它特殊过程(适用的话);灭菌类产品的包装材料及方法;质量体系、质量手册;医疗器械CE认证流程:步骤一、确定并分析出口器械,确定它是否在欧盟的3个医疗器械指令的范围内。
因为CE认证过程比较复杂,因此寻找合适的医疗器械咨询公司配合如上海沃华,将会缩短产品进入欧洲市场的时间和减少认证成本。
步骤二、确认适用的基本要求指令规定,任何医疗器械必须满足相关指令中所规定的预期用途,所以对制造商来说,首先要做的而且是最重要的事情就是确认所有的适用于其产品的基本条件。
步骤三、确认任何有关的欧洲协调标准协调标准是由欧洲标淮委员会(CEN)和欧洲电气技术委员会 (CENELEC)制定的公布在欧盟官方杂志上的标准,对于某种医疗器械来说,可能有多种协调标准适用于它,因此在确认哪些协调标准适用于某种产品对应十分仔细。
医疗器械CE认证要点解析

医疗器械CE认证要点解析CE 认证包括哪 四方面?CE 认证是一个完善的安全保障系统,并非仅仅是将一个样品拿到试验室检验通过而已。
因为 CE 标志是一个安全标志,所以,一个通过CE认证的产品必须确保自产品 的设计,生产,包装,说明书的编写,到运输,销售,产品的整个有效使用寿命 中,以及使用后产品的回收,等等所有环节中,均符合欧洲的健康、安全、与 环境保护之相关法律中所规定的基本要求。
因此,一家制造商欲想使其产品通过 CE认证,通常要满足如下4方面的要求:1.产品投放到欧洲市场前,在产品上加贴CE标签。
2.产品投放到欧洲市场后,技术文件(Technical Files)必须存放于欧盟境 内供监督机构随时检查。
3.对被市场监督机构发现的不合CE要求的产品、或者使用过程中出现事故但是已加贴CE标签的产品,必须采取补救措施。
(比如从货架上暂时拿 掉,或从市场中永久地撤除)。
4.已加贴 CE标签之产品型号在投放到欧洲市场后,若遇到欧盟有关的法律更改或变化,其后续生产的同型号产品也必须相应地加以更改或修正,以便符合欧盟新的法律要求。
CE 认证程序1. 确认出口国家2. 确认产品类别及欧盟相关产品指令3. 指定“欧盟授权代表( 欧盟授权代理 ) ”(Authorized Representative)4. 确认认证所需的模式(Module)5. 采用 " 自我声明 " 模式还是 " 必须通过第三方认证机构"6. 建立技术文件 (Technical Files) 及其维护与更新1、确认出口国家若出口至欧洲经济区EEA包括欧盟EU及欧洲自由贸易协议EFTA的 30 个成员国 中的任何一国,则可能需要CE认证。
2、确认产品类别及欧盟相关产品指令若产品属于这里所列22 类中的任何一个,一般地讲,则需要进行 CE认证。
若一个产品同时属于一个以上的类别,则必须满足所有类别相对应的产品指令中所列出的要求。
医疗器械CE认证医疗器械(MDD)指令

医疗器械CE认证|医疗器械(MDD)指令医疗器械产品要顺利通过CE认证,需要做好三方面的工作。
其一,收集与认证产品有关的欧盟技术法规和欧盟(EN)标准,通过消化、吸收、纳入企业产品标准。
其二,企业严格按照以上产品标准组织生产,也就是把上述技术法规和EN 标准的要求,贯彻到企业产品的设计开发和生产制造的全过程。
第三,企业必须按ISO9000+ISO13485标准建和维护质量体系,并取得ISO9000+ISO13485认证。
(奥咨达医疗器械咨询)医疗器械CE认证应遵循的欧盟技术法规和EN标准对于目前欧盟已发布的18类工业产品指令,从这些指令的结构看,它们可分为垂直指令和水平指令。
垂直指令是以具体产品为对象,如医疗器械指令;水平指令适用于各种产品系列,如电磁兼容性指令,它适用于全部电器及电子零部件产品。
对于医疗器械,适用的指令有第十四项、第一项和第五项,即:93/42/EEC 医疗器械指令、73/23/EEC低电压(LVD)指令89/336/EEC电磁兼容性(EMC)指令。
