重组质粒的构建
重组质粒构建流程

重组质粒构建流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by the editor. I hope that after you download them, they can help yousolve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you!In addition, our shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts,other materials and so on, want to know different data formats and writing methods, please pay attention!重组质粒构建是生命科学研究中常见的实验技术之一,通过将感兴趣的基因片段插入到质粒中,可以实现对基因的进一步研究和应用。
重组质粒的构建经验 [技巧]
![重组质粒的构建经验 [技巧]](https://img.taocdn.com/s3/m/f026746187c24028915fc3fa.png)
重组质粒的构建经验 [技巧]重组质粒的构建经验~~~昨天我在版中我看很多谷友询问重组质粒的构建问题,有些谷友说构建质粒需要一个月,甚至更长时间,这让我联想我刚做分子生物学时候的曲折。
重组质粒构建是常用的分子生物学手段,其实只是最基本的方法,一般一个星期同时构建三二个组质粒是没有问题的。
在国内先进的实验中,也大都是由实验员搞定。
但是其中还是有些基本的技巧需要掌握。
在这里将我的心得分享于大家,这也是我本人几年来一线工作时的经验积累,以期能为谷友提供借鉴,让大家在实验中少走弯路。
所涉及内容如下: 1) 克隆基因的酶切位点问题 2) 载体酶切的问题 3) 连接片段浓度比的问题在阐明上述问题同时,本人尽可能举些实验中的问题案例予以说明。
一、克隆基因的酶切位点问题 1、克隆位点选择的问题。
首先要对目标基因进行酶切位点扫描分析,列出其所含酶切位点清单。
然后对照质粒多克隆位点,所选择的克隆位点必须是目标基因所不含的酶切位点。
这是常识,不赘述。
2、保护碱基数目的问题。
在设计PCR引物时,引入酶切位点后,常常要加入保护碱基,这是大家所熟知的。
但是保护碱基数量多少,可能被新手所忽视。
这种忽视碰可能会大大影响后续的实验进展。
一般情况下,普通的内切酶只加入两个保护碱基,其内切反应就可以正常进行;而有一类,仅仅只加入两个保护碱基,其内切反应就不能正常进行,这是因为内切酶不能正常结合DNA片段上。
如NdeI就属这类,需要加入至少6个保护碱基,常用的HindIII也要三个。
下面是我提供这类酶的列表及其所需最少的保护碱基数,相信下列将有助于大这家的实验设计。
NcoI 4 NdeI 6 NheI 3 NotI 8 PmeI 6SacI 3 SalI 3 SmaI 3 HindIII 3 BstI 8 SphI 4XhoI 3 XbaI 3 SmaI 4 案例分析一:本人最初曾选用NdeI克隆位点,未注意到保护碱基数目的问题,设计PCR引物时,引入NdeI酶切位点后,只加上两个保护碱基,一个月内没有进展,始终不能成功构建重组载体。
重组质粒的构建

重组质粒的构建重组质粒的构建是基因工程的核心步骤之一,其目的是将目的基因插入到质粒载体中,以实现目的基因的稳定表达和克隆化。
以下是重组质粒构建的主要步骤:1.目的基因获取首先需要获取目的基因。
目的基因可以从基因文库、PCR、基因组测序等方法中获取。
根据需要选择合适的方法,将目的基因克隆到质粒载体中。
2.载体质粒选择选择适合的质粒载体是重组质粒构建的关键步骤之一。
根据目的基因的特点和表达要求,选择适合的质粒载体。
常见的质粒载体有pET、pUC、pBluescript等。
3.限制性酶切限制性酶切是重组质粒构建的重要步骤之一。
通过限制性酶切,将目的基因和质粒载体分别切开,露出粘性末端,以便于连接反应。
4.连接反应将切好的目的基因和质粒载体的粘性末端连接在一起,形成重组质粒。
连接反应需要使用T4DNA连接酶或其它连接酶进行催化。
连接反应需要在适宜的温度和pH 条件下进行一定时间,以确保重组质粒的正确构建。
5.转化宿主细胞将连接反应得到的重组质粒转化到宿主细胞中。
常见的宿主细胞有细菌、酵母、昆虫等。
转化方法有多种,如电穿孔法、化学转化法等。
转化后需要在适宜的培养条件下进行培养,以获得大量的重组质粒。
6.克隆筛选克隆筛选是重组质粒构建的重要步骤之一。
通过克隆筛选,可以确定重组质粒是否正确构建。
常见的克隆筛选方法有蓝白斑筛选、酶切法等。
7.序列验证最后需要对重组质粒进行序列验证,以确保目的基因的正确插入和序列的准确性。
序列验证可以通过Sanger测序等方法进行。
重组表达质粒的构建——原核表达载体选择
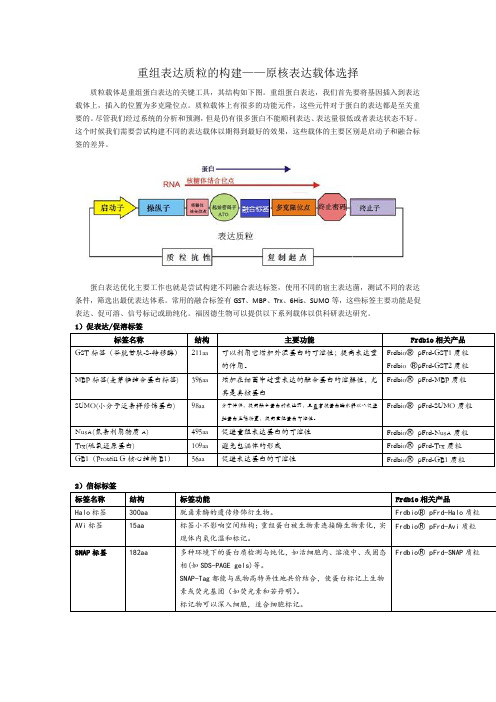
