稳定性同位素内标与质谱检测
质谱标准工作曲线和内标法 理论说明

质谱标准工作曲线和内标法理论说明1. 引言1.1 概述在化学和生物分析领域中,质谱标准工作曲线和内标法是常用的方法,用于定量分析和质量控制。
质谱标准工作曲线是一种建立样品中目标分析物浓度与其质谱信号响应之间关系的方法。
而内标法则是通过引入稳定同位素的化合物来校正实验过程中的变异性,以提高分析结果的准确性和可靠性。
1.2 文章结构本文将详细阐述质谱标准工作曲线和内标法的原理、步骤、优缺点以及应用领域差异等内容。
首先,我们将对质谱标准工作曲线进行定义与原理的介绍,并探讨构建标准曲线的步骤和相应的曲线拟合与评估方法。
其次,我们会解释内标法的概念与作用,并分享选择和优化内标的方法。
进一步,我们将介绍如何进行内标校正计算并解读结果。
最后,我们将比较质谱标准工作曲线和内标法之间的优缺点,并说明它们在不同应用领域下的选用依据。
此外,我们还会通过实验操作流程示例案例的讲解,更加直观地说明这两种方法的应用。
1.3 目的本文的目的是帮助读者对质谱标准工作曲线和内标法有一个全面的理解。
我们将从理论层面出发,解释它们原理和操作步骤,并分析其优缺点以及适用领域。
通过深入了解这些方法,读者可以更好地应用于实际工作中,提高分析结果的准确性和可靠性。
2. 质谱标准工作曲线:2.1 定义与原理:质谱标准工作曲线是一种用于定量分析的方法,通过建立目标物质的浓度与其对应峰面积或峰高的关系曲线来推断样品中目标物质的浓度。
这个曲线通常是在质谱仪中进行绘制和评估的。
该方法基于以下原理:当已知一个物质(即内标)与需要定量分析的物质具有相似的化学特性和相近的化学反应,且能够在样品预处理过程中稳定存在时,我们可以利用内标来纠正可能由样品前处理过程引起的变异。
通过构建一系列内标浓度不同、但相对恒定的样品,并测量它们产生的响应信号,我们可以获得内标响应与内标浓度之间的关系。
2.2 构建标准曲线的步骤:构建质谱标准工作曲线一般包括以下步骤:a) 准备一系列不同含量(浓度)已知目标物质的溶液。
3_种常用碳青霉烯类抗生素血药浓度UPLC-MS

3种常用碳青霉烯类抗生素血药浓度UPLC-MS/MS检测方法的建立Δ秦怡1*,张瑞霞2,吕雅瑶2,翁莉莉1,张弋2 #(1.天津医科大学一中心临床学院,天津 300192;2.天津市第一中心医院药学部,天津 300192)中图分类号 R917;R978.1文献标志码 A 文章编号 1001-0408(2024)03-0343-05DOI 10.6039/j.issn.1001-0408.2024.03.14摘要目的建立3种临床常用碳青霉烯类抗生素——厄他培南(ETP)、亚胺培南(IPM)、美罗培南(MEM)血药浓度检测的超高效液相色谱-质谱联用(UPLC-MS/MS)法。
方法血浆样品经甲醇沉淀蛋白后,以3种抗生素的稳定性同位素(ETP-D4、IPM-D4、MEM-D6)为内标,采用ACQUITY UPLC BEH C18(2.1 mm×50 mm,1.7μm)色谱柱分离;流动相为98%乙腈+2%水+0.1%甲酸和98%水+2%乙腈+0.1%甲酸,梯度洗脱;流速为0.3 mL/min;柱温为40 ℃;采用正离子、多反应监测模式进行扫描分析。
结果该方法专属性良好,在ETP、IPM、MEM 0.2~200、0.1~100、0.1~100μg/mL范围内线性良好(r2≥0.993),批内、批间精密度和准确度良好(RE均≤5.14%,RSD均≤11.15%),基质效应、提取回收率较一致(RSD≤12.99%)。
结论本实验建立了一种可以同时定量ETP、IPM、MEM血药浓度的UPLC-MS/MS法,该方法样品前处理简单、检测时间短、所需样品量少,可满足临床需求。
关键词碳青霉烯类抗生素;超高效液相色谱-质谱联用;血药浓度;厄他培南;亚胺培南;美罗培南Establishment of UPLC-MS/MS method for the determination of plasma concentration of three common carbapenem antibioticsQIN Yi1,ZHANG Ruixia2,LYU Yayao2,WENG Lili1,ZHANG Yi2(1. First Central Clinical College of Tianjin Medical University,Tianjin 300192,China;2. Dept. of Pharmacy,Tianjin First Central Clinical Hospital,Tianjin 300192, China)ABSTRACT OBJECTIVE To establish a UPLC-MS/MS method for the determination of plasma concentration of three carbapenem antibiotics,i.e. ertapenem (ETP),imipenem (IPM)and meropenem (MEM).METHODS After protein precipitation with methanol,the plasma samples were separated by ACQUITY UPLC BEH C18column (2.1mm×50mm,1.7μm)using stable isotopes of three antibiotics (ETP-D4,IPM-D4,MEM-D6)as the internal standard. The mobile phases were 98%acetonitrile +2% water +0.1%formic acid and 98%water +2%acetonitrile +0.1%formic acid,by gradient elution. The flow rate was 0.3mL/min and the column temperature was 40 ℃. Scanning analysis was performed in the positive ion and multiple reaction monitoring mode. RESULTS The method had good specificity,good linearity (r2≥0.993)in the range of 0.2-200,0.1-100and 0.1-100μg/mL