启动子生物信息学分析软件
常用分子生物学软件简介

常用分子生物学软件简介常用分子生物学软件一、基因芯片:1、基因芯片综合分析软件。
ArrayVision 7.0一种功能强大的商业版基因芯片分析软件,不仅可以进行图像分析,还可以进行数据处理,方便protocol的管理功能强大,商业版正式版:6900美元。
Arraypro 4.0Media Cybernetics公司的产品,该公司的gelpro, imagepro一直以精确成为同类产品中的佼佼者,相信arraypro也不会差。
phoretix?Array Nonlinear Dynamics公司的基因片综合分析软件。
J-express挪威Bergen大学编写,是一个用JAVA语言写的应用程序,界面清晰漂亮,用来分析微矩阵(microarray)实验获得的基因表达数据,需要下载安装JAVA运行环境JRE1.2后(5.1M)后,才能运行。
2、基因芯片阅读图像分析软件ScanAlyze 2.44,斯坦福的基因芯片基因芯片阅读软件,进行微矩阵荧光图像分析,包括半自动定义格栅与像素点分析。
输出为分隔的文本格式,可很容易地转化为任何数据库。
3、基因芯片数据分析软件Cluster斯坦福的对大量微矩阵数据组进行各种簇(Cluster)分析与其它各种处理的软件。
SAMSignificance Analysis of Microarrays 的缩写,微矩阵显著性分析软件,EXCEL软件的插件,由Stanford大学编制。
4.基因芯片聚类图形显示TreeView 1.5斯坦福开发的用来显示Cluster软件分析的图形化结果。
现已和Cluster成为了基因芯片处理的标准软件。
FreeView是基于JAVA语言的系统树生成软件,接收Cluster生成的数据,比Treeview增强了某些功能。
5.基因芯片引物设计Array Designer 2.00DNA微矩阵(microarray)软件,批量设计DNA和寡核苷酸引物工具二、RNA二级结构。
人HO-1基因启动子区生物信息学分析

