Makekml.exe 使用方法
Makecert使用方法

Makecert.exe(证书创建工具)使用方法概述证书创建工具生成仅用于测试目的的X.509 证书。
它创建用于数字签名的公钥和私钥对,并将其存储在证书文件中。
此工具还将密钥对与指定发行者的名称相关联,并创建一个X.509 证书(该证书将用户指定的名称绑定到密钥对的公共部分)。
使用说明✧Makecert.exe 包含基本选项和扩展选项。
基本选项是最常用于创建证书的选项。
扩展选项提供更多的灵活性。
✧绝不要将此工具生成的证书私钥存储在 .snk 文件中。
如果需要存储私钥,则应使用密钥容器。
警告:应使用证书存储来安全地存储证书。
此工具使用的 .snk 文件以不受保护的方式存储私钥。
创建或导入 .snk 文件时,在使用期间应注意保证其安全,并在使用后将其移除。
✧此工具会自动随Visual Studio 一起安装。
若要运行此工具,请使用开发人员命令提示(或Visual Studio 命令提示)。
使用方法命令语法(在命令提示符中输入):示例1.下面的命令创建一个由默认测试根颁发的测试证书并将其写入testCert.cer。
makecert testCert.cer2.下面的命令创建一个由默认测试根颁发的证书并将其保存到证书存储中。
makecert -ss testCertStore3.下面的命令创建一个由默认测试根颁发的证书并将其保存到证书存储中。
它将证书显式放入currentuser 存储中。
makecert -ss testCertStore -sr currentuser4.下面的命令使用主题的密钥容器和证书主题的X.500 名称创建一个测试证书,并将其写入textXYZ.cer。
makecert -sk XYZ -n "CN=XYZ Company" testXYZ.cer5.下面的命令创建一个由默认测试根颁发的证书和一个 .pvk 文件,并将此证书同时输出到存储和该文件。
makecert -sv testCert.pvk -ss testCertStore testCert.cer6.下面的命令创建一个由默认测试根颁发的证书和一个密钥容器,并将此证书同时输出到存储和该文件。
Make工程管理器

第12章 make工程管理器 (1)12.1 make工程管理器简介 (1)12.1.1 了解make工程管理器 (1)12.1.2 了解makefile文档 (2)12.2 编写makefile (2)12.2.1 makefile的格式 (2)12.2.2 makefile的五部分 (3)12.2.2.1 伪目标 (4)12.2.2.2 变量 (4)12.2.2.3 隐式规则 (7)12.2.2.4 模式规则 (7)12.2.2.5 注释 (8)12.3 命令行选项和参数 (8)12.4 使用autoTools工具集 (8)12.4.1 什么是autoTools (8)12.4.2 autoTools使用流程 (9)12.4.3 使用make命令 (12)12.5 小结 (14)第12章 make工程管理器课时安排:4节课程目标:本课程完成后,您将具备以下能力:1、理解make工程管理器。
2、掌握makefile格式。
3、应用编写makefile文档。
4、应用autoTools生成makefile文档。
课程讲解:在本章中同学们会了解有关make工程管理的介绍及使用,一步步学习写makefile文档,最后了解autoTools系列工具的功能和使用,掌握autoTools自动生成makefile文档的步骤。
12.1 make工程管理器简介12.1.1 了解make工程管理器所谓工程管理器是指管理工程的工具。
假如我们面对是一个上百个文件的代码构成的项目,假如其中几个文件进行了修改,按照之前所学的gcc编译工具,就不得不把项目内所有的文件重新编译一遍,因为编译器并不知道哪些文件是最近更新的,但那些没有改动的源代码根本不需要重新编译,而只须把它们重新链接进去即可。
所以,人们就希望有这样一款能够自动识别更新的文件代码并管理项目的软件,所以make工程管理器也就应运而生了。
make工程管理器是个“自动编译管理器”,这里的“自动”是指它能够根据文件时间戳自动发现更新过的文件而减少编译的工作量,同时,它通过读入makefile文件的内容来自动执行大量的编译工作。
科箭操作手册

科箭操作手册摘要:一、引言二、科箭操作手册的简介1.科箭操作手册的版本和适用范围2.科箭操作手册的目的和结构三、科箭操作手册的使用方法1.安装和启动科箭操作手册2.科箭操作手册的基本操作3.科箭操作手册的高级操作四、科箭操作手册的注意事项1.使用科箭操作手册的安全措施2.维护科箭操作手册的方法五、科箭操作手册的常见问题及解决方法六、科箭操作手册的附录1.术语解释2.软件更新和历史版本正文:一、引言科箭操作手册是一本介绍如何使用科箭操作系统的指南,它包含了科箭操作系统的详细介绍和操作方法,可以帮助用户更好地理解和使用科箭操作系统。
二、科箭操作手册的简介科箭操作手册的版本和适用范围:科箭操作手册的最新版本是V1.0,适用于所有使用科箭操作系统的用户。
科箭操作手册的目的和结构:科箭操作手册的目的是帮助用户更好地理解和使用科箭操作系统,它的结构包括概述、安装和启动、基本操作、高级操作、注意事项、常见问题及解决方法和附录等章节。
三、科箭操作手册的使用方法安装和启动科箭操作手册:在安装科箭操作系统后,用户可以通过“开始”菜单启动科箭操作手册。
科箭操作手册的基本操作:科箭操作手册提供了多种基本操作,如打开、关闭、复制、粘贴等。
科箭操作手册的高级操作:科箭操作手册的高级操作包括创建、编辑、格式化等。
四、科箭操作手册的注意事项使用科箭操作手册的安全措施:在使用科箭操作手册时,用户需要注意保护自己的信息安全,不要泄露自己的账号和密码。
维护科箭操作手册的方法:用户需要定期对科箭操作手册进行维护,以确保其正常运行。
五、科箭操作手册的常见问题及解决方法科箭操作手册的常见问题包括启动失败、操作不正常等,用户可以通过查阅科箭操作手册的附录来获得解决方法。
make命令与makefile文件的写法

什么是make file?或许很多Win odws的程序员都不知道这个东西,因为那些Win dows的I DE都为你做了这个工作,但我觉得要作一个好的和pr ofessi onal的程序员,makefil e还是要懂。
这就好像现在有这么多的HT ML的编辑器,但如果你想成为一个专业人士,你还是要了解H TML的标识的含义。
特别在Unix下的软件编译,你就不能不自己写makef ile了,会不会写mak efile,从一个侧面说明了一个人是否具备完成大型工程的能力。
因为,makefil e关系到了整个工程的编译规则。
一个工程中的源文件不计数,其按类型、功能、模块分别放在若干个目录中,makefil e定义了一系列的规则来指定,哪些文件需要先编译,哪些文件需要后编译,哪些文件需要重新编译,甚至于进行更复杂的功能操作,因为makef ile就像一个Shell脚本一样,其中也可以执行操作系统的命令。
makefil e带来的好处就是——“自动化编译”,一旦写好,只需要一个ma ke命令,整个工程完全自动编译,极大的提高了软件开发的效率。
make是一个命令工具,是一个解释ma kefile中指令的命令工具,一般来说,大多数的IDE都有这个命令,比如:Delphi的make,V isu alC++的nmake,Linux下G NU的mak e。
可见,makefil e都成为了一种在工程方面的编译方法。
现在讲述如何写makefi le的文章比较少,这是我想写这篇文章的原因。
当然,不同产商的ma ke各不相同,也有不同的语法,但其本质都是在“文件依赖性”上做文章,这里,我仅对GN U的make进行讲述,我的环境是Re dHat Linux 8.0,make的版本是3.80。
makefile用途和用法

