中英文对照欧洲医疗器械法规资料MDR2017_745 Part 1
中文版欧盟mdr法规

中文版欧盟mdr法规欧盟医疗器械法规(Medical Device Regulation, MDR)是欧洲联盟针对医疗器械领域制定的一份重要法规。
它于2017年5月发布,并于2020年5月正式生效。
本文将根据各项规定,为您详细介绍中文版欧盟MDR法规的内容。
第一部分:导言这一部分主要介绍了欧盟MDR法规的背景和目的,以及适用范围和基本定义。
MDR法规的目标是确保欧盟市场上的医疗器械的安全性、有效性和可靠性,以保障公众的健康和安全。
第二部分:一般要求1. 医疗器械分类和命名MDR法规对医疗器械进行了新的分类,以便更好地管理和监督不同风险等级的产品。
同时,对医疗器械的命名也有明确规定,以避免混淆和误导。
2. 表演的规定MDR法规要求制造商对其产品进行临床评价,并根据风险等级制定相应的技术文件和技术规格。
此外,MDR还提出了对制造商质量管理体系的要求,以确保其产品符合法规的要求。
3. CE标志的规定MDR法规明确了CE标志的使用规定,要求制造商在产品上正确使用并保持其可见性。
这是制造商将其产品引入欧盟市场的必要步骤。
第三部分:具体要求1. 临床评价和临床试验MDR法规对医疗器械的临床评价和临床试验进行了详细规定。
制造商需要对产品进行充分的临床评估,以确保其安全性和有效性符合要求。
2. 医疗器械监督和报告MDR法规明确了各方在市场监督和产品报告方面的责任。
制造商需要建立有效的市场监督和报告机制,并及时识别、评估和报告任何可能危害人类健康的事件。
3. 医疗器械技术文献和标签MDR法规要求制造商提供充分的技术文献和产品标签,以便医疗专业人员和最终用户正确使用和了解产品。
第四部分:认证和授权1. 医疗器械的市场准入MDR法规规定了医疗器械的市场准入程序,制造商需要满足一系列要求,包括提交技术文件、进行质量控制审核等。
只有通过认证的产品才能进入欧盟市场。
2. 第三方评估机构MDR法规对第三方评估机构的审核和授权进行了规定,确保其独立性和专业性。
欧盟医疗器械2017∕745 法规(MDR)(附录 XI 基于产品合规性验证的符合性评估 )

附录XI基于产品合规性验证的符合性评估1.基于产品合规性验证的符合性评估的目的是确保器械符合已发布EC 型式检验证书中所说明的形式,并满足本法规中的适用规定要求。
2.如已根据附录X 颁发EC 型式检验证书,制造商可申请表第A 部分所述流程(生产质量保证),或本附录第B 部分所述流程(产品验证)。
3.通过豁免上述第1 节和第2 节,本附录所述程序加上附录II 和III 所述技术文件也可适用于IIa 类器械制造商。
第A 部分:生产质量保证4.制造商应确保实施批准的相关器械生产质量管理体系,并按照第 6 节的规定进行最终验证,且接受第7 节所述的监管。
5.制造商应履行第4 节所规定的义务,并根据第19 条和附录IV 起草并保存符合性评估流程所涵盖器械型号的EU 符合性声明。
通过发布EC 符合性声明,制造商应确定并声明有关器械是否符合EC 型式检验证书中所说明的型式,以及是否满足本法规中的适用规定要求。
6.质量管理体系6.1制造商应向公告机构提出申请表,评估自己的质量管理体系。
申请表应当包括:- 附录IX 第2.1 节所列的所有要素;- 附录II 和III 所述批准型式的技术文件;- 附录X 第4 节所述EC 型式检验证书副本;若提出申请表后,EC 型式检验证书由同一公告机构颁发,则技术文件及其更新信息和所颁发证书的参考资料应包含在申请中。
6.2质量管理体系的实施应确保器械是否符合EC 型式检验证书中所说明的型式,以及是否满足本法规中的适用于各阶段器械的规定要求。
制造商为其质量管理体系而采用的所有要素、要求和规定,必须以系统和有序的方式记录在质量手册、书面政策和程序之中,例如质量程序、质量计划和质量记录。
该文件尤其应包括对附录IX 第2.2 节(a)、(b)、(d)和(e)所列所有要素的适当说明。
6.3附录IX 第2.3 节第一和第二段的规定适用。
若质量管理体系可确保器械符合EC 型式检验证书中所说明的形式,并满足本法规中的适用规定要求,则欧盟公告机构应出具欧盟质量保证证书。
MDR-EU-2017-745-欧盟医疗器械法规 -附录 16

附录16ANNEX XVI非条款1(2)中所涉及的预期医学目的的产品类别清单:LIST OF GROUPS OF PRODUCTS WITHOUT AN INTENDED MEDICAL PURPOSE REFERRED TO IN ARTICLE1(2)1.隐形眼镜或置于眼睛内部或表面的其他产品。
1.Contact lenses or other items intended to be introduced into or onto the eye.2.通过侵入型手术完全或部分导入人体的产品,目的是矫正人体某些部位的解剖学位置或者固定人体某些部位,不包括纹身类和穿孔类产品。
2.Products intended to be totally or partially introduced into the human bodythrough surgically invasive means for the purpose of modifying the anatomy or fixation of body parts with the exception of tattooing products and piercings.3.用于面部、其他表皮或者黏膜填充的物质或者组合物质,方式有皮下注射、黏膜下注射或表皮内注射等,不包括用于纹身的物质。
3.Substances,combinations of substances,or items intended to be used for facialor other dermal or mucous membrane filling by subcutaneous,submucous or intradermal injection or other introduction,excluding those for tattooing.4.用于减少、移除或者破坏脂肪组织的设备,比如用于抽脂、脂解或进行抽脂手术。
中文版欧盟mdr法规

