无菌检查方法验证告报告2015
无菌验证总结报告

关闭 无菌 缓冲 罐入 口 阀, 自动 CO P3 运行 待机 24 小时。 从培 养基 进无 菌罐 开始 计时
1、使用氮气置换装置会产生不良品
2、高速下生产,可能会产生不良品 2、可能瓶或盖的杀菌不足 2、在下一次测试中对速度的影响再作确认
1、氮气置换装置设计上不易清洁,特别在SIP时灭菌不彻底
1、拆卸氮气置换装置,经清洗液清洗后用PAA浸泡灭菌,再用杀菌釜高压灭菌
无菌测试总结报告
报告人:翁建波
2006年12月27日
报告大纲
◆ sidel无菌线验证流程 ◆ 各步骤测试方法与目的 ◆ 各步骤进展状况 ◆ 问题分析与讨论
◆ 验证流程
> T1准备阶段 > T2灌装测试 (灌水试验) > T3无菌区杀菌测试 ( 钢片试验) > T4整机无菌测试 (分段测试) > T5包材杀菌测试 (挑战测试) > T6商业无菌验证 > T7机械效率测试
×
× 24h后
×
×
×
× ×
2974 8 0.27
2049 0 0.00
120 1 0.83
335 1 0.30
2126 7 0.33
2291 12 0.52
低速下没有不良品,高速下有不良品 1、跟速度相关的因素是瓶的预杀菌和盖的杀菌时间以及瓶内部杀菌时瓶底部PAA的喷射 2、在储盖箱、盖提升机、盖 压力 整列机以及盖滑道上涂抹检测都检出有与变质品类似的芽孢菌 3、因此分析盖杀菌很可能不足 1、对整个储盖箱、盖提升机、盖整列机以及盖滑道用SU626碱性泡沫擦拭清洁,然后用事 百得擦拭抑菌,最后用PAA擦拭和熏蒸 2、调整瓶盖进入盖杀菌通道的方向和改变部分喷嘴的喷射角度
公式分析
♦ ♦ ♦ ♦ ♦ ♦ ♦
中国药典2015版无菌检查法与美、欧、日药典的差异分析与讨论_潘友文

中国药典2015版无菌检查法与美、欧、日药典的差异分析与讨论潘友文(罗氏/基因泰克,美国南旧金山,94080)2016年07月29日摘要目的:分析中国药典2015版无菌检查法与美、欧、日药典无菌检查法的之间的差异性,为评价不同药典中无菌检查法的等效性提供参考。
方法:对无菌检查法的主要实验步骤和参数进行一对一比较,对有差异的步骤和参数进行科学论证和评价。
结果:中国药典2015版无菌检查法与美、欧、日药典无菌检查法的主要参数和步骤是一致的,但中国药典无菌检查法还需要做阳性对照和厌氧需氧菌的额外培养。
并且,中国药典用大肠埃希菌代替美、欧、日药典中的铜绿假单胞菌参与无菌检查法的适用性试验。
结论:各药典的无菌检查法是等效的。
在不影响方法等效性的前提下,中国药典2015版无菌检查法在阳性对照和培养方法上还可以进一步简化。
关键词:无菌检查法;中国药典;美国药典;欧洲药典;日本药典Gap Assessment and Discussion on Sterility Tests in Chinese Pharmacopoeia 2015, United States Pharmacopoeia, European Pharmacopoeia, and Japanese PharmacopoeiaYouwen Pan (Genentech, a Member of Roche, South San Francisco, USA 94080)Abstract Objective:Gap assessment and discussion on the sterility test methods in Chinese Pharmacopoeia 2015 edition (CP2015), United States Pharmacopoeia (USP), European Pharmacopoeia (EP), and Japanese Pharmacopoeia (JP). Method:The test procedures and key parameters in the sterility test methods in different pharmacopoeia were compared step by step and the differences were identified. The identified differences are scientifically evaluated for their impact to the equivalence of the methods. Result:The sterility test method in CP2015 is largely harmonized with that in USP, EP and JP except for a few differences. Positive control and extra incubation bacteria are required in CP2015 only, and Escherichia coli is used in method suitability test in CP2015 while Pseudomonas aeruginosa is used in USP/EP/JP. Conclusion:The Sterility Test Methods in CP2015, USP, EP, and JP are equivalent. The method in CP2015 could be simplified more without compromising the validity, accuracy and reliability of the method.Key words:sterility test;Chinese Pharmacopoeia 2015;United States Pharmacopoeia;European Pharmacopoeia; Japanese Pharmacopoeia无菌检查法是用于检查药典规定的无菌物品是否被微生物污染的检测方法。
无菌检验方法验证

YOUR SITE HERE
LOGO
无菌检验阳性结果调查
实验室调查:一个合适的调查程序和记录是必要的
YOUR SITE HERE
LOGO
无菌检验阳性结果调查
若产品不合格,必须通过该批生产过程回顾找出污染 原因和污染源 –人 – 物料 – 环境 – 设备 – 方法和工艺
YOUR SITE HERE
LOGO
方法二:用实验确定是否具有抑菌性的方法 在规定量的供试液中加入实验用微生物10~100个,
如微生物生长良好,即说明供试品对该种微生物无抑 菌活性。如已用药典规定的各试验菌试验,结果均能 生长良好,说明供试品无抑菌性,可用常规方法检验 之。将这些试验记录在案——就是证明该供试品无抑 菌性的验证试验部分;如加入的微生物不生长或生长 缓慢,说明供试品有抑菌性,将这些试验记录在案— —就是证明该供试品有抑菌性的验证试验部分;如加 入的微生物中有某一种菌最不能生长或生长最缓慢, 这一种菌就是该样品的敏感菌。
LOGO
验证试验和对验证结果技术评价的判断为:如平行3 次的试验中,各管微生物均生长良好,说明供试品 已消除了抑菌活性。可以确定按该方法进行该品种 的无菌检查;如有供试品管的微生物生长微弱、缓 慢或不生长,说明供试品仍有抑菌活性,应考虑进 一步增加冲洗量或中和剂的用量以消除抑菌活性, 并重新验证,直至验证证明已充分消除或有效降低 抑菌活性。
条件 无菌检查应在环境洁净度10000级下的局部洁净 度100级的单向流空气区域内或隔离系统中进行, 其全过程应严格遵守无菌操作,防止微生物污 染,防止污染的措施不得影响供试品中微生物 的检出。
无菌检查方法的验证