makefile用途和用法Makefile是一种用于自动化构建和管理软件项目的工具,它由一系列规则和命令组成。
Makefile的主要作用是根据项目中源代码的依赖关系,判断哪些文件需要重新编译,然后执行相应的编译和链接操作,以生成最终的可执行文件或库文件。
Makefile的用法如下所示:1. 定义规则:Makefile中的规则由目标、依赖和命令组成。
目标是待生成的文件或操作的名称,依赖是生成目标所依赖的文件或操作,命令是生成目标的具体操作步骤。
2. 指定规则:使用“target: prerequisites”格式指定规则,其中target是目标文件,prerequisites是该目标所依赖的文件。
3. 编写命令:在每个规则中,使用Tab键缩进的命令行来定义生成目标的操作步骤。
命令行可以是任何Shell命令,包括编译器和链接器的命令。
4. 依赖关系:通过指定目标文件所依赖的源代码文件或其他目标文件,Makefile可以自动判断哪些文件需要重新编译。
5. 变量定义:Makefile中可以定义变量来存储常用的参数和选项,以便在各个规则中复用。
通过编写Makefile,可以实现以下几个方面的功能:1. 自动编译:Makefile可以根据源代码文件和依赖关系,自动判断哪些文件需要重新编译,从而避免重复编译不必要的文件,提高编译效率。
2. 自动链接:Makefile可以自动处理目标文件之间的依赖关系,并根据链接选项生成最终的可执行文件或库文件。
3. 构建工程:通过Makefile,可以定义多个目标文件和相应的规则,从而一次性构建整个项目,减少手动操作的工作量。
4. 管理项目:Makefile可以定义清理规则,用于删除生成的目标文件和临时文件,保持项目目录的清洁。
Makefile是一个强大的工具,可以大大简化软件项目的构建和管理过程,提高开发效率和可维护性。
通过合理使用Makefile,可以更好地组织和管理项目代码,实现自动化构建和持续集成。
科箭操作手册

科箭操作手册
【原创版】
目录
1.科箭操作手册概述
2.科箭操作手册的内容
3.科箭操作手册的使用方法
4.科箭操作手册的优点
5.科箭操作手册的适用对象
正文
科箭操作手册是一款专业的操作指南,旨在帮助用户更好地理解和使用科箭软件。
本文将从以下几个方面详细介绍科箭操作手册。
首先,科箭操作手册概述。
科箭操作手册是一本全面的指南,涵盖了科箭软件的各个方面,包括功能、操作流程和技巧。
它不仅是一本初级用户入门的教程,也是一本进阶用户深入学习的参考书。
其次,科箭操作手册的内容。
科箭操作手册的内容详尽且系统,它从基础的软件安装、账号注册开始,逐步介绍了科箭软件的各项功能,包括数据管理、流程设计、报表生成等,让用户能够全面地了解和掌握科箭软件的使用。
再次,科箭操作手册的使用方法。
用户可以通过阅读手册,按照其中的步骤和指导进行操作,也可以在实际操作中遇到问题时,查阅手册寻找解决方案。
此外,手册中还提供了一些常见问题的解答,帮助用户快速解决使用过程中可能遇到的问题。
然后,科箭操作手册的优点。
科箭操作手册的最大优点在于它的全面性和系统性,能够帮助用户全面地了解和掌握科箭软件的使用。
此外,手册的内容更新及时,能够及时反映软件的更新和升级,保证用户使用的是
最新的操作方法和技巧。
最后,科箭操作手册的适用对象。
科箭操作手册适用于所有使用科箭软件的用户,无论是初学者还是进阶用户,都能够从中获得帮助。
symchk.exe的用法 -回复

symchk.exe的用法-回复symchk.exe是Windows操作系统中一个非常有用的工具,用于验证和修复动态链接库(DLL)和可执行文件(EXE)的符号。
它可以帮助开发人员和系统管理员在调试和排除系统问题时定位错误,并提供正确的符号文件,以便更好地理解和分析代码。
在本文中,我将一步一步地介绍symchk.exe的用法,并说明如何使用该工具来进行符号验证和修复。
首先,让我们了解一下符号文件的概念。
在编译源代码时,编译器会生成一个二进制文件,其中包含编译后的机器代码。
同时,编译器会生成一个符号文件,其中包含了代码中变量和函数的名称、类型信息和调用堆栈等。
符号文件通常具有.PDB(Program Database)扩展名,并与二进制文件相对应。
这些符号文件对于调试和分析代码非常重要,因为它们提供了有关二进制文件内部结构和数据的详细信息。
一、安装Debugging Tools for Windows要使用symchk.exe工具,我们首先需要安装“Debugging Tools for Windows”。
这是一组开发工具,可以在Windows系统上进行高级调试和诊断。
您可以在微软的官方网站上找到Debugging Tools for Windows的安装程序,并按照说明进行安装。
安装完成后,我们可以在系统的安装目录下找到symchk.exe工具的可执行文件。
二、使用symchk.exe进行符号验证1. 打开命令提示符或PowerShell窗口,并导航到包含symchk.exe文件的目录。
2. 使用以下命令来验证一个二进制文件的符号:shellsymchk.exe <binary_path> /s <symbol_path> /v`<binary_path>`是要验证的二进制文件的路径,`<symbol_path>`是包含符号文件的路径。
`/v`选项用于输出验证过程的详细信息。
nmake install 用法

nmake install 用法nmake install 是一种软件安装工具,主要用于在Windows系统上编译和安装软件。
它是Microsoft Visual Studio中的一部分,是在Windows平台上最常用的构建工具之一。
本文将详细介绍nmake install的用法,帮助读者了解如何使用它来编译和安装软件。
1. nmake install简介nmake install是nmake命令的一种参数组合,它在执行完nmake build命令后,会自动将编译后的文件复制到指定的目录中。
一般来说,nmake install命令会在使用了nmake build命令编译源代码后执行,用于将编译生成的可执行文件、库文件、配置文件等复制到系统中适当的位置,以便用户可以方便地使用和操作。
2. nmake install的基本用法nmake install命令的基本用法非常简单,只需要在命令行中输入“nmake install”即可。
但在执行之前,我们需要在当前目录下创建一个名为Makefile的文件,并在其中定义安装规则和相关参数。
2.1 创建Makefile文件在执行nmake install之前,我们需要先创建一个名为Makefile的文件。
在该文件中,我们需要定义一些规则和变量,以指定编译结果的安装位置和安装方式。
首先,我们可以定义两个变量,分别用于指定编译结果的安装目录和待安装文件的位置。
例如:```INSTALL_DIR = C:\Program Files\MySoftwareFILE_TO_INSTALL = bin\MyApp.exe```在上述例子中,INSTALL_DIR变量指定了待安装文件的目标目录,而FILE_TO_INSTALL变量则指定了待安装的文件的位置。
接下来,我们需要定义规则,以指定在执行nmake install时需要执行的操作。
一般来说,我们需要在Makefile文件中定义一个名为install的规则。
makeimage使用方法

makeimage使用方法(实用版3篇)目录(篇1)1.引言2.makeimage 的使用方法3.示例4.结论正文(篇1)【引言】在计算机科学领域,图像处理技术越来越受到关注。
其中,makeimage 是一个用于创建图像的工具,它可以帮助用户轻松地生成各种类型的图像。
然而,对于许多新手来说,如何使用 makeimage 还是一个谜。
因此,本文将介绍 makeimage 的使用方法,以帮助您更好地掌握这个工具。
【makeimage 的使用方法】makeimage 的使用方法非常简单。
首先,您需要确保您的计算机上已经安装了 makeimage。
安装完成后,您可以通过以下步骤来使用它:1.打开命令行界面(Windows 系统下为 CMD,Mac 和 Linux 系统下为终端)。
2.输入以下命令:```makeimage -f format image_name```其中,`-f`选项表示图像格式,如`png`、`jpg`等;`format`选项表示要创建的图像的格式;`image_name`选项表示要创建的图像的名称。
例如,如果您想创建一个名为“example.png”的 PNG 格式图像,您需要输入以下命令:```makeimage -f png example.png```3.按 Enter 键。
此时,makeimage 将根据您提供的信息创建一个新的图像文件。
【示例】让我们通过一个简单的示例来演示如何使用 makeimage。
假设您想要创建一个名为“example.png”的 PNG 格式图像,并且该图像的大小为200x200 像素,颜色为蓝色。
您可以按照以下步骤操作:1.打开命令行界面。
2.输入以下命令:```makeimage -f png -s 200x200 -c blue example.png```其中,`-s`选项表示图像的大小,`-c`选项表示图像的颜色。
3.按 Enter 键。
kml文件生成工具使用说明