㊃生物信息学㊃d o i:10.3969/j.i s s n.1671-8348.2021.09.031网络首发h t t p s://k n s.c n k i.n e t/k c m s/d e t a i l/50.1097.R.20201229.1633.017.h t m l(2020-12-30)人HO-1基因启动子区生物信息学分析*王凡,徐世明,古同男,王宏娟ә(首都医科大学燕京医学院生物化学与分子生物学教研室,北京101300) [摘要]目的分析人血红素加氧酶-1(h HO-1)基因启动子区序列特点,转录因子结合位点种类及C p G岛位置㊂方法利用美国国立生物技术信息中心(N C B I)数据库获取h HO-1基因序列和启动子序列,S o f t B e r-r y分析h HO-1基因核心启动子位置,P R OMO和G e n e R e g u l a t i o n分析转录因子结合位点种类,M e t h P r i m e r 2.0和C p G F i n d e r预测C p G岛位置㊂结果 h HO-1基因位于22q12.3,全长13112b p㊂转录起始点上游约1995b p范围内为启动子区域,h HO-1启动子区含79种转录因子结合位点和1个C p G岛㊂结论 h HO-1基因启动子能预测出多种转录因子结合位点,可为更深入地研究h HO-1基因的调控机制及其在抗逆方面的潜在作用提供理论基础㊂[关键词]血红素加氧酶-1;启动子;转录因子结合位点;C p G岛[中图法分类号] Q527;Q754[文献标识码] A[文章编号]1671-8348(2021)09-1581-05 B i o i n f o r m a t i c a n a l y s i s o f h u m a n h e m e o x y g e n a s e1g e n e p r o m o t e r r e g i o n*WA N G F a n,X U S h i m i n g,G U T o n g n a n,WA N G H o n g j u a nә(T e a c h i n g a n d R e s e a r c h i n g S e c t i o n o f B i o c h e m i s t r y a n d M o l e c u l a r B i o l o g y,Y a n j i n g M e d i c a l C o l l e g e,C a p i t a l M e d i c a l U n i v e r s i t y,B e i j i n g101300,C h i n a)[A b s t r a c t]O b j e c t i v e T o a n a l y z e t h e s e q u e n c e c h a r a c t e r i s t i c s,k i n d s o f t r a n s c r i p t i o n f a c t o r s b i n d i n g s i t e s a n d l o c a t i o n o f C p G i s l a n d o f h u m a n h e m e o x y g e n a s e1(h HO-1)g e n e p r o m o t e r r e g i o n.M e t h o d s T h e s e q u e n c e s o f h HO-1g e n e a n d i t s p r o m o t e r w e r e o b t a i n e d f r o m t h e N C B I d a t a b a s e.T h e p o s i t i o n o f h HO-1 g e n e c o r e p r o m o t e r w a s a n a l y z e d b y S o f t B e r r y.T h e t y p e o f t r a n s c r i p t i o n f a c t o r b i n d i n g s i t e s w a s a n a l y z e d b y P R OMO a n d G e n e r e g u l a t i o n.T h e p o s i t i o n o f C p G i s l a n d w a s p r e d i c t e d b y M e t h P r i m e r2.0a n d C p G F i n d e r. R e s u l t s T h e h HO-1g e n e w a s l o c a t e d a t22q12.3,a n d t h e f u l l l e n g t h w a s13112b p.T h e p r o m o t e r r e g i o n w a s a b o u t l o c a t e d i n t h e1995b p u p s t r e a m o f t r a n s c r i p t i o n i n i t i a t i o n s i t e.T h e r e m i g h t b e79s p e c i e s o f t r a n s c r i p-t i o n f a c t o r b i n d i n g s i t e s a n d1C p G i s l a n d s i n t h e p r o m o t e r r e g i o n o f t h e h HO-1g e n e.C o n c l u s i o n T h e h HO-1g e n e p r o m o t e r c a n p r e d i c t m a n y k i n d s o f t r a n s c r i p t i o n f a c t o r b i n d i n g s i t e s,w h i c h p r o v i d e s a t h e o r e t i c a l b a s i s f o r f u r t h e r r e s e a r c h o n t h e r e g u l a t i o n m e c h a n i s m o f h HO-1g e n e a n d i t s p o t e n t i a l r o l e o f s t r e s s r e s i s t a n c e.[K e y w o r d s]h e m e o x y g e n a s e-1;p r o m o t e r;t r a n s c r i p t i o n f a c t o r b i n d i n g s i t e;C p G i s l a n d人血红素加氧酶-1(h HO-1)是一种细胞保护酶,在内体广泛分布㊂血红素可被h HO-1催化降解成胆绿素㊁C O和亚铁离子[1]㊂研究表明,h HO-1与呼吸系统㊁消化系统㊁心血管系统㊁泌尿系统㊁神经系统等疾病发生㊁发展密切相关[1-2]㊂有报道称h HO-1启动子区(G T)n双核苷酸重复多态性与肿瘤关系密切,较短的(G T)n双核苷酸重复可能对食管鳞状上皮细胞癌的发生起抑制作用,而较长的(G T)n双核苷酸重复可能会增加患食管鳞状上皮细胞癌[2],口腔癌和心血管等疾病[3]的风险㊂本研究利用生物信息学方法,通过对h HO-1基因启动子转录因子结合位点及C p G 岛的预测分析,探讨h HO-1启动子区与相关疾病的关系,旨在为今后研究h HO-1的生物学功能及调控机制提供有价值的信息㊂1材料与方法1.1材料h HO-1基因及启动子序列信息的获取:美国国立生物技术信息中心(N C B I)数据库(h t t p s://w w w.n c-b i.n l m.n i h.g o v/)㊂T A T A盒预测网站:S o f t b e r r y(h t-t p://l i n u x1.s o f t b e r r y.c o m/b e r r y.p h t m l?t o p i c=t s s w &g r o u p=p r o g r a m s&s u b g r o u p=p r o m o t e r)㊂转录因子结合位点预测网站:P R OMO(h t t p://a l g g e n.l s i. u p c.e s/c g i-b i n/p r o m o_v3/p r o m o/p r o m o i n i t.c g i?d i r D B=T F_8.3),Ge n e r e g u l a t i o n(h t t p://g e n e-r e g-1851重庆医学2021年第50卷第9期*基金项目:北京市自然科学基金项目(7132033);首都医科大学燕京医学院科研基金项目(19p y01,18q d k y04,19q d k y01)㊂作者简介:王凡(1989-),讲师,硕士,主要从事生物化学与分子生物学研究㊂ә通信作者,E-m a i l:w h j1006@163.c o m㊂u l a t i o n .c o m /)㊂C p G 岛位置预测网站:C pG F i n d e r (h t t p ://w w w.s o f t b e r r y .c o m /b e r r y .p h t m l ?t o pi c =c p g f i n d e r &g r o u p =p r o g r a m s &s u b g r o u p =pr o -m o t e r ),M e t h P r i m e r (h t t p ://w w w.u r o g e n e .o r g /c g i -b i n /m e t h p r i m e r /m e t h p r i m e r .c g i )㊂1.2 方法1.2.1 h HO -1基因及其启动子序列的获取在N C B I 中搜索人H O -1基因,进入G e n B a n k 获取h H O -1基因序列㊂在S e q u e n c e t e x t v i e w 中选取转录起始点上游3000b p 的序列,将序列上传至N u c l e o t i d e B L A S T 中进行比对,推测出启动子序列长度㊂在S o f t -b e r r y 中上传启动子序列,预测T A T A 盒㊂1.2.2 h HO -1启动子转录因子结合位点预测登录P R OMO 网站,在S e l e c t S pe c i e s 选项中选择O n l y h u m a nf a c t o r s 和O n l y hu m a n s i t e s ㊂点击S e a r c h S i t e s 按钮,将启动子序列上传至对话框中,设置M a x i m u m m a t r i x d i s s i m i l a r i t y r a t e 参数为5㊂登录G e n e r e gu l a t i o n 网站,选择A l i B a b a 2.1程序,设置M i n m a t .c o n s e r v a t i o n 参数为80%,上传序列预测即可㊂1.2.3 h HO -1启动子C pG 岛预测登录M e t h P r i m e r 网站,将W i n d o w 值设置为200,其他数据为默认值(O b p /E x p =0.6,G C %=50),上传启动子和外显子1及部分内含子1序列进行预测㊂用C p G F i n d e r 预测C p G 岛时使用默认设置预测即可,具体数值设置同上㊂2 结 果2.1 h HO -1基因及其启动子序列特点G e n b a n k 中显示h HO -1基因位于22q12.3,包含5个外显子,登录号为N C _000022,基因全长13112b p,其转录出的m R N A 全长为1554n t ,基因蛋白质编码区(C D S )编码288个氨基酸㊂本研究选取转录起始点上游3000b p 以内的DN A 序列进行比对㊂N u c l e o t i d e B L A S T 比对结果显示,所选取D N A 序列的第1006位脱氧核苷酸与信息库中h HO -1基因启动子序列的第1位脱氧核苷酸比对上,可以确定h HO -1启动子区位于转录起始点上游1995b p 以内,G e n e b a n k 登录号为A F 145047.1(图1)㊂Ⅱ类基因的启动子一般在转录起始位点上游-25~-30b p 附近存在T A T A 盒,富含A T 序列,负责基因转录起始位点的定位,是R N A 聚合酶的结合位点之一㊂S o f t b e r r y 预测结果显示,在h HO -1启动子序列第1963b p 处(即转录起始点上游33b p 处)存在T A T A 盒,见图2㊂图1 h HO -1启动子区N u c l e o t i d e B L A S T比对结果图2 h HO -1启动子区T A T A 盒2.2 h HO -1启动子区转录因子结合位点预测结果2.2.1 P R OMO 预测结果P R OMO 3.0.2预测系统使用的是T R A N S F A C 数据库8.3版本,经预测并通过手工去重后得到57种转录因子结合位点(图3),图中显示出各种转录因子及其与启动子的结合位置,其中G R -a l ph a ㊁P a x -5㊁p 53㊁G R -b e t a ㊁T F I I -I ㊁F O X P 3㊁R X R -a l ph a ㊁C /E B P b e -t a ㊁S T A T 4㊁T F I I D ㊁A P -2a l p h a A ㊁WT 1等转录因子结合位点的重复次数较高㊂2.2.2 A l i B a b a 2.1预测结果A l iB a b a 2.1在线预测软件使用的是T R A N S F A C4.0版本,经预测并通过手工去重后得到31种转录因子结合位点:A P -1㊁A P -2a l p h a ㊁c -M yc ㊁C /E B P ㊁C /E B P a l -p h a ㊁C /E B P b e t a ㊁C O U P ㊁E 1㊁E R ㊁G A T A -1㊁G L O ㊁G C N 4㊁H b ㊁H N F -1C ㊁H N F -3㊁Id 3㊁I R F -1㊁M E B -1㊁M y o D ㊁N F -A T c 3㊁N F -1㊁N F -k a p pa B ㊁N F -m u E 1㊁P i t -1a ㊁R A P 1㊁R A R -a l p h ㊁S p 1㊁T 3R -a l ph a ㊁T E C 1㊁U S F ㊁Y Y 1㊂将该结果与P R O M O 3.0.2预测结果合并,经去重后共得到79种转录因子结合位点:A P -1㊁A P -2a l ph ㊁A R ㊁c -E t s -1㊁c -E t s -2㊁c -F o s ㊁c -J u n ㊁c -M y b ㊁c -M y c ㊁C /E B P ㊁C /E B P a l -ph a ㊁C /E B P b e t a ㊁C O U P ㊁E 1㊁E 2F -1㊁E l k -1㊁E L F -1㊁E R ㊁E R -a l p h a ㊁F O X O 4㊁F O X P 3㊁G A T A -1㊁G A T A -2㊁G C N 4㊁G C F ㊁G L O ㊁G R ㊁G R -a l p h a ㊁G R -b e t a H b ㊁H N F -1C ㊁H N F -3㊁H N F -3a l ph a ㊁I d 3㊁I k -1㊁I R F -1㊁I R F -2㊁L E F -1㊁M E B -1㊁2851重庆医学2021年第50卷第9期M y o D ㊁N F -1㊁N F -A T 1㊁N F -A T c 3㊁N F -k a p pa B ㊁N F -m u E 1㊁N F -Y ㊁N F I /C T F ㊁p53㊁P a x -5㊁P E A 3㊁P i t -1a ㊁P P A R -a l p h a ʒR X R -a l ph a ㊁P R -A ㊁P R -B ㊁P X R -1ʒR X R -a l p h a ㊁R A P 1㊁R A R -a l p h ㊁R A R -b e t a ㊁R X R -a l p h a ㊁S p1㊁S R Y ㊁S T A T1b e t a ㊁S T A T4㊁T 3R -a l ph a ㊁T 3R -b e t a 1㊁T B P ㊁T E C 1㊁T F I I -I ㊁T F I I D ㊁U S F ㊁U S F 1㊁U S F 2㊁V D R ㊁W T 1㊁W T 1I ㊁W T 1-K T S ㊁W T 1I -K T S ㊁W T 1-d e l 2㊁I -d e l 2㊁Y Y 1㊂预测结果中包括了两大类反式作用因子的结合位点㊂一类是通用转录因子(T F ),其中T F I I 作用因子又包含T F I I A ㊁T F I I B ㊁T F I I D 和T F I I E ,这些转录因子涉及R N A 聚合酶㊁顺式作用元件和反式作用因子的相互作用,参与基因表达调控㊂第二类是特异转录因子,能对基本转录因子起增效作用,如S P 1转录因子与启动子上G C 盒结合后可使转录效率提高㊂2.3 h HO -1启动子区C pG 岛预测结果2.3.1 M e t h P r i m e r 预测结果C p G 岛主要位于基因的启动子和第一外显子区域,是富含C pG 二核苷酸的一些区域,约有60%以上的基因启动子区含有C p G 岛㊂C p G 岛不仅是基因的一种标志,而且还参与基因表达的调控㊂M e t h P r i m e r预测结果显示h HO -1启动子区C p G 岛长307b p,位于1857~2163b p;跨越启动子区末端,外显子1及部分内含子1上游序列,具体细节见图4㊂图3 P R OMO 3.0.2转录因子结合位点预测结果图4 M e t h P r i m e r C pG 岛预测结果3851重庆医学2021年第50卷第9期2.3.2 C pG F i n d e r 预测结果在C p G F i n d e r 预测结果中同样也预测到h HO -1存在C p G 岛,预测结果显示h HO -1启动子区C pG 岛长259b p ,位于1870~2128b p (图5);同样跨越启动子区末端,外显子1及部分内含子1上游序列,长度略短于M e t h P r i m e r 的C pG 岛预测结果㊂图5 C p G F i n d e r C pG 岛预测结果3 讨 论h HO -1是一种具有免疫调节活性的防御酶,可被多种因素调控进而影响表达结果[4]㊂其C 端被裂解后,可转移到细胞核内,参与由氧化应激介导的转录调节作用[5]㊂此外血红素经h HO -1催化降解的产物分别具有抗炎[6]㊁抗增殖[7]和促血管舒张活性[8]等作用㊂目前,关于h HO -1启动子的研究主要在启动子多态性与疾病关系方面的研究较多㊂本研究从h HO -1启动子转录因子结合位点入手,探讨其在抗逆方面的潜在作用㊂通过在N C B I 数据库中进行检索比对,获得了h HO -1转录起始位点上游1995b p 的启动子区域㊂S o f t b e r r y 的预测结果与理论情况相符㊂经预测汇总后共得到79种转录因子结合位点㊂其中有多个转录因子已被证明与肿瘤㊁炎症㊁热应激有关,此外还有些转录因子是信号通路里的重要组分㊂如A P -1转录因子结合位点,属于激活蛋白家族成员结合位点,在生长因子㊁细胞因子㊁胁迫㊁病原菌等因素刺激下,A P -1被活化进而调节基因表达[9]㊂A P -1在恶性肿瘤及自身免疫病当中也有很重要的调控作用[10]㊂促氧化因子和促炎因子可刺激A P -1的表达上调,各种损伤性刺激可使A P -1上游的信号分子J N K 和E R K 分别磷酸化c -J u n 和c -F o s ,c -J u n 和c -F o s 被磷酸化后进入细胞核进而形成二聚体,与A P -1转录因子结合位点结合启动HO -1基因表达[11]㊂因此,h HO -1表现出的多种抗逆作用,可以通过其启动子含有众多转录因子结合位点来解释㊂N F -κB 是二聚体转录因子,属于R e l 家族成员㊂当机体受到损伤性刺激时,N F -κB 进入细胞核,与HO -1启动子上的N F -κB 转录因子结合位点结合,启动HO -1基因转录,以应对损伤刺激[12]㊂E 26转录因子1(E T S 1)属于外转录间隔区(E T S )家族成员,E T S 主要参与细胞的生长与分化及器官的形成,在人的血管生成过程中起到重要作用㊂E T S 1可促进血管内皮细胞的迁移,在血管内皮受损后修复时E T S 1表达量有所上调[13]㊂转录因子E T S 1㊁c -J u n 和L E F 1能协同作用于孕丸X 受体基因的启动子区域,并增强该基因的表达[14]㊂转录因子L E F 1的功能与E T S 1相似,L E F 1可促进血管内皮细胞的增殖及其在基质中的侵袭能力[15]㊂由此可见,一个基因可受到多种转录因子的调控,且转录因子之间存在协同作用㊂上述转录因子结合位点在h HO -1启动子区都已预测出来,但E T S 1转录因子与h HO -1启动子之间的关系还未见报道,推测h HO -1可能与血管内皮细胞再生有一定关系㊂此外,在h HO -1启动子区未证实的转录因子结合位点还有多种,如c -E t s -2㊁c -M y b ㊁E L F -1㊁G A -T A -1㊁I d 3㊁L E F -1㊁M E B -1㊁N F -A T 1等㊂在今后的研究中,可通过构建荧光素报告载体的方法证实其他转录因子结合位点,为更深入地了解h HO -1基因功能及转录调控机制奠定基础㊂D N A 甲基化是一种表观遗传修饰,可在组织特异性基因表达㊁X 染色体失活㊁基因组印记㊁细胞增殖及衰老㊁胚胎发育等生物学进程中起重要作用㊂哺乳动物的D N A 甲基化主要发生在转录起始点附近的C p G 二核苷酸胞嘧啶残基上,即C p G 岛㊂一般情况下,正常细胞的C p G 岛由于被保护而处于非甲基化的状态㊂在肿瘤细胞中,抑癌基因的C p G 岛表现为高甲基化,肿瘤抗原表达缺失,抑癌基因表达下调,最终导致肿瘤发生[16-17]㊂本研究预测结果显示,h HO -1基因的C p G 岛位于启动子区末端,外显子1及部分内含子1上游序列,h HO -1表现出的多种功能可能与其C p G 岛的甲基化状态有一定关系㊂针对C p G 岛序列特点可以设计用于D N A 甲基化分析的P C R 引物,这还有待今后进一步实验分析㊂随着生物信息学的不断发展,关于启动子功能元件的预测将更准确可靠,可为研究基因的功能及转录调控提供更多有价值的信息㊂参考文献[1]王凡,王宏娟,徐世明.血红素加氧酶-1在感染性疾病中的作用及研究进展[J ].重庆医学,2019,48(20):3541-3544.[2]王献伟,黄金娜,王爱玲,等.血红素加氧酶-1启动子基因多态性与食管鳞癌的相关研究[J ].河北医药,2014,36(5):668-670.4851重庆医学2021年第50卷第9期[3]C H A N G K W,L E E T C,Y E H W I,e t a l.P o l y-m o r p h i s m i n h e m e o x y g e n a s e-1(HO-1)p r o-m o t e r i s r e l a t e d t o t h e r i s k o f o r a l s q u a m o u s c e l l c a r c i n o m a o c c u r r i n g o n m a l e a r e c a c h e w e r s[J].B r J C a n c e r,2004,91(8):1551-1555.[4]R Y T E R S W,C HO I A M.H e m e o x y g e n a s e-1/C a r b o n m o n o x i d e:f r o m m e t a b o l i s m t o m o l e c u-l a r t h e r a p y[J].A m J R e s p i r C e l l M o l B i o l, 2009,41(3):251-260.[5]L I N Q,W E I S S,Y A N G G,e t a l.H e m e o x y g e n-a s e-1p r o t e i n l o c a l i z e s t o t h e n u c l e u s a n d a c t i-v a t e s t r a n s c r i p t i o n f a c t o r s i m p o r t a n t i n o x i d a-t i v e s t r e s s[J].J B i o l C h e m,2007,282(28): 20621-20633.[6]O T T E R B E I N L E,B A C H F H,A L AM J,e t a l.C a r b o n m o n o x i d e h a s a n t i-i n f l a mm a t o r y e f f e c t s i n v o l v i n g t h e m i t o g e n-a c t i v a t e d p r o t e i n k i n a s e p a t h w a y[J].N a t M e d,2000,6(4):422-428.[7]S I N G H N,A HM A D Z,B A I D N,e t a l.H o s t h e m e o x y g e n a s e-1:F r i e n d o r f o e i n t a c k l i n g p a t h o g e n s?[J].I U B M B L i f e,2018,70(9):869-880. [8]WA Z A A A,H AM I D Z,A L I S,e t a l.A r e v i e w o n h e m e o x y g e n a s e-1i n d u c t i o n:i s i t a n e c e s s a r ye v i l[J].I nf l a mm R e s,2018,67(7):579-588.[9]周长春,刘芝华,齐军.A P-1和肿瘤的关系研究进展[J].世界华人消化杂志,2006,14(1):1-5.[10]T R O P-S T E I N B E R G S,A Z A R Y.A P-1e x p r e s-s i o n a n d i t s c l i n i c a l r e l e v a n c e i n i mm u n e d i s o r-d e r s a n d c a n c e r[J].AM J M E D S C I,2017,353(5):474-483.[11]马小华,游晓星,吴移谋.HO-1在免疫调节中的作用及其相关信号转导通路研究进展[J].中南医学科学杂志,2011,39(6):705-709.[12]L I Q,G U O Y,O U Q,e t a l.G e n e t r a n s f e r o f i n-d u c i b le n i t r i c o x i d e s y n t h a s e af f o r d s c a r d i o p r o-t e c t i o n b y u p r eg u l a t i n gh e m e o x y g e n a s e-1vi a an u c l e a r f a c t o r-κB-d e p e n d e n t p a t h w a y[J].C i r-c u l a t i o n,2009,120(13):1222-1230.[13]P A I N E A,E I Z-V E S P E R B,B L A S C Z Y K R,e ta l.S i g n a l i n g t o h e m e o x y g e n a s e-1a n d i t s a n t i-i n f l a mm a t o r y t h e r a p e u t i c p o t e n t i a l[J].B i o-c h e m P h a r m a c o l,2010,80(12):1895-1903.[14]P H E L A N D,W I N T E R G M,R O G E R S W J,e ta l.A c t i v a t i o n o f t h e A h r e c e p t o r s i g n a l t r a n s-d u c t i o n p a t h w a y b y b i l i r u b i n a n d b i l i ve r d i n[J].A r c hB i o c h e m B i o p h y s,1998,357(1):155-163.[15]Z E L E N A Y S,C HO R A A,S O A R E S M P,e t a l.H e m e o x y g e n a s e-1i s n o t r e q u i r e d f o r m o u s e r e g u l a t o r y T c e l l d e v e l o p m e n t a n d f u n c t i o n[J].I n t I mm u n o l,2006,19(1):11-18.[16]MO O R E D,L E T,F A N G.D N A M e t h y l a t i o na n d I t s B a s i c F u n c t i o n[J].N e u r o p s y c h o p h a r-m a c o l o g y,2013,38(1):23-38.[17]L I A N G G N,W E I S E N B E R G E R D J.D N A m e t h y-l a t i o n a b e r r a n c i e s a s a g u i d e f o r s u r v e i l l a n c e a n d t r e a t m e n t o f h u m a n c a n c e r s[J].E p i g e n e t i c s,2017, 12(6):416-432.(收稿日期:2020-07-28修回日期:2020-12-29)(上接第1580页)[10]D U T T A T.T h e l e f t v e n t r i c u l a r e j e c t i o n f r a c t i o n:n e w i n s i g h t s i n t o a n o l d p a r a m e t e r[J].H o s p P r a c t(1995),2019,47(5):221-230.[11]陈文宇,吴光龙,洪静文.实时三维超声心动图在行经皮冠状动脉介入治疗的左心室室壁瘤患者心功能评估中的应用价值[J].临床合理用药, 2019,12(4C):12-16.[12]T U R T O N E W,E N D E R J.3D r o l e o f e c h o c a r-d i o g r a p h y i n c a r d i a c s u r ge r y:s t r e n g t h s a n d l i m i t a t i o n s[J].C u r A n e s t h e s i o l o g y R e p o r t s, 2017,7(3):291-298.[13]L A N G R M,B I E R I G M,D E V E R E U X R B,e ta l.R e c o mm e n d a t i o n s f o r c h a mb e r q u a n t i f ic a-t i o n:a r e p o r t f r o m t h e a m e r i c a n s o c i e t y o f e c h-o c a r d i o g r a p h y's g u i d e l i n e s a n d s t a n d a r d s c o m-m i t t e e a n d t h e c h a m b e r q u a n t i f i c a t i o n w r i t i n gg r o u p[J].J A m S o c E c h o c a r d i o g r,2005,18(12):1440-1463.[14]I N C I A R D I R,G A L D E R I S I M,N I S T R I S,e t a l.E c h-o c a r d i o g r a p h i c a d v a n c e s i n h y p e r t r o p h i c c a r d i o m y o p-a t h y:t h r e e d i m e n s i o n a l a n d s t r a i n i m a g i n g e c h o c a r-d i o g r a p h y[J].E c h o c a r d i o g r a p h y,2018,35(5):716-726.[15]O R V A L H O J S.R e a l-t i m e t h r e e-d i m e n s i o n a l e c h-o c a r d i o g r a p h y:f r o m d i a g n o s i s t o i n t e r v e n t i o n[J].P e d i a t r C a r d i o l,2017,47(5):1005-1019.(收稿日期:2020-07-07修回日期:2021-01-03)5851重庆医学2021年第50卷第9期。
bim启动子的调控及生物信息学分析