of ETP,IPM and MEM,and good intra-batch and inter-batch precision and accuracy (all RE≤5.14%,all RSD≤11.15%),the matrix effect and extraction recovery were consistent (RSD≤12.99%). CONCLUSIONS This study establishes the UPLC-MS/MS method to simultaneously quantify the plasma concentration of ETP,IPM and MEM. The method has the advantages of simple pretreatment, short detection time and small sample quantity to meet clinical requirement.KEYWORDS carbapenem antibiotics; UPLC-MS/MS; plasma concentration; ertapenem; imipenem; meropenem碳青霉烯类抗生素具有抗菌谱广、抗菌活性强、耐药率低的特点,已成为治疗重症感染的主要选择。
比较蛋白质组学研究中的稳定同位素标记技术

进展评述比较蛋白质组学研究中的稳定同位素标记技术刘新1,2 应万涛1,2 钱小红1,23(1军事医学科学院放射与辐射医学研究所 北京 100850;2北京蛋白质组研究中心 北京 102206)摘 要 比较蛋白质组学是指在蛋白质组学水平上研究正常和病理情况下细胞或组织中蛋白质表达变化,以期发现具有重要功能的生物标识物,为疾病的早期诊断提供依据。
近年来它正成为蛋白质组学研究的热点和发展趋势。
比较蛋白质组学的研究方法和策略有多种,本文就最近几年来稳定同位素标记技术(体内代谢标记技术和体外化学标记技术)在比较蛋白质组学研究中的进展进行综述。
关键词 比较蛋白质组学 稳定同位素标记 体内代谢标记 体外化学标记Application of Stable Isotope Labeling in Comparative ProteomicsLiu X in1,2,Y ing Wantao1,2,Qian X iaohong1,23(1Beijing Institute of Radiation Medicine,Beijing100850;2Beijing Proteome Research Center,Beijing102206)Abstract C omparative proteomics is the research of protein expression changing between normal and pathological cell or tissue on the proteome level.P otential biomarkers w ould be discovered from the research by comparative proteomics, which will be helpful to the diagnosis and therapy of diseases.In the recent years,it has been becoming the hot spot of the proteomics research and many strategies used in comparative proteomics have been developed.During those approaches,the strategies based on stable is otopic labeling coupled with mass spectrometry have been extensively used and lots of success ful applications have been reported.In contrast to the traditional radioactive is otope labeling method,stable is otope labeling technique was not radioactive and the operation is simple.Metabolic labeling in viv o and chemical labeling in vitro are tw o parts of stable is otope labeling technique,which both have various advantages and disadvantages.This paper reviewed the progress of stable is otope labeling technique in comparative proteomics.K ey w ords C omparative proteomics,S table is otope labeling,Metabolic labeling in viv o,Chemical labeling in vitro随着人类基因组精确图谱的公布,基因组功能的阐明已经成为生命科学研究中一项极重要的任务[1]。
质谱介绍及质谱图的解析(2)

质谱介绍及质谱图的解析(2)5. 傅⾥叶变换分析器在⼀定强度的磁场中,离⼦做圆周运动,离⼦运⾏轨道受共振变换电场限制。
当变换电场频率和回旋频率相同时,离⼦稳定加速,运动轨道半径越来越⼤,动能也越来越⼤。
当电场消失时,沿轨道飞⾏的离⼦在电极上产⽣交变电流。
对信号频率进⾏分析可得出离⼦质量。
将时间与相应的频率谱利⽤计算机经过傅⾥叶变换形成质谱。
其优点为分辨率很⾼,质荷⽐可以精确到千分之⼀道尔顿。
四、串联质谱及联⽤技术1. 串联质谱两个或更多的质谱连接在⼀起,称为串联质谱。
最简单的串联质谱(MS/MS)由两个质谱串联⽽成,其中第⼀个质量分析器(MS1)将离⼦预分离或加能量修饰,由第⼆级质量分析器(MS2)分析结果。
最常见的串联质谱为三级四极杆串联质谱。
第⼀级和第三级四极杆分析器分别为MS1和MS2,第⼆级四极杆分析器所起作⽤是将从MS1得到的各个峰进⾏轰击,实现母离⼦碎裂后进⼊MS2再⾏分析。