Kml文件生成工具使用说明
如果没有1:2000图,将线位展到Google earth上将会是一个不错的选择。
在Google earth上,你可以合理组织安排作业路线,可以合理布设基础控制网,可以了解线位走向及所穿越的地形地物。
想将线位展到Google earth上,只要完成以下几步便可轻松完成:一.生成逐桩坐标,将逐桩坐标通过LGO坐标匹配转换为精确经纬度坐标,然后将经纬度坐标换算成十进制。
二.运行kml文件生成工具,选定需要生成的区域(从第二行开始,必须选定A~E五列,注释一般为空),如下图
所示。
选定生成区域后,点击工具-宏,出现如下界面,
点击执行,出现如下提示:
选择确定,提示kml文件已经成功保存在C盘。
三.在C盘里找到xls2GE.kml文件,双击即可运行Google earth并显示所展点位。
姬同昌
2011-10-8。
Makekml.exe 使用方法
使用软件Makekml.exe将工参表和路测数据导入
到Google Earth中
一、将工参表导入到Google Earth中
1.首先将文本格式的工参表转换为Excel格式。
(可直接修改后缀名或者将文本内容复制粘贴到Excel表中)
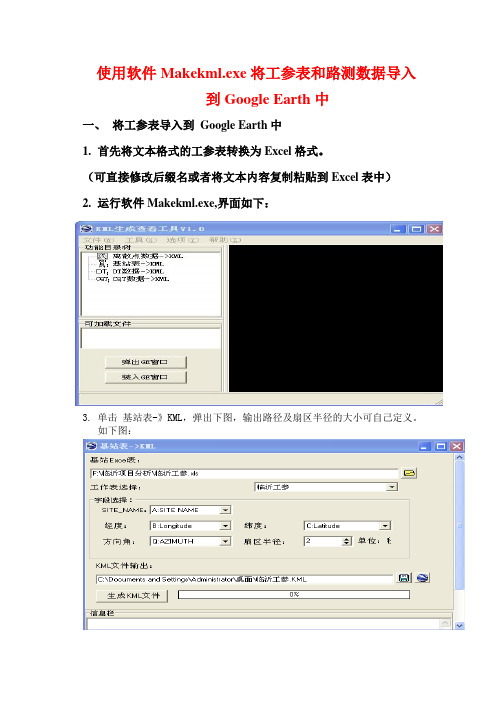
2.运行软件Makekml.exe,界面如下:
3.单击基站表-》KML,弹出下图,输出路径及扇区半径的大小可自己定义。
如下图:
4.点击生成KML文件,等待一会完成。
找到生成的KML文件用Google earth 打开,即可在Google earth中查看基站信息了。
二、将路程数据导入到Google earth中。
过程大概和工参表的导入过程一样,只需单击DT数据》KML
不过需要说明的一点是:鼎力的log 后缀名是.rcu, 需要在Pioneer 软件中将.rcu转换为.txt格式。
大致的步骤如下:
1.运行Pioneer软件,点击编辑》数据》导入出现如下界面
2.空白处右击选择第三项,打开原文件*****.rcu
3.然后点击编辑》数据》导出,出现以下界面,勾选相关参数
4.点击OK后,弹出对话框,设置保存路径。
至此.rcu成功转换为.txt
5.最后参照工参表的导入过程即可完成将路测数据转换成能用
Google earth 打开的KML文件。
工参表和路测数据在Google earth 中同时显示如下图:。
MATLAB生成可执行文件

MATLAB生成可执行文件(.exe文件)2021-07-28 14:59:25| 分类:matlab学习要将用Matlab语言编写的函数文件编译成可独立执行的*.exe文件(即可离开Matalab环境的执行程序),首先要安装和配置好Matlab Compiler,一般来讲,在安装Matlab时就已经安装了相应版本的Matlab Compiler。
只是不同版本的Matlab,其编译器的利用方式有必然的差别,这一点要引发必然的注意。
在肯定安装好Matlab Compiler后,还需要对Compiler进行适当的配置,方式是在Matlab命令窗口输入:Mbuild –setup然后按照提示执行相应的操作,利用者可按照自己计算机中现有编译器的情况选择适合的编译器,如VC++ 6.0、VC++7.0、Bland C的编译器等,目前Matlab好象还不支持VC++8.0(我计算机安装的就是VC++2021,Matlab就无法识别)。
固然,若是你的计算机里根本就没有安装其他任何语言的编译器,也可选择Matlab自带的Lcc编译器,其实这个编译器对大多数用户已经够用了(我就是选择的Matlab自带的Lcc编译器)。
配置好编译器后,自然就是对自己编写的M文件进行编译了。
将M文件编译为独立可执行文件的语法是:>>mcc –m fun1.m fun2.m…..其中fun1就是最后的可执行文件的名称。
另外,也可通过采用命令开关-o指定编译最终目标文件的名称,如mcc –m main.m –o mrank_main,就是将编译后的文件指定为mrank_main.exe。
编译后的生成文件按照编译器的版本不同而不同。
具体的可参阅相关资料。
若是要在没有安装matlab的计算机上执行编译后的程序,首先要将MATLAB701toolboxcompilerdeploywin32中的MCRinstaller.exe安装到该计算机上(7.0 以前的版本是mglinstaller.exe)。
CMake使用教程
CMake使⽤教程CMake是⼀个⽐make更⾼级的编译配置⼯具,它可以根据不同平台、不同的编译器,⽣成相应的Makefile或者vcproj项⽬。
通过编写CMakeLists.txt,可以控制⽣成的Makefile,从⽽控制编译过程。
CMake⾃动⽣成的Makefile不仅可以通过make命令构建项⽬⽣成⽬标⽂件,还⽀持安装(make install)、测试安装的程序是否能正确执⾏(make test,或者ctest)、⽣成当前平台的安装包(make package)、⽣成源码包(make package_source)、产⽣Dashboard显⽰数据并上传等⾼级功能,只要在CMakeLists.txt中简单配置,就可以完成很多复杂的功能,包括写测试⽤例。
如果有嵌套⽬录,⼦⽬录下可以有⾃⼰的CMakeLists.txt。
总之,CMake是⼀个⾮常强⼤的编译⾃动配置⼯具,⽀持各种平台,KDE也是⽤它编译的,感兴趣的可以试⽤⼀下。
准备活动:(1)安装cmake。
下载地址:根据⾃⼰的需要下载相应的包即可,Windows下可以下载zip压缩的绿⾊版本,还可以下载源代码。
(2)运⾏cmake的⽅法。
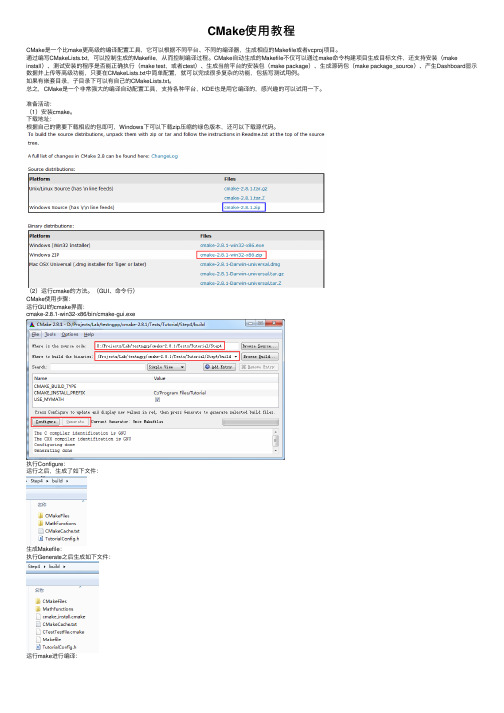
(GUI、命令⾏)CMake使⽤步骤:运⾏GUI的cmake界⾯:cmake-2.8.1-win32-x86/bin/cmake-gui.exe执⾏Configure:运⾏之后,⽣成了如下⽂件:⽣成Makefile:执⾏Generate之后⽣成如下⽂件:运⾏make进⾏编译:编译完成后,在build⽬录⽣成Tutorial.exe,运⾏Tutorial.exe 25就可以看到运⾏结果:运⾏make install安装程序:运⾏make test进⾏测试:通过cmake tutorial学习CMake配置⽅法可以在源代码的Tests/Turorial⽬录中找到这个⼿册对应的代码。
1、Step1。
(如果不知道如何使⽤cmake,以及如何使⽤编译产⽣的Turorial.exe,可先看下前⾯“CMake使⽤步骤”的说明,它以Step4为例详细介绍了使⽤过程,Step1的配置可能不够完全,⽐如⽆法运⾏make install,⽆法运⾏make test,但可以参考。
make工程管理器及其Makefile 及其使用

