SWISSMODEL蛋白质结构预测教程
5.5计算方法预测三级结构-02-同源建模法SWISS-MODEL

《生物信息学》第五章:蛋白质结构预测与分析(第二部分) 计算方法预测三级结构:同源建模法SWISS-MODEL预测蛋白质三级结构的首选方法是同源建模法(homolog modeling)。
该方法基于原理:相似的氨基酸序列对应着相似的蛋白质结构。
比如三个蛋白质,它们在序列水平上十分相似,解析出的结构也十分相似。
第四个蛋白质的序列和前面三个也高度相似,那么就可以比着前三个结构的样子“画”出第四个的样子。
所以同源建模法的关键就是找到一个好的模板。
好的模板要求,在序列水平上模板(template)要与目标(target)蛋白质具有超过30%的一致度。
同源建模法操作流程如下(图1):图1. 同源建模法操作流程1. 确定模板:找到与目标蛋白质同源的已知蛋白质结构作为模版(目标序列与模版序列间的一致度要≥30%)。
2. 序列比对:为目标序列与模板序列创建序列对比。
模板可以选取多个,通过做多序列比对,各取所长,让模板序列中与目标序列相似的片段尽可能多的覆盖整个目标序列,同时要尽量避免没有模板参考的断口。
3. 计算模型:通过序列比对,将目标序列里的氨基酸替换到模板结构里对应的氨基酸所在的空间位置上。
这一步通过同源建模软件来实现。
4.换模板或修正序列比对,重新构建模型,再次评估。
SWISS-MODEL()它能帮助完成上述步骤中从模板选取到创建序列比对,再到计算模型,以及最后的质量评估的全部过程。
你需要做的只是:输入目标序列,点Build Model(创建模型)(图2左)。
大约三到五分钟之后就会返回结果。
如果这种自动挡模式不能满足你的要求,可以通过点击Search For Templates切换成手动挡,以便指定模板。
也可以直接把做好的目标序列与模板序列之间的序列比对按照指定格式黏贴到输入框里,再点击Build Model(创建模型)(图2右)。
这时,SWISS-MODEL会根据输入的特定格式的序列比对,识别出哪个是目标序列,哪个是模板,并自动从PDB数据库下载模板结构,最后根据输入的比对计算结构模型。
蛋白质三级结构预测(swiss-model同源建模)
利用同源建模预测蛋白质的三级结构首先声明一下,以下纯属个人观点,方法步骤仅供参考,不可作为规范标准,结果出来之后请自行分析结果。
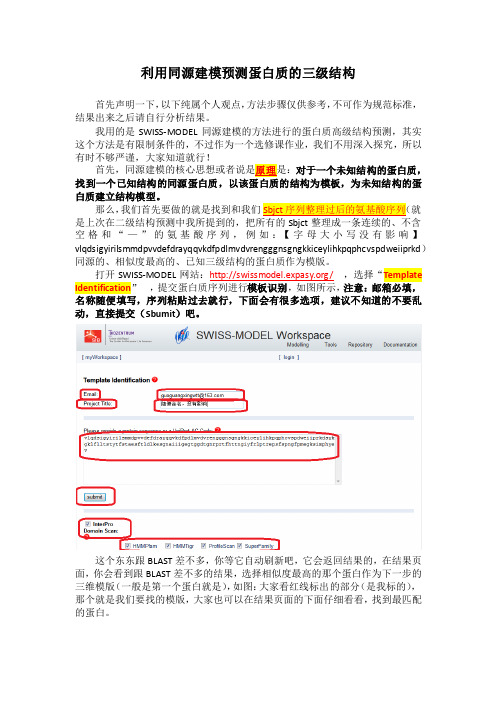
我用的是SWISS-MODEL同源建模的方法进行的蛋白质高级结构预测,其实这个方法是有限制条件的,不过作为一个选修课作业,我们不用深入探究,所以有时不够严谨,大家知道就行!对于一个未知结构的蛋白质,白质建立结构模型。
那么,我们首先要做的就是找到和我们空格和“—”的氨基酸序列,例如:【字母大小写没有影响】vlqdsigyirilsmmdpvvdefdrayqqvkdfpdlmvdvrengggnsgngkkiceylihkpqphcvspdweiiprkd)同源的、相似度最高的、已知三级结构的蛋白质作为模版。
打开SWISS-MODEL网站:/,选择“Template Identification,提交蛋白质序列进行模板识别,如图所示,注意:邮箱必填,名称随便填写,序列粘贴过去就行,下面会有很多选项,建议不知道的不要乱动,直接提交(Sbumit)吧。
这个东东跟BLAST差不多,你等它自动刷新吧,它会返回结果的,在结果页面,你会看到跟BLAST差不多的结果,选择相似度最高的那个蛋白作为下一步的三维模版(一般是第一个蛋白就是),如图:大家看红线标出的部分(是我标的),那个就是我们要找的模版,大家也可以在结果页面的下面仔细看看,找到最匹配的蛋白。