现在出现了多种质量分析器组成的串联质谱,如四极杆-飞⾏时间串联质谱(Q-TOF)和飞⾏时间-飞⾏时间(TOF-TOF)串联质谱等,⼤⼤扩展了应⽤范围。
离⼦阱和傅⾥叶变换分析器可在不同时间顺序实现时间序列多级质谱扫描功能。
MS/MS最基本的功能包括能说明MS1中的母离⼦和MS2中的⼦离⼦间的联系。
根据MS1和MS2的扫描模式,如⼦离⼦扫描、母离⼦扫描和中性碎⽚丢失扫描,可以查明不同质量数离⼦间的关系。
母离⼦的碎裂可以通过以下⽅式实现:碰撞诱导解离,表⾯诱导解离和激光诱导解离。
不⽤激发即可解离则称为亚稳态分解。
MS/MS在混合物分析中有很多优势。
在质谱与⽓相⾊谱或液相⾊谱联⽤时,即使⾊谱未能将物质完全分离,也可以进⾏鉴定。
MS/MS可从样品中选择母离⼦进⾏分析,⽽不受其他物质⼲扰。
MS/MS在药物领域有很多应⽤。
⼦离⼦扫描可获得药物主要成分,杂质和其他物质的母离⼦的定性信息,有助于未知物的鉴别,也可⽤于肽和蛋⽩质氨基酸序列的鉴别。
同位素内标法——超高效液相色谱

基金项目:广西科技重大专项(编号:桂科A A 22068085)作者简介:石金娥,女,梧州学院正高级工程师,博士.通信作者:奚广生(1967 ),男,梧州学院教授,博士.E Gm a i l :1790341091@q q .c o m 收稿日期:2023G07G18㊀㊀改回日期:2023G12G11D O I :10.13652/j .s p j x .1003.5788.2023.80672[文章编号]1003G5788(2024)03G0089G06同位素内标法 超高效液相色谱/串联质谱法测定六堡茶中苦参碱和氧化苦参碱残留量D e t e r m i n a t i o n t h e r e s i d u e s o fm a t r i n ea n do x y m a t r i n e i nL i u p a o t e ab yU P L C GM S /M Sw i t h i s o t o pe i n t e r n a l s t a n d a r dm e t h o d 石金娥1,2S H IJ i n e 1,2㊀张洪禹3Z HA N G H o n g y u 3㊀辛若竹4X I N R u o z h u 4㊀丁㊀梅4D I N G M e i 4㊀奚广生1,2X IG u a n g s h e n g1,2(1.梧州学院食品与制药工程学院,广西梧州㊀543002;2.梧州学院六堡茶现代产业学院,广西梧州㊀543002;3.梧州学院体育健康学院,广西梧州㊀543002;4.梅河口市食品药品检验检测中心,吉林梅河口㊀135000)(1.S c h o o l o f F o o da n dP h a r m a c e u t i c a lE n g i n e e r i n g ,W u z h o uC o l l e g e ,W u z h o u ,G u a n gx i 543002,C h i n a ;2.L i u p a oT e a M o d e r nI n d u s t r y C o l l e g e ,W u z h o uC o l l e g e ,W u z h o u ,G u a n gx i 543002,C h i n a ;3.S c h o o l o f P h y s i c a lE d u c a t i o na n d H e a l t h ,W u z h o uC o l l e g e ,W u z h o u ,G u a n gx i 543002,C h i n a ;4.M e i h e k o uC e n t e r f o rF o o da n dD r u g Co n t r o l ,M e i h e k o u ,J i l i n 135000,C h i n a )摘要:目的:以广西六堡茶为例,建立超高效液相色谱 质谱/质谱(U P L C GM S /M S )分析方法测定苦参碱和氧化苦参碱残留量.方法:样品经乙腈水溶液(80%乙腈+0.2%氨水)超声提取,流动相为0.1%甲酸 水和0.1%甲酸 甲醇溶液,梯度洗脱,内标法定量.结果:苦参碱和氧化苦参碱在0.1~80.0n g /m L 质量浓度范围内呈线性,相关系数均>0.9996,检出限均为1.0μg /k g ,定量限均为3.0μg /k g ,满足茶叶中苦参碱和氧化苦参碱残留量的监管判定要求.通过加标验证,回收率均为92.0%~104.7%,相对标准偏差(R S D )均<5.4%.结论:试验方法结合净化管净化(147.7m g P S A ,15.1m g G C B ,887.2m g 硫酸镁),以K i n e t e x2.6μm B i p h e n yl 100Å色谱柱(100mmˑ3.0mm )分离,解决了苦参碱和氧化苦参碱提取回收率低㊁前处理复杂的问题,使苦参碱和氧化苦参碱出峰良好,抗干扰性强.关键词:六堡茶;苦参碱;氧化苦参碱;U P L C GM S /M SA b s t r a c t :O b je c t i v e :T a k i n g G u a n g x iL i u p a o t e aa sa ne x a m p l e ,a n U P L C GM S /M S a n a l y s i s m e t h o d w i t h r e s i d u a la m o u n t s of m a t r i n e a n dO x y m a t r i n ew a s e s t a b l i s h e d .M e t h o d s :T h e p r e pa r e d s a m p l e sw e r e s ub j ec t ed t o o p t i m i ze d a c e t o n i t r i l e a q u e o u s s o l u t i o n (80%a c e t o n i t r i l e +0.2%a mm o n i aw a t e r )u l t r a s o n i c e x t r a c t i o n,m o b i l e p h a s e :f o r m i c a c i dw a t e r a n d f o r m i c a c i dm e t h a n o l (b o t h0.1%),g r a d i e n t e l u t i o