无菌检查方法的验证无菌检查方法是为了检查药典要求无菌的制剂及其他制品是否无菌而建立的试验方法,是作为无菌产品批放行的重要依据及药监部门对无菌产品质量监管的一个重要项目。
因此如何确保无菌检查方法的准确可靠至关重要,而检查方法的验证是保证检查结果的公正、科学和准确的基础,因此各个主要国家的GMP或药典都对无菌检查方法的验证提出了严格的要求,2010版中国药典对分析方法验证和检查的要求也有大幅度的提高,同时也是GMP检查中检查的重点和容易发现问题的区域。
验证要求与方法无菌检查方法验证一般分为前验证和再验证两种。
前验证,也称预验证,指在无菌分析方法正式使用前,按照预定验证方案进行的验证。
如果没有充分的理由,任何检查方法必须进行前验证。
再验证,指某一检查方法经过验证并在使用一段时间后进行的,旨在证实已验证状态没有发生飘移而进行的重新验证及对检查方法进行修订、改变时进行的验证.通常一个无菌产品的检查流程为:首先基于产品的剂型、溶解度等性质,按照药典的要求确定是否需要进行前处理;然后根据产品的特性是否有抑菌性,确定是否需要增加去除产品抑菌性的方法;最后验证整个检查方法中用到的一切及试验过程中的每一个环节包括样品的预处理方式、检查过程、培养条件等均不影响样品中微生物的生长。
这里,验证的重点环节包括:前处理方法直接影响后续步骤的效果和重现性,应是验证的重点.供试品中抑菌活性的去除是当前验证工作的重点,尤其强调应充分验证供试品本身对微生物生长的影响.具体的验证方法如下:菌种的选择无菌检查方法验证中通常选择以下6种试验中常用的控制菌的标准菌株,它们分别代表不同类型的菌种:枯草芽孢杆菌[CMCC(B)63 501]代表药品中常见的污染菌——芽孢杆菌、金黄色葡萄球菌 [CMCC(B)26 003]代表革兰阳性菌、生孢梭菌 [CMCC(B)64 941]代表厌氧菌、大肠埃希菌[CMCC(B)44 102]代表革兰阴性菌、白色念珠菌[CMCC(F)98 001]代表酵母菌、黑曲霉菌[CMCC(F)98 003]代表霉菌。
直接接种无菌检查法验证方案及报告

贵州金玖生物有限责任公司验证方案及报告验证方案名称:直接接种无菌检查方法验证验证方案编号:验证完成日期:有效期:验证方案申请人: 日期: 年月日验证方案审核人: 日期: 年月日验证方案审批人: 日期: 年月日1 概述无菌检查法是为了检查药典要求无菌的制剂及其他制品是否无菌而建立的检查法,是作为批准无菌产品放行的检验或监督部门对无菌产品质量监督中的一个重要项目。
它是根据用于实验的培养基中是否有微生物生长来判定样品的无菌性,液体培养基变浑浊一般表明样品受微生物的污染。
基于微生物污染的不均匀性,使无菌检查法结果的可信度受许多因素制约,如抑菌因素、检查法、检验量、检查用的培养基质量、操作环境、无菌技术等。
检验方法的验证是现代质量保证体系中关系到质控技术、方法、手段的科学性、准确性的重要组成部分,是保证检验结果的公正、科学、准确的基础。
2 验证目的对本公司所采用的直接接种无菌检查法进行分析验证,以证明所采用的方法适合于本公司产品的无菌检查。
3 实验原理直接接种法是通过加入一定的抑菌中和剂,以方便而快捷地中和抑菌剂,消除抑菌作用,从而消除抑菌作用对无菌检查的影响。
本实验通过设计对照试验对直接接种法所加入的抑菌剂的中和效果进行验证,从而得出直接接种法无菌检验的有效性。
4 验证范围实用于本公司所采用的直接接种法进行的无菌检验过程验证。
56 职责7 验证内容建立样品组、对照组及菌种活性检查组,接种菌株到指定培养基,培养24~72小时,比较观察菌落的生长状况,得出结论。
8 验证指示物本实验所用菌种为购于贵州食品药品监督管理局标准菌株:大肠埃希菌、金黄色葡萄球菌、白色念球菌及生孢梭菌。
9 实验过程(1)培养基的配制用于大肠埃希菌、金黄色葡萄球菌的营养琼脂培养基用于白色念球菌的改良马丁培养基用于生孢梭菌的流体硫乙醇酸盐培养基(2)供试品的制备术泰舒TM生物多糖冲洗胶液供试品:直接取成品10ml,加pH7.0氯化钠—蛋白胨缓冲液至100ml,混匀,作为供试品。
无菌检查方法验证告报告2015

无菌检查方法验证告报告2015
无菌检查方法验证报告2015
1. 目的
本报告旨在介绍无菌检查方法验证报告2015,该报告是根据企业生产的产品特点,结合国内外相关法规要求,制定的一种无菌检查方法。
2. 范围
本报告适用于企业生产的一系列产品,这些产品都具有相同的特点,需要进行无菌检查。
3. 无菌检查方法验证过程
在本报告中,我们介绍了如何对无菌检查方法进行验证的过程。
首先,我们需要对试验样品进行灭菌处理,然后将其放入无菌环境中进行试验。
在此过程中,我们需要严格控制各种影响因素,以确保试验结果的可靠性。
最后,我们需要对试验结果进行分析和总结,以确定该无菌检查方法的可行性。
4. 结果与分析
在本报告中,我们介绍了无菌检查方法验证的结果。
通过试验,我们发现该无菌检查方法可以有效地检测出产品中的微生物,并且试验结果稳定可靠。
因此,我们可以认为该无菌检查方法可以用于企业的生产过程中。
5. 结论
综上所述,本报告介绍了一种有效的无菌检查方法验证报告2015。
该方法可以有效地检测出产品中的微生物,并且具有较高的可靠性。
2015年版中国药典微生物限度检查方法验证方案

2015年版中国药典微生物限度检查方法验证方案佛山市华普生药业有限公司文件编码:TS-VP-4201-00页数:1/56 人工牛黄甲哨唑胶囊微生物限度检查方法验证方案人工牛黄甲硝唑胶囊微生物限度检查方法验证方案起草部门:QC组签名:日期:审核部门:QC组签名:日期:部门:质量部签名:日期:批准质量负责人签名:日期:质量部颁发本文件根据需要应分发于以下部门:01 质量部下表用于记录修订/变更主要内容及历史。
佛山市华普生药业有限公司文件编码:TS-VP-4201-00页数:2/56 人工牛黄甲哨唑胶囊微生物限度检查方法验证方案文件编号修订原因修订日期TS-VP-4201-00 按GMP(2010年修订版)要求新制定2015.10.22佛山市华普生药业有限公司文件编码:TS-VP-4201-00页数:3/56 人工牛黄甲哨唑胶囊微生物限度检查方法验证方案目录1. 概述2. 验证目的和范围3. 组织及职责4. 验证进度计划表5. 验证所需要的仪器设备及相关文件的确认6. 验证所需要的菌种、培养基、检验样品的确认7.验证项目和验证方法7.1试验菌株7.2需氧菌总数检查、霉菌和酵母菌总数检查方法验证的菌液制备7.3需氧菌总数检查、霉菌和酵母菌总数检查方法验证--常规倾注平皿法7.4需氧菌总数检查方法验证—离心沉淀-薄膜过滤法7.5控制菌检查方法验证—离心沉淀-薄膜过滤法8.偏差与漏项控制佛山市华普生药业有限公司文件编码:TS-VP-4201-00页数:4/56 人工牛黄甲哨唑胶囊微生物限度检查方法验证方案9.验证报告会审佛山市华普生药业有限公司文件编码:TS-VP-4201-00页数:5/56 人工牛黄甲哨唑胶囊微生物限度检查方法验证方案1. 概述我公司生产品种人工牛黄甲硝唑胶囊,产品微生物限度检查项目为需氧菌总数、霉菌和酵母菌总数、以及大肠埃希菌检查和沙门菌检查。
参照《中国药典》2015版四部附录1105:微生物计数法,以及1106:控制菌检查法的规定,本公司对该产品的微生物限度检查方法予以验证。
2015年版药典 无菌检查法