这里还有一点要作说明,就是上图标出的代码是PDB编号,前四个表示PDB- Code,最后一位表示Chain-ID,具体什么意思,大家有兴趣就去了解一些吧。
接下来,去NCBI串串门吧,在NCBI中搜索上面查到的蛋白的PDB号,一般输入前四位就行啦,注意:搜索蛋白库(Protein)。
找到以后,以FASTA格式显示。
接下来,我们再回到SWISS-MODEL,接下来就是重点和难点啦,在线提交序列进行同源建模分析,这个在线提交不是大家想象的那么容易,这个耗费了我大部分的时间,说到这里我就想画个圈圈诅咒它,大家注意啦~~~~~~~~~~~SWISS-MODEL 是一个自动化的蛋白质比较建模服务器,该服务器提供用户三种模式可选择:Automatic mode(简捷模式): 用于建模的氨基酸序列或是Swiss-Prot/TrEMBL (/sprot )编目号(accession)可以直接通过web界面提交。
Swissmodel蛋白三维结构预测

Swissmodel蛋⽩三维结构预测swiss-model蛋⽩三维结构预测蛋⽩三维结构模型预测LHCGR基因NM_000233,c.547G>A,p.G183R蛋⽩质三维结构预测⽅法概览⽐较建模法⽐较建模⼜称同源建模,原理简单,是基于计划相关的序列具有相似的三维结构且进化过程中三维结构⽐序列保守的原理,利⽤计划相关模板结构信息建模。
基本步骤:1)将⽬标序列作为查询序列,在已知蛋⽩结构数据中搜索,确定和识别出⼀个同源模板。
2)将⽬标序列和模板结构进⾏⽐对。
3)以模板结构⾻架为模型,建⽴⽬标蛋⽩质⾻架模型。
4)对模型结果进⾏评价,确定模板的实⽤性。
查找蛋⽩序列LHCGR基因,NM_000233;exon14;c.547G>A(p.G183R);699aaNCBI数据库:https://w /查找蛋⽩序列LHCGR基因,NM_000233;exon14;c.547G>A(p.G183R);699aaSWISS-MODEL同源建模法--swiss-model:⽬前应⽤最⼴泛的⽐较建模法蛋⽩结构预测⼯具https://查询到的氨基酸序列复制到输⼊框,开始建模输⼊蛋⽩全序列构建模型,共2个结果页⾯解读:(1)⾸先看搜到的模板是否覆盖当前变异位点;(2)检查搜到的模板与输⼊序列⼀致度是否>30%(2)如果可⽤,再看swissmodel评分⾼低情况。
GMQE:可信度范围为0-1,值越⼤表明质量越好QMEAN4:区间-4-0,越接近0,评估待测蛋⽩与模板蛋⽩的匹配度越好。
模型下载下载后的蛋⽩结构预测模型可以⽤Swiss-pdbviewer查看ATCG⽤Swiss-pdbviewer打开pdb模型⽂件,在controlpanel⾥去掉氨基酸⾻架和side显⽰,添加ribn显⽰在controlpanel的colR ⾥选择添加ribbon(colR->ribbon)。
Color栏下选择SecondaryStructureSuccession根据⼆级机构进⾏着⾊进⾏着⾊。
蛋白质三级结构预测精品课件

Swissmodel基于同源建模的方法 是目前三级结构预测当中基于同源建模方法做的最好 的一个之一 优点是:如果能得到结果往往很可靠 缺点是:很多时候得不到结果
例:给定的example 进行三级结构预测
主页
选用 名字,然后提交
等待结果,结果发到邮箱中
三级结构可视化(Pdbviewer)
作业:对给定的seq1 进行三级结构预测,并用Pdbview可 视化结果