2.解决问题的方法解决这个问题通常我们根据坚实的相关专业知识,提出可能的转录因子,逐个研究分析。
但是这种做法存在较大的盲目性和偶然性,研究时可能会费时、费力。
生物信息学的出现为我们提供了一个较合理的解决问题的方法。
生物信息学是-I'1新兴学科,随着基因组计划的进展而发展壮大。
它储存、处理、分析大量的生物数据序列及结构资料并从中得到有意义的信息,为生物学研究做出一些预测,提供可能性。
生物信息学为基因调控的研究提供了一个新的方法一转录因子DNA结合位点预测分析。
通过分析启动子区域序列找到可能的转录因子结合位点,进而找到可能参与基因调控表达的转录因子,为进一步的实验指明方向。
3.转录因子数据库的建立转录因子在基因的表达调控过程中起了重要的作用,转录因子及其DNA结合位点的研究也成为生物信息学的一个热点。
随着实验数据的大量积累,多个转录因子数据库应运而生,上个世纪九十年代出现了国际著名的转录因子数据库Transfac,存贮真核生物转录因子及其DNA结合位点。
目前Transfac数据库中收录转录因子记录4219条,包括了转录因子相关信息、位点序列、转录因子结合序列矩阵等多方面信息。
转录因子结合位点序列的丰富积累使转录因子DNA结合位点得以合理的描述,进而开发出多种转录因子结合位点预测分析的算法和工具。
4.转录因子DNA位点描述方法目前对转录因子DNA结合位点的描述可以分为三类:Pattern方法、Profile方法及HiddenMarkov模型方法。
Pattem方法是描述转录因子DNA结合位点每一位鬣所有可能出现的碱基;而Profile不仅描述每一位置可能出现碱基的种类,而且指出每一碱基出现的可能性大小。
显然使用Profile描述方法比使用Pattern描述包括更多的信息因而也更准确。
但是Profile描述方法也存在很多的不足,它只是对每一位置分别进行研究而没有考虑相邻碱基之问的关系,由此人们又找到了一种更合理的位点描述方法HiddenMarkov模型。
启动子生物信息学分析软件