中文版欧盟mdr法规一、欧盟MDR法规概述1.背景介绍近年来,随着医疗器械技术的不断发展,欧盟为了加强医疗器械监管,提高产品质量和安全性,于2017年5月5日发布了新的医疗器械法规(MDR,Medical Devices Regulation),取代了原有的医疗器械指令(MDD,Medical Devices Directive)。
新法规将于2021年5月26日正式生效,对欧洲市场的医疗器械产品进行全面监管。
2.法规适用范围欧盟MDR法规适用于所有在欧盟市场上销售的医疗器械,包括体外诊断医疗器械。
法规将医疗器械分为三类:一类为低风险产品,二类为中等风险产品,三类为高风险产品。
不同风险类别的产品在合规要求上有所不同。
二、欧盟MDR法规的主要变化1.监管要求与原有指令相比,MDR法规对医疗器械的监管要求更为严格。
新法规要求企业必须确保产品安全有效,并在整个供应链中建立完善的质量管理体系。
2.合规认证流程MDR法规对认证流程进行了调整,增加了认证机构的审核要求和产品的技术评估。
此外,新法规还要求企业进行上市前申请(MDR-CE认证),以确保产品符合欧盟市场准入要求。
3.产品标签要求根据MDR法规,所有医疗器械产品必须贴有CE标志,并附有产品说明书和标签。
此外,企业还需在标签上注明产品类别、规格型号、生产日期等信息。
4.责任与义务新法规明确了各相关方的责任和义务。
企业需对产品安全性负主体责任,政府部门负责监管和执法,认证机构负责产品认证,医疗机构负责产品使用。
三、我国应对欧盟MDR法规的策略1.产品分类与评估企业应对产品进行准确分类,并根据风险类别进行评估。
对于高风险产品,需提前准备技术文档,以确保产品符合MDR法规要求。
2.技术文档准备企业需准备完整的技术文档,包括产品设计、生产工艺、原材料采购、质量控制等方面的信息。
此外,还需关注产品说明书和标签的设计,确保符合法规要求。
3.合规测试与认证在产品合规测试方面,企业应选择具备资质的实验室进行检测,确保产品符合MDR法规的相关技术要求。
中英文对照新版欧洲医疗器械法规资料MDR2017_745 Part 6

Article 61 Clinical evaluation
In this case, the notified body shall check that the PMCF plan is appropriate and includes post market studies to demonstrate the safety and performance of the device.在这种情况下,公告 机构检查发现 PMCF计划很合适且其中包括上 市后研究,以证明器械的安全性能。 In addition, clinical investigations need not be performed in the cases referred to in paragraph 6. 此外,临床研究无需在第6段所所述的情况下 执行。
Article 61 Clinical evaluation
5. A manufacturer of a device demonstrated to be equivalent to an already marketed device not manufactured by him, may also rely on paragraph 4 in order not to perform a clinical investigation provided that the following conditions are fulfilled in addition to what is required in that paragraph: 依据第4段规定,制造商生产出的器械经证实等 同于已投放市场的器械(不属于同一制造商生 产),除此条所要求的内容外,若以下条件均 满足,则无需进行临床研究:
MDR EU 2017 745 Table of Contents
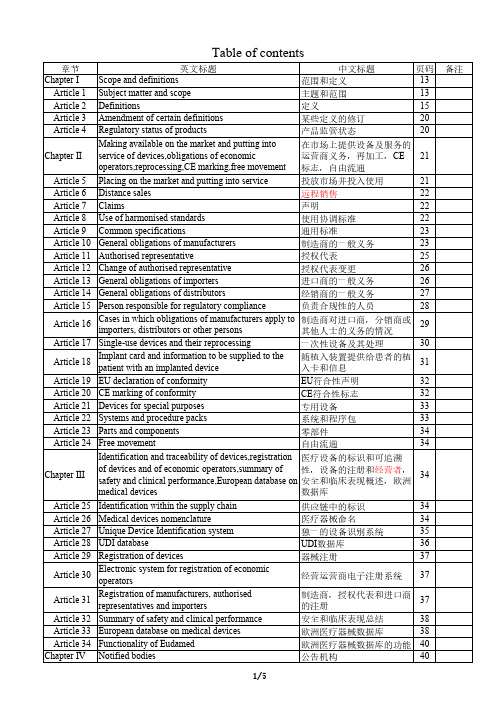
章节 Chapter I
Article 1 Article 2 Article 3 Article 4
英文标题 Scope and definitions Subject matter and scope Definitions Amendment of certain definitions Regulatory status of products
22
Article 8 Use of harmonised standards
使用协调标准
22
Article 9 Common specifications
通用标准
23
Article 10 General obligations of manufacturers
制造商的一般义务
23
Article 11 Authorised representative
安全和临床表现总结
38
欧洲医疗器械数据库
38
欧洲医疗器械数据库的功能 40
公告机构
40
备注
1/5
Article 35 Authorities responsible for notified bodies
负责公告机构的主管机构 40
Article 36 Requirements relating to notified bodies
EU符合性声明
32
CE符合性标志32Fra bibliotek专用设备
33
系统和程序包
33
零部件
34
自由流通
34
Chapter III
Identification and traceability of devices,registration 医疗设备的标识和可追溯
MDR-IVDR-欧盟立法-中文翻译版

MDR-IVDR-欧盟立法-中文翻译版简介本文是对欧盟医疗器械法规(MDR)和欧盟体外诊断器械法规(IVDR)的中文翻译版。
这两个法规是欧洲联盟针对医疗器械和体外诊断器械的法规框架。
它们于2017年发布,将于2021年生效。
这些法规对在欧洲市场销售的医疗器械和体外诊断器械的制造商、进口商和分销商有着重要影响。
MDR第一章:总则第一章包括了MDR的目的、范围和定义。
它强调了MDR 的主要目标是保护公共健康,确保医疗器械的安全和有效性。
此外,该章节还明确了哪些产品属于医疗器械的范畴,并给出了相关的定义。
第二章:一般要求第二章列出了制造商在生产医疗器械时必须遵守的一般要求。
这些要求包括但不限于:•标记和标识要求:医疗器械必须标明相关的信息,以便于识别和跟踪。
•说明书和标签要求:医疗器械必须提供符合规定的说明书和标签,以便用户正确使用。
•报告和注册要求:制造商必须按照规定提交相关报告,并将医疗器械注册到相应的数据库中。
第三章:验证与验证要求第三章明确了制造商必须进行验证和验证的要求。
这些要求包括但不限于:•技术文件:制造商必须编制技术文件,以证明医疗器械符合相关的要求。
•临床评估:制造商必须进行临床评估,并提供相应的数据来支持医疗器械的安全性和有效性。
•验证和验证的要求:制造商必须进行相应的验证和验证,以确保医疗器械符合相关的要求。
第四章:符合性评估第四章规定了制造商进行符合性评估的要求。
这些要求包括但不限于:•符合性审查:制造商必须通过符合性审查证明医疗器械符合相关的要求。
•合规证书:制造商必须获得合规证书,以证明其医疗器械符合相关的要求。
IVDR第一章:总则第一章包括了IVDR的目的、范围和定义。
它强调了IVDR的主要目标是保护公共健康,确保体外诊断器械的安全和有效性。
此外,该章节还明确了哪些产品属于体外诊断器械的范畴,并给出了相关的定义。
第二章:一般要求第二章列出了制造商在生产体外诊断器械时必须遵守的一般要求。
欧盟mdcg医疗器械法规中英文对照