灵敏度检査 菌 种 培养基灵敏度检查所用的菌株传代次数不得超过 5 代(从 菌 种 保 存 中 心 获 得 的 干 燥 菌 种 为 第 0 代 ),并 采 用 适 宜的菌种保藏技术进行保存,以保证试验 菌株的 生物 学特 性。 金 黄 色 葡 萄 球 菌 (Staphylococcus aureus ) C C M C C (B) 26 003〕 铜 绿 假 单 胞 菌 (Pseudomonas aeruginosa ) C C M C C ( B) 10 104〕 枯 草 芽 孢 杆 菌 仏 5)〔C M C C ( B ) 6 3 501〕 生 抱 梭 菌 sporogenes) C C M C C ( B ) 64 941〕 白 色 念 珠 菌 albicans) C C M C C ( F ) 98 001〕 黑 曲 霉 n ig e r) C C M C C ( F ) 98 003〕 苗 液 制 备 接 种 金 黄 色 葡 萄 球 菌 、铜 绿 假 单 胞 菌 、枯草 芽孢杆菌的新鲜培养物至胰酪大豆胨液体培养基中或胰酪大 豆 胨 琼 脂 培 养 基 上 ,接 种 生 孢 梭 菌 的 新 鲜 培 养 物 至 硫 乙 醇 酸 盐 流 体 培 养 基 中 ,30〜 35°C培 养 18〜 2 4 小 时 ;接 种 白 色 念 珠菌的新鲜培养物至沙氏葡萄糖液体培养基中或沙氏葡萄糖 琼 脂 培 养 基 上 ,20〜 25°C培 养 24〜 4 8 小 时 ,上 述 培 养 物 用 PH7. 0 无 菌 氣 化 钠 -蛋 白 胨 缓 冲 液 或 0. 9% 无 菌 氣 化 钠 溶 液 制 成 每 l m l 含 菌 数 小 于 lOOcfu( 菌 落 形 成 单 位 )的 菌 悬 液 。接 种黑曲霉的新鲜培养物至沙氏葡萄糖琼脂斜面培养基上, 20〜25*C培 养 5〜7 天 ,加 人 3〜 5 m l含 a 0 5 % (m l/m l) 聚山 梨 酯 8 0 的 pH7. 0 无 菌 氣 化 钠 -蛋 白 胨 缓 冲 液 或 0. 9 % 无 菌 氣 化 钠 溶 液 ,将 孢 子 洗 脱 。然 后 ,采 用 适 宜 的 方 法 吸 出 孢 子 悬 液 至 无 菌 试 管 内 ,用 含 0.05% ( m l/m l) 聚 山 梨 醋 8 0 的 pH7. 0 无 菌 氣 化 钠 -蛋 白 胨 缓 冲 液 或 0. 9 % 无 菌 氣 化 钠 溶 液 制 成 每 l m l 含 孢 子 数 小 于 lOOcfu的 孢 子 悬 液 。 菌 悬 液 若 在 室 温 下 放 置 ,应 在 2 小 时 内 使 用 ;若 保 存 在 2〜8°C可 在 2 4 小 时 内 使 用 。黑 曲 霉 孢 子 悬 液 可 保 存 在 2 〜 8°C,在 验 证 过 的 贮 存 期 内 使 用 。 培 养 基 接 种 取 每 管 装 量 为 12ml的硫乙醇酸盐流体培 养 基 7 支 ,分 别 接 种 小 于 lO O c fu 的 金 黄 色 葡 萄 球 菌 、铜 绿 假 单胞菌、生 孢 梭 菌 各 2 支 ,另 1 支 不 接 种 作 为 空 白 对 照 ,培 养 3 天 ;取 每 管 装 量 为 9 m l的 胰 酪 大 豆 胨 液 体 培 养 基 7 支 ,分 别接种小 于lOOcfu的 枯 草 芽 孢 杆 菌 、白 色 念 珠 菌 、黑 曲 霉 各 2 支,另 1 支不接种作为空白对照,培 养 5 天 。逐 日观 察结果。 结 果 判 定 空 白 对 照 管 应 无 菌 生 长 ,若 加 菌 的 培 养 基 管 均 生 长 良 好 ,判 该 培 养 基 的 灵 敏 度 检 查 符 合 规 定 。
无菌检查方法验证报告

无菌检查方法验证报告目录1. 目的 (3)2. 适用范围 (3)3. 接受标准 (3)4. 抽样计划 (3)5. 设备、试剂和菌种信息 (3)6. 试验步骤 (4)7. 供试品检查 (6)8. 结论 (6)9. 再验证周期 (6)10. 附件清单 (6)1.目的支据中国药典2020版无菌检查法的要求,对产品的无菌检查法进行验证,以确定一种无菌检查的方法能有效的消除该产品对微生物的抑制作用,从而证实该方法的有效性和实用性。
2.适用范围适用于本司产品的无菌检查。
3.接受标准与阳性对照比较,含供试液各容器中的试验菌(金黄色葡萄球菌、大肠埃希菌、生孢梭菌、白色念珠菌、枯草芽孢杆菌和黑曲霉)均生长良好,则可照此检查方法和检查条件进行供试品的无菌检查。
如含供试品的任一容器内的试验菌生长微弱、缓慢或不生长,则说明供试品的该检验量在该检验条件下有抑菌作用,需进一步改进检验条件再进行方法验证试验。
4.抽样计划随机抽取灭菌后的样品24份。
具体信息见表一:表一样品信息确认人/日期:复核人/日期:5.设备、试剂和菌种信息5.1设备信息详见表二表二设备信息确认人/日期:复核人/日期:5.2试剂信息详见表三:表三试剂信息确认人/日期:复核人/日期:5.3菌种信息详见表四:表四菌种信息确认人/日期:复核人/日期:5.3.1菌液的制备用接种环取金黄色葡萄球菌、大肠埃希菌、枯草芽孢杆菌的第三代培养物用0.9%无菌氯化钠溶液制成浓度≤100cfu/mL的菌悬液;用接种环取生孢梭菌的第三代培养物用0.9%无菌氯化钠溶液制成浓度≤100cfu/mL的菌悬液;用接种环取白色念珠菌的第四代培养物用0.9%无菌氯化钠溶液制成浓度≤100cfu/mL的菌悬液;用接种环取黑曲霉的第三代培养物,向培养物中加入10mL0.9%无菌氯化钠溶液,将孢子洗脱,转移孢子悬液至无菌试管内,用0.9%无菌氯化钠溶液制成浓度≤100cfu/mL的孢子悬液。
以上操作均在微生物检验室阳性菌室的生物安全柜中进行。
无菌检验方法验证报告