9、 人的价值,在招收诱惑的一瞬间被决定 。20.9.1 920.9.1 9Saturd ay , September 19, 2020 10、低头要有勇气,抬头要有低气。1 0:56:24 10:56:2 410:56 9/19/20 20 10:56:24 AM 11、人总是珍惜为得到。20.9.1910:56:2410:5 6Sep-2 019-Sep -20 12、人乱于心,不宽余请。10:56:2410 :56:241 0:56Saturday , September 19, 2020 13、生气是拿别人做错的事来惩罚自 己。20. 9.1920. 9.1910:56:2410 :56:24September 19, 2020 14、抱最大的希望,作最大的努力。2 020年9 月19日 星期六 上午10 时56分 24秒10 :56:242 0.9.19 15、一个人炫耀什么,说明他内心缺 少什么 。。202 0年9月 上午10 时56分 20.9.19 10:56September 19, 2020 16、业余生活要有意义,不要越轨。2 020年9 月19日 星期六 10时56 分24秒 10:56:2 419 September 2020 17、一个人即使已登上顶峰,也仍要 自强不 息。上 午10时5 6分24 秒上午1 0时56 分10:56:2420.9. 19
讲稿9-蛋白质结构预测

第九章蛋白质结构预测第一节概述蛋白质研究的核心内容:收集大量的蛋白质分子结构的信息,建立结构与功能之间关系的数据库,奠定蛋白质结构与功能之间关系的理论研究基础。
●验证蛋白质设计的假设证明是新结构改变了原有生物功能三维空间结构的测定。
●晶体学的技术制备出单晶体需纯蛋白质(几毫克~几十毫克)进行繁杂的数据收集、计算和分析。
蛋白质的晶体状态与自然状态不尽相同,应考虑的问题。
●NMR(核磁共振)技术可以分析液态下的肽链结构,绕过了结晶、X-射线衍射成像分析等难点,直接分析自然状态下的蛋白质的结构。
直接模拟出:蛋白质的空间结构、蛋白质与辅基和底物结合的情况酶催化的动态机理。
有效地分析蛋白质的突变。
蛋白质的分子结构蛋白质的一级结构(primary structure)多肽链的氨基酸残基的排列顺序。
蛋白质二级结构(secondary structure)多肽链借助于氢键沿一维方向排列成具有周期性的结构的构象,是多肽链局部的空间结构(构象),主要有α-螺旋、β-折迭、转角等。
超二级结构(supersecondary structure)相邻的二级结构单元组合在一起,彼此相互作用,排列形成规则的、在空间结构上能够辨认的二级结构组合体,充当三级结构的构件(block building),基本形式:αα、βαβ和βββ等。
结构域(domain)在超二级结构的基础上形成的,通常由50-300AA残基组成,在三维空间可以明显区分和相对独立,具有一定的生物功能。
结构域的亚单位:模体或基序(motif),三级结构(tertiary structure)整条多肽链的三维结构,包括骨架和侧链在内的所以原子的空间排列。
四级结构(quaternary structure)在亚基和亚基之间通过疏水作用等次级键结合成为有序排列的特定的空间结构。
亚基通常由一条多肽链组成,有时含两条以上的多肽链,单独存在时一般没有生物活性。
第二节蛋白质结构测定1. 一级结构测定或蛋白质顺序分析:(1)应用化学裂解法和蛋白酶水解法将多肽链专一性裂解;(2)逐一测定每个纯化的小肽段的顺序;(3)根据肽段氨基酸顺序中的重迭区确定小肽段的排列次序;(4)完成整条多肽链的顺序分析。
5 蛋白质三级结构预测

• 令代表核心折叠C中的环到序列S中空位的 映射,显然是通过线索化而确定的。
令f(t)是进行比对的得分函数,其定义如下:
f(t) = g1 (v,t) + g2 (u,v,t) + g3 (,t)
• g1 (v,t) 评价氨基酸残基v所处的位置 • g2 (u,v,t) 评价残基u和v的相对位置,如果u和v 键合,则得 分高; • g3肖 飞