n ,a n d i n t e r n a l s t a n d a r dm e t h o d q u a n t i f i c a t i o n .R e s u l t s :I tw a s l i n e a r i nt h er a n g eo f0.1~80.0n g/m L ,r w a s gr e a t e rt h a n 0.9996,t h e d e t e c t i o n l i m i t s o f m a t r i n e a n d o x y m a t r i n ew e r e1.0μg /k g,a n dt h e q u a n t i t a t i v el i m i t s w e r e 3.0μg /k g ,w h i c hm e t t h e r e g u l a t o r y d e t e r m i n a t i o n r e qu i r e m e n t s f o r t h e r e s i d u e o fm a t r i n e a n do x i d a t i v em a t r i n e i nt e a .T h r o u gh t h es p i k e v e r i f i c a t i o n ,t h er e c o v e r y r a t e w a si nt h er a n g e o f 92.0%~104.7%,a n d t h e r e l a t i v e s t a n d a r dd e v i a t i o n (R S D )w a sl e s s t h a n5.4%.C o n c l u s i o n :M e t h o dc o m b i n e dw i t h p u r i f i c a t i o n t u b e p u r i f i c a t i o n (147.7m g P S A ,15.1m g G C B ,887.2m g m a g n e s i u ms u l f a t e ),u s i n g K i n e t e x 2.6μm B i p h e n yl100Åc h r o m a t o g r a p h i c c o l u m n (100mmˑ3.0mm )s e pa r a t i o ns o l v e s t h e p r ob l e m so f l o w e x t r ac t i o nr e c o v e r y r a t ea n dc o m p l e x p r e Gt r e a t m e n t o f m a t r i n ea n do x y m a t r i n e ,s ot h a tt h es y mm e t r i c a l p e a ks h a p ea n ds t r o n g a n t i Gi n t e r f e r e n c ea b i l i t y o f m a t r i n ea nd o x ym a t r i n e .K e yw o r d s :L i u p a o t e a ;m a t r i n e ;o x y m a t r i n e ;U P L C GM S /M S 六堡茶为黑茶的一种,以 红㊁浓㊁陈㊁醇 著称.与其他茶树一样,六堡茶茶树每年也会受小绿叶蝉㊁茶尺蠖㊁茶毛虫㊁茶丽纹象甲等虫害的侵袭[1-3].茶园中常用苦参碱㊁印楝素㊁鱼藤酮㊁藜芦碱等植物源农药防治小绿叶蝉㊁茶尺蠖等虫害[4-7],植物源农药已成为茶园病虫害绿色防控的新趋势[8-10].其中苦参碱和氧化苦参碱属于喹98F O O D &MA C H I N E R Y 第40卷第3期总第269期|2024年3月|诺里西啶类生物碱,为高效㊁广谱植物源杀虫剂[11],联合使用更具速效和持久毒杀效果[12-15].但一定剂量的苦参碱和氧化苦参碱会导致神经㊁肝损伤等[16].G B/T19630 2019中有机产品可以施用苦参碱和氧化苦参碱等植物源农药,但G B2763 2021中却未对茶叶中苦参碱和氧化苦参碱的施用残留量及检测方法做出任何规定.目前,有关苦参碱检测的研究较多,而氧化苦参碱的研究却鲜见报道[17],主要涉及的检测技术有G C 技术[18]㊁L C技术[19]㊁G C/M S技术[20-21]㊁H P L CGM S/M S 技术等[22-23],多采用外标法或基质外标法为主要定量方法,且多数方法回收率不高[24-26].研究拟结合苦参碱和氧化苦参碱的性质,通过优化色㊁质谱条件及样品前处理过程,采用同位素内标法定量,建立U P L CGM S/M S法测定六堡茶中苦参碱和氧化苦参碱残留量的测定方法,为茶叶中苦参碱和氧化苦参碱残留量检测以及风险评估提供依据.1㊀材料与方法1.1㊀材料与仪器1.1.1㊀材料与试剂甲醇㊁甲酸㊁乙腈㊁甲酸铵:色谱纯,美国M R E D A 公司;浓氨水㊁无水硫酸钠:化学纯,北京化工厂;Q E C h E R S提取盐(1.5g N a A c,6g M g S O4)㊁Q u E C h E R S净化管1(15.1m g G C B,147.7m g P S A,887.2m g M g S O4)㊁Q u E C h E R S净化管2(200m g G C B,400m g C18,400m g P S A,1200m g M g S O4):美国A g i l e n t公司;0.2μm滤膜:美国P a l l公司;苦参碱㊁氧化苦参碱标准物质:纯度ȡ98.9%,美国C e r i l l i a n t公司;苦参碱GD3㊁氧化苦参碱GD3:纯度ȡ98%,加拿大T r c 公司;六堡茶样品:不同年份㊁不同季节的六堡茶16批次(2012年春茶㊁2012年秋茶㊁2012年老树茶㊁2014年夏茶㊁2014年秋茶㊁2016年社前茶㊁2016年明前茶㊁2018年春茶㊁2018年秋茶㊁2020年春茶㊁2020年秋茶㊁2020年老茶婆㊁2020年金花茶㊁2022年春茶㊁2022年秋茶㊁2022年老茶婆),市售.1.1.2㊀主要仪器设备超高效液相色谱 串联质谱仪:T S Q E n d u r a型,美国T h e r m oF i s h e r公司;涡旋混合器:I K A型,欧莱博科学仪器有限公司;超声波清洗器:D QG110E型,瑞莱博科技有限公司;离心机:A l l e g r a64型,美国B e c k m a n公司;氮吹仪:T T LGD C型,联泰科技发展有限公司.1.2㊀方法1.2.1㊀标准溶液配制(1)标准储备液:以甲醇为溶剂分别配制苦参碱㊁氧化苦参碱㊁苦参碱GD3㊁氧化苦参碱GD3标准储备液(1m g/m L),于-18ħ避光保存.