/seq_tools/promoter.html2. PlantCARE(plant cis-acting regulatory elements), a database of plant cis-acting regulatory elementshttp://bioinformatics.psb.ugent.be/webtools/p lantcare/html/3. promoter 2.0 prediction serverhttp://www.cbs.dtu.dk/services/Promoter/4.启动子分析网址:1 /seq_tools/promoter.html2 http://alggen.lsi.upc.es/recerca/menu_recerca.html3 http://www.cbs.dtu.dk/services/Promoter/4 /~molb470/ ... s/solorz/index.html5 /molbio/proscan/http://bip.weizmann.ac.il/toolbo ... ters.html#databases/seq_tools/promoter.html.sg/promoter/CGrich1_0/CGRICH.htm/pub/programs.html#pmatch.hk/~b400559/arraysoft_pathway.html#Promoterhttp://www.dna.affrc.go.jp/PLACE/signalup.htmlhttp://intra.psb.ugent.be:8080/PlantCARE/http://www.cbs.dtu.dk/services/Promoter//molbio/proscan//molbio/signal//thread-41571-1-1.htm常用启动子分析网址:http://bip.weizmann.ac.il/toolbox/seq_analysis/promoters.html#databas es/seq_tools/promoter.html.sg/promoter/CGrich1_0/CGRICH.htm/pub/programs.html#pmatch.hk/~b400559/arraysoft_pathway.html#Promoter http://www.dna.affrc.go.jp/PLACE/signalup.htmlhttp://intra.psb.ugent.be:8080/PlantCARE/http://www.cbs.dtu.dk/services/Promoter//molbio/proscan//molbio/signal/首先就是想直接查找有没有人做过这条基因的启动子,在pubmed中输入genename+promoter接着就想看看有没有数据库可以直接给出启动子序列的,很幸运竟然发现一个极好的启动子搜索讲义网站,如下,.il/workshops/bgu/promoterworkshop.html第一步就是要找到基因确定基因所在基因组区域,其中列出很多网站,不过偶还是习惯genbank,在gene栏中search某个基因,不要搞错基因种属!进入后即可看到该基因的详细条目,别眼花,就点击右侧link栏的Map viewer 链接,进入即可看到该基因在染色体上的形象定位,鼠标悬停在基因的起始位点时,即可在浏览器下方的状态栏中显示该位点在染色体上的明确定位,比如110997788,结合给出的基因跨度,比如110778899-117708899,即可大概确定该启动子在基因组中的大概定位,即110778899-110997788;第二步搞清楚基因组状态,我没搞太清楚,不过其中给的一个链接来查出启动子所在克隆(查出克隆号可以购买)/genome/guide/mouse/该链接中的clonefinder工具可以做到,只要提交你要查找的基因officialname就可以返回一个clonelist;第三步搜索启动子,其中可以用启动子数据库和启动子预测软件,当然如果启动子数据库中有最好,但很失望给出的数据库均不能查到!只好用启动子预测软件,使用了几个在线预测工具后觉得下面这个速度贼快,推荐http://www.cbs.dtu.dk/services/Promoter/我把该基因的dna序列submit之后返回了很多个PolII识别位点,到底哪个是呢?我个人理解启动子应该是翻译起始位点附近,所以在这个dna序列中定位翻译起始位点即可找到最近的Highly likely prediction,那么怎么定位呢?利用blast2这个利器,只要把dna和mrna序列粘贴进去提交就ok,正好在翻译起始位点上游几百bp有个识别位点,ok!启动子序列就是翻译起始位点上游大概1kb长度的序列了!直接用ensemble数据库的话,可以直接知道基因外显子和起始位点的位置,然后直接可以查到之前的序列,再选3k-4k的长度预测就比较方便了。
人SIRT1基因启动子及蛋白的生物信息学分析