重组表达质粒的构建——原核表达载体选择质粒载体是重组蛋白表达的关键工具,其结构如下图。
重组蛋白表达,我们首先要将基因插入到表达载体上,插入的位置为多克隆位点。
质粒载体上有很多的功能元件,这些元件对于蛋白的表达都是至关重要的。
尽管我们经过系统的分析和预测,但是仍有很多蛋白不能顺利表达、表达量很低或者表达状态不好。
这个时候我们需要尝试构建不同的表达载体以期得到最好的效果,这些载体的主要区别是启动子和融合标签的差异。
蛋白表达优化主要工作也就是尝试构建不同融合表达标签,使用不同的宿主表达菌,测试不同的表达条件,筛选出最优表达体系。
常用的融合标签有GST、MBP、Trx、6His、SUMO等,这些标签主要功能是促表达、促可溶、信号标记或助纯化。
福因德生物可以提供以下系列载体以供科研表达研究。
1)促表达/促溶标签2)信标标签3)纯化标签我们选择表达载体的时候不但要考虑蛋白怎么表达成功,更要考虑蛋白怎么纯化出来,纯化的问题主要是考虑纯化标签和酶切位点的选择,下表我们列举了常见的纯化标签和酶切位点。
4)酶切位点以上为原核表达常用的标签和酶切位点,其性质也都作了简要的介绍,各专业网站或专业书籍已对此做详尽解释,科研工作者可根据具体实验设计方案,组合设计以上标签和酶切位点的使用。
特别值得注意的是,选用和设计蛋白酶切位点的时候首要考虑的是序列内部有没有蛋白酶位点,同时要考虑酶切的效率和蛋白酶试剂成本。
一般商业化载体,在标签蛋白与载体多克隆位点之间都设计有酶切位点。
标签可设计在N-端也可在C-端,设计在N-端的优势是,可通过标签高效翻译起始位点带动插入蛋白的表达,可溶性标签的高效表达更可促进蛋白的可溶性表达;同时,大部分的蛋白内切酶的切割位点在C-端,所以标签设计在N-端可将标签切割完全。
在设计标签序列与酶切位点的时候还要考虑N-端稳定性原则,也就是所谓宿主细胞的N-端规则(N-end rule),这个要避免;同时,还应该检查是否引入了可与别的蛋白相互作用的序列或者蛋白酶切位点。
4重组表达质粒的构建——基因的克隆

重组表达质粒的构建——基因的克隆长片段基因在大肠杆菌中表达往往比较困难,作为抗原使用的重组蛋白可以考虑选择抗原性好的区段原核表达,前文已作阐述。
对整个蛋白结构研究,必须全长表达该蛋白,此时最好考虑真核表达系统,特别是含有跨膜区的蛋白。
选定要克隆的区段,需先富集纯化之后才方便插入载体,常用的富集方法是PCR或者质粒繁殖复制。
为了防止在PCR扩增过程中引入碱基错误或者碱基缺失,PCR扩增基因时候必须使用高保真Taq酶。
为了满足科研工作者不同实验需求,福因德生物将高保真Taq酶优化为即用型Mix,使用时直接加引物和模板就可以扩增。
除此之外,福因德生物还开发出LA Taq、S-Taq Mix以及SYBR荧光定量PCR Mix(需要更高品质的可选用SYBR PCR SuperMix)。
原核重组表达常用克隆技术主要有以下几种:1)酶切连接这个是目前应用最为广泛的的克隆技术,主要优点是技术稳定;缺点是周期长、步骤多,任何一个环节产生的误差都会影响克隆构建的成败。
如用酶切连接的策略进行载体批量构建,不同载体和不同外源基因尽可能选用相同的上下游酶切位点,比如,批量克隆基因到某个载体上,可一次性大量双酶切将载体线性化后保存备用,每次构建载体只需酶切外源基因片段,载体可直接取用,不必每次都酶切,省时省力(此处需特别留意的是基因内部不能有与上述所用冲突的酶切位点)。
2)TA克隆TA克隆必须使用商业的线性化载体,线性载体3´末端有一个T碱基,与PCR扩增产物3´末端A正好匹配。
这种克隆策略最大的优点是载体使用方便,扩增产物可以直接克隆到载体上,不需要酶切位点等冗余序列;缺点是:必须依赖商业化载体,载体选择受限;扩增外源片段所使用的Taq酶也必须是可以在3´末端加A,这种Taq酶的保真度不高;外源片段插入之后还必须鉴定方向。
目前,这种构建表达载体的策略已经逐渐被淘汰。
3)TOPO克隆TOPO克隆载体利用DNA拓扑异构酶I识别序列中的CCCTT松弛双螺旋并重新连接,同时兼具限制性内切酶和连接酶的功能。
重组质粒的构建

连接酶催化DNA连接的最佳反应温度是37℃
▪ DNA重组技术中的核心是DNA片段之间的体外连接方案
DNA连接酶及连接机制
1 ) ATP 通 过 磷 酸 基 团 与 T4 DNA连接酶中的亮氨酸形成 磷酸-氨基键,从而形成酶ATP复合物;
限制性内切酶
具备多个限制酶的 识别位点(多克隆位 点) ,以便外源DNA的 插入与截取。
多种酶切口 单一酶切口 多克隆位点
或具备特异的重组 位点
11
载体的选择与改造应具备的条件
3.具有合适的筛选标记
具有遗传表型或筛选 标记,以区别阳性重组分子 和阴性重组分子,主要有抗 药性基因、酶基因、营养缺 陷型及形成噬菌斑的能力等。
二、基础知识
2. 重组DNA技术的基本过程