欧盟mdcg医疗器械法规中英文对照全文共3篇示例,供读者参考篇1The European Union Medical Device Regulation (MDR) provides a comprehensive regulatory framework to ensure the safety and effectiveness of medical devices marketed in the EU. The MDR replaces the existing Medical Device Directive (MDD) and introduces several new requirements to enhance the oversight of medical devices throughout their lifecycle.In this article, we will provide a side-by-side comparison of key provisions in the MDR in both English and Chinese to help stakeholders understand the changes and implications of the new regulation.Title/标题欧盟医疗器械法规/ European Medical Device RegulationScope/范围The MDR applies to medical devices and in vitro diagnostic medical devices intended for use by humans for the purpose of diagnosis, prevention, monitoring, treatment or alleviation ofdisease./ 欧盟医疗器械法规适用于用于诊断、预防、监测、治疗或减轻疾病目的的医疗器械和体外诊断医疗器械。
欧盟医疗器械2017∕745 法规(MDR)(附录 XIV 临床评价和上市后临床跟踪)

附录XIV临床评价和上市后临床跟踪第A 部分:临床评价1.如需计划、不断进行并记录临床评价,制造商应:(a) 建立并更新临床评价计划,该计划至少应包括:- 标识通用安全与性能要求(需要相关临床数据的支持);- 器械的预期用途说明;- 明确预期使用者以及明确适应症和禁忌症;- 对患者预期临床益处的详细说明以及相关和指定的临床结果参数;- 检验临床安全性的定性和定量方面的方法说明,并明确说明对剩余风险和副作用的确定方法;- 根据医学中最先进的技术确定器械各种适应症和预期用途的收益风险比的可接受性所使用的参数的指示性清单和说明;- 如何解决特定方面(如药物、非活性动物或人体组织的使用)相关的风险利益问题的指示;- 一份用于指示从探索性研究(如首次人体研究、可行性研究、先导研究)到验证性研究(如关键的临床研究)进展过程的临床研发计划,以及符合本附录第B 部分所述的PMCF,此PMCF 需列出里程碑并说明潜在验收标准;(b) 确定器械相关的可用临床资料及其预期用途,以及通过系统的科学文献检索找到临床证据的缺口;(c) 通过评估临床数据在构建器械安全性和性能方面的适用性,对全部相关临床数据做出评价;(d) 根据临床研发计划,通过合理设计的临床研究,生成解决现存问题所需的任何新的或额外的临床数据;(e) 分析所有相关临床数据,以便得出器械的安全和临床性能(包括临床益处)方面的结论。
2.临床评价应深入且客观,并同时兼顾有利和不利数据。
其深度和程度应与所述器械的性质、分类、预期用途、制造商有关该器械的声明和风险相称。
3.临床评价只能基于可证明与所述器械同等的相关器械的临床数据。
在证明同等性的过程中应考虑以下技术、生物和临床特点:- 技术特点:具有类似设计的器械;在类似条件下使用;具有类似规格和特性,包括物理化学特性,如能源强度、拉伸强度、粘度、表面特征、波长、软件算法等;使用类似部署方法(如相关);具有类似工作原理和关键性能要求。
MDREU_2017_745欧盟医疗器械最新法规(中英对照版)

MDREU_2017_745欧盟医疗器械最新法规(中英对照版)公报中⽂版⽴法L117第60卷2017年5⽉5⽇内容I ⽴法法案法规★欧洲议会和理事会于2017年4⽉5⽇签发的关于医疗器械第2017/745号法规,修订了第2001/83/EC号指令,第178/2002号(EU)法规和第1223/2009号(EU)法规,并废除了理事会第90/385/EEC号和第93/42/EEC号指令(1) (1)★欧洲议会和理事会于2017年4⽉5⽇签发的关于体外诊断医疗器械第2017/746号(EU)法规并废除了第98/79/EC号指令和理事会第2010/227/EU号决议 (176)________________(1)EEA相关性⽂本。
以浅⾊字体打印标题的法案均为涉及农业⽇常管理的法案,⼀般在有限期内有效。
所有其他法案的标题均以粗体打印,并以星号开头。
I(⽴法法案)法规欧洲议会和理事会于2017年4⽉5⽇签发的关于医疗器械第2017/745号法规,修订了第2001/83/EC号指令,第178/2002号(EU)法规和第1223/2009号(EU)法规,并废除了理事会第90/385/EEC号和第93/42/EEC号指令(EEA相关性⽂本)欧洲议会和欧盟委员会,考虑到“欧盟运作条约”,特别是其中第114条和第168(4)(c)条规定,并考虑到欧盟委员会提案,于⽴法草案转交各国议会后,考虑到欧洲经济和社会委员会之意见(1),在咨询地区委员之后,根据⼀般⽴法程序运作(2),鉴于:(1)理事会第90/385/EEC号指令(3)和理事会第93/42/EEC号指令(4)构成有关医疗器械(不包括体外诊断医疗器械)的欧盟监管框架。
但需要对该指令进⾏⼤幅修订,以便建⽴稳健、透明、可预测和可持续的医疗器械监管框架,以确保⾼⽔平的安全和健康,同时为创新提供⽀持。
Council Directive 90/385/EEC (3) and Council Directive 93/42/EEC (4) constitute the Union regulatoryframework for medical devices, other than in vitro diagnostic medical devices. However, a fundamentalrevision of those Directives is needed to establish a robust, transparent, predictable andregulatory framework for medical devices which ensures a high level of safety and health whilst supporting innovation. (2)本法规旨在确保区域内医疗器械市场的平稳运作,在为患者和使⽤者提供⾼⽔平健康保护的基础上,同时考虑到活跃于本⾏业的中⼩型企业利益。
中文版欧盟mdr法规

中文版欧盟mdr法规一、欧盟MDR法规的背景与意义随着医疗器械行业的快速发展,欧盟为了加强医疗器械监管,提高市场准入门槛,保障患者用械安全,于2017年5月25日颁布了全新的医疗器械法规(MDR,Medical Devices Regulation),并于2021年5月26日正式生效。
MDR法规对欧洲市场的医疗器械制造商、进口商和分销商提出了更为严格的要求,旨在提高医疗器械的整体质量、安全和有效性。
二、欧盟MDR法规的主要内容与要求1.监管范围:MDR法规扩大了监管范围,包括体外诊断医疗器械、定制式医疗器械、生物制品等。
此外,法规还涉及到器械原材料、生产过程、产品包装、标签等方面的要求。
2.制造商责任:MDR法规强调制造商在产品全生命周期内的责任,要求制造商对产品的安全性、有效性进行持续评估,并确保产品符合法规要求。
3.产品分类与认证:MDR法规对医疗器械产品进行了更为详细的分类,并根据风险等级实施不同的认证要求。
制造商需按照法规规定,对产品进行分类,并按照相应认证程序获取CE标志。
4.临床评估与合规性检查:MDR法规要求制造商在产品上市前进行临床评估,以证明产品安全性、有效性与预期目标一致。
同时,法规还强化了合规性检查,对不合规产品进行严格的市场监督与执法。
5.市场监督与执法:MDR法规加大了对市场监督的力度,要求各成员国加强对医疗器械市场的监管,并对违法企业进行严厉处罚。
三、我国应对欧盟MDR法规的策略与措施1.了解与关注法规动态:企业应及时关注欧盟MDR法规的最新动态,了解法规要求,确保产品合规。
2.产品分类与认证策略:企业应按照MDR法规对产品进行准确分类,并按照相应认证程序获取CE标志。
3.强化技术文档准备与管理:企业需充分准备技术文档,包括产品说明书、设计输入、设计输出、生产过程控制等,并确保文档的完整、真实、可靠。
4.建立良好的供应链管理体系:企业应加强与供应商的沟通与合作,确保供应链的合规性。
欧盟mdcg医疗器械法规中英文对照