激光生物学报ACTA LASER BIOLOGY SINICAVol. 32 No. 4Aug . 2023第32卷第4期2023年8月收稿日期:2023-04-18;修回日期:2023-05-11。
基金项目:国家自然科学基金项目(31670178);湖南省自然科学基金项目(2021JJ 30918)。
作者简介:王道,主管技师,主要从事女性生殖系统疾病的基础研究。
* 通信作者:宋甜,助理研究员,主要从事衣原体感染疾病的诊断研究。
E-mail:****************.cn 。
人SIRT1基因启动子及蛋白的生物信息学分析王 道1,陈建林1,刘文彬2,刘 丹1,宋 甜1*(1. 中南大学湘雅二医院妇产科,长沙 410011;2. 湖南师范大学生命科学学院,长沙 410081)摘 要:为深入分析人SIRT1基因启动子及蛋白的结构和功能,本文采用生物信息学方法分析人SIRT1基因5′端启动子、启动子区Motif 、转录因子结合位点、甲基化CpG 岛、单核苷酸多态性、进化关系、理化性质、二级和三级结构、保守结构域、突变和翻译后修饰位点、互作蛋白及生物学功能。
TSSW 和Neural Network Promoter Prediction 数据库预测其区间分别存在3个、2个启动子。
MEME 数据库预测启动子区存在3个Motif 。
EMBOSS 、MethPrimer 和CpG Finder 数据库都发现CpG 岛聚集分布于1 600 ~ 2 200 bp 区。
PROMO 和AliBaba 2.1数据库预测其启动子区域共同转录因子为22个。
JASPAR 软件获得6个与正负链相结合的转录因子。
SNP Function Prediction 数据库还发现不同种族等位基因频率存在差异。
人SIRT1基因位于染色体10q 21.3上,广泛分布在不同组织中。
人SIRT 1蛋白由747个氨基酸组成,属于亲水、不稳定的蛋白质,在不同物种间具有较高的保守性。
生物信息学实验一

生物信息学实验一简介:生物信息学实验一是生物信息学实验课程的第一部分,旨在介绍生物信息学的基本概念、工具和技术,以及生物信息学在生物学研究中的应用。
本实验将引导学生通过实际操作,学习并掌握生物信息学的基本原理和操作技巧。
实验设备和材料:- 计算机或笔记本电脑- 生物信息学软件(例如NCBI BLAST、UCSC Genome Browser等)- 相关数据库和工具(例如GenBank、KEGG等)实验目的:1. 了解生物信息学的基本概念和应用领域;2. 学习生物信息学的常用工具和技术;3. 掌握生物序列分析、基因注释和比对等基本操作;4. 学会使用生物信息学软件和数据库进行数据查询和分析;5. 培养科学研究的数据处理和解读能力。
实验步骤:1. 确定研究对象:选择一个感兴趣的生物学问题或基因序列进行研究。
2. 数据获取:使用生物信息学工具和数据库,获取与研究对象相关的生物序列数据。
3. 序列分析:使用生物信息学软件对序列数据进行分析,包括碱基组成、氨基酸序列、启动子分析等。
4. 基因注释:通过比对算法和数据库,对序列进行基因功能注释,确定基因的命名、结构和功能信息。
5. 比对分析:使用比对工具进行序列比对,比较两个或多个序列之间的相似性和差异性。
6. 数据解读:根据分析结果,结合相关文献和知识,对实验数据进行解读和分析,得出科学结论。
实验注意事项:1. 在进行实验前,先了解所要使用的工具和软件的基本操作方法和原理;2. 实验过程中注意数据安全和保密,不得将数据泄露或用于非科研目的;3. 在进行数据分析和解读时,务必准确、客观地进行,不得造假或歪曲实验结果;4. 注意数据的备份和存储,以防止数据丢失或损坏;5. 尊重他人的研究成果和知识产权,合理引用和参考相关文献。
实验结果与讨论:本实验所得的结果可以根据具体的研究对象和实验数据来展开讨论和分析。
例如,如果研究对象是某个基因序列,可以讨论其结构和功能,与其他基因的关联性,以及在哪些生物过程中有重要作用等。
常用分子生物学软件简介

常用分子生物学软件一、基因芯片:1、基因芯片综合分析软件。
ArrayVision 7.0一种功能强大的商业版基因芯片分析软件,不仅可以进行图像分析,还可以进行数据处理,方便protocol的管理功能强大,商业版正式版:6900美元。
Arraypro 4.0Media Cybernetics公司的产品,该公司的gelpro, imagepro一直以精确成为同类产品中的佼佼者,相信arraypro也不会差。
phoretix™Array Nonlinear Dynamics公司的基因片综合分析软件。
J-express挪威Bergen大学编写,是一个用JAVA语言写的应用程序,界面清晰漂亮,用来分析微矩阵(microarray)实验获得的基因表达数据,需要下载安装JAVA运行环境JRE1.2后(5.1M)后,才能运行。
2、基因芯片阅读图像分析软件ScanAlyze 2.44,斯坦福的基因芯片基因芯片阅读软件,进行微矩阵荧光图像分析,包括半自动定义格栅与像素点分析。
输出为分隔的文本格式,可很容易地转化为任何数据库。
3、基因芯片数据分析软件Cluster斯坦福的对大量微矩阵数据组进行各种簇(Cluster)分析与其它各种处理的软件。
SAMSignificance Analysis of Microarrays 的缩写,微矩阵显著性分析软件,EXCEL软件的插件,由Stanford大学编制。
4.基因芯片聚类图形显示TreeView 1.5斯坦福开发的用来显示Cluster软件分析的图形化结果。
现已和Cluster成为了基因芯片处理的标准软件。
FreeView是基于JAVA语言的系统树生成软件,接收Cluster生成的数据,比Treeview增强了某些功能。
5.基因芯片引物设计Array Designer 2.00DNA微矩阵(microarray)软件,批量设计DNA和寡核苷酸引物工具二、RNA二级结构。
RNA Structure 3.5RNA Sturcture 根据最小自由能原理,将Zuker的根据RNA一级序列预测RNA二级结构的算法在软件上实现。
生物信息学中的基因组注释方法介绍

生物信息学中的基因组注释方法介绍随着基因组测序技术的快速发展,生物信息学在基因组研究中的应用越来越广泛。
基因组注释是基因组研究的重要环节,它可以帮助我们理解基因的功能和调控机制。
本文将介绍生物信息学中常用的基因组注释方法。
1. 基因预测基因预测是基因组注释的第一步。
它通过分析基因组序列中的开放阅读框(ORF)来预测潜在的基因。
常用的基因预测软件包括GeneMark、Glimmer和Augustus等。
这些软件根据基因的编码特征、保守序列和启动子等信息来预测基因的存在和位置。
2. 基因结构注释基因结构注释是对基因的内部结构进行注释,包括外显子、内含子和剪接变异等信息。
这可以通过比对已知基因组和转录本序列来实现。
常用的基因结构注释工具有BLAST、BLAT和Exonerate等。
这些工具可以将基因组序列与已知基因组或转录本序列进行比对,以识别外显子和内含子的位置。
3. 功能注释功能注释是对基因的功能进行注释,包括基因的功能分类、蛋白质结构域和功能区域等信息。
功能注释可以通过比对已知蛋白质数据库,如Swiss-Prot和TrEMBL,来实现。
常用的功能注释工具有BLAST、InterProScan和Pfam等。
这些工具可以将基因的编码蛋白质序列与已知蛋白质序列进行比对,并通过功能域和保守序列的分析来注释基因的功能。
4. 转录本组装转录本组装是对基因组中的转录本进行注释,包括外显子和内含子的组装以及剪接变异的分析。
常用的转录本组装工具有Cufflinks、StringTie和Trinity等。
这些工具可以根据RNA测序数据将转录本的外显子和内含子进行组装,并通过比对转录本序列与基因组序列来分析剪接变异。
5. 转录因子结合位点预测转录因子结合位点是转录因子与DNA结合的特定区域,它在基因调控中起着重要的作用。
转录因子结合位点预测可以通过比对转录因子结合位点数据库,如JASPAR和TRANSFAC,来实现。
Softberry网站部分功能介绍
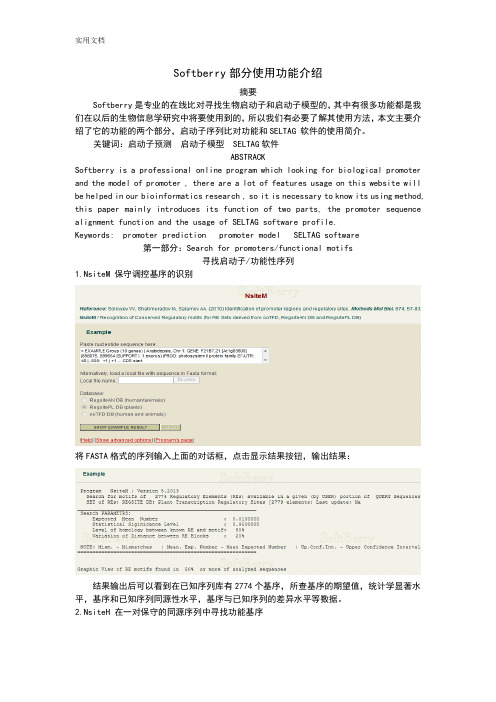
Softberry部分使用功能介绍摘要Softberry是专业的在线比对寻找生物启动子和启动子模型的,其中有很多功能都是我们在以后的生物信息学研究中将要使用到的,所以我们有必要了解其使用方法,本文主要介绍了它的功能的两个部分,启动子序列比对功能和SELTAG 软件的使用简介。
关键词:启动子预测启动子模型 SELTAG软件ABSTRACKSoftberry is a professional online program which looking for biological promoter and the model of promoter , there are a lot of features usage on this website will be helped in our bioinformatics research , so it is necessary to know its using method, this paper mainly introduces its function of two parts, the promoter sequence alignment function and the usage of SELTAG software profile.Keywords: promoter prediction promoter model SELTAG software第一部分:Search for promoters/functional motifs寻找启动子/功能性序列1.NsiteM 保守调控基序的识别将FASTA格式的序列输入上面的对话框,点击显示结果按钮,输出结果:结果输出后可以看到在已知序列库有2774个基序,所查基序的期望值,统计学显著水平,基序和已知序列同源性水平,基序与已知序列的差异水平等数据。
2.NsiteH 在一对保守的同源序列中寻找功能基序在对话框分别输入两条要比较的序列,然后点击下面的显示结果按钮,输出结果:在结果可以看到两条序列比对的结果,期望值,统计学显著水平,最小保守水平,同源水平,变异水平的数据。
生物信息学常用工具,作用及操作流程