蛋白质三级结构预测的方法
1
2 3
方法比较
同源建模(比较建模)
基础 - 相似的序列结构相近 - PDB结构数据库的快速增长 - 结构基因组学的启动 - 发散进化 特点 - 相对精确可靠
• 假设待预测三维结构的目标蛋白质为U (Unknown),利用同源模型化方法建立结 构模型的过程包括下述6个步骤: (1)搜索结构模型的模板(T) (2)序列比对 U T (3)建立骨架 (4)构建目标蛋白质的侧链 (5)构建目标蛋白质的环区 (6)优化模型
至于最后建立三维结构模型则是非常困难的
• 线索化的主要思想: 利用氨基酸的结构倾向(如形成二级结构 的倾向、疏水性、极性等),评价一个序 列所对应的结构是否能够适配到一个给定 的结构环境中。
• 建立序列到结构的线索的过程称为线索化, 线索技术又称折叠识别技术。 • 线索化或者折叠识别的目标是为目标蛋白质 U寻找合适的蛋白质模板,这些模板蛋白质 与U没有显著的序列相似性,但却是远程同 源的。
新的趋势 混合预测方法 在比较建模法和折叠识别法中使用从头预 测法来预测部分难以找到模板的片断 在从头预测法中使用二级结构预测的结果 和其他已知结构信息辅助建模
• Meta-predictor 使用多个预测方法 对收集的结果进行综合比较和分析 改进收集的结果
蛋白质数据库及其结构预测攻略

蛋白质数据库及其结构预测攻略一、蛋白质结构层次一般情况下,蛋白质的结构分为4 个层次:▪初级结构——氨基酸序列;▪二级结构——а螺旋(alpha-helix),β折叠(β-sheets),β转角,无规则卷曲(random coil)▪三级结构——三维结构,由模体(motif)和结构域(domain)组成;▪四级结构——亚基之间的互作。
二、蛋白质数据库:1. 蛋白质一级数据库1.1序列数据库:UniProt包含三大蛋白质序列数据库,Swiss-Prot,TrEMBL 和PIR,分为三个层次:第一层叫UniParc,收录了所有UniProt 数据库子库中的蛋白质序列,量大,粗糙。
第二层是UniRef,他归纳了UniProt 几个主要数据库并且是将重复序列去除后的数据库。
第三层是UniProtKB,他有详细注释并与其他数据库有链接,分为Swiss-Prot(最有用的)和TrEMBL。
1.2蛋白质结构数据库PDBPDB存储生物大分子3D 结构。
这些生物大分子除了蛋白质以外还包括核酸以及核酸和蛋白质的复合物。
只有通过实验方法获得的3D 结构才会被收入其中。
PDB文件是一堆数字字母,那是每个原子的坐标,一般用用可视化软件VMD打开,免费的,这里不作具体说明。
2. 蛋白质二级数据库2.1结构域家族Pfam数据库Pfam 主页上的搜索工具可以查找某条序列上有哪些结构域。
2.2结构分类数据库CATHCATH是四种结构分类层次的首字母。
根据PDB编号搜索,可以获得各层次具体的结构分类信息以及各种结构相关分析信息、聚类分析。
2.3结构分类数据库SCOP2在搜集、整理、分析PDB数据中已知的蛋白质三维结构的基础上,详细描述了一直结构的蛋白质在结构、进化事件与功能类型三个方面的关系,主要依赖人工验证。
三、蛋白质结构研究1.二级结构1.1已知PDB-输入检索号-sequence- view sequence& DSSP image1.2未知预测网址如下:输入氨基酸序列,等待大概半小时。
蛋白质三级结构预测ppt课件

15
完整版课件
Байду номын сангаас
16
完整版课件
17
完整版课件
18
完整版课件
19
作业:对给定的seq1 进行三级结构预测,并用Pdbview可 视化结果
完整版课件
20
感谢亲观看此幻灯片,此课件部分内容来源于网络, 如有侵权请及时联系我们删除,谢谢配合!