(1)目的基因的获得 (2)载体的选择与制备 (3)目的基因与载体的结合(DNA分子的体外连接) (4)重组DNA导入受体 (5)重组体筛选和鉴定 (6)目的基因表达 (7)基因产物的分离纯化
DNA重组操作过程
ab
剪切
载体
重组
B
引入宿 主细胞
Ab
抗性筛选
T载体
▪ 一般采用的方法是先把载体用某种限制性内切酶消化成平 头,再在70℃或72℃下在只加入dTTP的反应体系中用Taq DNA聚合酶处理半小时(也有人报道处理1~2小时能提高克隆效 率,这样加T反应会更彻底)。也可以用末端转移酶来完成加T 反应。载体自连、PCR产物串连可以忽略。
▪ 如果使用ddTTP,效果会更好。
实验二 重组质粒的构建
一、实验目的
▪通过本实验学会琼脂糖凝胶DNA回收和重组 DNA连接的方法。
重组质粒的构建、转化和重组子的筛选与鉴定

重组质粒的构建、转化和重组子的筛选与鉴定姓名:郑小煜学号:201100140069班级:11级生技班同组者:赵莉、高瑞【实验目的】1、学习在实现DNA的体外重组过程中,正确选择合适的载体和限制性内切酶,并利用限制性核酸内切酶对载体和目的DNA进行切割,产生利于连接的合适末端。
2、通过对DNA的酶切,学习设计构建重组DNA分子的基本方法,掌握载体和外源目的DNA酶切的操作技术。
3、学习利用T4 DNA连接酶把酶切后的载体片段和外源目的DNA片段连接起来,构建体外重组DNA 分子的技术,了解并掌握几种常用的连接方法。
4、掌握利用CaCl2制备感受态细胞的方法。
5、学习并掌握热击法转化E.coli的原理和方法。
6、在使用红白菌落法筛选获得重组子的基础上,本实验学习通过对重组子进行重组质粒DNA的抽提鉴定,以进一步确定重组质粒中含有外源目的DNA片段,验证重组子是期望的重组子的方法。
7、通过学习掌握重组DNA分子鉴定的基本方法。
8、掌握α互补筛选法筛选重组子的方法。
并鉴定体外导入目的DNA片段的大小。
9、学习用试剂盒提取重组质粒DNA的方法。
10、复习琼脂糖凝胶电泳的原理及方法。
【实验原理】外源DNA与载体分子的连接即为DNA重组技术,这样重新组合的DNA分子叫做重组子。
重组的DNA分子在DNA连接酶的作用下,有Mg2+、ATP存在的连接缓冲系统中,将分别经限制性内切酶酶切的载体分子和外源DNA分子连接起来。
将重组质粒导入感受态细胞中,将转化后的细胞在选择性培养基中培养,可以通过α互补筛选法筛选出重组子,并可通过酶切电泳及PCR检验的方法进行重组子的鉴定。
1、限制性核酸内切酶及酶切反应体外构建重组DNA分子,首先,要了解目的基因的酶切图谱,选用的限制性内切酶不能目的基因内部有专一的识别位点,即当用一种或两种限制性核酸内切酶切割外源供体DNA时,能得到完整的目的基因。
其次,要选择具有相应的单一酶切位点的质粒或者噬菌体等载体分子作为克隆的载体。
构建重组质粒基本方法

构建重组质粒基本方法重组质粒是一种重要的遗传工程工具,用于将外源基因导入到宿主细胞中,从而实现特定基因的表达与功能研究。
构建重组质粒的基本方法可以概括为:选择质粒骨架、引物设计与合成、PCR扩增外源基因片段、DNA连接与重组、质粒扩增与提取、质粒鉴定与筛选,以下分别进行详细介绍。
一、选择质粒骨架在构建重组质粒时,首先需要选择一个合适的质粒骨架。
质粒骨架是指一个可复制的质粒DNA分子,常见的质粒骨架有pUC、pBR322、pET等。
质粒骨架上通常包含有宿主细胞可以识别的起始子和起始子附近的终止子,用于启动和终止转录过程,同时还包含选择标记基因,如抗生素抗性基因,以及其他在分子克隆中常用的诸如多克隆位点、限制酶切位点等。
二、引物设计与合成在构建重组质粒时,需要利用引物来扩增并克隆外源基因片段。
引物一般是两条DNA可控引物,其中一条是正向引物,另一条是反向引物。
引物的设计需要注意以下几点:引物的长度通常为15-30个碱基对,引物应该具有合适的Tm值,并且在引物双链的末端至少有2个碱基对是纯G或纯C。
引物可以使用商业引物合成公司合成。
三、PCR扩增外源基因片段使用引物扩增外源基因片段是构建重组质粒的一个关键步骤。
PCR反应一般包括DNA模板、引物、dNTPs和DNA聚合酶。
根据需要,可以使用特异性引物对目标基因进行PCR扩增,然后通过凝胶电泳检查PCR产物长度和纯度,并使用PCR产物进行下一步处理。
四、DNA连接与重组将PCR扩增得到的外源基因片段与质粒骨架进行连接和重组。
连接通常通过使用限制酶切和连接酶来实现。
限制酶切是利用限制酶切剪切DNA,生成具有互补粘性末端的DNA片段,然后将外源基因片段与质粒骨架进行连接。
连接酶可以使DNA片段之间的末端骨架参与phosphodiester结合反应,从而形成连体分子。
五、质粒扩增与提取将重组质粒转化到宿主细胞中,通过培养和培养基筛选来扩增质粒。
质粒扩增一般在含有抗生素的琼脂糖平板上进行,抗生素可以选择对宿主细胞有毒作用但不对重组质粒有毒的抗生素。
重组质粒构建流程

重组质粒构建流程导言在分子生物学研究中,质粒是一种重要的工具,可用于携带外源DNA,转导到靶细胞内进行表达或操纵基因。