(2)混合标准使用液:取苦参碱和氧化苦参碱标准储备液,用50%的甲醇溶液(含0.1%甲酸)配制,得到质量浓度分别为10,100μg/L的苦参碱和氧化苦参碱混合标准使用液.(3)内标使用液:取苦参碱GD3和氧化苦参碱GD3标准储备液,用50%甲醇溶液(含0.1%甲酸)配制,得到苦参碱GD3质量浓度为10μg/m L,氧化苦参碱GD3质量浓度为1μg/m L.(4)混合标准工作溶液:分别吸取10μg/L的混合标准使用液10,50,100μL及100μg/L的混合标准使用液50,100,200,500,800μL于样品瓶中,加内标使用液10μL,用含0.1%甲酸的甲醇溶液(V甲酸ʒV甲醇为1ʒ1)定容至1.0m L,混匀.配制的混合标准工作溶液质量浓度分别为0.1,0.5,1.0,5.0,10.0,20.0,50.0,80.0μg/L.其中苦参碱GD3质量浓度为100μg/L,氧化苦参碱GD3质量浓度为10μg/L.1.2.2㊀样品前处理㊀称取0.5g(精确至0.01g)试样于50m L玻璃离心管中,加入50μL内标混合使用液,加入10m L含0.2%氨水的乙腈水溶液(V氨水ʒV乙腈为4ʒ1),涡旋60s,超声10m i n,加入5g无水N a2S O4,再次涡旋2m i n,离心5m i n(9500r/m i n,4ħ).吸取5.0m L上清液于Q u E C h E R S净化管1中,涡旋2m i n,离心5m i n (5000r/m i n,4ħ).将2.00m L上述液体转移至另一支10m L玻璃离心管中,氮吹近干(40ħ水浴),加入1.0m L含0.1%甲酸的50%甲醇溶液溶解残渣,涡旋1m i n,过0.22μm滤膜,上机测定.1.2.3㊀液相色谱条件㊀色谱柱为K i n e t e x 2.6μm B i p h e n y l100Å(100mmˑ3.0mm)或等效柱;柱温35ħ;流动相A泵为水相(0.1%甲酸),B泵为甲醇相(0.1%甲酸),流速0.35m L/m i n;进样量5μL;洗脱程序见表1.表1㊀流动相配比及洗脱条件T a b l e1㊀M o b i l e p h a s e r a t i o a n de l u t i o n c o n d i t i o n s保留时间/m i n流动相A/%流动相B/%0.09551.060406.015856.15958.05958.195511.095509安全与检测S A F E T Y&I N S P E C T I O N总第269期|2024年3月|1.2.4㊀质谱条件㊀电离方式为电喷雾离子源正离子模式,多反应监测模式采集;电喷雾电压5500V;气帘气压力207k P a;辅助气压力483k P a;雾化气压力345k P a;离子源温度500ħ;碰撞气6m L/m i n;离子对㊁保留时间等参数见表2.1.2.5㊀线性范围测定㊀按优化后的参数和条件对标准工作液系列进行测定,以苦参碱标准溶液中待测组分峰面积与相应氧化苦参碱内标物峰面积的比值为纵坐标,苦参碱标准溶液中待测组分浓度与相应氧化苦参碱内标物浓度的比值为横坐标,绘制内标 校准工作曲线.表2㊀去簇电压㊁离子对及碰撞能量值†T a b l e2㊀D e c l u s t e r i n g v o l t a g e,i o n p a i r s,a n d c o l l i s i o ne n e r g y v a l u e s化合物保留时间/m i n母离子(m/z)产物离子(m/z)驻留时间/m s去簇电压/V碰撞能量/e V 苦参碱2.76249.2148.3∗100.0100.042150.3100.0100.042苦参碱GD32.75252.2151.3100.090.047氧化苦参碱2.95265.2247.3∗100.089.035148.2100.089.040氧化苦参碱GD32.93268.2250.3100.080.039㊀㊀㊀㊀†㊀ ∗ 为定量离子.1.2.6㊀定量限及检出限测定㊀向基质中添加一定浓度的目标物及内标物,以3倍信噪比(S/N=3)对应的目标物浓度计算检出限,以10倍信噪比(S/N=10)对应的目标物浓度计算定量限.1.2.7㊀回收率和精密度测定㊀向基质中定量添加目标物及内标物,按建立的方法测定6次,并计算方法回收率和精密度.2㊀结果与分析2.1㊀前处理条件优化2.1.1㊀提取溶剂选择㊀因苦参碱和氧化苦参碱为碱性水溶性化合物[23],考虑茶叶基质(含鞣酸等酸性物质)的特殊性,仅对中性和碱性提取体系进行考察.分别对水复溶+1%氨水乙腈提取㊁水复溶+含0.2%氨水的乙腈水溶液(V氨水ʒV乙腈为4ʒ1)提取和直接用含0.2%氨水的乙腈水溶液(V氨水ʒV乙腈为4ʒ1)提取3种方式进行回收率考察.试验发现,直接用80%乙腈水溶液(含0.2%氨水)对茶叶样品进行提取的回收率最高,可能是水的前期参与会使茶叶溶出大量的酸性物质,这些酸性物质会与苦参碱发生反应[25],导致回收率较低;但直接用80%乙腈水溶液(含0.2%氨水)提取时,因过量的氨水可以中和茶叶基质中的酸性物质,保护了苦参碱,从而有利于提取效率的提升.当直接用80%乙腈水溶液(含0.2%氨水)提取时,苦参碱回收率为93.8%~103.7%,氧化苦参碱回收率为92.0%~104.7%,结果均满意.2.1.2㊀净化方法优化㊀分别选用无水硫酸钠和Q u E C h E R S盐为提取盐,用80%乙腈水溶液(含0.2%氨水)对加标样品进行提取,选择Q u E C h E R S净化管1㊁Q u E C h E R S净化管2进行净化,考察净化方式对回收率的影响.