编码:无菌检验方法验证报告药业有限公司1. 概述:无菌检查法系用于检查药典要求无菌的药品、原料、辅料及其他品种是否无菌的一种方法。
本公司的无菌检查只是针对于灭菌注射用水的无菌检查。
灭菌注射用水灭菌工艺采用121C 30min过度杀菌法,按照2005年版《中国药典》的规定,注射剂的无菌检查应采用薄膜过滤法。
制定的《无菌检查法标准操作规程》(SOP4.14.038-B)应经过验证后,才能批准使用。
2. 验证目的:验证所采用的方法和条件是否适合于供试品的无菌检查。
即确认供试品在该检验量、该检验条件下无抑菌活性或其抑菌活性以被充分消除到可以忽略不计。
3. 验证范围:适用于灭菌注射用水无菌检查法的验证。
4. 验证人员及职责质量部负责该验证方案的起草及组织实施;验证小组负责验证方案的审批;质量部参与验证的实施及监督。
5. 文件准备和培训检查验证所需的各类文件资料,应齐全;相关的文件草案是否已具备。
6. 验证条件6.1. 无菌检查室空气净化系统已经过验证并合格,仪器安装完成;仪表量器经过校验合格,且在有效期内。
6.2. 供试品:3批,批号:批号:批号:6.3. 培养基及试剂:试剂试液:氯化钠:临用前配成0.9%浓度的溶液,配制记录见附件1 冲洗液:0.1%蛋白胨水溶液,配制记录见附件2。
O Q6・3・2・培养硫乙醇酸盐流体培养基生产厂家:批号: 改良马丁培养基生产厂家:批号: 营养肉汤培养基生产厂家:批号: 改良马丁琼脂培养基生产厂家:批号: 蛋白胨生产厂家:批号: 培养基配制记录见附件3。
6.4.验证用菌株:金黄色葡萄球菌【CMCC ( B) 26003】铜绿假单胞菌【CMCC (B) 10104】枯草芽抱杆菌【CMCC (B) 63501】生抱梭菌【CMCC ( B) 64941】白色念珠菌【CMCC (F) 98001】黑曲霉【CMCC (F) 98003】购自:各验证用菌种传代记录见附件4。
6.5.无菌检验仪器及相关设备:压力蒸汽灭菌器型号:生产厂家:校验日期:有效期:生化培养箱(细菌培养)型号:生产厂家:校验日期:有效期:生化培养箱(霉菌培养)型号:生产厂家:校验日期:有效期:7. 验证内容:7.1. 培养基无菌性检查:每批培养基随机取不少于5支,培养14天,应无菌生长试验菌液的制备7.2.1. 接种金黄色葡萄球菌、铜绿假单胞菌、枯草芽抱杆菌的新鲜培养物至10ml营养肉汤培养基中,置30- 35C培养18-24小时,分别取上述培养物1ml加9ml 0.9%的无菌氯化钠溶液10倍递增稀释制成每1ml含菌数小于100 CFU的菌悬液。
纯化水需氧菌总数计数方法的验证2015

目录一、引言1、概述2、验证的目的二、验证方法合格标准三、主要材料确认1、仪器设备确认2、辅助材料确认3、人员确认4、商品培养基及灭菌培养基确认5、菌种及菌液的制备四、样品制备1、稀释液2、供试液制备3、操作方法五、方法验证1、需氧菌总数的计数方法验证2、计算菌回收率公式六、验证结果及评价七、再验证周期八、验证结论批准一、引言1、概述:根据《中国药典》2015年版三部通则“1105 非无菌产品微生物限度检查:微生物计数法”(以下简称《药典》)的要求,对内控标准中有“微生物限度”检查项的产品进行该项验证。
根据《中国药典》2015年版三部纯化水进行方法学确认。
2、验证目的:确保药品微生物限度检查实验的准确性,在建立药品的微生物限度检查法时,应进行需氧菌总数计数方法的验证,以确认所采用的方法适合于该样品的需氧菌总数的测定。
二、验证方法合格标准1.前提条件:培养基适用性实验应符合规定,即:被检R2A培养基上菌落平均数与对照培养基上菌落平均数比值应为0.5~2,且菌落形态大小应与对照培养基上的菌落一致。
2.计数方法适用性试验:在3次独立的平行试验中,若试验组的菌回收率(试验组的平均菌落数减去供试品对照组的平均菌落数的值与菌液对照组的平均菌落数的比值)均在0.5~2范围内,照该供试液制备方法和计数法测定供试品的需氧菌总数。
三、主要材料确认1、仪器、设备确认检查结果:检查人:复核人:日期:2、辅助材料确认2.1移液器、接种环、酒精灯、记号笔、酒精棉球、试管架、试管、玻璃平皿、止血钳等2.2需灭菌物品见《微生物限度检查法验证记录》2.3一次性无菌用具见下表检查结果:检查人:复核人:日期:3、人员确认化验员经设备操作、微生物检验技术和实验室生物安全等方面的培训,考核合格后上岗。
4、商品培养基及灭菌培养基4.1 所用培养基均不得有结块、吸潮、异味、颜色发生改变等现象。
培养基检查结果,如下检查结果:检查人:复核人:日期:4.2已灭菌培养基的检查结果及结论见《微生物限度检查法验证记录》。
无菌药品GMP检查指南 2015