在许多应用中,需要从头开始构建特定的质粒来满足实验需求。
本文将介绍重组质粒的构建流程,包括质粒设计、DNA片段的合成、连接和转化等步骤。
质粒设计重组质粒的构建首先需要进行质粒设计。
在设计过程中,需要考虑以下几个方面:质粒拓扑结构、宿主细胞、选择标记、启动子、终止子等。
其中,质粒拓扑结构是质粒构建的基础,可以选择环状质粒或线性质粒;宿主细胞是质粒的宿主细胞,需要考虑宿主细胞的特性和适用范围;选择标记是用于筛选携带外源DNA的宿主细胞,可以选择抗生素抗性标记、荧光蛋白标记等;启动子和终止子则是用于调控外源DNA的表达水平。
DNA片段的合成在质粒构建中,需要合成一系列DNA片段,包括载体骨架、选择标记、启动子、基因、终止子等。
DNA片段的合成可以通过多种方法进行,包括化学合成、PCR扩增、酶切和连接等。
在合成过程中,需要确保DNA片段的正确性和纯度,以保证后续的连接和转化效率。
连接连接是质粒构建的关键步骤,通过连接不同的DNA片段来构建目标质粒。
连接的方法包括酶切和连接、PCR扩增和连接、重组DNA技术等。
在连接过程中,需要确保连接效率和准确性,避免产生错误连接或杂交产物。
此外,对于大片段DNA的连接,还需要考虑连接的稳定性和转化效率。
质粒的放大和提取连接完成后,需要将质粒放大到足够的数量,并提取纯净的质粒DNA。
放大的方法可以选择细菌发酵、真菌发酵等,根据质粒的特性选择合适的宿主细胞进行放大。
质粒提取的方法包括碱裂解法、隐式裂解法等,确保提取的质粒DNA的纯度和完整性。
质粒的转化最后一步是将构建好的质粒转化到目标宿主细胞中,进行表达或操纵基因。
转化的方法可以选择化学转化、电转化、热激转化等,根据宿主细胞的特性和实验需求选择合适的转化方法。
在转化过程中,需要考虑转化效率、亲和性和表达水平等因素,确保转化的质粒可以稳定存在和表达。
重组表达质粒的构建

重组表达质粒的构建重组表达质粒的构建1.原核表达载体选择质粒载体是重组蛋⽩表达的关键⼯具,其结构如下图。
重组蛋⽩表达,我们⾸先要将基因插⼊到表达载体上,插⼊的位置为多克隆位点。
质粒载体上有很多的功能元件,这些元件对于蛋⽩的表达都是⾄关重要的。
尽管我们经过系统的分析和预测,但是仍有很多蛋⽩不能顺利表达、表达量很低或者表达状态不好。
这个时候我们需要尝试构建不同的表达载体以期得到最好的效果,这些载体的主要区别是启动⼦和融合标签的差异。
蛋⽩表达优化主要⼯作也就是尝试构建不同融合表达标签,使⽤不同的宿主表达菌,测试不同的表达条件,筛选出最优表达体系。
常⽤的融合标签有GST、MBP、Trx、6His、SUMO等,这些标签主要功能是促表达、促可溶、信号标记或助纯化。
福因德⽣物可以提供以下系列载体以供科研表达研究。
我们选择表达载体的时候不但要考虑蛋⽩怎么表达成功,更要考虑蛋⽩怎么纯化出来,纯化的问题主要是考虑纯化标签和酶切位点的选择,下表我们列举了常见的纯化标签和酶切位点。
以上为原核表达常⽤的标签和酶切位点,其性质也都作了简要的介绍,各专业⽹站或专业书籍已对此做详尽解释,科研⼯作者可根据具体实验设计⽅案,组合设计以上标签和酶切位点的使⽤。
特别值得注意的是,选⽤和设计蛋⽩酶切位点的时候⾸要考虑的是序列内部有没有蛋⽩酶位点,同时要考虑酶切的效率和蛋⽩酶试剂成本。
⼀般商业化载体,在标签蛋⽩与载体多克隆位点之间都设计有酶切位点。
标签可设计在N-端也可在C-端,设计在N-端的优势是,可通过标签⾼效翻译起始位点带动插⼊蛋⽩的表达,可溶性标签的⾼效表达更可促进蛋⽩的可溶性表达;同时,⼤部分的蛋⽩内切酶的切割位点在C-端,所以标签设计在N-端可将标签切割完全。
在设计标签序列与酶切位点的时候还要考虑N-端稳定性原则,也就是所谓宿主细胞的N-端规则(N-end rule),这个要避免;同时,还应该检查是否引⼊了可与别的蛋⽩相互作⽤的序列或者蛋⽩酶切位点。
重组质粒的构建

重组质粒的构建实验流程—质粒构建实验操作一、LB培养基配置LB培养基用于一般细菌培养,特别用于分子生物学试验中大肠杆菌的保存和培养。
其中蛋白胨、酵母膏粉提供氮源、维生素和生长因子,NaCl维持均衡的渗透压,葡萄糖提供碳源,琼脂是培养基的凝固剂。
【试剂】胰蛋白胨(Tryptone)、酵母提取物(Yeast Extract)、NaCl、琼脂(Agar)【实验步骤】1、LB固体培养基配方(配置100ml培养基)胰蛋白胨(Tryptone) 1g酵母提取物(Yeast Extract)0.5gNaCl 1g琼脂(Agar)1.5g单蒸水100ml蛋白胨很易吸潮,在称取时动作要迅速,另外,称药品时严防药品混杂,一把药匙用于一种药品、或在称取一种药品后,洗净、擦干,再称取另一种药品,瓶盖也不要盖错。
2、液体培养基除不加琼脂外,其余同固体培养基一样。
3、包扎用报纸封住瓶口,再用皮筋捆扎好,用记号笔注明培养基名称、组别、日期。
4、灭菌将上述培养基以1.05kg/cm2、121.3℃、20min高压蒸汽灭菌。
如因特殊情况不能及时灭菌,则应放入4℃冰箱内暂存。