支持这些指令的欧盟标准是:(只专注于医疗器械领域)(1)EN60601-1医用电气设备第一部分:安全通用要求;(2)EN60601-1-1医用电气设备第一部分:安全通用要求及第一号修正;(3)EN60601-2-11医用电气设备第二部分:γ射束治疗设备安全专用要求;(4)EN60601-1-2医用电气设备第一部分:安全通用要求1.2节并行标准电磁兼容性——要求和测试。
其中第(1)、(2)、(3)项标准是伽玛刀低电压(LVD)测试的依据:第(4)项标准是伽玛刀电磁兼容性(EMC)测试的依据。
按照欧盟医疗器械CE认证程序和内容如下:1)企业向认证机构提出认证申请,并填写认证询价单交认证机构;2)认证机构向申请认证企业提出报价单,企业签字确认即完成合约;3)企业向认证机构提交ISO9000+ISO13485质量体系文件即质量手册和程序文件,供认证机构进行体系文件审核;质量体系审核前,企业应有至少三个月的质量体系运行记录,并完成1-2次内部质量体系审核。
无源医疗器械CE技术文档和CE设计档案材料指南(中英文 也是极好的)

无源医疗器械技术文件和设计文档指南Whereas the term “Technical File“ is used for Medical Devices of class I, class IIa and class IIb, the term “Design Dossier“ is used for the class III products.标题中的“技术文件”适用于I类,IIa类,IIb类医疗器械,“设计文档”适用于III类医疗器械。
Technical Files are retained in the premises of the manufacturer or the Authorized Representative for potential review of Competent Authorities and Notified Body.Part B of the Technical File may be available at the manufacturer only.技术文件是保留在制造商或授权代表单位的主管部门和认证机构。
部分技术文件B部分只保留在制造商处。
Whereas Design Dossiers have to be submitted to the Notified Body for review prior to CE-Marking of the product (use form Application for CE Conformity Assessment (Product)MED_F_03.03). We will assign a project manager who will entrust one or more further experts with the review of particular modules. All experts are at your disposal directly or indirectly through the project manager. After successful review, the Notified Body issues a design examination certificate according to Annex II.4 of the Council Directive certifying compliance with the relevant provisions of Annex I of the MDD.设计档案材料已被提交到公告机构用于需要CE认证前的产品审查(用CE合格评定(产品)MED_F_03.03规定的格式)。
医疗器械进入欧盟医疗器械市场要求-20210601170202

医疗器械进入欧盟医疗器械市场要求-20210601170202WORD格式能够任意编写入欧盟的有关知识一、欧盟国家(共31家)1、2004年5月1日前:法国、德国、意大利、荷兰、比利时、卢森堡、丹麦、爱尔兰、英国、希腊、西班牙、葡萄牙、奥地利、芬兰、瑞典;2、2004年5月1日加入:波兰、匈牙利、捷克、斯洛伐克、斯洛文尼亚、爱沙尼亚、拉脱维亚、立陶宛、塞浦路斯、马耳他;3、欧洲自由贸易协会EFTA:挪威、列支敦士登、冰岛、瑞士;4、新入欧盟:罗马利亚、保加利亚;5、瑞士不要求产品携带CE标志。