蛋白质三级结构预测(Swissmodel)
完整版课件
1
Swissmodel基于同源建模的方法 是目前三级结构预测当中基于同源建模方法做的最好 的一个之一 优点是:如果能得到结果往往很可靠 缺点是:很多时候得不到结果
完整版课件
2
例:给定的example 进行三级结构预测
完整版课件
3
主页
完整版课件
4
完整版课件
5
选用 Automated mode
完整版课件
6
完整版课件
7
输入序列、Email、姓名、序列名字,然后提交
完整版课件
8
等待结果,结果发到邮箱中
完整版课件
9
完整版课件
10
三级结构可视化(Pdbviewer)
完整版课件
11
完整版课件
12
完整版课件
13
完整版课件
14
完整版课件
蛋白质结构预测技术的使用教程

蛋白质结构预测技术的使用教程蛋白质是生物体内构成细胞的基本组成部分之一,其结构决定了蛋白质的功能和相互作用。
蛋白质结构预测是一项重要的生物信息学研究领域,它的发展为科学家们提供了洞察蛋白质功能和疾病发生机制的重要工具。
本文将介绍蛋白质结构预测技术的使用教程,帮助读者了解并使用这一技术。
1. 背景知识在开始使用蛋白质结构预测技术之前,我们首先需要了解一些基本的背景知识。
蛋白质的结构可以分为四个层次:一级结构是指氨基酸序列;二级结构是指α-螺旋和β-折叠等部分;三级结构是指蛋白质立体构型的整体形态;四级结构是指多个蛋白质相互作用形成的功能性蛋白质复合体。
蛋白质结构的理解对于揭示蛋白质的功能和相互作用关系至关重要。
2. 蛋白质结构预测的方法目前,蛋白质结构预测的方法可以分为两大类:实验方法和计算方法。
实验方法包括晶体学、核磁共振、电子显微镜等技术,但由于其昂贵和耗时的特点,计算方法逐渐成为蛋白质结构预测的主要手段。
计算方法可以进一步分为基于比对的方法和基于物理模拟的方法。
基于比对的方法使用已知结构的蛋白质作为模板,通过比对计算目标蛋白质的结构。
基于物理模拟的方法则通过模拟生物物理过程,推测蛋白质的结构。
3. 常用的蛋白质结构预测软件在选择蛋白质结构预测软件之前,我们需要考虑软件的准确性、计算速度和易于使用等因素。
以下是几个常用的蛋白质结构预测软件:- I-TASSER:I-TASSER 是一种基于比对的蛋白质结构预测软件,它使用蛋白质序列与数据库中已知结构的蛋白质进行比对,并通过模型构建和优化来预测蛋白质的结构。
- ROSETTA:ROSETTA 是一种基于物理模拟的蛋白质结构预测软件,它使用分子力学和能量函数来模拟蛋白质的结构。
ROSETTA 在模拟过程中考虑了蛋白质的氨基酸序列、二级结构和溶液环境等因素。
- PHYRE2:PHYRE2 是一种基于比对的蛋白质结构预测软件,它通过比对目标蛋白质序列与数据库中已知结构的蛋白质,然后通过从比对到的结构中挑选最佳模板来预测目标蛋白质的结构。
SWISS-MODEL蛋白质结构预测教程

SWISS-MODEL 蛋白质结构预测SWISS-MODEL是一项预测蛋白质三级结构的服务,它利用同源建模的方法实现对一段未知序列的三级结构的预测。
该服务创建于1993年,开创了自动建模的先河,并且它是讫今为止应用最广泛的免费服务之一。