make工程管理器是一种能够自动识别更新了文件代码的工具,同时又不需要重复输入冗长的命令行,当文件较多是比较实用
Autoconf和Automake等是这样的工具可以自动生成Makefile文件
1:Make命令和Makefile
要使用make,必须编写一个叫Makefile的文件,它描述了软件包中各个文件之间的关系,提供了更新每个文件的命令
Makefile中还可以使用环境变量,使用环境变量的方法相对比较简单
Makefile中还有一些规则称为隐晦规则,运行make工具对目标文件寻找传统的更新方法,而避免指定任何命令。可以编写没有命令行的规则或根本不编写规则,这样make工具将根据存在的源文件类型或要生成的文件类型决定使用何种隐含规则
Makefile中常见的隐含规则:
在程序目录中创建一个名为makefile的文本文件,将当前工作目录切换到makefile所在的目录,目前这个makefile支持在当前目录中的调用,不支持当前目录和makefile所在不是同一目录的名字
2:Makefile的规则
最基本的编写规则的方法是从最终的源程序文件开始一个一个地查看源文件,把它们要生成的目标文件作为目标,而C源文件和源文件包含的头文件作为依赖文件生成规则,但是我们必须去分析源码文件的嵌套关系,实际上我们可以让gcc做这个工作,当使用gcc的时候,可以用-MM(输出一个Make兼容的相关列表)参数,它可以为每一个输入的C语言源文件输出一个依赖规则,把gcc生成的目标文件作为Makefile规则的目标文件,而把生成这个目标文件的C语言源文件和所有应该被引用的头文件作为依赖,gcc只输出规则的依赖关系,不含有命令部分,用户需要写入需要的命令或什么都不写,make会使用隐含规则
Make使用说明