由表3可知,当以无水硫酸钠为提取盐表3㊀净化方法对回收率的影响T a b l e3㊀E f f e c t o f d i f f e r e n t p u r i f i c a t i o nm e t h o d s o nr e c o v e r y r a t e%净化方法苦参碱氧化苦参碱Q u E C h E R S提取盐+Q u E C h E R S净化管23320无水硫酸钠+Q u E C h E R S净化管24228无水硫酸钠+Q u E C h E R S净化管18685Q u E C h E R S提取盐+Q u E C h E R S净化管19387Q u E C h E R S净化管1净化时,苦参碱和氧化苦参碱的回收率较好,其余组合回收率均较低,可能是Q u E C h E R S净化管2中大量的石墨化炭黑(G C B)对苦参碱和氧化苦参碱有较强的吸附[24],导致苦参碱和氧化苦参碱的回收率偏低.2.2㊀仪器条件优化2.2.1㊀色谱条件优化㊀分别以0.1%甲酸水 甲醇㊁0.1%甲酸水 0.1%甲酸 甲醇㊁0.1%甲酸水 0.1%甲酸 乙腈㊁5mm o l/L甲酸铵溶液(含0.1%甲酸) 0.1%甲酸 甲醇为流动相,考察不同配比的流动相对目标物的洗脱效果,结果见表4.由表4可知,以甲醇为主体的流动相使得苦参碱和氧化苦参碱的响应值和分离效果均较好,且甲酸浓度增加后,响应效果更显著;甲酸铵的存在同样能得到较高的响应值和较好的分离效果,但从配制便捷的角度考虑,优先选择0.1%甲酸水 0.1%甲酸甲醇为流动相.2.2.2㊀质谱条件优化㊀按试验条件进样分析,进一步验证所选离子对的响应值和稳定性,最终确定了4种目标物的离子对,苦参碱和氧化苦参碱及内标物溶液的M R M色谱图如图1所示.19|V o l.40,N o.3石金娥等:同位素内标法 超高效液相色谱/串联质谱法测定六堡茶中苦参碱和氧化苦参碱残留量表4㊀流动相对苦参碱和氧化苦参碱响应值的影响T a b l e 4㊀E f f e c t s o f d i f f e r e n t f l o w p h a s e so n t h e r e s po n s e v a l u e s o fm a t r i n e a n do x ym a t r i n e 流动相苦参碱氧化苦参碱0.1%甲酸水 甲醇1.755ˑ1052.171ˑ1050.1%甲酸水 0.1%甲酸 甲醇1.927ˑ1052.420ˑ1050.1%甲酸水 0.1%甲酸 乙腈1.019ˑ1051.289ˑ1055mm o l /L 甲酸铵溶液(含0.1%甲酸) 0.1%甲酸 甲醇1.904ˑ1052.644ˑ1052.3㊀方法的校准曲线和检出限由表5可知,苦参碱和氧化苦参碱在0.1~80.0n g/m L 质量浓度范围内线性关系良好,相关系数均>0.9996.以3倍信噪比(S /N =3)对应的加标量为检出限,10倍信噪比(以S /N =10)对应的加标量为定量限,测得苦参碱和氧化苦参碱的检出限均为1.0μg /k g ,定量限均为3.0μg /k g.2.4㊀方法的精密度和加标回收率茶叶样品按5个浓度水平加标,分别测定6次,其回收率及相对标准偏差见表6.由表6可知,苦参碱的相对图苦参碱㊁氧化苦参碱及内标物的色谱图F i g u r e 1㊀M u l t i r e a c t i o nm o n i t o r i n g c h r o m a t o g r a m s o fm a t r i n e ,o x y m a t r i n e a n d t h e i r i s o t o pe i n t e r n a l s t a n d a r d s o l u t i o n s (50n g/m L )表5㊀校准曲线及线性范围T a b l e 5㊀C a l i b r a t i o n c u r v e a n d l i n e a r r a n ge 目标物出峰时间/m i n回归方程相关系数线性范围/(n gm L -1)苦参碱㊀㊀2.76Y =4.31349x +0.180630.999670.1~80.0氧化苦参碱2.95Y =0.64038x +0.001200.999610.1~80.0标准偏差为1.7%~4.8%,回收率为93.8%~103.7%;氧化苦参碱的相对标准偏差为2.1%~5.3%,回收率为92.0%~104.7%;均满足G B /T27404 2008的要求,说明试验方法准确可靠.2.5㊀样品测试对采购的16种不同年份㊁不同季节的六堡茶进行测定,结果显示,苦参碱和氧化苦参碱均未检出,说明在苦参碱和氧化苦参碱类生物类农药考察中,六堡茶具有较高的安全性.3㊀结论通过对色谱㊁质谱条件及样品前处理过程的不断优化,建立了一种内标法测定六堡茶中苦参碱和氧化苦参碱残留量的方法.该方法缩短了茶叶中苦参碱和氧化苦参碱的前处理周期,提高了检测效率,且该方法具有良好的线性范围和较高的回收率,灵敏度㊁准确度㊁精密度均满足G B /T27404 2008要求.对市售六堡茶进行摸底,结果显示六堡茶中均未检出苦参碱和氧化苦参碱.后续29安全与检测S A F E T Y &I N S P E C T I O N 总第269期|2024年3月|表6㊀精密度及回收率结果T a b l e6㊀P r e c i s i o na n d r e c o v e r y m e a s u r e m e n t r e s u l t s(n=6)目标物本底值/(μg k g-1)加入量/(μg k g-1)平均检出量/(μg k g-1)回收率/%相对标准偏差/%苦参碱㊀㊀0.010.9494.04.00.055.12102.43.20.01010.37103.74.80.05046.8893.81.70.0100103.01103.02.9氧化苦参碱0.010.9292.04.20.055.23104.64.90.0109.8298.25.30.05052.34104.73.80.0100102.45102.42.1将以六大茶类为研究对象,确认该方法在六大茶类中的适用性,同时对市售六大茶类在施用苦参碱和氧化苦参碱杀虫剂的安全性进行考察,以期该方法有更广泛的适用性.参考文献[1]张勇.安徽省郎溪县茶树病虫害统防统治与绿色防控融合示范研究[J].农家参谋,2022(17):58G60.ZHANG Y.Demonstration study on the integration of integrated control and green control of tea plant diseases and pests in Langxi County, Anhui Province[J].The Farmers Consultant,2022(17):58G60.[2]汪敏.茶树病虫害检测及防治信息挖掘与可视化分析[D].合肥:安徽农业大学,2020:1G3.WANG M.Tea tree diseases and insect pests detection and centrol information mining and visual analysis[D].Hefei:Anhui Agricultural University,2020:1G3.