灭菌后,将锥形瓶放入烘箱烘干,烘干后,4℃保存。
5、LB固体培养基倒板○1配置:如上述配方配置100ml的LB固体培养基。
○2抗生素的加入:将凝固的培养基放入微波炉内加热至完全融化,然后置于55℃的水浴中,待培养基温度降至55℃时(手可触摸)加入抗生素,以免温度过高导致抗生素失效,并充分摇匀。
○3倒板:一般10ml倒1个板子,培养基倒入培养皿后,打开盖子,在紫外下照10—15min。
○4保存:将培养皿倒置放于4℃保存,一个月内使用。
二、质粒的提取(protocol)1、大肠杆菌的培养:从平板培养基上挑选单菌落接种至5ml的含有抗生素的LB液体培养基中,37℃过夜培养。
【注:培养液不宜过量,过量时会因菌量太大而溶菌不充分,纯化时会影响质粒的纯度。
】2、5ml菌液倒入5ml离心管中,10,000xg,1min离心,弃上清。
实验二 重组质粒的构建

实验二重组质粒的构建一、实验步骤1.提取质粒DNA2.酶切(EcoRI+SpeI)注意事项:①酶量,反应时间及体积: One unit of enzyme is defined asthe quantity needed to cut 1μg of DNA in 50ul in onehour。
反应时间的选择。
一般酶切鉴定30分钟就可以了,如果酶减少,可延长反应时间(16h);反应体系不应太小,常规的酶切一般要维持在10-50ul。
酶的体积不要超过总体积的10%(甘油应在5%以下)。
②酶的使用:酶应永远放冰上,应是最后一个被加入到反应体系中,用完后及时放回冰箱③DNA的制备:待切割的DNA应当已去除酚,氯仿,乙醇,EDTA, 去污剂或过多盐离子等④缓冲液:不同酶需不同离子强度缓冲液,使用前应将缓冲液完全溶解并充分混匀。
⑤混匀:很重要,注意不可振荡⑥反应温度:通常37 ℃⑦终止反应:终止液,热失活,酚/ 氯仿抽提⑧星号活性:在非理想条件下,内切酶切割与识别位点相似但不完全相同的序列。
3.: 酶切产物纯化4.连接二、实验结果与分析1.酶切结果检测图1 EcoRI和SpeI双酶切GST-T及proB载体结果1%琼脂糖凝胶电泳检测图注:M:DL5000DNAmarker;1号泳道;未酶切proB 质粒DNA;10、11号泳道:EcoRI和SpeI双酶切proB载体结果;16、17泳道:EcoRI和SpeI双酶切GST-T载体结果分析:①由图1可知,1号泳道的未酶切proB质粒DNA未显示有条带,其原因可能是所点的未酶切proB标准品浓度过低,条带亮度过低难以识别。
②10和11泳道中大小约为5000bp的较亮条带是proB载体,但有轻微拖尾现象,条带呈圆弧型,应该是因为点样量较大。
经EcoRI和SpeI双酶切后,环状的质粒DNA被分成两个部分,分别为约5000bp和40bp的片段,其中40bp的小片段因分子量小,迁移速度快而跑出了琼脂糖凝胶,在图中无法看到。
重组构建质粒

重组质粒的构建关键词:重组质粒构建重组质粒构建是常用的分子生物学手段,其实只是最基本的方法,一般一个星期同时构建三二个组质粒是没有问题的。
在国内先进的实验中,也大都是由实验员搞定。
但是其中还是有些基本的技巧需要掌握。
在这里将我的心得分享于大家,这也是我本人几年来一线工作时的经验积累,以期能为谷友提供借鉴,让大家在实验中少走弯路。
所涉及内容如下:1) 克隆基因的酶切位点问题2) 载体酶切的问题3) 连接片段浓度比的问题在阐明上述问题同时,本人尽可能举些实验中的问题案例予以说明。
一、克隆基因的酶切位点问题1、克隆位点选择的问题。
首先要对目标基因进行酶切位点扫描分析,列出其所含酶切位点清单。
然后对照质粒多克隆位点,所选择的克隆位点必须是目标基因所不含的酶切位点。
这是常识,不赘述。
2、保护碱基数目的问题。
在设计PCR引物时,引入酶切位点后,常常要加入保护碱基,这是大家所熟知的。
但是保护碱基数量多少,可能被新手所忽视。
这种忽视碰可能会大大影响后续的实验进展。
一般情况下,普通的内切酶只加入两个保护碱基,其内切反应就可以正常进行;而有一类,仅仅只加入两个保护碱基,其内切反应就不能正常进行,这是因为内切酶不能正常结合DN段上。
如NdeI就属这类,需要加入至少6个保护碱基,常用的HindIII也要三个。
下面是我提供这类酶的列表及其所需最少的保护碱基数,相信下列将有助于大这家的实验设计。
NcoI 4NdeI 6NheI 3 NotI 8 PmeI 6 SacI 3 SalI 3 SmaI 3 HindIII 3 BstI 8 SphI 4 XhoI 3 XbaI 3SmaI 4案例分析一:本人最初曾选用NdeI克隆位点,未注意到保护碱基数目的问题,设计PCR引物时,引入NdeI酶切位点后,只加上两个保护碱基,一个月内没有进展,始终不能成功构建重组载体。
后查文献得知症结所在,在NdeI序列后加上六个保护碱基后,迎刃而解。
重组质粒的构建