用于分析DNA、RNA以及蛋白质一级结构1、VecScreen用于分析未知序列的长度、载体序列的区域、判断可能使用的克隆载体。
操作过程:NCBI→Resource List (A-Z)→V→VecScreen→输入序列→Run VecScreen→获得结果2、RepeatMasker用于分析未知序列的重复序列情况,输出重复序列的区域、包含的所有重复序列的类型、重复序列的总长度及Masked Sequence。
操作流程:RepeatMasker Home Page→RepeatMasking→输入文本→选择参数→submit sequence→Results→选择所需结果3、使用CpGPlot工具,分析未知序列的CpG岛的长度、区域、GC数量及Obs/Exp 值。
EMBL→service→Search “cpg”→EMBOSS cpgplot→输入序列→选择参数→submit→得到结果4、Neural Network Promoter Prediction和Splice Site Prediction用于预测未知序列的启动子,获得可能的启动子序列及相应的位置。
Neural Network Promoter PredictionBDGP: Home→Analysis Tools→Promoter Prediction→输入序列→选择参数→submit →得到结果Splice Site PredictionSplice Site Prediction→输入序列→选择参数(物种)→submit→得到结果这两个都是bdgp里边的,sp这个直接能进去操作。
5、ORF finder用于分析未知序列开放阅读框的预测,寻找潜在的蛋白质编码片段,并进行六框翻译(概念性翻译)。
操作流程NCBI→Resource List (A-Z)→ORF finder→输入序列→选择参数→submit→获得结果→选择符合要求的形式的结果6、GENSCAN,用于未知序列综合分析,预测来自各种生物的基因组序列中基因的位置和外显子结构,并对其进行概念性翻译。
Matlab的计算生物学和生物信息学技术

Matlab的计算生物学和生物信息学技术随着生物技术的发展,计算生物学和生物信息学在生命科学研究中发挥着越来越重要的作用。
Matlab作为一种强大的数值计算和数据可视化工具,在计算生物学和生物信息学领域中扮演着重要角色。
本文将探讨Matlab在这两个领域的应用,并介绍一些常见的技术和方法。
一. 介绍计算生物学和生物信息学是将计算机科学与生物学相结合,利用计算模型和算法解决生命科学中的复杂问题。
计算生物学主要关注生物系统的结构、功能和动力学等方面的研究,而生物信息学则关注利用大数据和生物信息来探索生物学的奥秘。
Matlab作为一种功能强大、易于使用的科学计算软件,为生物学家们提供了一个灵活而高效的工具。
二. Matlab在计算生物学中的应用1. 基因调控网络分析基因调控网络是生物系统中基因之间相互作用的图模型。
Matlab提供了一系列的图论和网络分析工具,可以用于构建和分析基因调控网络。
生物学家们可以利用Matlab计算网络的网络参数,如最短路径、中心性指标等,从而深入理解基因调控网络的结构和功能。
2. 蛋白质结构预测蛋白质结构预测是计算生物学中的一个重要问题,它涉及到蛋白质的三维结构和功能的预测。
Matlab提供了一些蛋白质结构预测的工具包,如Rosetta和Protein Data Bank等,可以帮助生物学家们预测蛋白质的结构和运动。
3. 数据挖掘和模式识别生物学研究中常常需要处理大量的生物数据。
Matlab提供了一系列的数据处理和分析工具,如数据挖掘和模式识别工具包,可以帮助生物学家们从生物数据中提取有价值的信息。
例如,生物学家们可以利用Matlab进行基因表达数据的聚类分析和差异表达基因的识别,从而揭示基因调控网络中的重要节点。
三. Matlab在生物信息学中的应用1. DNA序列分析DNA序列是生物信息学中的重要数据之一。
Matlab提供了一些DNA序列分析工具包,如Bioinformatics Toolbox和Genomic Analysis Toolbox等,可以帮助生物学家们进行DNA序列的序列比对、启动子预测和基因预测等分析。
DNA序列的生物信息学分析

DNA序列的生物信息学分析生物信息学是对生物学数据进行处理、分析和解释的跨学科领域。
在生命科学和医学研究中,生物信息学分析是至关重要的工具,可用于理解基因序列、蛋白质结构、基因组功能等方面。
DNA序列是生物信息学分析的核心内容之一,本文将围绕DNA序列的生物信息学分析展开。
DNA序列是基因组的基本单位,可以采集并以文本文件的形式储存。
生物信息学分析DNA序列的主要方法包括序列比对、基因注释、基因功能预测、DNA变异分析等。
这些方法可以通过多种工具和软件实现,其中一些常用的工具包括BLAST、GeneMark、MAFFT、Clustal等。
下面将详细介绍这些方法和工具。
1. 序列比对序列比对是将两个或多个序列进行对齐,以确定它们之间的相似性、差异性和同源性的过程。
序列比对可以用于DNA序列、蛋白质序列和RNA序列的比较。
在DNA序列的比较中,序列的相似性和差异性信息可以用于确定物种的进化关系、DNA序列的保守区域、功能区域和突变位点等。
常用的序列比对工具包括BLAST、Clustal、T-Coffee等。
BLAST是最常用的序列比对工具之一,可以在不同数据库中比对DNA、蛋白质和RNA序列。
BLAST通过在一个“查询序列”中搜索与“数据库序列”相似的区域来实现序列比对。
比对得分是基于匹配度、错配和间隙数目确定的。
BLAST比对结果提供了比对得分、查询和数据库序列的保守区域、匹配、错配和间隙数目等信息。
2. 基因注释基因注释是为基因序列赋予功能或信息的过程。
这个过程通常包括基因位置、外显子、内含子、启动子、终止子、基因名称、编码蛋白质等信息的确定。
在基因组中注释基因是理解整个基因组结构和功能的重要步骤。
常用的基因注释工具包括GeneMark、Glimmer等。
GeneMark是一个广泛使用的基因预测工具之一,可以预测基因的位置、方向和外显子结构。
GeneMark使用了马尔可夫模型和基因富含偏好等方法来预测基因位置,并根据之前预测的结果来增加预测准确性。
生物信息学作业1---开放读码框与预测序列启动子(何婷,学号;1302008)