欧盟mdcg医疗器械法规中英文对照EU MDR/IVDR Regulations - Key Changes and Impacts on Medical Device ManufacturersIntroduction:The European Union's Medical Device Regulation (MDR) and In Vitro Diagnostic Medical Device Regulation (IVDR) represent a major overhaul of the regulatory framework for medical devices in Europe. These new regulations aim to strengthen patient safety, ensure transparency and improve the performance of medical devices in the EU market. Understanding the key changes and impacts of these regulations is crucial for medical device manufacturers looking to market their products in the EU.Key Changes in the EU MDR/IVDR:1. Classification: The MDR/IVDR introduces new classification rules for medical devices based on risk, taking into account new technology and scientific advancements. Manufacturers will need to reclassify their devices under the new rules to ensure compliance.2. Notified Bodies: The MDR/IVDR establishes stricter requirements for Notified Bodies, including increased competence and resource requirements. Manufacturers willneed to work with designated Notified Bodies for conformity assessment and certification.3. Unique Device Identification (UDI): The MDR/IVDR requires all medical devices to carry a unique device identifier (UDI) for traceability and post-market surveillance purposes. Manufacturers will need to implement UDI systems and comply with the labeling requirements.4. Clinical Evidence: The MDR/IVDR introduces new requirements for clinical evaluation, including a more robust process for demonstrating safety and performance based on clinical data. Manufacturers will need to ensure their devices meet the new clinical evidence requirements.5. Post-Market Surveillance: The MDR/IVDR places greater emphasis on post-market surveillance and vigilance, requiring manufacturers to actively monitor the safety and performance of their devices throughout their lifecycle. Manufacturers will need to establish and maintain post-market surveillance systems.Impacts on Medical Device Manufacturers:1. Compliance Costs: The implementation of the MDR/IVDR will require significant investment in resources, expertise, andinfrastructure to ensure compliance with the new regulations. Manufacturers will need to budget for these additional costs.2. Market Access: The MDR/IVDR will impact market access for medical devices in the EU, as manufacturers will need to demonstrate compliance with the new regulations to obtain CE marking and market their products. Non-compliance could result in market withdrawal.3. Time to Market: The MDR/IVDR introduces new requirements for conformity assessment and certification, which could lead to delays in getting products to market. Manufacturers will need to factor in additional time for compliance with the new regulations.4. Competitiveness: Manufacturers that proactively adapt to the new regulatory requirements of the MDR/IVDR will have a competitive advantage in the EU market. Those that fail to comply could risk losing market share to competitors.Conclusion:The EU MDR/IVDR regulations represent a significant shift in the regulatory landscape for medical devices in Europe. Understanding the key changes and impacts of these regulations is essential for medical device manufacturers looking to navigatethe new regulatory environment. By proactively adapting to the new requirements, manufacturers can ensure compliance, maintain market access, and stay competitive in the EU market.。
中英文对照新版欧洲医疗器械法规资料MDR2017_745 Part 7

2017.10.29
SECTION 1 Post-market surveillance
Article 83 Post-market surveillance system of the manufacturer
Article 86 Periodic safety update report
1. Manufacturers of class IIa, class IIb and class III devices shall prepare a periodic safety update report (‘PSUR’) for each device and where relevant for each category or group of devices summarising the results and conclusions of the analyses of the post-market surveillance data gathered as a result of the post-market surveillance plan referred to in Article 84 together with a rationale and description of any preventive and corrective actions taken. Throughout the lifetime of the device concerned, that PSUR shall set out:IIa、 IIb和III类器械制造商应针对各器械或类别或器械组编制 定期安全性更新报告(PSUR),总结根据从第84条所述 的上市后监管计划收集的数据分析结果和结论,并对采 取的任何预防和纠正措施提供理由和说明。在该器械的 整个生命周期内,PSUR应列出:
REGULATION(EU) 2017∕745 欧盟医疗器械法规MDR(附录 VIII分类规则)

附录VIII分类规则第I 章分类规则的具体定义1.使用持续时间1.1.“短暂”是指预期正常连续使用不超过60 分钟。
1.2.“短期”是指预期正常连续使用60 分钟到30 天之间。
1.3.“长期”是指预期正常连续使用超过30 天。
2.侵入性器械和有源器械2.1.“身体孔口”是指身体的任何天然开口,以及眼球的外表面,或者任何永久性人工开口,如造口。
2.2.“外科侵入性器械”是指(a) 侵入性器械从身体表面穿透进身体,包括外科手术时通过身体孔口的粘膜穿透;(b) 一种不通过身体孔口穿透的器械2.3.“可重复使用的外科器械”是指通过切割、钻、锯、刮、削、夹、收缩、剪切或类似方式用于外科使用的器械,不连接到任何有源医疗器械,制造商预期可通过适当的处理之后再次使用,如实施清洁、消毒和灭菌。
2.4.“有源治疗器械”是指任何有源器械,无论是单独使用或与其他器械联合使用,以支持、更改、替换或恢复生物学功能或结构,以期疾病、损伤或残障得到治疗或缓解。
2.5.“用于诊断和监测的有源器械”是指任何有源器械,无论是单独使用或与其他器械组合使用,用于为检测、诊断、监测或治疗生理病症、健康状况、疾病或先天畸形。
2.6.“中央循环系统”是指以下血管:肺动脉、升主动脉、弓主动脉、动脉分岔的降主动脉、冠状动脉、颈总动脉、颈外动脉、颈内动脉、脑动脉、头臂干、心静脉、肺静脉、上腔静脉、下腔静脉。
2.7.“中枢神经系统”是指脑、脑膜和脊髓。
2.8.“损伤的皮肤或粘膜”是指皮肤或粘膜呈现病理变化或带来疾病或伤口变化的区域。
第II 章实施规则3.实施规则3.1.分类规则的使用应基于器械的预期目的。
3.2.若相关器械将与其他器械共同使用,分类规则应分别适用于各器械。
医疗器械和附录XVI 所列产品的附件,应根据其自身因素进行分类,独立于它们所适用的器械。
3.3.驱动某一器械或影响器械使用的软件,应与该器械归为同一类别。
若该软件独立于任何其他器械,则应按照其本身进行分类。
MDR-EU-2017-745-欧盟医疗器械法规 -附录 17