Make 使用说明Make是一种代码维护工具.在大中型项目中,他将根据程序各个模块的更新情况,自动的维护和生成目标.能很好的做到”不多”,”不重””不漏”.不多:他只更新那些需要更新的文件,而不懂那些并不过时的东西不重:是指当make对某个文件进行更新时,即使有很多文件过时,他也将只更新一次,当然,这一次就足够了不漏:是指他不会漏下任何应该更新的文件.1.make的基本使用make在使用时有一系列的规则,他将根据这些规则来解释他的配置脚本,以达到它的设计目的.这个配置脚本的缺剩名是makefile ,Makefile.也可以用其它文件名.我们这里所讲的make的应用就是怎样写出高效,简洁,正确的makefile文件.Make的使用形式为Make [option] [macrodef][target]Option指出make的工作行为,make的具体选项有:-c dir make在开始运行后的工作目录为指定目录-e 不允许在makefile中队环境的宏赋值-f filename 使用指定的文件作makefiel-k 执行命令出错时,放弃改目标去维护其它目标-n 按实际运行时的执行顺序现实命令,包括用@开头的命令,但不振的执行-p 现实makefile中所有的宏定义,河内部规则-r 忽略内部规则-s 执行但不显示命令,常用来检查makefile的正确性-S 执行make时出错即退出-t 修改每个目标文件的创建日期,但又不正的创建-V 打印make的版本号-x 将所有的宏定义输出的shell环境中-i忽略运行make中的错误,而又不退出make在使用make维护目标的时候,为了调试错误,常使用 make 2 >; errofile.这样错误信息就都写入了errofile2.makefile 的基本书写make使用的依据是依赖关系.如工程example ,他依赖于main.o e1.o e2.o,而main.o 依赖于main.c l.h,e1.o依赖于e1.c l.h ,e2.o 依赖于e2.c.这样就形成一棵树.及双亲节点,依赖于孩子节点.当make 进行维护时.他呼对这棵树.进行一次后序遍历.如果发现孩子节点形成的时间晚于双亲节点形成的时间.便开始维护.原理十分简单.现在,我们开始,我们的主要内容: 如何书写makefilemakefile 是供make 使用的一个配置脚本.他是按一定的规则写成的.它的组成是以一个要维护的目标为一个单元,每个单元的书写形式如下:要维护的目标列表要维护的目标属性分割符(一般为:) 依赖的文件列表命令行的属性 ;第一条命令tab键命令1…..需要说明的是,第一条命令可以加; 跟在依赖文件列表后,也可以和其它的命令另起一行写.一定要记得加tab键.否则make 不认识.当一条命令太长一行放不下是使用/l来换行.注释行用#开头.这样一来.我们前面举的例子就可以写成这样的makfileexample : main.o e1.o e2.ogcc -o example main.o e1.o e2.omain.o :main.c l.hgcc -c main.ce1.o :e1.c l.hgcc -c e1.c l.he2.o:e2.cgcc -c e2.c可以用cat -e -v-t makefile 其中-v-t可以让tab键显示^I ,是makefile 的末尾显示$,这样可以检验,makefile的正确性.可见,书写一个简单的makefile文件并不困难.然而在大中型项目中.要通过makfile 做大量的维护与调试.这样会是程序员增加很大的工作量.因此,make 引入了一些属性变量,和宏变量,来控制make .甚至可以像c预言的条件编译那样.通过一些shell语句来控制.接下来.我们将一一讲解.3.makfile中的属性控制2.2.1分割符分割符一般用: 但也用::,:!:^,:-一下举例说明一下这几个分割符的作用一般目标文件只能出现一次.只有使用::时才可以出现多次如:a.o::a.c#命令1a.o::b.c#命令2意思是:如果a.c更新.使用命令1.b.c更新使用命令2:^ 将指定的依赖文件和目标文件一有的依赖文件结合起来:- 清除目标文件原有的依赖文件,将指定文件为目标文件的唯一依赖文件:! 对每个更新过的依赖文件都执行一遍命令菜单如:a.o :b.ca.o:^ c.c此时a.o 的依赖文件是b.c 和c.ca.o:- a.c此时 a.o 的依赖文件只剩下a.c了2.2.2 命令行属性命令好属性较简单只有三种+ 当运行条件成熟(依赖文件过时) 始终执行,即使 make 有 -n -q -t 选项- 当此行出错时,忽略继续运行make@ 不显示本行命令(make 执行使命令行在屏幕上打印)2.2.3 目标文件属性目标属性比较多.这里举几个常用的例子.IGNORE 类似于命令行中的减号,在本目标维护出错时.并不马上退出make 而是继续运行.SILENT 类似于命令行中的@ ,维护本目标时不在屏幕上显示.PRECIOUS 保留中间文件.LIBARY 说明维护的目标是一个库.SYMBOL 说明维护的目标是指定入口的库成员.LIBRARYM 说明维护的是库的一个成员在使用目标文件的属性时有两种方法.IGNORE main.o e1.o 和main.o .IGNORE : ….……e1.o .INGORE :…..…….效果是相同的,在实际使用时,可以根据个人的具体情况使用.2.2.3 伪目标在make 中有两类目标,一类是实目标.一类是伪目标.伪目标不要求make生成世纪的目标文件,只是完成一些辅助性的工作,如打印,删除等.伪目标有两种.一种是任意给出名字如: lp:pr *.c/lp将所有的源文件分页打印.由于系统中没有lp 文件,他总是认为是过时的,因此这个命令总被执行.另一种方式是使用linux提供的伪目标.ERRO :依赖文件命令行作用时,一旦遇到错误时,执行命令行.INCLUDE :file1 file2作用是: 将文件包含进来,注意:这里的包含不是指针的转移而是拷贝.INCLUDE /usr.INCLUDE file1make 首先在当前目录下寻找file1,如没有则在/usr下找.另一种使用方法是:/INCLDE <;dir/filename>;.IMPORT 将一些宏载入 .如:.IMPORT shell 这样makefile 便可以使用shell 里的宏了..EXPORT 和.IMPORT恰恰相反.是将makefile里定义的宏,输出到环境.不赘述.当需要维护几个目标时可使用这样的伪目标:all: main.o main1.o ….重要说明是: 命令行属性>;>;目标文件属性>;>;make 命令行属性我们在各种应用中已熟悉宏的概念.不赘述.在make 中使用宏也要先定以后使用.宏名可以是任意数字.字母下划线的组合. 不过不能用数字开头.宏的定义方式有3种= 直接将后面的字符串赋给宏:= 后面跟字符串常量,将他的值赋给宏+= 宏原来的值加上一个空格,将它的内容赋给宏宏的应用各是有两种:$(宏名) 或 ${宏名}宏名可以嵌套使用.如h2=mylib.aindex=2则在makefile 中调用$(h$(index)) ,就等于调用 mylib.a.使用shell 环境的宏.不需要定义.只要用.IMPORT 在如就行.3.2 内定义宏控制宏为了控制make 的属性,定义了一系列的内定义宏.分为两类.控制宏用于控制make 的属性.属性宏代表一些特殊的值.控制宏:.DIREPATR 表示路径中用于分割目录与文件的分割符一般为 /.MAKDIR 调用make的绝对路径名.NULL 空字符串.PWD 运行make 时活动目录的绝对目录名.SHELL 运行是的shell 名另一类宏.我们成为属性宏.和目标文件的属性书写和意义都相同.用于设置这些属性.如.IGNORE=yes将整个makefile里的目标文件都设为.IGNORE.其它的属性宏.和目标文件属性里面的相同.3.3 动态宏引入动态宏的目的是,简化makefile的书写.他利用了很多一用的代码.但给makefile 带来了不可读性$@ 当前目标文件的名字$% 同$@,不过若维护的目标是库时.$@指库名 $% 指库成员$>; 适用于lib(member)的情况$>;指库名如$@$* 目标文件,去掉后缀$&; 当前目标文件.在所有依赖文件$^ 当前目标文件在本单元中的依赖文件$? 当情目标文件在所有单元中比目标新的依赖文件$<; 当前目标文件在本单元中比目标文件新的文件这些宏只能用在命令行中.make 提供可与此对应的动态宏用在依赖文件中.他们分别是在上面的宏上在加一个$$$@ 在依赖文件列表中,代表目标文件的名字等等.这样,我们原先的makefile便可以写成:example:main.o e1.o e2.ogcc -c $?main.c e1.c:$$*.c l.he2.c :$$*.cgcc -o $*.c3.4 宏的修改一个宏定义以后.可以用make的某些功能.对其进行修改1,如:file=/usr/l/main.c 引入修改描述符:d :仅展开目录b :仅展开文件名,不包括扩展名f :展开全名这样$(file:d)等于main . 其它的类推2,替换宏中的字符串规则宏名: s /远字符串/新字符串file =file1.o file2.o file3.ofile:s/file/fun则file=fun1.o fun2.o fun3.o3.还有宏名:^前缀宏名:+后缀file= a b cfile=^";1";则file=1a 1b 1c4.内部规则4.1make 的内部规则是系统或者后呼吁定义的一些规则.他在定义时.不必指出全名.只是定义了一些最常用的依赖关系和依赖命令.和c 编程有关的就以下.这几种情况与c编程又过的文件扩张名是.o 目标文件.c c语言源文件.C c++语言源文件.h 头文件与c 相关的规则主要有.c:$(CC)$CFLAGS)-O $@ $<;.c .o:$(CC)$CFLAGS)-c $@ $<;这里面使用到了一些宏.以上两句话的意思是对.c 文件缺剩是的编译.和编译成.o 文件.有必要说一些宏的定义宏名初始值说明AR Ar 库管理命令ARFLAGS -ruv 库管理命令选项CC cc C编译命令CGLAGS -o C编译命令选项C++C CC C++编译命令C++FLAGS -O C++编译命令MAKE make MAKE命令MAKEFLAGS NULL MAKE命令选项LIBSUFFIX .a 库的扩展名A .a 库的扩展名有了内部规则.我们的makefile 会更加简练如原先的makefile 我们可以写成example:main.o e1.o e2.ogcc -o example main.o e1.o e2.omain.o e1.o :l.h4.2修改和定义内部规则修改内部规则.可以有两种办法:一是修改内部规则用到的宏.而是直接修改内部规则的命令行如我们前面提到的规则:.c .o:$(CC)$(CFLAGS)-c $@ $<;我们只要定义 CC=6788 其命令行就变成了 6788 -O -c $@ $<; 原因是makefile 内部的宏定义大于make 的默认定义至于写该命令行,直接在makefile 中写就行了.他会踢换掉默认的定义如.c .o:$(CC) -c $@ $<;就会以下面这个定义为主对于一些没有的后缀名可以自己定义,各时如:new roman 后缀名1 后缀名2 ….SUFFIXES .n 就定义了一个新的后缀n .从目标文件生成文字表文件的命令是:nm 目标文件 >; 名字表文件现在我们定义一个内部规则,即.o 文件依赖于通明的.o 文件.SUFFIXES .n.o.nnm $<; >; $@这样的话.make 会根据.o文件的更新,自动的维护.n 文件5.库的使用5.1库的建立和维护在开发大中型软件时.常把一些编译好的模块同意放到一个库中.在编译时可以节省时间和空间.库中的文件一般成为库的成员.成员的表示形式一般为库名(库成员名) 如: mylib(myrea.o) 用make 维护库时,我们将库名作为目标文件格式如下:库名: 库名(库成员名1) 库名(库成员名2) 库名(库成员名3)make 遇到这样的命令行 ,自动吧库名当成目标文件,并赋予.LIBRARY属性例如:mylib.a :mylib.a(myread.o) …也可以这样定义mylib.a .LIBRAAY :myread.o …还有一种定义方式是:库名: ((rentry))make 遇到这种情况,把库名置成.LIBRARY 把entery 置成 .SYMBOL.然后根据entry 找到相应的文件以后的处理和直接些文件相同make 从.o 文件维护库的命令是ae -ruv 库名目标文件名如维护一个名为mylib的库.makefile 需要这样写mylib:mylib(f1.o)gcc -c fi.car -ruv muylib f1.orm -f f1.omylib:mylib(f2.o)gcc -c f2.car -ruv muylib f2.orm -f f2.o……利用动态宏我们可以将他该成这样:mylib:mylib(f1.o f2.o…..)gcc -c ($?:b).car -ruv $@ $?rm -f $@我们也可以利用内部规则,来简化所有makefie 中库的维护.o.a:$(AR) $(ARFLAGS) $@ $?rm -f $?然后库的makfile 写成mylib: mylib(f1.o) mylib(f2.o)………还有一条内部过则是:.amake $@即make 将自动维护所有的库5.2 利用库进行连接使用库进行连接时.只需要将库名.o文件名说明就行.形式如下:库名(库成员名) 或库名(((成员entery))例如想在有一可执行程序eampl2.exe 依赖于mylib 中的f1.o ,在makefile 中可以这么写:eampl2.exe:mylib(f1.o)#连接命令行(一般是ld)make 读到这样的规则就知道mylib 是一个库.有.LIBRAAY的属性f1.0 是库的成员函数有.LIBRAYM的属性eampl2.exe 依赖于f1.o在这里也有意跳内部规则.o.e$(LD) $(LDFLAGES) -o $@ $<; $(LDLIB)其中宏的定义是LD 连接命令ldLDFLAGEA ld 的选项]LDLIB 库名列表因此只要把你自己定义的库,加到库名列表中,在使用内部规则就行了.如上例可以写成: LDLIB+=mylib.a # 将自定义的库加到库名列表中去eaple2.exe:mylib(f1.o)7.条件编译make 中还引入了一些shell 语法.以帮助进行类似于c 语言的于编译的功能.下面是在linux核心的makfile 中抄来的一段.我们可以看一下他是如何进行条件维护的ifeq (.config,$(wildcard .config))include .configifeq (.depend,$(wildcard .depend))include .dependdo-it-all: Version vmlinuxelseCONFIGURATION = dependdo-it-all: dependendifelseCONFIGURATION = configdo-it-all: configendif类似于c 语言的 if else 语句.不同的是他是比较两个值是否相等,来判断是否满足条件.因为在makefile中.很多值是由字符串组成的,所以这个于就十分有用.还有这样的语句:ifdef CONFIG_PCIDRIVERS := $(DRIVERS) drivers/pci/pci.aendif这个语句够简单,我多说什么了.如何使用make在shell的提示符号下,若键入"make";,则它会到目前的目录下找寻Makefile这个文件。
MAKER使用指南说明书