二、医疗器械CE认证的通用要求1、基本要求(总要求)(1)安全性(任何风险与器械提供的益处相比较,必须在能够接受的范围内,故亦称风险剖析);(2)风险的可预防性或被除去性,起码应赐予警示(报警系统或戒备报警系统);(3)性能切合性(产品的基本要求);(4)器械性能和安全的效期(器械的安全和性能必须在器械的使用寿命内得到保证。
);(5)器械的储藏和运输(应保证器械在合理的运输、储藏条件下不受影响)。
2、基本要求的详细包括如下14条:1)器械设计和生产必须保证:按照其预定和条件使用,器械不会损害医疗环境、患者安全、操作者或其他人员的安全和健康;使用时的潜在危险与患者受益相比较能够为人们所接受,但应具有高水平的防备办法。
2)生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守安全准则、生产者应:首先应尽可能降低甚至避免危险。
其次,对无法避免的危险采取适合的防备举措,包括安装报警装置;最后,见告用户所提供防备举措的弱点及其可能带来的危险。
(3)器械必须取得生产者希望获得的功能。
器械设计制造和包装应有利于第一条(2)(A)D多规定的各项功能的发挥。
(4)在生产线者确定的器械使用寿命期内,在正常使用可能出现的压力,第1、2、3款所指的各项性能应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康。
5)器械的设计、生产和包装应当保证,器械的性能在运输和储藏过程中只要遵守有关规定不会发生根本逆变。
制药机械的CE认证的认证程序以及具体问题

CE认证是一个完善的安全保障系统,并非仅仅是一个样品拿到试验室检验通过而已。
一个通过CE认证的产品必须确保自产品的设计、生产、包装、说明书到运输、销售等产品的整个有效使用的寿命中,以及使用后产品的回收等环节均符合欧洲的健康、安全与环境保护之相关法律中所规定的基本要求,确保产品始终对于使用者及环境都是安全的,因此一家制造商预想其产品通过CE认证,通常要满足如下4个方面的要求:(1)产品投放到欧洲市场前,在产品上加贴CE标志。
(2)产品投放到欧洲市场后,技术文件(TechnicalFiles)必须存放于欧盟境内供监督机构随时检查。
(3)对被市场监督机构发现的不符合CE要求的产品,或者使用过程中出现事故但是已加贴CE标签的产品,必须采取补救措施。
(4)已加贴CE标签之产品型号在投放到欧洲市场后若遇欧盟有关的法律更改或变化,其后续生产的同类型号产品也必须相应地加以更改或修正,以便符合欧盟新的法律要求。
认证流程制药机械企业实施CE认证过程中的若干具体问题1.通常一家制药机械企业生产的产品有若干种,而企业在实施CE 认证前应统一规划,分段实施,因此在选择产品的品种上可选择那些适合欧洲市场的产品作为突破口,在设计制造和市场上积累经验,逐步推广。
2.一般产品的CE认证都是由欧洲代理商要求所为,并由其提出做出哪几项指令,因此针对性会比较强,当然现在越来越多的制造商已经认识到CE的重要性而主动提出认证。
3.CE认证所涉及工作量很大。
首先,被认证方自身应理解和掌握CE有关工业机器的EN(欧盟)标准。
这对判断和应用标准以及与认证公司打交道有好处,其次有利于不符合项的整改。
有些情况下还会对机器的局部结构进行调整或重新设计。
4.为了减少安全元器件的测试费用和数量,要想方设法到市场上寻找有CE标准的安全元器件或零部件。
例如:变压器、接触器、继电器、开关、步进驱动器、步进电机、接线桩头、接近开关。
电源插座、电机等。
5.认证的时间具体分硬件(产品本身)和软件(产品的技术资料),认证一般最少要一个月,多则几个月,甚至半年一年,这与认证公司无关,主要视产品的易难程度和被认证方对不符合项整改情况及进度而定。
医疗CE认证