重组质粒的构建、转化和重组子的筛选与鉴定崔文强201300140012同组者:陈斌,郝书平【摘要】重组质粒的构建需要对DNA分子进行切割,并连接到合适的载体上进行体外重组。
这就需要正确选择合适的载体和限制性内切酶,并利用限制性核酸内切酶对载体和目的DNA进行切割,产生利于连接的合适末端。
然后利用T4DNA连接酶把酶切后的载体片段和外源目的DNA片段连接起来,构建体外重组DNA分子。
接着将外源重组DNA引入受体细胞,所以首先要用CaCl2法制备感受态细胞,通过复制和表达,实现遗传信息的转移,使受体细胞出现新的遗传性状,再用红白斑筛选重组子,最后检验导入片段的大小。
本次实验涉及的操作方法有很多,比如利用CaCl2制备感受态细胞的方法、热击法转化E.coli的方法、重组DNA分子鉴定的基本方法、用试剂盒提取重组质粒DNA的方法和琼脂糖凝胶电泳的方法等等。
【关键词】重组质粒;限制性内切酶;DNA连接酶;感受态细胞;红白斑筛选;琼脂糖凝胶电泳【引文】外源DNA与载体分子的连接即为DNA重组技术,这样重新组合的DNA分子叫做重组子。
重组的DNA分子在DNA连接酶的作用下,将分别经限制性内切酶酶切的载体分子和外源DNA分子连接起来。
将重组质粒导入感受态细胞中,将转化后的细胞在选择性培养基中培养,可以通过α互补筛选法筛选出重组子,并可通过酶切电泳检验的方法进行重组子的鉴定。
本实验采用单酶切法,即只能用一种限制性核酸内切酶切割目的DNA片段,酶切后的片段两端将产生相同的黏性末端或平末端。
选择具有相同限制性核酸内切酶识别位点的合适载体,在构建重组分子时,除了形成正常的重组子外,还可能出现目的DNA片段以相反方向插入载体分子中,或目的DNA串联后再插入载体分子中,甚至出现载体分子自连,重新环化的现象,因此重组效率较低。
重组DNA转化宿主细胞后,并非所有的受体细胞都能被导入重组DNA分子,一般仅有少数重组DNA分子能进入受体细胞,同时也只有极少数的受体细胞在吸纳重组DNA分子之后能良好增殖。
分子生物学重组质粒的制备

实验一感受态细胞的制备一、实验材料大肠杆菌DH5a 、LB培养基、0.1M CaCl2,50ml大Ep管,冰,80%甘油。
二、操作步骤氯化钙法1.将单个菌落或液体菌种(1:50)接种于20mL LB培养液的试管中,37℃振荡(200r/min)培养过夜。
2.取过夜2ml菌体加入100mLLB培养液37℃振荡培养至光密度(OD600)值在0.4-0.5之间。
(约需1h-2h).3.预冷0.1M CaCl2,和2个50ml大Ep管。
4.将细菌培养物分别倒入预先冰浴的无菌50mL大Ep管中,冰浴30min.5.于4℃下离心10min,(4000r/min),弃去上清夜,放于冰浴中。
6.加10mL冰冷的0.1mol/L MgCl2溶液混匀,冰浴30min。
7.4℃下离心10min,(4000r/min),弃去上清夜(吸尽),放于冰浴中。
8.加入2ml 0.1 mol/L CaCl2轻混。
9.分装于1.5ml离心管中,(每支200uL),-80℃储存备用。
1)转化用:200ul/Ep2)保存:180ul+20ul甘油(80%)注意:1.为了获得高感受态细胞,应选用处于生长对数期的细胞,OD600值不应高于0.6,并且在整个试验过程中均需将细胞置于冰上。
2.细胞在冰冻后即获得了感受性,在-80℃几小时后感受性达到最高值,几个月不会改变。
实验二质粒DNA的小量提取一、试剂及溶液LB培养基、葡萄糖(1)用于碱法提取质粒DNA的溶液a.溶液Ⅰ(GET)缓冲液(50ml):葡萄糖0.49g,0.5M的EDTA(pH8.0)1ml,1M的Tris-HCl(pH8.0)1.25ml。
b. 溶液Ⅱ(变性液):(现用现配)c. (50ml)]:60mL的5M的KAc,11.5mL冰醋酸,28.5mL H2O. PH4.8。
(2)缓冲液TBE缓冲液(10Χ):称取Tris 108g,硼酸55g,5M EDTA(Ph8.0)40ml,用H2O定容到1000mL。
实验报告构建重组质粒(3篇)

第1篇一、实验目的1. 学习DNA体外重组技术的原理和方法。
2. 掌握重组质粒的构建、转化、筛选和鉴定技术。
3. 熟悉实验操作步骤和注意事项。
二、实验原理DNA体外重组技术是指将外源DNA片段与载体DNA片段在体外进行连接,形成重组DNA分子的技术。
通过该技术,可以将外源基因导入宿主细胞,实现基因表达和功能研究。
三、实验材料1. 载体:pUC19质粒2. 目的基因:基因片段3. 限制性核酸内切酶:EcoRI、BamHI4. DNA连接酶:T4 DNA连接酶5. DNA聚合酶:Klenow片段6. 琼脂糖凝胶电泳试剂7. DNA标记物8. 转化试剂:CaCl2、SDS、乙醇9. 受体细胞:大肠杆菌DH5α四、实验步骤1. 目的基因的扩增(1)设计引物:根据目的基因序列设计一对引物,引物长度一般为20-25bp,5'端添加EcoRI和BamHI酶切位点。
(2)PCR扩增:以基因片段为模板,引物为引物,进行PCR扩增。