无菌药品GMP检查指南2015年10月目录一、 .............................................................. 目的5本指南的主要目的是为检查员在实施无菌药品生产企业检查时提供指导。
检查组应参照本指南的要求检查无菌药品生产质量管理情况,评价企业无菌保证的能力,以确定企业是否符合《药品生产质量管理规范(2010年修订)》(以下简称GMP)的要求。
5二、 ................................................ 适用范围及检查依据5本指南适用于无菌药品的GMP检查,包括无菌制剂生产全过程和无菌原料药的灭菌和无菌生产过程。
(5)无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,通常包括大容量注射剂、小容量注射剂、粉针剂、冻干粉针剂、眼用制剂、耳用制剂、埋植剂、供雾化器用的液体制剂、冲洗剂、外用制剂、无菌原料药等。
无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
本指南适用于对上述不同生产工艺及不同类型的无菌药品的检查。
5检查过程中,检查员应依据《药品生产质量管理规范(2010年修订)》及其附录来确定检查缺陷所涉及的条款。
(5)三、 .............................................. 无菌药品生产工艺概述5无菌药品按生产工艺可分为最终灭菌产品和非最终灭菌产品两类,部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
无菌药品、直接接触药品的包装材料应尽可能采用热力灭菌方式进行最终灭菌。
采用湿热灭菌方法进行最终灭菌的,通常标准灭菌时间F0值应当大于8分钟,流通蒸汽处理不属于最终灭菌。
最终灭菌产品中的微生物存活概率(即无菌保证水平,SAL)不得高于10-6。
(5)(一) .................................................. 最终灭菌工艺6产品的无菌保证水平不能仅依赖最终灭菌。
灭菌注射用水无菌检查方法适用性-报告

试验结果评价:
评价人:
日期:
年月日
6.3.3.3 第三次试验结果
试验时间: 年 月 日
培养温度:硫乙醇酸盐流体培养基置
℃;胰酪大豆胨液体培养基置
℃。
试验结果见表 4。 表 4 无菌检查方法适用性第三次试验结果
6
试验菌
培养对象 培养时间
大肠埃希菌菌
金黄色 葡萄球菌 生孢梭菌
试验管 对照管 试验管 对照管 试验管 对照管 阴性对照
003〕,上述标准菌株均源自
,工作用菌株为第三
代。
6.2.2、菌液制备:
6.2.2.1 接种金黄色葡萄球菌的新鲜培养物至胰酪大豆胨液体培养基(TSB 培
养基)中,30~35℃培养 18~24h 培养后,用 0.9%无菌氯化钠溶液制成每 1ml 含
菌数小于 100cfu 的混悬液。
6.2.2.2 接种生孢梭菌的新鲜培养物至硫乙醇酸盐流体培养基(+琼脂)中,
30~35℃培养 18~24h 培养后,用 0.9%无菌氯化钠溶液制成每 1ml 含菌数小于
100cfu 的混悬液。
6.2.2.3 接种大肠埃希菌的新鲜培养物至胰酪大豆胨液体培养基(TSB 培养基)
中,30~35℃培养 18~24h 培养后,用 0.9%无菌氯化钠溶液制成每 1ml 含菌数小
于 100cfu 的混悬液。
试验菌
培养对象 培养时间
硫乙醇酸盐流体培养基观察结果
1
2
3
4
5
硫乙醇酸盐流体培养基( - ) 胰酪大豆胨液体培养基观察结果
1
2
3
4
5
枯草芽孢杆菌
试验管 对照管
白色念珠菌
试验管 对照管
无菌检验技术—无菌检查方法验证

• 3.3无菌检查的范围 • 凡进入人体无菌部位(人体的血液循环பைடு நூலகம்统、肌肉、皮下组织)或接触创
伤、溃疡面等部位而发生作用的制品或要求无菌的材料、无菌器具均应 进行无菌检查。对于规定灭菌的药物,包括注射剂及用于体腔、严重烧 伤、溃疡及眼科用药等,必须严格无菌,即在规定检验量的供检品中不 得检出活微生物。
任务3 无菌检验方法验证 知识点3 无菌检查方法
• 3 无菌检查方法 • 3.1无菌检查法概念 • 无菌检查法是指检查《中国药典》要求应无菌的药品、医疗器具、原料、 辅料及其他要求无菌的品种是否染有活菌的一种方法(供试品符合无菌检查 法的规定,仅进表明了供试品在该检查条件下未发现被微生物污染)。 • 为了保证药品的卫生质量,保证药品在临床上的安全使用和保障人民的健 康,《中国药典》规定:注射用药、灭菌的外用制剂等均需做无菌检查。
• 的制剂。心脏瓣膜以及固定金属板和有机器材等。
• (3)冲洗剂,即用于冲洗开放性伤口或腔体的冲洗剂。
• (4)眼用制剂。
• (5)用于烧伤、创伤或溃疡的制剂。包括:用于烧伤或严重创伤的局部用 散剂、凝胶剂、软膏剂、乳膏剂;用于烧伤、创伤或溃疡的气雾剂和喷 雾剂等。
• (6)用于手术、耳部伤口或耳膜穿孔的滴耳剂或洗耳剂。 • (7)用于手术或创伤的鼻用制剂。 • (8)用于止血并可被组织吸收的制剂。如明胶发泡剂、凝血酶等,用于止
• 3.2验证目的 • 确认所用的无菌检查方法适用于“所有无菌产品”的无菌检查。 • 确保检查结果的准确性、可靠性、准确性和重现性以及检查方法的完整 性。 • 通过对不同批次产品无菌检查的比较,对医用即无菌产品的生产全过程 进行质量监控。 • 3.3验证范围 • “所有无菌产品”的无菌检查。
• 3.4验证条件 • 3.4.1验证用菌株 • 金黄色葡萄球菌 • 菌种传代次数铜绿假单胞菌 • 枯草芽孢杆菌 • 生孢梭菌 • 白色念珠菌
无菌、微生物限度检查及方法验证

此法为实验室工作用菌种的最常用的保藏方法,仅能用于工作用菌种的短期保藏。
8
琼脂斜面低温保藏法保藏法(厂家常用)
保藏芽胞液对
产芽胞的微生物宜制成芽胞菌悬液后再保藏, 无菌操作,取1ml孢子液接入茄形瓶或培养皿内,轻轻转动使菌液均匀分布于培养基表面。 将培养皿在适当的温度下培养。一般需要7天或更长时间 在层流台下,取3~5ml氯化钠磷酸盐缓冲液(pH7.0)加入培养皿内 用无菌玻璃L棒轻轻刮下菌苔,注意不要划破培养基。 用无菌注射器或无菌吸管吸取菌悬液,收集于试管中。 将收集的芽胞液放在80~90℃水浴中加热15分钟,冷却。 做好标记,放至2~8℃冰箱中保藏。原始芽胞液可保存1年。 每次使用前用平皿法重新标定其浓度。
微生物实验室的质量管理
设施—洁净室、净化工作台、生物安全柜。 设备—高压蒸汽灭菌器、电热恒温干烤箱、培养箱、冰箱。 实验仪器—全封闭薄膜过滤装置、电动匀浆仪、电热恒温水浴箱、离心机、显微镜,及刻度吸管、试管、三角烧瓶、培养皿等。 建立主要设施、设备、仪器的明细记录,内容包括名称、型号、生产厂家、购买日期。 质量保证档案:购买发票、安装验收或开箱验收记录、调试记录、计量检定或运行校验合格证、指定专人负责、制订标准操作规程(SOP)、建立使用登记册,维修、保养记录、改造或更新记录、直至报废记录。
记录温湿度是否在规定的范围内,每次实验时,对操作室和层流台做微生物沉降菌落计数并记录;如发现问题应找原因,及时报修和报告并将报修原因和结果记录归档。如遇停电,应立即停止实验,重新进入无菌室前,至少开启机房运转1小时以上。
实验室使用管理
微生物实验室的质量管理
平时实验室内应尽可能减少人员的走动或活动,通向洁净室的门要关闭或安装自动闭门器使其保持关闭状态。
枯草芽孢杆菌 [CMCC(B)63501] 金黄色葡萄球菌 [CMCC(B)26003] 生孢梭菌 [CMCC(B)64941] 铜绿假单胞菌 [CMCC(B)10104] 大肠埃希菌 [CMCC(B)44102] 乙型副伤寒沙门菌 [CMCC(B)50094] 白色念珠菌 [CMCC(B)98001] 黑曲霉 [CMCC(B)9 003]
注射液无菌检查的方法学验证方案