医疗CE认证医疗CE认证指令适用范围很广,包括除有源植入性和体外诊断器械之外的几乎所有的医疗器械,如无源性医疗器械;以及有源性医疗器械,如核磁共振仪、超声诊断和治疗仪、输液泵等。
医疗产品要顺利通过CE认证,需要做好三方面的工作。
其一,收集与认证产品有关的欧盟技术法规和欧盟(EN)标准,通过消化、吸收、纳入企业产品标准。
其二,企业严格按照以上产品标准组织生产,也就是把上述技术法规和EN标准的要求,贯彻到企业产品的设计开发和生产制造的全过程。
第三,企业必须按ISO9000+ISO13485标准建和维护质量体系,并取得ISO9000+ISO13485认证。
医疗CE认证应遵循的欧盟技术法规和EN标准对于目前欧盟已发布的18类工业产品指令,从这些指令的结构看,它们可分为垂直指令和水平指令。
垂直指令是以具体产品为对象,如医疗器械指令;水平指令适用于各种产品系列,如电磁兼容性指令,它适用于全部电器及电子零部件产品。
对于医疗器械,适用的指令有第十四项、第一项和第五项,即:93/42/EEC医疗器械指令、73/23/EEC低电压(LVD)指令89/336/EEC电磁兼容性(EMC)指令。
支持这些指令的欧盟标准是:(1)EN60601-1医用电气设备第一部分:安全通用要求;(2)EN60601-1-1医用电气设备第一部分:安全通用要求及第一号修正;(3)EN60601-2-11医用电气设备第二部分:γ射束治疗设备安全专用要求;(4)EN60601-1-2医用电气设备第一部分:安全通用要求1.2节并行标准电磁兼容性——要求和测试。
其中第(1)、(2)、(3)项标准是伽玛刀低电压(LVD)测试的依据:第(4)项标准是伽玛刀电磁兼容性(EMC)测试的依据。
医疗CE认证程序、内容欧盟把医疗产品分为四类,即:第Ⅰ类、第Ⅱa类、第Ⅱb类、第Ⅲ类。
第Ⅰ类产品要加贴CE标志,可采取自行宣告的方式。
即厂商编制产品的技术文件档案,同时自行按有关EN标准对产品进行测试或委托有能力的试验室进行测试合格。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
产品信息
▪名称(包括商标名称) ▪分类(按照医疗器械指令的要求) ▪描述与说明 ▪预期用途、适应症、警告或禁忌症 ▪型号与规格
27
基本要求检查表
▪技术文档中最重要的文件之一 ▪来源于医疗器械指令的附录ANNEX I ▪证明产品符合医疗器械指令的直接证据
28
如何填写基本要求检查表
▪识别产品适用的标准 ▪ 判断每个条款是否适用 ▪ 如果适用,填入相关的标准 ▪ 找到支持性的证据
2020/3/2
Module 3 – CE技术文档编写
技术文档-第一部分
包括与符合性评价程序有关的,最重要 技术数据: ▪制造商信息 ▪欧洲代表信息 ▪质量系统所涉及的全部制造场所的清 单; ▪产品说明,包括 ✓2020产/3/2 品的识别(型号/编号),
Module 3 – CE技术文档编写
技术文档-第一部分
还包括: ▪适用于产品的协调标准清单 ▪产品的总体制造和检查方案; ▪风险分析报告 ▪基本要求检查表 ▪临床报告 ▪产品标签、使用说明、患者信息、广
2020/3/2
告材料
Module 3 – CE技术文档编写
技术文档-第一部分
还包括: ▪适用于产品的协调标准清单 ▪产品的总体制造和检查方案; ▪风险分析报告 ▪基本要求检查表 ▪临床报告 ▪产品标签、使用说明、患者信息、广
44
检测、测试验证报告
▪生物性能报告 ▪灭菌验证报告 ▪包装验证报告 ▪稳定性研究报告
45
检测、测试验证报告
▪物理性能试验 ▪化学性能试验 ▪EMC测试报告 ▪电气安全测试报告 ▪。。。。。。
46
生物性能报告
▪生物相容性试验
▪适用的标准:EN ISO 10993系列
▪根据不同的产品类型选择不同的生物相容性 试验
32
如何填写基本要求检查表
▪如果不知道选择哪个标准怎么办??
▪标准本身会帮助您!!
33
如何填写基本要求检查表
▪如果不知道选择哪个标准怎么办??
▪标准本身会帮助您!!
34
如果不知道产品适用的标准怎么办??