Exercise 1. Using MAKER for Genome AnnotationIf you are following this guide for your own research project, please make the following modifications:1. In this exercise, SNAP was used for gene prediction. When you are working on your owngenome, we recommend that you use Augustus. The instructions for using Augustus is in appendix.2. In the exercise, you will be using 2 CPU cores. When you are working on your own genome,you should use all CPU cores on your machine. When you run the command:"/usr/local/mpich/bin/mpiexec -n 2", replace 2 with number of cores available on yourmachine.3. The steps for Repeatmodeler and Repeatmasker are optional in the exercise, but requiredwhen you work on your own genome.The example here is from a workshop by Mark Yandell Lab (/ ) Further readings:1. Yandel Lab Workshop. /MAKER/wiki/index.php/MAKER_Tutorial_for_WGS_Assembly_and_Annotation_Winter_School_2018 .2. MAKER protocol from Yandell Lab. It is good reference. https:///pmc/articles/PMC4286374/3. Tutorial for training Augustus https:///simonlab/bioinformatics/programs/augustus/docs/tutorial2015/training.html4. Maker control file explained: /MAKER/wiki/index.php/The_MAKER_control_files_explainedPart 1. Prepare working directory.1. Copy the data file from /shared_data/annotation2018/ into /workdir/$USER, and de-compress the file. You will also copy the maker software directory to /workdir/USER. The maker software directory including a large sequence repeats database. It would be good to put it under /workdir which is on local hard drive.mkdir /workdir/$USERmkdir /workdir/$USER/tmpcd /workdir/$USERcp /shared_data/annotation2019/maker_tutorial.tgz ./cp -rH /programs/maker/ ./cp -rH /programs/RepeatMasker ./tar -zxf maker_tutorial.tgzcd maker_tutorialls -1Part 2. Maker round 1 - Map known genes to the genomeRun everything in "screen".Round 1 includes two steps:Repeat masking;Align known transcriptome/protein sequences to the genome;1. [Optional] Build a custom repeat database. This step is optional for this exercise, as it is avery small genome, it is ok without repeat masking. When you work on a real project, you can either download a database from RepBase (https:///repbase/, license required), or you can build a custom repeat database with your genome sequence.RepeatModeler is a software for building custom databases. The commands for building a repeat database are provided here.cd example_02_abinitioexport PATH=/programs/RepeatModeler-2.0:$PATHBuildDatabase -name pyu pyu_contig.fastaRepeatModeler -pa 4 -database pyu -LTRStruct >& repeatmodeler.logAt the end of run, you would find a file "pyu-families.fa". This is the file you can supply to "rmlib=" in the control file.2. Set environment to run Maker and create MAKER control files.Every steps in Maker are specified by the Maker control files. The command "maker -CTL" will create three control files: maker_bopts.ctl, maker_exe.ctl, maker_opts.ctl.by.exportPATH=/workdir/$USER/maker/bin:/workdir/$USER/RepeatMasker:/programs/snap:$PATH export ZOE=/programs/snap/Zoeexport LD_LIBRARY_PATH=/programs/boost_1_62_0/libcd /workdir/$USER/maker_tutorial/example_02_abinitiomaker -CTL3. Modify the control file maker_opts.ctl.Open the maker_opts.ctl file in a text editor (e.g. Notepad++ on Windows, BBEdit on Mac, or vi on Linux). Modify the following values. Put the modified file in the same directory“example_02_abinitio”.genome=pyu_contig.fastaest=pyu_est.fastaprotein=sp_protein.fastamodel_org=simplermlib= #fasta file of your repeat sequence from RepeatModeler. Leave blank to skip.softmask=1est2genome=1protein2genome=1TMP=/workdir/$USER/tmp #important for big genome, as the default /tmp is too smallThe modified maker_opts.ctl file instructs MAKER to do two things.a) Run RepeatMasker.The line “model_org=simple” tells RepeatMasker to mask the low complexity sequence (e.g.“AAAAAAAAAAAAA”.The line “rmlib=” sets "rmlib" to null, which tells RepeatMasker not to mask repeatsequences like transposon elements. If you have a repeat fasta file (e.g. output fromRepeatModeler) that you need to mask, put the fasta file name next to “rmlib=”The line “softmask=1” tells RepeatMasker to do soft-masking which converts repeats tolower case, instead of hard-masking which converts repeats to “N”. "Soft-masking" isimportant so that short repeat sequences within genes can still be annotated as part of gene.If you run RepeatMasker separately, as described in https:///darencard/bb10 01ac1532dd4225b030cf0cd61ce2 , you should leave rmlib to null, but set rm_gff to a repeat gff file.b) Align the transcript sequences from the pyu_est.fasta file and protein sequences from thesp_protein.fasta file to the genome and infer evidence supported gene model.The lines “est2genome=1” and “protein2genome=1” tell MAKER to align the transcriptsequences from the pyu_est.fasta file and protein sequences from the sp_protein.fasta file to the genome. These two files are used to define evidence supported gene model.The lines “est=pyu_est.fasta" and "protein=sp_protein.fasta" specify the fasta file names of the EST and protein sequences. In general, the EST sequence file contains the assembled transcriptome from RNA-seq data. The protein sequence file include proteins from closely related species or swiss-prot. If you have multiple protein or EST files, separate file names with ",".4. [Do it at home] Execute repeat masking and alignments. This step takes an hour. Run it in"screen". In the command: "mpiexec -n 2 " means that you will parallelize Maker using MPI, and use two threads at a time. When you work on a real project, it will take much longer, and you should increase this "-n" setting to the number of cores.Set Maker environment if it is new session:exportPATH=/workdir/$USER/maker/bin:/workdir/$USER/RepeatMasker:/programs/snap:$PATH export ZOE=/programs/snap/Zoeexport LD_LIBRARY_PATH=/programs/boost_1_62_0/libExecute the commands:cd /workdir/qisun/maker_tutorial/example_02_abinitio/usr/local/mpich/bin/mpiexec -n 2 maker -base pyu_rnd1 >& log1 &After it is done, you can check the log1 file. You should see a sentence: Maker is now finished!!!Part 3. Maker round 2 - Gene prediction using SNAP1. Train a SNAP gene model.SNAP is software to do ab initio gene prediction from a genome. In order to do gene prediction with SNAP, you will first train a SNAP model with alignment results produced in the previous step.If you skipped the step "4. [Do it at home] Execute Maker round 1", you can copy the result files from this directory: /shared_data/annotation2019/cd /workdir/qisun/maker_tutorial/example_02_abinitiocp /shared_data/annotation2019/pyu_rnd1.maker.output.tgz ./tar xvfz pyu_rnd1.maker.output.tgzSet Maker environment if it is new session:exportPATH=/workdir/$USER/maker/bin:/workdir/$USER/RepeatMasker:/programs/snap:$PATH export ZOE=/programs/snap/Zoeexport LD_LIBRARY_PATH=/programs/boost_1_62_0/libThe following commands will convert the MAKER round 1 results to input files for building a SNAP mode.mkdir snap1cd snap1gff3_merge -d ../pyu_rnd1.maker.output/pyu_rnd1_master_datastore_index.logmaker2zff -l 50 -x 0.5 pyu_rnd1.all.gffThe “-l 50 -x 0.5” parameter in maker2zff commands specify that only gene models with AED score>0.5 and protein length>50 are used for building models. You will find two new files: genome.ann and genome.dna.Now you will run the following commands to train SNAP. The basic steps for training SNAP are first to filter the input gene models, then capture genomic sequence immediately surrounding each model locus, and finally uses those captured segments to produce the HMM. You can explore the internal SNAP documentation for more details if you wish.fathom -categorize 1000 genome.ann genome.dnafathom -export 1000 -plus uni.ann uni.dnaforge export.ann export.dnahmm-assembler.pl pyu . > ../pyu1.hmmmv pyu_rnd1.all.gff ../cd ..After this, you will find two new files in the directory example_02_abinitio:pyu_rnd1.all.gff: A gff file from round 1, which is evidence based genes.pyu1.hmm: A hidden markov model trained from evidence based genes.2. Use SNAP to predict genes.Modify directly on the maker_opts.ctl file that you have modified previously.Before doing that, you might want to save a backup copy of maker_opts.ctl for round 1.cp maker_opts.ctl maker_opts.ctl_backup_rnd1Now modify the following values in the file: maker_opts.ctlRun maker with the new control file. This step takes a few minutes. (A real project could take hours to finish). You will use the option “-base pyu_rnd2” so that the results will be written into a new directory "pyu_rnd2".Again, make sure the log2 file ends with "Maker is now finished!!!".Part 4. Maker round 3 - Retrain SNAP model and do another round of SNAP gene predictionYou might need to run two or three rounds of SNAP . So you will repeat Part 2 again. Make sure you will replace snap1 to snap2, so that you would not over-write previous round.1. First train a new SNAP model.2. Use SNAP to predict genes.Modify directly on the maker_opts.ctl file that you have modified previously.Before doing that, you might want to save a backup copy of maker_opts.ctl for round 2.Now modify the following values in the file: maker_opts.ctlmaker_gff= pyu_rnd1.all.gffest_pass=1 # use est alignment from round 1protein_pass=1 #use protein alignment from round 1rm_pass=1 # use repeats in the gff filesnaphmm=pyu1.hmmest= # remove est file, do not run EST blast againprotein= # remove protein file, do not run blast againmodel_org= #remove repeat mask model, so not running RM againrmlib= # not running repeat masking againrepeat_protein= #not running repeat masking againest2genome=0 # do not do EST evidence based gene modelprotein2genome=0 # do not do protein based gene model.pred_stats=1 #report AED statsalt_splice=0 # 0: keep one isoform per gene; 1: identify splicing variants of the same genekeep_preds=1 # keep genes even without evidence support, set to 0 if no/usr/local/mpich/bin/mpiexec -n 2 maker -base pyu_rnd2 >& log2 &mkdir snap2cd snap2gff3_merge -d ../pyu_rnd2.maker.output/pyu_rnd2_master_datastore_index.logmaker2zff -l 50 -x 0.5 pyu_rnd2.all.gfffathom -categorize 1000 genome.ann genome.dnafathom -export 1000 -plus uni.ann uni.dnaforge export.ann export.dnahmm-assembler.pl pyu . > ../pyu2.hmmmv pyu_rnd2.all.gff ..cd ..cp maker_opts.ctl maker_opts.ctl_backup_rnd2maker_gff=pyu_rnd2.all.gffsnaphmm=pyu2.hmmRun Maker:/usr/local/mpich/bin/mpiexec -n 2 maker -base pyu_rnd3 >& log3 &Use the following command to create the final merged gff file. The “-n” option would produce a gff file without genome sequences:gff3_merge -n -dpyu_rnd3.maker.output/pyu_rnd3_master_datastore_index.log>pyu_rnd3.noseq.gff fasta_merge -d pyu_rnd3.maker.output/pyu_rnd3_master_datastore_index.logAfter this, you will get a new gff3 file: pyu_rnd3.noseq.gff, and protein and transcript fasta files. 3. Generate AED plots./programs/maker/AED_cdf_generator.pl -b 0.025 pyu_rnd2.all.gff > AED_rnd2/programs/maker/AED_cdf_generator.pl -b 0.025 pyu_rnd3.noseq.gff > AED_rnd3You can use Excel or R to plot the second column of the AED_rnd2 and AED_rnd3 files, and use the first column as the X-axis value. The X-axis label is "AED", and Y-axis label is "Cumulative Fraction of Annotations "Part 5. Visualize the gff file in IGVYou can load the gff file into IGV or JBrowse, together with RNA-seq read alignment bam files. For instructions of running IGV and loading the annotation gff file, you can read under "part 4" of this document:/doc/RNA-Seq-2019-exercise1.pdfAppendix: Training Augustus modelRun Part 1 & 2.In the same screen session, set up Augustus environment.cp -r /programs/Augustus-3.3.3/config/ /workdir/$USER/augustus_configexport LD_LIBRARY_PATH=/programs/boost_1_62_0/libexport AUGUSTUS_CONFIG_PATH=/workdir/$USER/augustus_config/export LD_LIBRARY_PATH=/programs/boost_1_62_0/libexport LC_ALL=en_US.utf-8export LANG=en_US.utf-8export PATH=/programs/augustus/bin:/programs/augustus/scripts:$PATHThe following commands will convert the MAKER round 1 results to input files for building a SNAP mode.mkdir augustus1cd augustus1gff3_merge -d ../pyu_rnd1.maker.output/pyu_rnd1_master_datastore_index.logAfter this step, you will see a new gff file pyu_rnd1.all.gff from round 1.## filter gff file, only keep maker annotation in the filtered gff fileawk '{if ($2=="maker") print }' pyu_rnd1.all.gff > maker_rnd1.gff##convert the maker gff and fasta file into a Genbank formated file named pyu.gb ##We keep 2000 bp up- and down-stream of each gene for training the modelsgff2gbSmallDNA.pl maker_rnd1.gff pyu_contig.fasta 2000 pyu.gb## check number of genes in training setgrep -c LOCUS pyu.gb## train model## first create a new Augustus species namednew_species.pl --species=pyu## initial trainingetraining --species=pyu pyu.gb## the initial model should be in the directoryls -ort $AUGUSTUS_CONFIG_PATH/species/pyu##create a smaller test set for evaluation before and after optimization. Name the evaluation set pyu.gb.evaluation.randomSplit.pl pyu.gb 200mv pyu.gb.test pyu.gb.evaluation# use the first model to predict the genes in the test set, and check theresultsaugustus --species=pyu pyu.gb.evaluation >& first_evaluate.outgrep -A 22 Evaluation first_evaluate.out# optimize the model. this step is very time consuming. It could take days. To speed things up, you can create a smaller test set# the following step will create a test and training sets. the test set has 1000 genes. This test set will be splitted into 24 kfolds for optimization (the kfold can be set up to 48, with processed with one cpu core per kfold. Kfold must be same number as as cpus). The training, prediction and evaluation will beperformed on each bucket in parallel (training on hh.gb.train+each bucket, then comparing each bucket with the union of the rest). By default, 5 rounds of optimization. As optimization for large genome could take days, I changed it to3 here.randomSplit.pl pyu.gb 1000optimize_augustus.pl --species=hh --kfold=24 --cpus=24 --rounds=3 --onlytrain=pyu.gb.train pyu.gb.test >& log &#train again after optimizationetraining --species=pyu pyu.gb# use the optionized model to evaluate again, and check the resultsaugustus --species=pyu pyu.gb.evaluation >& second_evaluate.outgrep -A 22 Evaluation second_evaluate.outAfter these steps, the species model is in the directory/workdir/$USER/augustus_config/species/pyu.Now modify the following values in the file: maker_opts.ctlmaker_gff= pyu_rnd1.all.gffest_pass=1 # use est alignment from round 1protein_pass=1 #use protein alignment from round 1rm_pass=1 # use repeats in the gff fileaugustus_species=pyu # augustus species model you just builtest= # remove est file, do not run EST blast againprotein= # remove protein file, do not run blast againmodel_org= #remove repeat mask model, so not running RM againrmlib= # not running repeat masking againrepeat_protein= #not running repeat masking againest2genome=0 # do not do EST evidence based gene modelprotein2genome=0 # do not do protein based gene model.pred_stats=1 #report AED statsalt_splice=0 # 0: keep one isoform per gene; 1: identify splicing variants of the same genekeep_preds=1 # keep genes even without evidence support, set to 0 if noRun maker with the new augustus model/usr/local/mpich/bin/mpiexec -n 2 maker -base pyu_rnd3 >& log3 &Create gff and fasta output files:Use the following command to create the final merged gff file. The “-n” option would produce a gff file without genome sequences:gff3_merge -n -dpyu_rnd3.maker.output/pyu_rnd3_master_datastore_index.log>pyu_rnd3.noseq.gff fasta_merge -d pyu_rnd3.maker.output/pyu_rnd3_master_datastore_index.logAfter this, you will get a new gff3 file: pyu_rnd3.noseq.gff, and protein and transcript fasta files. To make the gene names shorter, use the following commands:maker_map_ids --prefix pyu_ --justify 8 --iterate 1 pyu_rnd3.all.gff > id_map map_gff_ids id_map pyu_rnd3.all.gffmap_fasta_ids id_map pyu_rnd3.all.maker.proteins.fastamap_fasta_ids id_map pyu_rnd3.all.maker.transcripts.fasta。
EXE文件加密一机一码使用教程
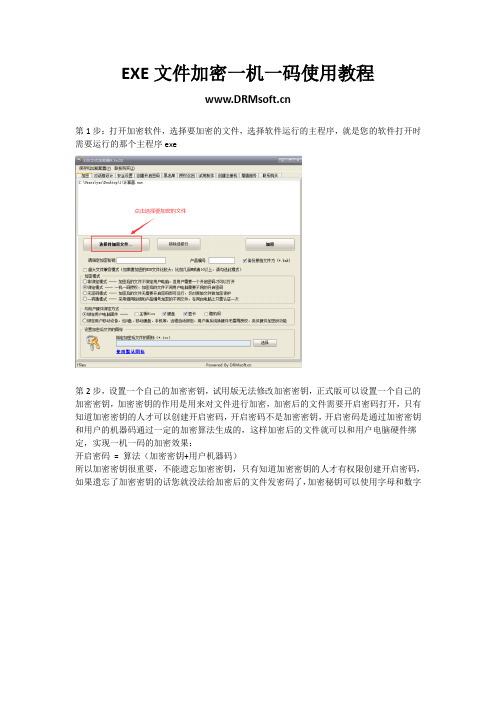
EXE文件加密一机一码使用教程第1步:打开加密软件,选择要加密的文件,选择软件运行的主程序,就是您的软件打开时需要运行的那个主程序exe第2步,设置一个自己的加密密钥,试用版无法修改加密密钥,正式版可以设置一个自己的加密密钥,加密密钥的作用是用来对文件进行加密,加密后的文件需要开启密码打开,只有知道加密密钥的人才可以创建开启密码,开启密码不是加密密钥,开启密码是通过加密密钥和用户的机器码通过一定的加密算法生成的,这样加密后的文件就可以和用户电脑硬件绑定,实现一机一码的加密效果:开启密码= 算法(加密密钥+用户机器码)所以加密密钥很重要,不能遗忘加密密钥,只有知道加密密钥的人才有权限创建开启密码,如果遗忘了加密密钥的话您就没法给加密后的文件发密码了,加密秘钥可以使用字母和数字的6位或以上的字符。
第3步设置文件编号,文件编号的作用是用来对文件进行分类,如果您有很多不同类别的文件需要加密,可以通过编号来区分不同的类别,文件编号会被加密到文件里面,用户无法修改,即使用户修改了加密后的文件名,他也无法修改文件编号,您可以通过编号知道用户打开的是哪个类别的文件,编号可以使用字母或数字,最好不要使用中文编号。
相同编号的文件可以用相同的开启密码打开,相同编号的文件只需要输入一次开启密码,不需要重复输入,如果您的文件不需要分类或者只有一个文件,那么就使用默认的文件编号就行,如果文件不需要分类可以直接用默认的编号P01,如果编号不填写的话加密系统会自动随机给文件生成一个编号。
第4步,选择加密模式非绑定模式,这个默认加密后的文件所有电脑可以使用一个相同的密码打开,非绑定模式打包后的文件所有电脑显示的机器码都是 00000-00000-00000 可以通过这个机器码和密钥生成一个公共的开启密码给不同的电脑使用;无密码模式,只对软件进行加密,加密后的文件不需要密码就可以打开,这个模式的目的只是为了防止用户修改你的原始文件。
mklink.exe用法及参数详解