Article6(2)Article7(1)Article114Article7Article8Articles94to97—Article9Article51Article8(1)Article10(1)Articles87(1)and89(2) Article8(2)Article10(2)Article87(10)andArticle87(11)firstsubparagraphArticle8(3)Article10(3)Article89(7)Article8(4)Article10(4)Article91Article9(1)Article11(1)Article52(3)—Article11(2)Article52(6)—Article11(3)Article52(4)and(5)—Article11(4)——Article11(5)Article52(7)Article9(2)Article11(6)Article52(8)Article9(3)Article11(8)Article11(3)Article9(4)Article11(12)Article52(12)Article9(5)Article11(7)—Article9(6)Article11(9)Article53(1)Article9(7)Article11(10)Article53(4)Article9(8)Article11(11)Article56(2)Article9(9)Article11(13)Article59Article9(10)Article11(14)Article4(5)and Article122third paragraph—Article12Article22—Article12a Article17Article13(1)(c)—Article9a(1)firstindentArticle13(1)(d)Article4(1)Article9a(1)secondindent—Article13(1)(a)Article51(3)(a)andArticle51(6)—Article13(1)(b)Article51(3)(b)andArticle51(6) Article10Article15Articles62to82Article10a(1),second sentence of Article10a(2)and Article10a(3)Article14(1),second sentence ofArticle14(2)andArticle14(3)Articles29(4),30and31Article10a(2),first sentence Article14(2)first sentenceArticle11(1)Article10b Article14a Articles33and34Article10c Article14b Article98Article11(1)Article16(1)Articles42and43Article11(2)Article16(2)Article36Article11(3)Article16(3)Article46(4)Article11(4)Article16(4)—Article11(5)Article16(5)Article56(5)Article11(6)Article16(6)Article56(4)Article11(7)Article16(7)Articles38(2)and44(2)Article12Article17Article20Article13Article18Articles94to97Article14Article19Article99Article15Article20Article109Article15a Article20a Article102Article16Article22—Article17Article23——Article21—(1)欧洲议会及理事会2004年3月31日颁布的法规中,第726条规定了人类使用以及动物使用的医药产品批准和监督流程,并建立了欧洲药品管理局。
欧盟mdcg医疗器械法规中英文对照

欧盟mdcg医疗器械法规中英文对照全文共3篇示例,供读者参考篇1The European Union's Medical Devices Regulation (MDR) is a comprehensive set of rules that govern the manufacture, distribution, and use of medical devices within the EU. The MDR aims to ensure that medical devices marketed in the EU are safe, effective, and of high quality.The MDR replaces the Medical Devices Directive (MDD) and brings significant changes to the regulatory framework for medical devices in the EU. It introduces new requirements for manufacturers, importers, authorized representatives, and distributors of medical devices, as well as stricter rules for clinical evaluation, post-market surveillance, and vigilance.One of the key aspects of the MDR is the introduction of the Unique Device Identification (UDI) system, which requires manufacturers to assign a unique identifier to each medical device. This will help improve traceability and facilitate the recall of faulty devices from the market.Another important change introduced by the MDR is the requirement for manufacturers to demonstrate compliance with a set of common specifications called Medical Device Coordination Group (MDCG) guidelines. These guidelines cover various aspects of medical device regulation, including clinical evaluation, classification, and conformity assessment.Overall, the MDR aims to enhance patient safety and ensure the quality of medical devices on the EU market. By introducing stricter requirements and improving transparency and oversight, the MDR seeks to address the shortcomings of the previous regulatory framework and adapt to the evolving technological landscape of the healthcare industry.In conclusion, the MDR represents a significant step forward in the regulation of medical devices in the EU. By setting high standards for the safety and performance of medical devices, the MDR aims to protect patients and healthcare professionals and promote innovation in the medical device industry. It is essential for manufacturers, importers, and distributors of medical devices to familiarize themselves with the new requirements of the MDR and ensure compliance to continue marketing their products in the EU market.篇2The Medical Devices Regulation (MDR) is a set of regulations established by the European Union (EU) to ensure that medical devices placed on the market meet high standards of safety and performance. The MDR replaces the previous Medical Devices Directive (MDD) and is intended to strengthen and harmonize the regulatory framework for medical devices in the EU.Below is a comparison of key provisions in the MDR and the MDD:1. Scope:- MDR: The scope of the regulation has been expanded to include a broader range of medical devices, including certain aesthetic and cosmetic devices.- MDD: The scope of the directive was more limited and did not cover certain types of products that are now included under the MDR.2. Classification:- MDR: The classification rules for medical devices have been revised to align with international standards, and there are now stricter requirements for classifying devices based on their potential risks.- MDD: The classification rules under the directive were less stringent and did not always reflect the level of risk associated with certain devices.3. Clinical Evaluation and Performance Studies:- MDR: There are new requirements for conducting clinical evaluations and performance studies to demonstrate the safety and efficacy of medical devices.- MDD: The requirements for clinical evaluations under the directive were less detailed and did not always require the same level of evidence.4. Post-Market Surveillance:- MDR: There are enhanced requirements for post-market surveillance, including the implementation of a Unique Device Identification (UDI) system to track devices throughout their lifecycle.- MDD: The post-market surveillance requirements in the directive were less detailed and did not always ensure timely reporting of adverse events.5. Notified Bodies:- MDR: Notified Bodies play a more active role in the assessment of medical devices and are subject to stricter requirements for designation and oversight.- MDD: Notified Bodies under the directive had more limited responsibilities and were not always subject to the same level of scrutiny.In conclusion, the MDR represents a significant shift in the regulation of medical devices in the EU, with a focus on increasing transparency, accountability, and patient safety. By harmonizing the regulatory framework and imposing stricter requirements on manufacturers, the MDR aims to ensure that only high-quality and safe medical devices are placed on the market. Manufacturers of medical devices should familiarize themselves with the new requirements under the MDR and take steps to ensure compliance to avoid any disruption in their business operations.篇3The European Union's Medical Device Coordination Group (MDCG) plays a crucial role in ensuring the safety and effectiveness of medical devices on the European market. The MDCG provides guidance and support to member states andstakeholders in implementing the EU medical device regulations to ensure harmonization and consistency across the EU.The MDCG was established under the Medical Device Regulation (MDR) to assist the European Commission and member states in harmonizing the implementation of the new regulations. The group is composed of representatives from member states, the European Commission, and other relevant stakeholders, such as industry associations and notified bodies.The MDCG provides guidance on various aspects of the MDR, including classification of medical devices, conformity assessment procedures, clinical evaluation, post-market surveillance, and vigilance. The group also publishes guidance documents and recommendations to help manufacturers, notified bodies, and other stakeholders understand and comply with the regulations.One of the key tasks of the MDCG is to provide guidance on the implementation of the Unique Device Identification (UDI) system, which is mandatory for all medical devices placed on the EU market. The UDI system aims to enhance traceability and transparency of medical devices throughout their lifecycle, from manufacturing to patient use.In addition to providing guidance on regulatory requirements, the MDCG also plays a role in coordinating surveillance and market monitoring activities to ensure the safety and performance of medical devices on the EU market. The group works closely with member states, the European Commission, and other stakeholders to exchange information and data on medical devices to identify potential risks and take appropriate regulatory actions.Overall, the MDCG plays a critical role in ensuring the safety, effectiveness, and quality of medical devices on the European market. By providing guidance, support, and coordination, the group helps to facilitate the smooth implementation of the EU medical device regulations and ensure the protection of public health and safety.欧盟的医疗器械协调组(MDCG)在确保欧洲市场上的医疗器械的安全性和有效性方面发挥着至关重要的作用。
中英文对照新版欧洲医疗器械法规 MDR Part