注射液无菌检查的方法学验证方案注射液的无菌性是医疗领域中最为重要的质量指标之一。
为了确保注射液的无菌性,需要进行方法学验证方案。
本文将介绍一种常见的注射液无菌检查方法学验证方案,确保验证结果准确可靠。
一、验证目的本验证方案旨在验证注射液无菌性检查的方法学准确性和可靠性。
通过验证,可以确保注射液无菌性检查的结果符合预期,并为其他类似检查提供参考。
二、验证对象本验证方案适用于各类注射液无菌性检查方法,包括但不限于常规培养法、膜过滤法、试管法等。
三、验证步骤1. 制定验证计划:明确验证的目标、方法和时间安排等。
2. 准备样品和试剂:收集需要验证的样品和所需试剂,并确认其质量符合要求。
3. 合理布局实验室:确保实验室环境符合无菌实验要求,避免干扰因素的存在。
4. 实验操作:a) 样品准备:按照标准操作程序,制备样品,确保操作无菌。
b) 检测方法操作:按照所选的检测方法,依次进行实验,确保操作无误。
c) 平行实验:为了验证结果的可重复性,进行平行实验,确保结果一致。
5. 结果分析:根据实验结果进行数据统计和分析,得出结论。
6. 结果确认:将验证结果与预期目标进行比较,确认验证是否合格。
7. 编写验证报告:将整个验证过程、操作方法和结果总结编写成验证报告。
四、验证要求1. 严格遵守无菌技术操作规范。
2. 注射液样品的选择应具有代表性和典型性。
3. 实验室环境应符合无菌要求,避免外界干扰。
4. 检测方法的选择应根据实际情况和需求进行。
5. 实验操作过程要规范、准确,确保每个步骤符合方法要求。
6. 结果分析要科学、客观,数据统计要准确无误。
7. 结果确认应根据验证目标和标准进行判断。
8. 验证报告要详细、完整,包括验证目的、方法、结果和结论等。
五、验证结果分析根据实验结果的分析,可以判断注射液无菌性检查方法是否准确可靠。
如果验证结果符合要求并且与预期目标一致,则说明所选的检查方法具有可靠性和精准性。
如果验证结果与预期目标不一致,则需要进一步分析原因,寻找解决方案。
4无菌检验方法验证

YOUR SITE HERE
LOGO
无菌检验方法验证
1.ONE
开展验证的思路和程序
首先了解供试品是否具有抑菌性,抑什么菌
2.TWO
考虑和选择出适当的方法已消除或减低供试品的抑菌性
3.three 4.four
用实验证明你所用的方法已消除抑菌性——能使人工加 入的各种菌生长良好。
将所作实验记录在案——即为确认该供试品在该检验量
条件 无菌检查应在环境洁净度10000级下的局部洁净 度100级的单向流空气区域内或隔离系统中进行, 其全过程应严格遵守无菌操作,防止微生物污 染,防止污染的措施不得影响供试品中微生物 的检出。
YOUR SITE HERE
LOGO
无菌检验方法验证
方法验证试验 当建立产品的无菌检查方法时,应进行
六、验证结论。
七、【检查】项下【无菌】或【微生物限度】具体方法确定。
八、实验员签名、验证报告批准人签名。
YOUR SITE HERE
LOGO
无菌检验阳性结果调查
无菌检查试验所用的设备及环境的微生物监控结果 不符合无菌检查法要求
回顾无菌测试过程,发现有可能引起微生物污染的 因素
阴性对照有菌生长
稀释:—— 验证降低供试品相对抑菌浓度的有效稀释 倍数。
离心:—— 验证所用转速、时间和微生物的分YO布UR情SIT况E H。ERE
LOGO
3、确定供试品是否有抑菌性、抑什么菌 方法一: 可通过查阅临床药物实验手册、文献资料 (特别是杂志“Drug”,每一种新药上市,都有专 门的综述)、申报资料中的药效学资料,关注其对不 同细菌的MIC情况、参考已经验证过的品种、抗菌 药物可根据抗菌谱确定其敏感菌、中成药如果抗菌谱 不清楚,至少需选择一株细菌和一株真菌来试验等方 法。也可直接通过验证实验确定之。
无菌试验分析报告范文