最后再说几点:
创建文件或目录链接仅限于 NTFS 文件系统,且链接只能针对同一个磁盘上的文件或目录;
硬链接只能创建对本分区文件的引用,即不可创建跨分区的文件硬链接;
通过硬链接创建的别名文件在统计大小时会按原文件的大小计算,而软链接和符号链接大小均为零;
硬链接在创建时会事先检测目标文件是否存在,即不允许空链接的存在。
再将这两个目录链接移动到其它目录下,则 dira 和 dirb 均可正常指向 c:\demo\tdir;
由此可见当创建目录链接时对目标目录使用绝对路径,D 和 J 两个参数实现的目录链接效果是一样的;
[3] 只能创建文件的硬链接,使用 /H 参数创建目录的硬链接时会提示拒绝访问;
之后将 dira、dirb 移动到其它目录下,则访问 dira 时会提示“位置不可用”,访问 dirb 时仍然正常指向 tdir;
而分别用 mklink /D dira c:\demo\tdir 和 mklink /J dirb c:\demo\tdir 创建 c:\demo\tdir 的符号链接和软链接,
?目录的软链接(JUNCTION) /J
该参数可以创建目录的软链接(联接),作用基本和符号链接类似[2],NT6系统的用户目录就是以这种形式存在的;
?文件的硬链接 /H
该参数可以创建文件的硬链接,即一个文件的多个别名[3],NT6系统WinSXS目录下的大部分文件是以这个形式存在的;
[1] 创建符号链接和软链接时允许目标文件或目录不存在,此时若访问链接文件会得到一个“位置不可用”的提示,
若之后重新将目标文件或目录补上(路径、名称一致即可),则该链接仍然有效;
[2] 目录符号链接和软链接的区别在于,软链接在创建时会自动引用目标目录的绝对路径,而符号链接允许相对路径的引用,
makecode基础操作