2017.09.15
Sterimd
1
主要内容
1. 法规立法和执行; 2. 适用范围; 3. 过渡期安排; 4. 法规结构。
Sterimd
2
一、REGULATION (EU) 2017/745法规
1. REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC. 欧洲议会和理事会于2017年4月5日 签发的关于医疗器械第2017/745号法规,修订 了第2001/83/EC号指令,第178/2002号(EU) 法规和第1223/2009号(EU)法规,并废除了 理事会第90/385/EEC号和第93/42/EEC号指令
• (e) cosmetic products covered by Regulation (EC) No 1223/2009;欧洲委 员会第1223/2009号法规所涵盖的美容产品;
Sterimd
7
• (f) transplants, tissues or cells of animal origin, or their derivatives, or products containing or consisting of them; however this Regulation does apply to devices manufactured utilising tissues or cells of animal origin, or their derivatives, which are non-viable or are rendered non-viable;动物源 的移植器官、组织或细胞或其衍生产品,或含有或由其组成的产品; 但本法规适用于使用非活性或活性动物来源的组织或细胞或其衍生产 品制造而成的器械。
最新MDR一类医疗器械全套技术文件模板(中英文对照)

最新MDR一类医疗器械全套技术文档模板(中英文对照)CE TECHNICALDOCUMENTATION Based on REGULATION (EU) 2017/745Product Name: {填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}{填写申请者的企业名称}ContentRationale for the qualification as a medical device and the risk class attributedProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:1.Rationale for the qualification as a medical device{填写申报产品可作为器械的合理的理由}【可从产品的预期用途,适应症方面考虑】(参考示例:According to the definition of medical device in REGULATION (EU) 2017/745 as below and the application of{填写申报产品名称}(参考示例:CRP mask), we consider that{填写申报产品名称}(参考示例:CRP mask)is a medical device.Define of medical device:“medical device” means any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:—diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease,—diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability,—investigation, replacement or modification of the anatomy or of a physiological or pathological process or state,—providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations,and which does notachieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.The following products shall also be deemed to be medical devices:—devices for the control or support of conception;—products specifically intended for the cleaning, disinfection or sterilisation of devices as referred to in Article 1(4) and of those referred to in the first paragraph of this point.)2.Rationale for the risk class attributed{填写申报产品风险分类的基本原理}(参考示例:The risk class is attributed according to Annex VIII CLASSIFICATION RULES in REGULATION (EU) 2017/745.{填写申报产品名称}(参考示例:CPR mask)is a{填写申报产品的类别}【根据产品实际情况填写申报产品所属类别如是属于非侵入、侵入还是有源】(参考示例:non-invasive)device. The rules of classification for{填写申报产品的类别}【根据产品实际情况填写申报产品所属类别如是属于非侵入、侵入还是有源】(参考示例:non-invasive)devices is in{填写申报产品分类所适用的分类法规}(参考示例:Rule1-Rule4), the details are as below.{填写申报产品分类所适用的分类法规原文}(4. NON-INVASIVE DEVICES4.1. Rule 1All non-invasive devices are classified as class I, unless one ofthe rules set out hereinafter applies.4.2. Rule 2All non-invasive devices intended for channelling or storingblood, body liquids, cells or tissues, liquids or gases for thepurpose of eventual infusion, administration or introduction intothe body are classified as class IIa:—if they may be connected to a class IIa, class IIb or class IIIactive device; or—if they are intended for use for channelling or storing blood or other body liquids or for storing organs, parts of organs or body cells and tissues, except for blood bags; blood bags are classified as class IIb.In all other cases, such devices are classified as class I.4.3. Rule 3All non-invasive devices intended for modifying the biological or chemical composition of human tissues or cells, blood, otherbody liquids or other liquids intended for implantation or administration into the body are classified as class IIb, unless thetreatment for which the device is used consists of filtration,centrifugation or exchanges of gas, heat, in which case they areclassified as class IIa.All non-invasive devices consisting of a substance or a mixtureof substances intended to be used in vitro in direct contact withhuman cells, tissues or organs taken from the human body orused in vitro with human embryos before their implantation oradministration into the body are classified as class III.4.4. Rule 4All non-invasive devices which come into contact with injuredskin or mucous membrane are classified as:—class I if they are intended to be used as a mechanical barrier,for compression or for absorption of exudates;—class IIb if they are intended to be used principally for injuriesto skin which have breached the dermis or mucous membraneand can only heal by secondary intent; —class IIa if they are principally intended to manage themicro-environment of injured skin or mucous membrane; and—class IIa in all other cases.This rule applies also to the invasive devices that come intocontact with injuredmucous membrane.)According to above rules, {填写申报产品名称}(参考示例:CPR mask)is classified as Class I according to{填写产品适用的分类规则}(参考示例:rule 1).)Description and specificationProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:1 product or trade name and a general description of the device including its intended purpose and intended users;1.1 Name of the DeviceProduct name: {填写申报产品名称}1.2 Product description{对产品进行简要描述}【可从产品的组成,是否含附件以及所属的类别,是否需要灭菌等方面,进行简要描述】1.3 Intended Purpose{填写申报产品的预期用途}1.4 Intended User{填写申报产品的目标使用者}(参考示例:hospital staff)2 The intended patient population and medical conditions to be diagnosed, treated and/or monitored and other considerations such as patient selection criteria, indications, contra-indications, warnings;2.1 Intended patient population{填写产品适用的患者人群}(参考示例:adult)2.2 Medical conditions{填写产品适用的疾病阶段或程度}(参考示例:The patient requires ventilation)2.3 Other considerations2.3.1 Patient selection criteria{填写申报产品适用的病人的选择标准}(参考示例:the years old of patient over18)2.3.2 Indications{填写申报产品的适应症}2.3.3 Contra-indications,{填写申报产品的禁忌症}2.3.4 Warnings{填写申报产品的警告}3 Principles of operation of the device and its mode of action, scientifically demonstrated if necessary3.1 Principles of operation{填写申报产品的操作原理}3.2 Mode of action{填写申报产品的作用方式}4 The rationale for the qualification of the product as a devicePlease refer to Document 1 “Rationale for the qualification as a medical device and the risk class attributed”.5 The risk class of the device and the justification for the classification rule(s) applied in accordance with Annex VIII;Please refer to Document 1 “Rationale for the qualification as a medical device and the risk class attributed”.6 An explanation of any novel features;{填写申报产品是否使用新技术的描述} (参考示例:This is a traditional technology. It has been used all over the world for many years. There are no novel features.)7 A description of the accessories for a device, other devices and other products that are not devices, which are intended to be used in combination with it;{对申报器械是否有附件,以及预期与其他非器械联合使用的简要描述} (参考示例:The actual device do not contain accessories. It is not used in combination with other devices.)8 A description or complete list of the various configurations/variants of the device that are intended to be made available on the market;{详细列出申报产品的所有的型号,并描述各型号之间的差异 }9 A general description of the key functional elements9.1 Its parts/components (including software if appropriate)9.1.1 Parts/ComponentsThe parts of {填写申报产品名称} are as below.