2. 载体的酶切(1)将pUC19质粒和PCR扩增的目的基因分别用EcoRI和BamHI进行酶切。
(2)回收酶切片段:琼脂糖凝胶电泳分离酶切片段,回收目的片段。
3. DNA连接(1)将回收的目的基因片段和载体片段混合,加入DNA连接酶,进行连接反应。
(2)将连接产物转化到大肠杆菌DH5α感受态细胞中。
4. 转化后细胞的培养与筛选(1)将转化后的细胞在含有氨苄青霉素的LB培养基中培养过夜。
(2)取适量菌液进行PCR检测,筛选出阳性克隆。
5. 阳性克隆的鉴定(1)提取阳性克隆的质粒DNA。
(2)用EcoRI和BamHI进行酶切,琼脂糖凝胶电泳检测酶切片段。
(3)将酶切片段与标记物进行对比,验证重组质粒的正确性。
五、实验结果与分析1. PCR扩增结果:在PCR产物中,可见一条与预期片段大小相符的条带。
2. 酶切结果:在琼脂糖凝胶电泳中,可见目的基因片段和载体片段的酶切片段。
3. 阳性克隆的鉴定:在琼脂糖凝胶电泳中,可见与预期片段大小相符的条带,证明重组质粒构建成功。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
我的质粒构建总结-
重组质粒构建是常用的分子生物学手段,其实只是最基本的方法,一般一个星期同时构建三二个组质粒是没有问题的。
在国内先进的实验中,也大都是由实验员搞定。
所以其中其中还是有基本的技巧需要掌握。
在这里决定将我的心得分享于大家,以期能提供借鉴,让大家在实验中少走弯路。
在本帖及之后继帖中将以一段PCR获得基因,以NdeI和HindIII位点克隆进入质粒为例来系统剖析重组质粒的构建中基本策略与技巧。
这作的经验积累与心得。
所涉及内容如下:
1) 克隆基因的酶切位点问题
2) 载体酶切的问题
3) 连接片段浓度比的问题
以上阐明上述问题同时,本人尽可能引入实验时会各种出现的问题予以说明。
一、克隆基因的酶切位点问题
1 对目标基因进行酶切位点扫描分析,列出其所含酶切位点清单。
对照质粒多克隆位点,所选择的克隆位点必须是目标基因所不含的酶切位点。
这是常识不赘述。
这里对NdeI和HindIII 为例。
2 设计PCR引物时的保护碱基数目。
这可能是初涉入未引起注意与重视问题。
NdeI需加入6个以上的保护碱基,而HindIII则要三个就可以。
一般情况下,普通的内切酶只加入两个保护碱基,其内切反应就可以正常进行;而有一类,只加入两个保护碱基,其内切反应就不能正常进行,这是因为内切酶不能正常结合DNA 片段上。
如NdeI就属这类。
下面是我提供这类酶的列表及其所需最少的保护碱基数:
NcoI 4
NdeI 6
NheI 3
NotI 8
PmeI 6
SacI 3
SalI 3
SmaI 3
HindIII 3
BstI 8
SphI 4
XhoI 3
XbaI 3
SmaI 4
案例分析:本人最初用NdeI酶,未注意到该问题,只与普通酶一样引入两个保护碱基,一个月内没有进展。
后查文献得知症结所在,加下六个后,迎刃而解。
大家引以为戒啊。
现在普通酶我都引入三个保护碱基,现在合成价格不贵,为保证酶切充分,连接顺利,不用
节约那点钱,再说若一次不成功,重要实验花费时间与金钱更多,孰利孰弊,不言自明。
呵呵。
二、载体酶切的问题
1单切鉴定。
这个问题实是简单,但我认为很有着重强调之必要。
现在大家手头的质粒都是转来转去的,其中的各酶切位点状况如何,是否能被有效地切开,在实验开始之前对质粒载体很有作单切鉴定之必要。
现在我每次构建之前,对所要用的酶切位点都作一一鉴定,比如要用到NdeI和HindIII,我就先对质粒该两个酶进行鉴定。
有效地切开后,再将引物发出合成;若不能,就按“一”中原则进行调换。
2 质粒双切后对照连接。
实验中这是连接步骤,但实质还是质粒酶切问题。
一般情况下,都在通用缓冲液中进行双酶切,但两种酶在通用缓冲液中中酶切效率不一样,可能导致部分只是单切缺口的质粒片段存在,这样,连接的对照实验在不加入外源片段时,质粒就会自连,长出菌斑,这种情况下,质粒酶切片段是不可能用于下一步真正连接实验。
案例分析:本人曾用XhoI和HindIII酶切位点构建重组质粒,对质粒进行双酶切后,直接就做连接,未上述两步鉴定,每次结果满板的菌斑。
但就是没有阳性。
后来对质粒进行单酶要鉴定后,发现XhoI酶切位点损坏。
又是一个月没有进展,浪费精力和药品。
血的教训啊。
因为当时没有注意到:单切质粒是一条带,双切质粒也是一条带,电泳行为上是一样的,分辨不出。
如果做上述任何一个鉴定就会知道问题出在那儿,呵呵。
案例分析:本实验室一个号称实验严谨的大博士,有KpnI和HindIII构建重组质粒,一个月未果,只得阴性斑,不得阳性斑,后怀疑KpnI酶失效。
迁怒KpnI,在我不知情下扔掉实验室所满管KpnI酶。
我得知后,问他做过上述两鉴定实验后,他支吾着说没有,反而责备本人不早说出问题原因所在。
呵呵,他不自责自己只是闷头做实验,不早问,反倒咬一口解铃人。
呵呵,你说冤不冤?版主你的类似的冤可能更多吧,辛苦了!