同源建模法预测蛋白质三级结构一般由四步完成:1.从待测蛋白质序列出发,搜索蛋白质结构数据库(如PDB,SWISS-PROT等),得到许多相似序列(同源序列),选定其中一个(或几个)作为待测蛋白质序列的模板;2.待测蛋白质序列与选定的模板进行再次比对,插入各种可能的空位使两者的保守位置尽量对齐;3.建模:调整待测蛋白序列中主链各个原子的位置,产生与模板相同或相似的空间结构——待测蛋白质空间结构模型;4.利用能量最小化原理,使待测蛋白质侧链基团处于能量最小的位置。
最后提供给用户的是经过如上四步(或重复其中某几步)后得到的蛋白质三级结构。
SWISS-MODEL工作模式SWISS-MODEL服务器是以用户输入信息的最小化为目的设计的,即在最简单的情况下,用户仅提供一条目标蛋白的氨基酸序列。
由于比较建模程序可以具有不同的复杂性,用户输入一些额外信息对建模程序的运行有时是有必要的,比如,选择不同的模板或者调整目标模板序列比对。
该服务主要有以下三种方式:?First Approach mode(简捷模式):这种模式提供一个简捷的用户介面:用户只需要输入一条氨基酸序列,服务器就会自动选择合适的模板。
或者,用户也可以自己指定模板(最多5条),这些模板可以来自ExPDB模板数据库(也可以是用户选择的含坐标参数的模板文件)。
如果一条模板与提交的目标序列相似度大于25%,建模程序就会自动开始运行。
但是,模板的可靠性会随着模板与目标序列之间的相似度的降低而降低,如果相似度不到50%往往就需要用手工来调整序列比对。
这种模式只能进行大于25个残基的单链蛋白三维结构预测。
?Alignment Interface(比对界面):这种模式要求用户提供两条已经比对好的序列,并指定哪一条是目标序列,哪一条是模板序列(模板序列应该对应于ExPDB模板数据库中一条已经知道其空间结构的蛋白序列)。
蛋白质结构预测及方法介绍2

随着蛋白质结构数据的积累,人们开始注意到一些较简单的序列与结构关系。
可以利用各种氨基酸的疏水值定位蛋白质的疏水区域,通过疏水氨基酸出现的周期性预测蛋白质的二级结构。
Lim等人很早就对α螺旋和β折叠归纳出了一套预测模式。
例如α螺旋的轮状结构特征,轮的一侧通常处于蛋白质的疏水核心,另一侧则常处于亲水表面,如图7.2所示。
因此,α螺旋中亲疏水氨基酸残基的出现位置也就有一定的规律性,亲水残基多出现在亲水侧面,而疏水残基则多出现在疏水侧面,反映在序列上就是一些特征的亲疏水残基间隔模式。
疏水性氨基酸的位置有助于推断蛋白质中二级结构的定位,通过显示疏水氨基酸的分布分析二级结构。
例如,图7.2 是利用HELICALWHEEL程序画出的蛋白质蜂毒素旋轮图。
图中各个氨基酸沿螺旋排布,相邻氨基酸之间的旋转角度为100o。
疏水性氨基酸L、I和V位于螺旋的一侧,而亲水性氨基酸则分布在另外一侧,显示这个螺旋的两亲特性。
根据蛋白质序列中疏水性氨基酸出现模式,可以预测局部的二级结构。
例如,当我们在一段序列中发现第i、i+3、i+4位是疏水氨基酸时,这一片段就被可以预测为α螺旋;当我们发现第i、i+1、i+4位为疏水氨基酸时,这一片段也可以被预测为α螺旋。
同样,对于β折叠,也存在着一些特征的亲疏水残基间隔模式,埋藏的β折叠通常由连续的疏水残基组成,一侧暴露的β折叠则通常具有亲水-疏水的两残基重复模式。
不过,由于β折叠受结构环境的影响较大,序列的亲疏水模式不及α螺旋有规则。