无菌试验分析报告范文一、实验目的无菌试验是为了确定某一物品是否符合无菌条件,即是否存在微生物污染。
本次实验旨在验证样品是否符合无菌要求,以保证其在医药行业的安全使用。
二、实验原理实验基于无菌技术,通过将待检样品与无菌培养基接触培养,观察是否有生长菌落出现来判断样品是否被微生物污染。
实验中使用的培养基为营养丰富的琼脂,可提供微生物生长所需营养物质。
三、实验过程1. 准备工作:消毒台面,戴上无菌手套,燃烧酒精灯。
2. 取样检测:使用无菌工具,将待检样品取出。
3. 涂抹法:将待检样品均匀涂抹于琼脂培养基表面,避免厚度不均。
4. 培养:将涂抹好的琼脂培养基培养皿放入恒温培养箱,以适宜的温度和时间进行培养。
5. 结果观察:培养结束后,观察培养皿上是否有生长菌落。
四、实验数据及结果分析经过培养结束后观察发现,检测样品所涂抹的琼脂培养基表面上无任何生长菌落的出现。
根据无菌试验的原理,样品没有出现菌落生长,说明未被微生物污染,符合无菌要求。
五、实验结论经过无菌试验,检测样品未出现菌落生长,符合无菌条件,可放心使用。
六、实验中遇到的问题及解决方法在实验过程中,由于琼脂培养基的涂抹量不均匀,导致实验结果可能不准确。
应注意仔细均匀涂抹样品,以确保实验结果的准确性。
七、改进方案为提高实验效果,建议在培养过程中,加入抗生素等抑菌剂,以防止培养过程中的外部微生物污染。
八、安全注意事项1. 在操作过程中戴上无菌手套,保持台面清洁。
2. 使用酒精灯时要小心火源,以免发生意外。
3. 处理样品时需注意避免扩散,以免造成污染。
结语通过本次实验,我们验证了样品的无菌质量,结果表明样品符合无菌要求,可安全使用。
无菌试验是确保产品和实验室环境安全的重要步骤,能够有效预防微生物污染,保证产品质量。
在今后的工作中,我们将进一步完善实验方法,提高实验的准确性和可靠性,为医药行业的无菌质量控制提供更高水平的支持。
中国药典2015版无菌检查方法适用性试验(直接接种法)
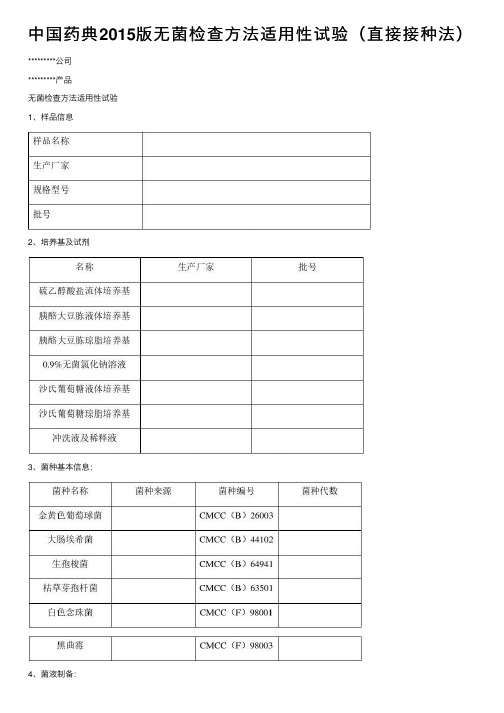
中国药典2015版⽆菌检查⽅法适⽤性试验(直接接种法)*********公司*********产品⽆菌检查⽅法适⽤性试验1、样品信息2、培养基及试剂3、菌种基本信息:4、菌液制备:2.1接种⾦黄⾊葡萄球菌、⼤肠埃希菌、枯草芽孢杆菌的新鲜培养物⾄胰酪⼤⾖胨培养基中或胰酪⼤⾖胨脂培养基上,30~35℃培养18~24⼩时,培养物⽤0.9%⽆菌氯化钠溶液制成每1ml含菌数⼩于100cfu的菌悬液。
2.2接种⽣孢梭菌的新鲜培养物⾄硫⼄醇酸盐流体培养基中,30~35℃培养18~24⼩时,培养物⽤0.9%⽆菌氯化钠溶液制成每1ml含菌数⼩于100cfu的菌悬液。
2.3接种⽩⾊念珠菌的新鲜培养物⾄沙⽒葡萄糖液体培养基或沙⽒葡萄糖琼脂斜⾯培养基上,20~25℃培养24~48⼩时,培养物⽤0.9%⽆菌氯化钠溶液制成每1ml 含菌数⼩于100cfu的菌悬液。
2.4接种⿊曲霉菌的新鲜培养物接种⾄沙⽒葡萄糖琼脂斜⾯培养基上,20~25℃培养5~7天,加⼊3~5ml含0.05%(ml/ml)聚⼭梨酯80的0.9%⽆菌氯化钠溶液,将孢⼦洗脱。
然后,采⽤适宜的⽅法吸出孢⼦悬液⾄⽆菌试管内,⽤含0.05%(ml/ml)聚⼭梨酯80的0.9%⽆菌氯化钠溶液制成每1ml含菌数⼩于100cfu的菌孢⼦悬液。
6、⽆菌检查直接接种法⽅法适⽤性试验6.1供试品制备:描述供试品制备过程(主要为每个样品的取样部位及⽤量)。
6.2试验组:取装量为 ml硫⼄醇酸盐流体培养基,接种规定的供试品量(同6.1),并接种⼩于100cfu的⾦黄⾊葡萄球菌;⼤肠埃希菌、⽣孢梭菌、枯草芽孢杆菌、⽩⾊念珠菌、⿊曲霉按照上述步骤操作,其中⼤肠埃希菌、⽣孢梭菌加⼊硫⼄醇酸盐流体培养基,枯草芽孢杆菌、⽩⾊念珠菌、⿊曲霉加⼊胰酪⼤⾖胨液体培养基。
6.3对照组:取装量为 ml硫⼄醇酸盐流体培养基3管,分别接种⼩于100cfu的⾦黄⾊葡萄球菌、⼤肠埃希菌、⽣孢梭菌;取相同装量的胰酪⼤⾖胨培养基3管,分别接种⼩于100cfu枯草芽孢杆菌、⽩⾊念珠菌、⿊曲霉。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
编号:FAL-YZ-003.1
无菌检查方法
验证报告
科技发展
目录
无菌检查方法验证报告
1.目的
通过对培养基无菌性检查、灵敏度检查,对产品的无菌检查方法适用性进行试验,证明该方法适用于产品无菌检查日常检测。
2.围
适用于本公司产品的无菌检查方法的建立和确认。
3.依据
中国药典(2015年版)
GB/T14233.2-2005 医用输液、输血、注射器具检验方法第2部分:生物学实验方法
4. 职责权限
5. 验证方法
实验前的准备
a仪器设备
b操作环境
微生物限度检查应在环境洁净度10000 级下的局部洁净度100 级的单向流空气区域进行。
检验全过程必须严格遵守无菌操作,防止再污染。
单向流空气区域、工作台面及环境应定期按《医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法》的现行国家标准进行洁净度验证。
c稀释液和试剂: PH7.0无菌氯化钠-蛋白胨缓冲液
d器具无菌注射器仪液器 YT-603集菌仪
5.1培养基的适用性检查
无菌检查用的硫乙醇酸盐流体培养基及胰胳大豆胨肉汤培养基等应符合培养基的无菌性检查及灵敏度检查的要求。
本检查可在供试品的无菌检查前或与供试品的无菌检查同时进行。
培养基:硫乙醇酸盐批号20140408 生产厂家:日水生物技术
胰胳大豆胨肉汤培养基批号20151023 生产厂家:尼赛欣合生物技术
5.1.1无菌性检查:每批培养基随机取不少于 5 支(瓶),培养 14 天,应无菌生长。
5.1.2灵敏度检查
菌种:培养基灵敏度检查所用的菌株传代次数不得超过 5 代(从菌种保存中心获得的冷冻干燥菌种为第 0 代),试验用菌种应采用适宜的菌种保存技术进行保存,以保证试验菌株的生物学特性。
金黄色葡萄球菌(Staphylococcus aureus)〔CMCC(B) 26 003〕