两星期前写了前两问题后,终于能抽时间写第三个问题,在做好前述两个方面工作后,这个问题相对简单。
三、连接时两片段浓度比问题
一般实验指导手册上都说质粒:片段=1:3(摩尔比),在实际操作中我以为在1:5甚至1:10为宜。
做好“一、二”,16℃10小时后,每次都能有效地连接上。
当然还有感觉态问题,我们以前自己做,现在懒得做了,都用“天为时代公司”的产品,还不错(注明我不是天为公司内线,呵呵)。
这里介绍一个估测处DNA浓度的方法:DNA可以用紫外法检测,也可以电泳对比marker 估测,在要求不是很精确情况下,大家不妨试试下面方法:
1.取一平皿。
2.薄薄倒一层含有EB的琼脂糖胶,凝固(4 ℃可以存一个星期)。
3.平皿背面可以画成小方格。
4.一小格中点1 ul样品。
5.另一小格格点1 ul DNA标准品(我一般用Takara 2000 DNA lander,1 ul相当60 ng) 6.凉干后,紫外灯下根据亮度就可以估测了。
OK,我连接时这么估测浓度,5分钟就要可以知道两片段浓度。
其实连接片段浓度比可以充许在一个范围内,1:5至1:10都可以,所以上述估测方法在这种情况下是行得能的。
2008-05-01
几个月的光阴。
几乎都耗在这上了。
也总结出些门道。
最终转化的成功。
取决于以下几方面:
1.PCR。
PCR条带特异性很重要。
哪怕是亮度低,只要能与其他条带分开,可以确定是目的条带就可。
SMEAR会影响转化的成功率。
引物的设计需要考虑的主要是酶切位点,保护碱基,长度。
当然特异性和DIMER不能忽视。
但前者更为重要。
在这需要注意引物的合成是有一定出错率的。
全世界现在用的技术都如此,出错率理论上各家都一样。
据说是千分之七。
不过我不知道是指一条引物还是一个碱基。
就我而言,应该是一个碱基的出错率。
2.酶切。
酶切的处理方法有两种:抽提、回收。
当然还可以直接加热灭活然后连接,没试过。
抽提的时候,酚仿醇容易降低连接效率,我采用的是用20lymda的移液器+白枪头。
宁可取少一点,也不要沾上它。
BOSS是个技术超群之人。
35的体系可以轻易取到34.
回收的话,比较废材料。
一般抽提能做4-5次的材料,回收一次就用了。
而且相对麻烦。
不过好处是可以定量(连接的时候应片段比质粒数量=10:1),而且看得见,踏实,方便失败分析。
(我发现引物错误就是酶切跑胶发现错误的带。
)
要特别注意的是保护碱基的设置。
保护碱基根据不同酶有不同要求。
我用的XhoI,保护碱基需要3个以上。
我一开始设少了,结果很难连上。
文献说2个保护碱基,20h NEB的XhoI 只能切去25%。
我采取的措施1.是超长时间酶切,适当增加酶量。
效果还可以。
2.构建T 载体,但T载体一般用TAQ来PCR,容易出突变,且耗时长。
3.连接。
说明书虽然说10分钟就能连好。
但是事实上不是的。
据说16度过夜连接是最好的。
4.转化。
很骄傲于我们实验室的10分钟转化大法。
当然我构建质粒时不敢用。
当连接体系加入到感受态细胞时,若体系过大,应补充CaCl2到30mM。
5.验证。
抽提一般长出来十多个属于正常(就我们的感受态),太多则应怀疑是载体未被切开。
验证手段有以下三种:
1.菌落PCR。
挑取单菌落,点于另一标记平板上,再把枪头于含适量水EP管中。
将EP管煮沸10分钟,作模板PCR。
得到与对照相异的相应片段。
2.提质粒。
通常在菌落PCR得到阳性结果后进行。
然而菌落本来少的时候。
直接提质粒可能省事。
质粒可以直接与空载体一起跑胶,比较大小以确定片段是否插入。
3.蛋白小量表达。
预测蛋白大小=将目的片段(到终止子)/3*106/1000+标签大小。
通过转化质粒到表达菌中,小量表达,SDS-PAGE电泳。
可以比较蛋白与预期蛋白SIZE。
4.(我不识数)。
测序。
如果你一切做得很好的话,阳性结果应该接近100%。