{列出申报产品组件 }9.1.2 Software{此处对申报产品的软件进行描述}.9.2 FormulationIt is consisted of components as section 9.1.1. It does not contain special formulation.9.3 Its compositionPlease refer to section 10.1.1.9.4 Functionality{对关键原件的功能进行描述}9.5 qualitative and quantitative composition.{对关键元件的定性和定量进行描述}9.6 Labelled pictorial representations(e.g. diagrams, photographs, and drawings), clearly indicating key parts/components, including sufficient explanation to underst and the drawings and diagrams;The picture of key parts/components for actual device are as below:{填写申报产品的关键部件的图片或图示}【提供带标记的图形指示关键部件/组件,如图表、照片、图纸等】10 A description of the raw materials incorporated into key functional elements and those making either direct contact with the human body or indirect contact with the body, e.g., during extracorporeal circulation of body fluids;{填写预期与人体间接接触和直接接触的材料说明}(示例见下文,包括不限于下列表格中的内容)11 Technical specifications{对申报产品的技术规格进行描述}【包括但不限于特征、尺寸、性能属性,通常出现在用户阅读的产品规范中,例如宣传册、目录、相似出版物等】Technical SpecificationsProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号} Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:1.Raw materials{填写原材料信息}2.Drawings of components{请详细列出各种组件的图片或图纸}【若有多个组件图片,请依次列出】3. Quality control procedures{对申报产品的质量控制进行简要描述}(参考示例:Our company has established EN ISO 13485 quality system. The manufacture process is continuously monitored.Firstly, the materials are monitored. All incoming suppliers shall be selected by the company in accordance with the requirements of the Supplier management control procedures. When incoming materials of the supplier are delivered to the factory, the quality department shall formulate incoming inspection specifications and conduct incoming inspection in accordance with the inspection requirements. The defective incoming materials shall be handled in accordance with the company's procedure document control procedure for nonconforming products.Secondly, final product is monitored. Before product delivery, the final inspection should be conducted. Meanwhile, deal with the unqualified products according to thecontrol procedure of unqualified products.Thirdly, ex-factory process is monitored.According to the inspection specification, the inspector shall carry out factory inspection on the products, make inspection records and mark the quality status as required, and deal with the unqualified products according to the control procedure of unqualified products.)Information to be supplied by the manufacturerProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:er Manual and Label{放入申报产品的使用手册和标签}(参考示例:Please refer to***User Manual and ***Label.){请放入申报产品的说明书}【对申报产品的说明书要求请参考MDR 法规第附录I第III章中对说明书,以及EN ISO 15223-1:2016标准中对说明书的要求】{请放入申报产品的标签}【对申报产品的标签的要求请参考MDR 法规第附录I第III章中对标签,以及EN ISO 15223-1:2016标准中对标签的要求】Reference to previous generations of the device andto similar devicesProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:1.An overview of the previous generation or generations of the device produced by the manufacturer, where such devices exist{若申报产品有前代产品的话,请对前代产品进行简要概述}【参考示例:The actual device is the First-generation products, we consider there is no previous generation 】2. An overview of identified similar devices available on the Union or international markets, where such devices exist.{对欧盟以及国际市场上的类似产品进行简要概述}Design and manufacturing informationProduct Name:{填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:rmation that allows the understanding of thedesign and manufacturing ofa device1.1Information that allows the understanding of thedesign of a device{填写申报产品的设计开发程序}(参考示例:The actual device are designed according to the Design and development Process as below.)1.2Information that allows the understanding of themanufacturing of a device1.2.1manufacturing processes{列出申报产品的生产程序}1.2.2Process validation{填写生产过程中的各种验证}.【应放入完整的测试数据或链接】(参考示例:The process validation are please refer to * * * Final product test report,* * * Packaging seal validation report)1.2.3 Continuous monitoring{对申报产品生产过程中的连续监测进行描述}(参考示例:Our company has established EN ISO 13485 quality system. The manufacture process is continuously monitored.Firstly, the materials are monitored. All incoming suppliers shall be selected by the company in accordance with the requirements of the Supplier management control procedures. When incoming materials of the supplier are delivered to the factory, the quality department shall formulate incoming inspection specifications and conduct incoming inspection in accordance with the inspection requirements. The defective incoming materials shall be handled in accordance with the company's procedure document control procedure for nonconforming products. Secondly, final product is monitored. Before product delivery, the final inspection should be conducted. Meanwhile, deal with the unqualified products according to the control procedure of unqualified products. Thirdly, ex-factory process is monitored.According to the inspection specification, the inspector shall carry out factory inspection on the products, make inspection records and mark the quality status as required, and deal with the unqualified products according to the control procedure of unqualified products.)1.2.4 Final product testing{列出最终产品的所有验证测试}2 Design calculations relevant to the intended use of the product{填写实现申报产品预期用途的设计原理}.3 Technology{对申报产品的当前技术生产工艺进行简要描述}.(参考示例:From the information of Section 2 of Document 5 Reference to previous generations of the device and to similar devices, the technology of actual device is mature. The design is safe and have been established for a number of years. Actual device have been performing as intended during that time such information is likely tobe sufficient to cover this requirement.)4 Identification of all sites4.1 Company addressThe company registration address:{填写公司注册地址} the manufacturing address:{填写生产地址}.4.2 Supplier addressThe supplier addresses of critical materials are as below:4.3 Sub-contractor address{填写分包商的地址}General safety and performance requirementsProduct Name: {填写申报产品名称}Model:{填写申报产品的具体型号}Document No.: {填写本文档编号}Edition: {填写本文档版本号}Drafted by: Date: {填写本文档编写日期}Checked by: Date: {填写本文档审核日期}Approved by: Date: {填写本文档批准日期}{填写申请者的企业名称}Revision records:1.General safety and performance requirements{放入一般安全和性能要求的符合性证明}(参考示例:Please refer to **** Checklist for General safety and performance requirements Compliance analysis)requirements Compliance analysis REGULATION (EU) 2017/745Conformity Assessment Procedure according to the following Annex of the Directive (pls. tick): Declare the conformity of their products by issuing the EU declaration of conformity referred to in Article 19(MDR regulation EU 2017/745) after drawing up the technical documentation set out in Annexes II and III.requirements Compliance analysisPage 3 of 104Page 4 of 104Page 5 of 104Page 6 of 104Page 7 of 104Page 8 of 104Page 10 of 104Page 11 of 104Page 12 of 104Page 14 of 104。
欧盟医疗器械2017∕745 法规(MDR)(附录 XII 由公告机构签发的证书)