铜绿假单胞菌(Pseudomonas aeruginosa) 〔CMCC(B) 10 104〕
枯草芽孢杆菌(Bacillus subtilis)〔CMCC(B)63 501〕
生孢梭菌(Clostridium sporogenes)〔CMCC(B) 64 941〕
白色念珠菌(Candida albicans)〔CMCC(F) 98 001〕
黑曲霉(Aspergillus niger) 〔CMCC(F) 98 003〕
5.1.3菌液制备
金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌、生孢梭菌、白色念珠菌、黑曲霉微生物培养基质控品,使用前注入1.1ml稀释液充分溶解后,在漩涡混合器上震荡混匀,制成10-100 cfu/0.1ml菌悬液,3-5天用完。
5.1.4培养基接种取每管装量为12ml 的硫乙醇酸盐流体培养基7支,分别接种小于100 cfu 的金黄色葡萄球菌、铜绿假单胞菌、生孢梭菌各2 支,另1支不接种作为空白对照,培养3 天;取每管装量为9ml的胰胳大豆胨肉汤培养基7支,分别接种小于100 cfu 的枯草芽孢杆菌、白色念珠菌、黑曲霉各2 支,另1 支不接种作为空白对照,培养5 天。
逐日观察结果。
5.1.5结果判定空白对照管应无菌生长,若加菌的培养基管均生长良好,判该
培养基的灵敏度检查符合规定。
见附件无菌培养基的适用性检查记录
5.2无菌检查方法验证试验
5.2.1当建立产品的无菌检查法时,应进行方法的验证,以证明所采用的方法适合于本产品的无菌检查。
若本产品的组分或原检验条件发生改变时,检查方法应重新验证。
验证时,按“中国药典2015版四部1101无菌检查法”的规定进行操作。
对每一试验菌应逐一进行验证。
5.2.2菌种及菌液制备金黄色葡萄球菌、铜绿假单胞菌、生孢梭菌、枯草芽孢杆菌、白色念珠菌、黑曲霉同培养基灵敏度检查。
5.2.3薄膜过滤法
5.2.3.1FAL胶无菌检测:使用YT-603集菌仪,滤膜孔经不大于0.45µm。
A样品组:取1ml医用胶缓慢滴入100ml氯化钠蛋白胨缓冲液中,使其固化成白色胶膜充分冲洗振荡1分钟,制成样品组供试液,取一幅二联式集菌培养器,将样品组供试液平均通过每只集菌培养器过滤,再用100ml氯化钠蛋白胨缓冲液冲洗,然后通过集菌仪每只集菌培养器加入100ml胰胳大豆胨肉汤培养基或硫乙醇酸盐培养基,后每只加入10~100cfu试验菌,做为样品组。
B中和剂对照组:取一幅二联式集菌培养器,将100ml氯化钠蛋白胨缓冲液平均通过每只集菌培养器过滤,然后通过集菌仪每只集菌培养器加入100ml胰胳大豆胨肉汤培养基、或硫乙醇酸盐培养基,后每只加入10~100cfu试验菌,做中和剂对照组。
C阳性对照组(菌液组):取一幅二联式集菌培养器,通过集菌仪每只集菌培养
器加入100ml胰胳大豆胨肉汤培养基、或硫乙醇酸盐培养基,后每只加入10~100cfu试验菌,做为阳性对照组。
D阴性对照:另取一幅二联式集菌培养器,通过集菌仪一只通过100ml胰胳大豆胨肉汤培养基,另一只通过100ml硫乙醇酸盐培养基,作为阴性对照。
5.2.3结果判断样品组与中和剂对照组微生物生长浓度相似,表明中和剂的中和方式有效消除了样品的抑菌性(即有效性),如对照组与阳性对照的微生物浓度相似,表明样品预处理,消除样品抑菌性的方法,检测程序和培养条件等均不影响微生物生长(即无毒性)。
阴性对照无菌生长。
否则实验无效。
6.验证结论
通过培养基无菌性检查、培养基灵敏度检查、无菌检查方法验证,结果表明,制定的无菌检查方法适用本公司的产品。
无菌检查方法验证试验记录
编号:ZJ/JL-8.2.4-4-10序号:样品名称:样品批号:检测日期:样品数量:
试验方法:依据《中国药典》2015年版四部 1101 无菌检查法:方法验证试验
□薄膜过滤法□直接接种法
硫乙醇酸盐流体培养基:(30~35℃培养3天)
注:“+”为生长,“-”为不生长
结论:
检验者:复核者:复核日期:
无菌培养基的适用性检查记录
编号:ZJ/JL-8.2.4-4-9
培养基:1. 硫乙醇酸盐流体培养基批号:_______________ 检验日期:
2. 胰酪大豆胨豆汤培养基批号:_______________ 完成日期:以上均为符合药典规定的干燥培养基。
一、培养基的无菌性检查
培养温度:硫乙醇酸盐流体培养基℃胰酪大豆胨豆汤培养基℃
注:“+”为生长,“-”为不生长
二、培养基的灵敏度检查
1. 菌液制备:
(1)金黄色葡萄球菌新鲜肉汤培养物
[CMCC(B)26003] 第_____代注入1.1ml稀释液充分深解后,在漩涡
混合器上震荡混匀.
至_______含菌量
_______CFU/0.1ml
(2)枯草芽孢杆菌新鲜肉汤培养物1ml
[CMCC(B)63501]第_____代注入1.1ml稀释液充分深解后,
在漩涡混合器上震荡混匀.
至_______含菌量
_______CFU/0.1ml
(3)铜绿假单胞菌新鲜肉汤培养物1ml
[CMCC(B) 10104]第_____代注入1.1ml稀释液充分深解后,
在漩涡混合器上震荡混匀. 至_______含菌量
_______CFU/0.1ml
(4)生孢梭菌新鲜硫乙醇酸盐培养物
[CMCC(B)64941]第_____代注入1.1ml稀释液充分深解后,
在漩涡混合器上震荡混匀.
至_______含菌量
_______CFU/0.1ml
(5)白色念珠菌新鲜改良马丁培养物1ml
[CMCC(F)98001]第_____代注入1.1ml稀释液充分深解后,
在漩涡混合器上震荡混匀.
至_______含菌量
_______CFU/0.1ml
(6)黑曲霉新鲜孢子洗脱液1ml [CMCC(F)98003]第_____代注入1.1ml稀释液充分深解后,
在漩涡混合器上震荡混匀.
至_______含菌量
_______CFU/0.1ml
2. 检查结果
细菌培养温度____℃ 3天霉菌及酵母菌培养温度____℃ 5天
注:“+”为生长,“-”为不生长
三、结论:
本品按《中国药典》2015年版四部1101无菌检查法培养基的适用性检查,结果_______________。
检验人:复核人:复核日期:。