▪技术文档中最重要的文件之一 ▪ 来源于医疗器械指令的附录I ▪ 证明产品符合医疗器械指令的直接证据
临床评估-WHAT
▪为了证实器械的临床安全性和性能,而对医 疗器械相关的临床数据进行的评定和分析
39
临床评估-WHEN
▪整个器械的生命周期,包括 ❖上市前(符合性评定过程) ❖上市后(医疗器械使用过程)
40
临床评估-HOW DETAIL
▪完整的和客观的(如应该同时考虑有利和不 利的数据) ▪可以用等价器械文献报告 ▪深度和广度应该是灵活的 ▪评估必须涉及到所有的临床声明,这包括产 品标签、和产品信息(特殊声明、禁忌症、 注意事项/警告)和说明书的适合性
– 有一个基本的索引来说明所有相关文件的存放地 – 只有索引存放在同一地点 – 相关的文件需存放在索引指定的地点 – 文件的最新版本能在指定的地点找到 – 定义每一个地点的归档系统
2020/3/2
Module 3 – CE技术文档编写 技术文档的目的
▪ 评估产品符合性的主要方法 ▪ 符合性的评估需基于生产者的符合性声明 ▪ 生产者编写的文件应以符合国家检测机构的要求为目的 ▪ 国家机构有权索要相关数据 ▪ 欧盟型式检验: 证书包括在技术文档中
2020/3/2
Module 3 – CE技术文档编写
技术文档的有效性
▪技术文档必须保存,以便在接受国家 机构检查时进行审查。 ▪至少有一份技术文档必须被保存在欧 盟区域之内。 ▪生产者及其欧盟代表负有责任,来决 定将技术文档存放在哪个欧盟国家。
2020/3/2
Module 3 – CE技术文档编写
綠色生活 讓環保融入您生活的各個層面
綠色產品 更低的環境影響, 帶來更高的市占率
綠色循環 負責的回收再利用
始於產品設計
德國萊因 綠色解決方案
綠色建築 打造環保生態建築
綠色諮詢 分享永續發展的 知識與經驗
綠色系統 維持生態平衡的流程
綠色交通 綠色交通,環保之路
57
35
风险管理报告
▪适用的标准:EN ISO 14971:2012 ▪贯穿产品的整个生命周期
36
37
术语和定义
▪危害(Hazard):损害的潜在源。 ▪损害(Harm):对人体健康的实际伤害或侵害 ,或是对财产或环境的侵害。 ▪危害处境(Hazard situation):人员、财产或 环境接触到一个或多个危害的境遇 ▪风险(Risk):损害发生38 概率与损害严重程度
2020/3/2
告材料
Module 3 – CE技术文档编写
技术文档-第二部分
包括所有的技术细节: ▪检测、测试和验证报告 ▪产品的详细说明(原理、设计历史) ▪配方、组成、原材料清单 ▪供货商清单 ▪设计图 ▪产品技术规范
2020/3/2
示例
例子: 技术文件的提供 1.产品概述 -产品的类型和预期用途 2.原材料、元件的文件提供 -技术要求-原材料的详细规定 -元件图纸-性能控制程序 3.半成品器械文件提供 -技术要求-图纸 -流程图-元件清单 18
技术文档的有效性
▪根据国家机构的要求,尽快递交技术 文档。 ▪文件持有人的姓名和地址不需要特别 提及。 ▪只有在审查时,才能系统地要求提供 文件。
2020/3/2
Module 3 – CE技术文档编写 技术文档的语言
▪如果欧盟指令未包括对文档语言的特 别规定时,生产者必须对成员国的需 求进行评估。 ▪可能不会出现更多状况来影响到相关 的翻译件。 (例如经过验证的)
▪技术文件的目的是证明产品符合医疗器械指 令的基本要求 ▪如果符合协调化标准,则可以认为符合指令 的基本要求
22
23
制造商信息
▪中英文名称和地址 ▪电话和传真 ▪联系人 ▪公司网页(如果有)
24
欧洲代表信息
▪名称和地址 ▪电话和传真 ▪联系人 ▪欧代协议
25
自我符合性声明
▪莱茵公司可以提供符合标准的模板文件
2020/3/2
Module 3 – CE技术文档编写
谁负责准备技术文档?