原则上,通过在序列中搜寻特殊的亲疏水残基间隔模式,就可以预测α螺旋和β折叠。
在Biou等人提出的点模式方法中,将20种氨基酸残基分为亲水和疏水残基,用八残基片段表征亲疏水间隔模式。
以一个二进制位代表一个残基,疏水为1,亲水为0,共八位。
这样,八残基片段的亲疏水模式就可用1个0~255的数值来表示。
α螺旋的特征模式对应的值为9,12,13,17,……,201,205,217,219,237;β折叠的特征模式则由连续的1或交替的01构成。
SWISS-MODEL

SWISS-MODEL服务器模板数据库ExPDB是由PDB中提取的:PDB文件被分成确定蛋白链和不确定蛋白链,去掉不确定蛋白链(理论模型或仅提供α-碳坐标的质量较差的数据文件)。SWISS-MODEL吸收了额外的有用信息,如可能的四级结构信息,明确的标志信息(经验力场能、ANOLEA平均势能得分)。对于某一目标序列,SWISS-MODEL搜索模板数据库ExPDB选择合适的模板。如果对某一目标序列找不到合适的模板,但可以找到几个模板序列,经过拼凑后覆盖目标序列,SWISS-MODEL的建模过程就分成几个部分,分别进行批处理。
2、比对
使用重复最小的方块算法,每个批处理最多能接受五个模板结构。去除不匹配模板(即那些与第一个模板相比有高α-碳均方差偏离的模板),产生结构比对。目标序列与模板序列进行局部两两序列比对,然后经过一次经验步骤改进比对。插入和缺失位置的选择由模板整体结构来决定,即比对产生的孤立的残基被移动到临近的无规则卷曲结构。
侧链建模
模型侧链的重建是基于模板结构中相应残基的最有利位置。以保守残基开头,模型侧链通过空间等位取代模板结构侧链的方式构。得分函数中着重体现氢键、二硫键,避免不利接触。
4、能量最小化
蛋白质结构几何结构的调整是建模中的最后一步。当连接刚性片段时,使用根据能量最小原理的GROMOS96力场算法进行调整。经验的力场用于发现模型构象中的错误。一般说来,能量最低化或分子动力学方法普遍不能提高模型的精度,在SWISS-MODEL中使用仅仅是使分子结构更加标准化。
同源建模方法预测蛋白结构一般包含以下四步:1、模板选择;2、目标序列上步骤直到找到一个令人满意的模型。SWISS-MODEL开发了几套不同的建模技术。SWISS-MODEL的预测方法可以被描述为以α-螺旋和β-折叠为基础的"刚性片段组装"。
蛋白质三级结构预测ppt课件

3
主页
可编辑课件PPT
4
可编辑课件PPT
5
选用 Automated mode
可编辑课件PPT
6
可编辑课件PPT
7
输入序列、Email、姓名、序列名字,然后提交
可编辑课件PPT
8
等待结果,结果发到邮箱中
可编辑课件PPT
9
可编辑课件PPT
10
三级结构可视化(Pdbviewer)
可编辑课件PPT
11
可编辑课件PPT
12
ቤተ መጻሕፍቲ ባይዱ
可编辑课件PPT
13
可编辑课件PPT
14
可编辑课件PPT
15
可编辑课件PPT
16
可编辑课件PPT
17
可编辑课件PPT
18
可编辑课件PPT
19
作业:对给定的seq1 进行三级结构预测,并用Pdbview可 视化结果
可编辑课件PPT
20
此课件下载可自行编辑修改,此课件供参考! 部分内容来源于网络,如有侵权请与我联系删除!感谢你的观看!