附录XII由公告机构签发的证书第I 章一般要求1.应用欧盟的其中一种官方语言起草证书;2.各证书均应仅参考一种符合性评估流程;3.证书应仅颁发给一家制造商证书中包含的制造商名称和地址应与在第30 条中所述的电子系统中注册的信息相同;4.证书适用范围的内容应明确说明所涵盖的器械:(a) EU技术文件评估证书、EU型式检验证书和EU产品验证证书应包含明确标识,包括器械名称、型号、类型、预期用途(制造商在使用说明中包含的并已通过符合性评估流程进行评定的预期用途)、风险分类以及第27(6)条所述的基本UDI - DI 号;(b) EU 质量管理体系证书和EU 质量保证证书应包括器械标识或器械组别、风险分类和IIb 类器械的预期用途;5.公告机构应能够应要求说明证书所涵盖的(单一)器械。
公告机构应说明能够确定证书所涵盖器械(包括其分类)的方法;6.如适用,证书应包含本证书所涵盖器械的上市记录,还需根据本法规颁发的另一证书;7.根据第52(7)条需要涉及公告机构的第I 类器械的EU 质量管理体系证书和EU 质量保证证书应包含一份声明,声明公告机构已审核质量管理体系涉及该段中要求的方面。
8.若本证书代替先前证书,即增补、修改或重新颁发证书时,新证书应包含先前证书的参考资料及其颁发日期以及变更标识。
第II 章证书的必需内容9.证书的必须内容9.1.1.公告机构名称、地址和标识号;9.1.2.制造商和授权代表(如适用)的名称和地址;9.1.3.证书的唯一标识号;9.1.4.第31(2)条所述的制造商单一注册号;9.1.5.颁发日期;9.1.6.失效日期;9.1.7.符合第I 部分第4 节规定的器械明确标识所需数据(如适用);9.1.8.如适用,参照在第I 章第8 节指定的先前证书;9.1.9.符合所进行符合性评估要求的本法规和相关附录参考资料;9.1.10.所进行的检验和试验,例如相关CS、协调标准、检验报告和审核报告的参考资料;9.1.11.涵盖器械上市所需技术文件相关部分或其他证书的参考资料(如适用);9.1.12.公告机构的监管信息(如适用);9.1.13.公告机构针对相关附录的符合性评估结论;9.1.14.证书有效性的条件或限制;9.1.15.符合相关国家法律要求且具有法律约束力的公告机构签名。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
• 1. This Regulation shall enter into force on the twentieth day following that of its publication in the Official Journal of the European Union. 本法 规应在《欧盟官方公报》上公布后第20天生 效。
• (c) advanced therapy medicinal products covered by Regulation (EC) No 1394/2007;欧洲委员会第1394/2007号法规所涵盖的前沿疗法医药产品;
• (d) human blood, blood products, plasma or blood cells of human origin or devices which incorporate, when placed on the market or put into service, such blood products, plasma or cells, except for devices referred to in paragraph 8 of this Article;人类血液或血液制品、人源的血浆或血细胞, 或者在投放市场或投入使用时,包含此类血液制品、血浆或细胞的器 械,但本条第8段所述的器械除外(8. 包含物);
• 2. It shall apply from 26 May 2020. 自2020年5 月26日起适用。
• 3. By way of derogation from paragraph 2(略)
Sterimd
4
二、Scope范围
• 1. This Regulation lays down rules concerning the placing on the market, making available on the market or putting into service of medical devices for human use and accessories for such devices in the Union. This Regulation also applies to clinical investigations concerning such medical devices and accessories conducted in the Union. 本法规规定了有 关欧盟境内供人类使用的医疗器械极其附件的市 场投放、市场提供或投入使用方面的规则。本法 规也适用于在欧盟进行的有关该医疗器械及其附 件临床研究。
Sterimd
5
Devices with a non-medical intended
purpose非医疗目的的器械
• Devices with both a medical and a nonmedical intended purpose shall fulfil cumulatively the requirements applicable to devices with an intended medical purpose and those applicable to devices without an intended medical purpose.具有医疗和非医 疗预期目的器械应逐渐的满足适用于具有 预期医疗目的器械要求和适用于无预期医 疗目的器械的那些要求。
EU MDR 2017/745医疗器械法规
2017.09.15
Sterimd
1
主要内容
1. 法规立法和执行; 2. 适用范围; 3. 过渡期安排; 4. 法规结构。
Sterimd
2
一、REGULATION (EU) 2017/745法规
1. REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC. 欧洲议会和理事会于2017年4月5日 签发的关于医疗器械第2017/745号法规,修订 了第2001/83/EC号指令,第178/2002号(EU) 法规和第1223/2009号(EU)法规,并废除了 理事会第90/385/EEC号和第93/42/EEC号指令
Sterimd
6
This Regulation does not apply to
• (a) in vitro diagnostic medical devices covered by Regulation (EU) 2017/746;欧盟第2017/746号法规所涵盖的体外诊断医疗器械
• (b) medicinal products as defined in point 2 of Article 1 of Directive 2001/83/EC. In deciding whether a product falls under Directive 2001/83/EC or under this Regulation, particular account shall be taken of the principal mode of action of the product;如第2001/83/EC号指令第1条 第2点中所定义的医疗产品。在确定产品是否属于第2001/83/EC号指 令或本法规的范围时,应特别考虑产品的主要作用模式。