[3]陈文义.常宁市茶树病虫害综合防治系统研究与设计[D].长沙:中南林业科技大学,2019:1G6.CHEN W Y.Research and design of integrated pest control system for tea plant in Changning city[D].Changsha:Central South University of Forestry and Technology,2019:1G6.[4]程长松,饶漾萍,李罡,等.性引诱剂与苦参碱组合防治茶毛虫效果[J].湖北植保,2022(1):30G32.CHENG C S,RAO Y P,LI Z,et al.Effect of sex attractant combined with matrine on controlling tea caterpillar[J].Hubei Plant Protection,2022(1):30G32.[5]吴庆丽,秦刚,黄艳飞,等.3种植物源农药对茶尺蠖的防治效果[J].农药,2020,59(5):379G381.WU Q L,QIN G,HUANG Y F,et al.Efficacy of three botanical insecticides against ectropis oblique prout[J].Agrochemicals,2020, 59(5):379G381.[6]龙同,乐群芬,杨国琼,等.几种杀虫剂对茶丽纹象甲的室内㊁田间药效比较试验[J].湖北农业科学,2019,58(24):109G112.LONG T,LE Q F,YANG G Q,et al.The laboratory and fieldcomparative effects of several pesticides against myllocerinus aurolineatus[J].Hubei Agricultural Sciences,2019,58(24):109G112.[7]王礼中,姚惠明,唐美君,等.几种新药剂对茶叶瘿螨的防治效果[J].中国茶业,2022,44(2):56G58.WANG L Z,YAO H M,TANG M J,et al.The control effects ofseveral new pesticides on calacarus carinatus[J].China Tea,2022,44 (2):56G58.[8]HOU R Y,JIAO W T,QIAN X S,et al.Effective extraction methodfor determination of neonicotinoid residues in tea[J].Journal of Agricultural and Food Chemistry,2013,61(51):12565G12571.[9]LI S C,LU M,SUN Z Q,et al.Optimization of osthole in thelactone ring:Structural elucidation,pesticidal activities,and controlefficiency of osthole ester derivatives[J].Journal of Agricultural and Food Chemistry,2021,69(23):6465G6474.[10]YAN S,HU Q,JIANG Q H,et al.Simple osthole/nanocarrierpesticide efficiently controls both pests and diseases fulfilling theneed of green production of strawberry[J].ACS Applied Materials&Interfaces,2021,13(30):36350G36360.[11]ISMAN M.Botanical insecticides:A global perspective[J].ACS Symp,2014,1172:21G30.[12]BLOOMQUIST J,JIANG S Y,JENNINA T W,et al.Insecticidalactivity and physiological actions of matrine,a plant naturalproduct[J].Advances in the Biorational Control of Medical and Veterinary Pests,2018,1289:175G186.[13]XU H,XU M,SUN Z Q,et al.Preparation of matrinic/oxymatrinicamide derivatives as insecticidal/acaricidal agents and study onthe mechanisms of action against tetranychus cinnabarinus[J]. Journal of Agricultural and Food Chemistry,2019,67:12182G12190.[14]ZOU J B,ZHAO L H,YI P,et al.Quinolizidine alkaloids withantiviral and insecticidal activities from the seeds of sophoratonkinensis gagnep[J].Journal of Agricultural and Food Chemistry,39|V o l.40,N o.3石金娥等:同位素内标法 超高效液相色谱/串联质谱法测定六堡茶中苦参碱和氧化苦参碱残留量2020,68(50):15015G15026.[15]PAN Q M,LI Y H,HUA J,et al.Antiviral matrineGtype alkaloidsfrom the rhizomes of sophora tonkinensis[J].Journal of Natural Products,2015,78(7):1683G1688.[16]郭秋平,金若敏.苦参碱和氧化苦参碱致小鼠肝毒性比较[J].