makecode基础操作
makecode是一款为初学者提供的编程工具,它可以帮助你用可视化的方式学习编程。
以下是 makecode 的基础操作:
第一步:打开 makecode 网站或软件。
第二步:选择你要编程的设备类型,比如 micro:bit。
第三步:打开代码编辑器,你可以在这里输入代码或使用可视化块来编程。
第四步:在代码编辑器中输入代码或使用可视化块来编程。
你可以在编程过程中使用 makecode 提供的帮助文档和示例代码。
第五步:点击“运行”按钮来测试你的代码。
如果出现错误,会有提示信息帮助你找到错误的位置。
第六步:当你完成编程后,你可以将代码下载到你的设备上进行运行。
以上就是 makecode 的基础操作,希望对初学者有所帮助。
- 1 -。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
使用软件Makekml.exe将工参表和路测数据导入
到Google Earth中
一、将工参表导入到Google Earth中
1.首先将文本格式的工参表转换为Excel格式。
(可直接修改后缀名或者将文本内容复制粘贴到Excel表中)
2.运行软件Makekml.exe,界面如下:
3.单击基站表-》KML,弹出下图,输出路径及扇区半径的大小可自己定义。
如下图:
4.点击生成KML文件,等待一会完成。
找到生成的KML文件用Google earth 打开,即可在Google earth中查看基站信息了。
二、将路程数据导入到Google earth中。
过程大概和工参表的导入过程一样,只需单击DT数据》KML
不过需要说明的一点是:鼎力的log 后缀名是.rcu, 需要在Pioneer 软件中将.rcu转换为.txt格式。
大致的步骤如下:
1.运行Pioneer软件,点击编辑》数据》导入出现如下界面
2.空白处右击选择第三项,打开原文件*****.rcu
3.然后点击编辑》数据》导出,出现以下界面,勾选相关参数
4.点击OK后,弹出对话框,设置保存路径。
至此.rcu成功转换为.txt
5.最后参照工参表的导入过程即可完成将路测数据转换成能用
Google earth 打开的KML文件。
工参表和路测数据在Google earth 中同时显示如下图:。