何婷学号:1302008 专业:病理学与病理生理学
1.使用Entrez信息查询系统检索与自己课题相关的基因核酸序列,预测开放读码框,并使用PromoterScan预测该序列中的启动子。
①查询HPV的核酸序列:输入网址:/,打开NCBI主页,在
检索窗口的选择数据库的下拉菜单选中Nucleotide项,在它右侧的文本输入栏输入检索词“HPV”,再点击“Search”按钮。
如下图所示:
搜索结果,如下图所示:
显示结果,如下图所示:
② 预测开放读码框:输入网址:/gorf/gorf.html ,打开NCBI
的ORF Finder 软件,输入HPV 的核酸序列的GI 号,最后点击“OrfFind ”按钮,如下图所示:
结果如下图所示:
点击“正链+1”,显示结果如下:
③使用PromoterScan预测该序列中的启动子:输入网址为
/molbio/proscan/,打开PromoterScan的在线操作页面,复制粘贴上述的HPV的核酸序列到指定的框中,点击submit按钮提交序列后,注意使用
软件时不需要设置任何参数,如下图所示:
输出结果为:
何婷学号:1302008 专业:病理学与病理生理学。
拟南芥盐胁迫响应启动子的生物信息学分析

因的启动子 , 得到 1 0个保守元件 。通过显著性分布分析表明 Mof l M t_ \ t_ 0显著分 布于上调 基因启 t_ \ oi 8 Mof1 i f i 动子中; oi lM t_ 0显著不分 布于盐胁 迫下调 基 因启 动子 中。Mof 1 件是 G—bx元件 ( B E( B M t_ \ oi 1 f f t一 元 i o AR AA R sos eEe et元件 ) 大量分布 于盐胁迫上调基 因启动子 中, epni l n) v m , 广泛参与盐胁迫过程中 A A依赖 的信 号转 导 B 途径 。Mof 8 Mof 1 别 类 似 于 A R t一 、 t一 0分 i i B E元 件 和 D E( eyrtn—R sos eEe et 元 件 , 由于 和 R D hda o i epni l n) v m 但 A R D E元件 的保守序列不尽相 同 , oi 8 M t_ 0很有可 能是 新 的盐胁 迫响应元件 。本研究 对于研究 拟 B E、 R M t_ \ oi 1 f f 南芥在盐胁迫应答过程中在转录水平上发生的调控过程具有重要帮助作 用. 关键词 : 南芥 ; 拟 全基 因组芯片 ; 盐胁迫 响应启动子
Ab t a t S lnt sr c :aii y—r s o sv t swe e a ay e t li l ii fr t o t r s i h s su y 2 a e p n i e mo i r n l z d wih mu tpe b on o ma i s f f c wa e n t i t d .6 7 s - ln t p—rg lt d g ne n 8 wn—r g l td g n swe e i e tf d fo t e mir a ry d t b u 5h i iy u e u ae e s a d 2 2 do e u a e e e r d n i e r m h c o ra a a a o t0. i a d 1 ih s l t te si a i o ss n h g a i y sr s n Ar b d p i.Th p te m 00 p p o trr go so l s ln t h ni e u sr a 1 0b r moe e in fal a iiy—r g l td g n s e ae e e u r tiv d fo TAI we e u e o ndn o s r e t s b ere e m r R r s d f rf ig c n ev d mo i y MEME s f r n 0 p ttv u h moiswe e i f ot e a d 1 u aie s c tf r wa ie t e d n i d.Mo i s i d n iid a —b x ee n ,wh c s o e o h i f t 1 wa n e tfe s a G f o lme t ih i n ft e ABA — rs o sv l me t h t e p n ie ee n s t a f n t n n ABA —d p nd n e e e p e so n e bo i te s,a d o e r p e e t n s ln t p—r g ltd u c i s i o e e e tg n x r si n u d ra itc sr s n v re r s n s i ai i u y e u ae
cazy注释结果解读

Cazy注释结果解读Cazy注释结果解读是生物信息学领域中的一项重要任务。
Cazy是一种用于基因组注释的软件,它可以识别基因组中的各种功能元件,并生成相应的注释结果。
这些注释结果对于研究基因组的结构和功能具有重要意义。
在解读Cazy注释结果时,我们需要对Cazy的注释结果进行详细的分析。
Cazy的注释结果包括基因、转录因子结合位点、调控元件、启动子、增强子、沉默子等。
我们需要对这些注释结果进行逐一分析,并结合基因组的结构和功能特征,来推断各个功能元件的作用和相互关系。
具体来说,我们可以采取以下步骤来解读Cazy注释结果:1. 确认注释结果的准确性:首先需要确认注释结果是否准确可靠。
可以通过比对注释结果与已知数据库中的数据,或者使用其他基因组注释软件进行验证,以确保注释结果的准确性。
2. 分析基因组结构:根据注释结果,可以分析基因组的整体结构,包括基因的数量、位置和排列顺序等。
同时,还可以发现基因组中的非编码区以及可能存在的重复序列等。
3. 研究基因功能:通过分析注释结果中各个功能元件的信息,可以研究基因的功能和作用。
例如,可以识别出基因的启动子、增强子和沉默子等调控元件,进一步研究它们对基因表达的调控作用。
4. 寻找基因之间的相互关系:通过比较不同基因之间的注释结果,可以发现它们之间的相互关系。
例如,可以识别出两个基因之间的共线性或共同调控关系等。
5. 建立基因组注释数据库:将注释结果整理成数据库形式,方便后续的数据分析和挖掘。
可以通过与其他数据库进行连接,实现数据共享和整合。
总之,解读Cazy注释结果需要具备一定的生物信息学知识和技能,同时还需要结合实验验证和后续数据分析等方法,才能更准确地理解基因组的结构和功能信息。
启动子预测软件汇总

启动子预测软件汇总Softberry:/all.htmUCSC: /EPD: /TFD: /TRRD: /mgs/gnw/trrdTransFac:/Jaspar: /cgi-bin/jaspar_db.pldbTSS: /PlantCare: :8080/PlantCARE/index.htmlPLACE: /htdocs/PLACE/PlantPromDB:/mendel.php?topic=plantprom二、程序Audic: /~audic/selfid.htmlConPro:/conpro/ConSite: /cgi-bin/CONSITE/consite/Dragon Promoter Finder: /promoter/promoter1_4/DPF.htm Eponine: :8080/eponine/FastM: Now available through Genomatix onlyFastM, Patch (PatSearch), Match (MatInspector)....(Transfac 6.0) - programs link:/FootPrinter:/~blanchem/FootPrinterWeb/FootPrinterInput2. plMcPromoter: /McPromoter.htmlPromFD (download): ftp:///pub/stormo/PromFDPromFind: /PromFind(download): :7780/perl/custom/index.cgi?dir=/public/molbio/dna/analysis/PromFindPromoterInspector:/cgi-bin/promoterinspector/promoterinspector.plPromoterScan 2::80/molbio/proscan/Promoter2.0:/services/Promoter/Regulatory Sequence Analysis Tools: /rsat/SignalScan: :80/molbio/signal/Tess:/tess/TFSearch: /research/db/TFSEARCH.htmlTSSG/TSSW/TSSP: /berry.phtmlArabidopsis, Yeast: /software/index1.htmPipmaker: /pipmaker/Vista:/vista/rVista:/三、在线工具/~lockwood/bindgene.html/:18080/EZRetrieve/index.jsp//~zhengjsh/OTFBS//~alggen/recerca/promo_v2/frame-promo.html/molbio/signal//tess//很多是针对特定特种的,如EPD主要针对真核生物,USCS主要针对哺乳动物如人、老鼠、猫、狗等,所以寻找启动子时应根据物种不同选择合适的工具。
newplace启动子分析