蛋白质三级结构预测(Swissmodel)
可编辑课件PPT
1
Swissmodel基于同源建模的方法 是目前三级结构预测当中基于同源建模方法做的最好 的一个之一 优点是:如果能得到结果往往很可靠 缺点是:很多时候得不到结果
可编辑课件PPT
2
例:给定的example 进行三级结构预测
可编辑课件PPT
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
SWISS-MODEL 蛋白质结构预测
SWISS-MODEL是一项预测蛋白质三级结构的服务,它利用同源建模的方法实现对一段未知序列的三级结构的预测。
该服务创建于1993年,开创了自动建模的先河,并且它是讫今为止应用最广泛的免费服务之一。
同源建模法预测蛋白质三级结构一般由四步完成:
1.从待测蛋白质序列出发,搜索蛋白质结构数据库(如PDB,SWISS-PROT等),得到许多相似序
列(同源序列),选定其中一个(或几个)作为待测蛋白质序列的模板;
2.待测蛋白质序列与选定的模板进行再次比对,插入各种可能的空位使两者的保守位置尽量对齐;
3.建模:调整待测蛋白序列中主链各个原子的位置,产生与模板相同或相似的空间结构——待测
蛋白质空间结构模型;
4.利用能量最小化原理,使待测蛋白质侧链基团处于能量最小的位置。
最后提供给用户的是经过如上四步(或重复其中某几步)后得到的蛋白质三级结构。
SWISS-MODEL工作模式
SWISS-MODEL服务器是以用户输入信息的最小化为目的设计的,即在最简单的情况下,用户仅提供一条目标蛋白的氨基酸序列。
由于比较建模程序可以具有不同的复杂性,用户输入一些额外信息对建模程序的运行有时是有必要的,比如,选择不同的模板或者调整目标模板序列比对。
该服务主要有以下三种方式:
∙First Approach mode(简捷模式):这种模式提供一个简捷的用户介面:用户只需要输入一条氨基酸序列,服务器就会自动选择合适的模板。
或者,用户也可以自己指定模板(最多5条),这些模板可以来自ExPDB模板数据库(也可以是用户选择的含坐标参数的模板文件)。
如果一条模板与提交的目标序列相似度大于25%,建模程序就会自动开始运行。
但是,模板的可靠性会随着模板与目标序列之间的相似度的降低而降低,如果相似度不到50%往往就需要用手工来调整序列比对。
这种模式只能进行大于25个残基的单链蛋白三维结构预测。
∙Alignment Interface(比对界面):这种模式要求用户提供两条已经比对好的序列,并指定哪一条是目标序列,哪一条是模板序列(模板序列应该对应于ExPDB模板数据库中一条已经知道其空间结构的蛋白序列)。
服务器会依据用户提供的信息进行建模预测。
∙Project mode(工程模式):手工操作建模过程:该模式需要用户首先构建一个DeepView工程文件,这个工程文件包括模板的结构信息和目标序列与模板序列间的比对信息。
这种模式让用户可以控制许多参数,例如:模板的选择,比对中的缺口位置等。
此外,这个模式也可以用于“first approach mode 简捷模式”输出结果的进一步加工完善。
此外,SWISS-MODEL还具有其他两种内容上的模式:
∙Oligomer modeling(寡聚蛋白建模):对于具有四级结构的目标蛋白,SWISS-MODEL提供多聚模板的模式,用于多单体的蛋白质建模。
这一模式弥补了简捷模式中只能提交单个目标序列,不能同时预测两条及以上目标序列的蛋白三维结构的不足。
∙GPCR mode(G蛋白偶联受体模式):是专门对7次跨膜G蛋白偶联受体的结构预测。
First Approach mode(简捷模式)的使用
在SWISS-MODEL主页中进行如下操作:
选择所需域值:
这里小数点后零的位数越多,在蛋白库中搜索时得到的结果与提交序列的同源性就越高。
SWISS-MODEL服务器的所有结果都将通过邮件的形式反馈用户,反馈结果的形式由用户自己选择,如图所示:
SWISS-MODEL服务器可以反馈给用户以下结果选项,spdbv模式、普通模式和简短模式。
这里,建议使用SWISS-MODEL默认的spdbv模式,这种模式可以用Swiss-PDBViewer程序打开,可以根据需要对结果进行多种操作。
结果:
以如下序列为例:
NIDRPKGLAFTDVDVDSIKIAWESPQGQVSRYRVTYSSPEDGIHELFPAPDGEEDTAELQ GLRPGSEYTVSVVALHDDMESQPLIGTQSTAIPA
域值选择:0.0000000001
结果选项:Swiss-PdbViewer mode
根据前面选项不同,可能会收到多封邮件。