生产者
谁必须保存技术文档?
生产者,欧盟代表
2020/3/2
Module 3 – CE技术文档编写 技术文档的格式和内容
▪程序的目的是为了使公共机构相信, 那些出现在市场上的产品是符合指令 所要求的条款的。 ▪必须避免过多的文书工作。
示例
4.最终产品-文件提供 -技术要求-图纸 -流程图-元件清单 -质量控制程序 5.包装和标示文件提供 -包装的技术要求 -所有标示的复印件和用户手册或指导手册
19
示例
6.设计验证 如果生产者能证明这些年来市场验证后是一 种安全设计且其性能按设计要求,则不需要 。 否则,按如下步骤提供文件: -设计合格检验 -关于预期用途的设计预测(如果有关,参 考其他产品) 7.风险分析
20
示例
8.符合基本要求和调查标准 -列出所有使用的调整标准或,如果使用不 完整,详细叙述如何满足基本要求。 9.临床数据(一般情况下,I类不要求) -通过临床与非临床数据验证预期用途和医 疗报告 -临床前数据和审查,关于器械预期用途的 临床数据 -特殊设计临床调查的测试报告
21
技术文档基本要求
41
临床评估-WHO
▪临床评估人的资质要求 ▪熟悉器械技术以及其应用 ▪熟悉研究方法(临床调查设计和生物统计学 ) ▪熟悉临床使用
42
临床评估方式
▪科学文献方式 ▪临床调查方式 ▪以上两种方式的结合
43
临床评估报告
▪临床评估报告应包括临床数据、评价阶段和 分析阶段、和关于器械的安全性和性能的结 论。例如: ▪器械使用的技术、预期用途、和任何关于器 械临床性能或者安全性的宣称 ▪临床数据的性质和范围 ▪证明器械的临床性能和安全性
▪通常由第三方检测机构出具
▪无灭菌验证报告
▪适用的标准: EN ISO 11135-1: 环氧乙烷灭菌 EN ISO 11137-1/-2: 辐照灭菌 EN ISO 17665-1:湿热灭菌
▪如果灭菌外包,还要提供: 分包方的灭菌CE证书 符合要求的灭菌外包协48 议
包装验证报告
55
产品标签和说明书
适用的标准 ▪标签:EN 980:2008 及相应的产品专标 ▪说明书:EN 1041:2008 及相应的产品专标 ,例如EN ISO 14630 ▪至少要准备英文版 ▪要有版本控制信息 ▪与风险分析的输出对56应
德國萊因綠色解決方案 – 協助您達成環保目標
綠色能源 利用自然,開發綠色能源
▪例如:带针注射器
尺寸
针管韧性和刚性
推拉力
器身密合性
残留容量
52
化学性能报告
▪不同的产品有不同的化学性能 ▪例如:带针注射器 酸碱度 可萃取金属含量 易氧化物 环氧乙烷残留(生物性5能3 )
可用性报告
▪EN 62366:2008
54
EMC测试报告 电气安全测试报告
▪EN 60601-1-2 ▪EN 60601-1
▪适用于灭菌产品
▪适用标准:EN ISO 11607-1/-2
▪包装验证通常包含:
密封性试验
剥离强度试验
材料阻菌性试验
跌落试验
49
运输验证报告
▪依据GB 4857系列标准 至少评价:堆码、跌落、震动性能
50
稳定性试验报告
▪加速稳定性试验 ▪实时(长期)稳定性试验
51
物理性能报告
▪不同的产品有不同的物理性能
Module 3 CE技术文档编写
1
Module 3 – CE技术文档编写 主要内容
•什么是技术文档 ? •技术文档的目标是什么 ? •谁负责准备技术文档 ? •由谁保管技术文档 ? •技术文档的内容