newplace启动子分析百合一直以其花姿优雅,气度不凡具有极高的观赏价值而著称。
其在颜色,图案和香味方面的吸引力而被顾客高度重视,在世界范围的切花行业中占有极高的市场份额。
然而它具有巨大的花药,同时伴有大量的花粉,如果花药不能及时去除,则花粉会污染花瓣和周围表面,沾染衣物甚至会导致人类对花粉过敏等症状。
因为消费者不愿购买有污染的百合花,且人工摘除花药又费时费力。
因此,控制花药的发育和减少花粉量的生产,同时又不影响百合花的美观一直是百合的分子育种的焦点。
本实验选取不产花粉的"无粉白"百合雄蕊和同一时期有粉百合雄蕊为实验材料,结合之前转录组分析数据通过荧光定量筛选鉴定基因,RT-PCR克隆得到C2基因的ORF序列,随即克隆其5'端序列,并在其启动子序列中找到与花药特异性相关的表达元件。
还通过同源克隆的方法克隆得到C4基因,该基因与控制雄性不育基因MMD1具有较高的同源性,并对该基因进行一系列的生物信息学分析。
具体研究结果如下:1、通过荧光定量PCR分析鉴定,发现C2基因在无粉百合雄蕊中的表达明显迟于其在有粉百合雄蕊中的表达,且表达量很低。
结合其在百合不同器官中表达量分析,C2基因在花药中表达最高。
这说明C2基因主要的作用位点在花药中。
2、在有粉可育雄蕊与无粉不可育雄蕊中分别通过RT-PCR克隆到C2基因ORF区,全长为774 bp,通过序列比对分析发现其ORF区没有突变,推测可能其发挥作用的区域在启动子部分。
3、在有粉可育雄蕊与无粉不可育雄蕊中分别通过染色体步移的方法克隆其启动子序列,且有粉可育材料中克隆得到序列619 bp,无粉不可育雄蕊中克隆得到片段395 bp。
通过启动子分析软件NewPlace预测发现启动子序列中含有TATA-box以及CAAT-box,其中TATA-box分别位于转录起始位点"A"位于起始密码子上游-53 bp和-54 bp处,CAAT-box位于起始密码子上游-132 bp和-167 bp处。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
/seq_tools/promoter.html2. PlantCARE(plant cis-acting regulatory elements), a database of plant cis-acting regulatory elementshttp://bioinformatics.psb.ugent.be/webtools/p lantcare/html/3. promoter 2.0 prediction serverhttp://www.cbs.dtu.dk/services/Promoter/4.启动子分析网址:1 /seq_tools/promoter.html2 http://alggen.lsi.upc.es/recerca/menu_recerca.html3 http://www.cbs.dtu.dk/services/Promoter/4 /~molb470/ ... s/solorz/index.html5 /molbio/proscan/http://bip.weizmann.ac.il/toolbo ... ters.html#databases/seq_tools/promoter.html.sg/promoter/CGrich1_0/CGRICH.htm/pub/programs.html#pmatch.hk/~b400559/arraysoft_pathway.html#Promoterhttp://www.dna.affrc.go.jp/PLACE/signalup.htmlhttp://intra.psb.ugent.be:8080/PlantCARE/http://www.cbs.dtu.dk/services/Promoter//molbio/proscan//molbio/signal//thread-41571-1-1.htm常用启动子分析网址:http://bip.weizmann.ac.il/toolbox/seq_analysis/promoters.html#databas es/seq_tools/promoter.html.sg/promoter/CGrich1_0/CGRICH.htm/pub/programs.html#pmatch.hk/~b400559/arraysoft_pathway.html#Promoter http://www.dna.affrc.go.jp/PLACE/signalup.htmlhttp://intra.psb.ugent.be:8080/PlantCARE/http://www.cbs.dtu.dk/services/Promoter//molbio/proscan//molbio/signal/首先就是想直接查找有没有人做过这条基因的启动子,在pubmed中输入genename+promoter接着就想看看有没有数据库可以直接给出启动子序列的,很幸运竟然发现一个极好的启动子搜索讲义网站,如下,.il/workshops/bgu/promoterworkshop.html第一步就是要找到基因确定基因所在基因组区域,其中列出很多网站,不过偶还是习惯genbank,在gene栏中search某个基因,不要搞错基因种属!进入后即可看到该基因的详细条目,别眼花,就点击右侧link栏的Map viewer 链接,进入即可看到该基因在染色体上的形象定位,鼠标悬停在基因的起始位点时,即可在浏览器下方的状态栏中显示该位点在染色体上的明确定位,比如110997788,结合给出的基因跨度,比如110778899-117708899,即可大概确定该启动子在基因组中的大概定位,即110778899-110997788;第二步搞清楚基因组状态,我没搞太清楚,不过其中给的一个链接来查出启动子所在克隆(查出克隆号可以购买)/genome/guide/mouse/该链接中的clonefinder工具可以做到,只要提交你要查找的基因officialname就可以返回一个clonelist;第三步搜索启动子,其中可以用启动子数据库和启动子预测软件,当然如果启动子数据库中有最好,但很失望给出的数据库均不能查到!只好用启动子预测软件,使用了几个在线预测工具后觉得下面这个速度贼快,推荐http://www.cbs.dtu.dk/services/Promoter/我把该基因的dna序列submit之后返回了很多个PolII识别位点,到底哪个是呢?我个人理解启动子应该是翻译起始位点附近,所以在这个dna序列中定位翻译起始位点即可找到最近的Highly likely prediction,那么怎么定位呢?利用blast2这个利器,只要把dna和mrna序列粘贴进去提交就ok,正好在翻译起始位点上游几百bp有个识别位点,ok!启动子序列就是翻译起始位点上游大概1kb长度的序列了!直接用ensemble数据库的话,可以直接知道基因外显子和起始位点的位置,然后直接可以查到之前的序列,再选3k-4k的长度预测就比较方便了。
启动子及转录因子结合位点数据库及预测工具(2009-05-14 23:54:56)转载▼忽然感觉很GUILTY的,BLOG里竟然不放一点点和研究有关的重要工具。
换了电脑之后才发现,很多有用的链接都没有COPY下来,于是,从头开始做吧。
这是Andrew给我的他的PAPER里的有关转录因子结合位点的数据库,还有其他网友整理的,都很有用,这个星期有空再核下几个重要基因的SNP。
PROMOTER FINDING AND ANAL YSIS PROGRAMS ON THE INTERNET--------------------------------------------------------------------------------TRANSPLORER (TRANScription exPLORER)Dnanalyze (TF mapping)Dragon Promoter Finder 1.2 (TSS finder and promoter region analysis)FunSiteP 2.1HCtata (TATA signal prediction)McPromoter Ver.3MatInspector (Search for TF binding sites)ModelGenerator and ModelInspectorNNPP2.1 (TSS finder)PromoterInspector (Strand non-specific promoter region finder)Promoter2.0 (TSS finder)Promoter Scan II (Promoter region prediction)RGSiteScanSignal Scan (Search for Eukaryotic Transcriptional Elements)TESS (Search for Transcription Elements)TFSEARCH (Predicts TF binding sites based on TRANSFAC data)TRANSFAC (TF database and a number of associated programs)TSSG and TSSWPROMOTER 2.0 http://www.cbs.dtu.dk/services/Promoter/通常确定启动子的算法可以分成两种,一种根据启动子区各种转录信号,如TATA 盒、CCAA T 盒,结合对这些保守信号及信号间保守的空间排列顺序的识别进行预测。
如PROMOTER 2.0, 用神经网络方法确定TATA 盒、CCAAT盒、加帽位点(cap site) 和GC 盒(GCbox) 的位置和距离, 识别含TATA 盒的启动子。
PROMOTER SCAN /molbio/proscan/根据转录因子结合部位在基因组中分布的不平衡性,将转录因子结合部位分布密度与TATA 盒的权重矩阵(weight matrix) 结合起来,从基因组DNA中识别出启动子区[3 ] 。
但上述程序预测的假阳性率较高,PROMOTER 210 每23kb 出现一个假阳性;PRO2MOTER SCAN 平均每19kb 出现一个假阳性。
PromoterInspector http://www.genomatix.de/products/PromoterInspector/PromoterInspector2.ht ml另一种方法根据启动子区序列的特征进行预测。
Promo2terInspector 从一组训练序列中提取出启动子区的环境特征,并将外显子、内含子和3’端非翻译区的特征与启动子区加以区分,从而在基因组中确定启动子位置FirstEF /tools/FirstEF/近来还有一些程序将上述方法与CpG 岛(CpG islands) 信息相结合。
CpG岛是一段200 bp 或更长的DNA 序列,核苷酸G + C 的含量较高,并且CpG双核苷酸的出现频率占G+ C 含量的50 %以上。
许多脊椎动物的启动子区都与CpG岛的位置重合。
FirstEF ( http :/ / rulai1cshl1org/ tools/ FirstEF/ ) 搜索通过5’UTR 定位技术构建的第一外显子数据库,识别第一剪切点(first splicing donor site) ,结合CpG 岛信息,确定启动子区。
这种方法使预测的敏感性和特异性都明显提高。
该程序预测含CpG岛的启动子的敏感性和特异性都高于90 % ,预测不含CpG岛的启动子的精确性相对略低。
TRRD 数据库http://wwwmgs.bionet.nsc.ru/mgs/dbases/trrd4/ 收录了真核基因调控区结构和基因表达方式的信息,每个条目对应一个基因。
应用权重矩阵数据库搜索转录因子结合部位的程序包括SIGNAL SCAN /molbio/signal/MatInspector http://www.genomatix.de/products/index.html转录因子搜索程序( transcriptional factor search ,TF2 SEARCH ) http://www.cbrc.jp/research/db/TFSEARCH.html等等。
尽管基于PWM 的搜索比较敏感,但它最大的缺点就是假阳性率过高,在预测的结果中有很多结合部位并不真正具有生物学功能。