中国药理学与毒理学志,2016,30(7):736G740.GUO Q P,JIN R parison of liver toxicity of matrine and oxymatrine in mice[J].Chin J Pharmaco Toxicol,2016,30(7): 736G740.[17]赵岩,杨丹,邸子真,等.LCGMS/MS法测定苦参提取物中苦参碱和氧化苦参碱的质量浓度[J].现代生物医学进展,2021,21 (14):2792G2796.ZHAO Y,YANG D,DI Z Z,et al.LCGMS/MS determination of the concentration for matrine and oxymatrine in sophora flavescens extract[J].Progress in Modern Biomedicine,2021,21(14):2792G2796.[18]孙扬,徐应明,秦冬梅,等.苦参碱在黄瓜和土壤中的检测方法及其残留动态研究[J].农业环境科学学报,2010,29(4): 686G691.SUN Y,XU Y M,QIN D M,et al.Residue detection and degradation of matrine in cucumber and soil[J].Journal of AgroGEnvironment Science,2010,29(4):686G691.[19]门磊,张梦莹,胡文忠.高效液相色谱法同时测定复方木鸡颗粒中金雀花碱㊁苦参碱㊁槐果碱和槲皮苷的含量[J].沈阳药科大学学报,2020,37(2):131G135.MEN L,ZHANG M Y,HU W Z.Simultaneous determination of cytosine,matrine,sophocarpine and quercitrin in Fufang Muji granules by HPLC[J].Journal of Shenyang Pharmaceutical University,2020,37(2):131G135.[20]吴惠勤,张春华,黄晓兰,等.气相色谱 串联质谱法同时检测尿液中15种有毒生物碱[J].分析测试学报,2013,32(9): 1031G1037.WU H Q,ZHANG C H,HUANG X L,et al.Simultaneous determination of15toxic alkaloids in urine by gas chromatographyGtandem mass spectrometry[J].Journal of Instrumental Analysis,2013,32(9):1031G1037.[21]陈红平,刘新,汪庆华,等.液相色谱 串联质谱法与气相色谱 串联质谱法测定茶叶中苦参碱残留量[J].分析测试学报, 2010,29(12):1162G1167.CHEN H P,LIU X,WANG Q H,et al.Determination of matrine residue in tea using liquid chromatographyGtandem mass spectrometry or gas chromatographyGtandem mass spectrometry[J]. Journal of Instrumental Analysis,2010,29(12):1162G1167.[22]周鹏,黄芊,欧阳立群.超高效液相色谱 串联质谱法测定茶叶中9种天然植物源农药残留量[J].质谱学报,2020,41(5): 490G501.ZHOU P,HUANG Q,OUYANG L Q.Determination of nine botanical pesticide residues in tea by UHPLCGMS/MS[J].Journal of Chinese Mass Spectrometry Society,2020,41(5):490G501.[23]荆辉华,向俊,蒋登辉,等.蜂蜜中苦参碱与氧化苦参碱的快速检测[J].食品与机械,2022,38(7):57G63.JING H H,XIANG J,JIANG D H,et al.Rapid deterrination of matrine and oxymatrine in honey[J].Food&Machinery,2022,38 (7):57G63.[24]李丽.液相色谱串接质谱仪测定有机茶叶中的苦参碱与氧化苦参碱[J].化工设计通讯,2022,48(1):96G99.LI L.Determination of matrine and oxymatrine in organic tea by liquid chromatography coupled with mass spectrometer[J].Chemical Engineering Design Communications,2022,48(1): 96G99.[25]沈沛霖,钱圆,卫严冰,等.超高效液相色谱 串联质谱法分析柑橘及土壤中苦参碱残留[J].浙江农业科学,2018,59(3): 501G503.SHEN P L,QIAN Y,WEI Y B,et al.Determination of matrine residues in citrus and soil by ultra performance liquid chromatography tandem mass spectrometry[J].Zhejiang Agricultural Science,2018,59(3):501G503.[26]刘颖,石璐,魏永辉,等.超高效液相色谱 串联质谱法测定蔬菜水果中苦参碱残留量[J].食品安全质量检测学报,2022, 13(9):2871G2878.LIU Y,SHI L,WEI Y H,et al.Determination of matrine residues in vegetables and fruits by ultra performance liquid chromatographyGtandem mass spectrometry[J].Journal of Food Safety and Quality,2022,13(9):2871G2878.(上接第88页)[13]中华人民共和国国家质量监督检验检疫总局.食品中诱惑红㊁酸性红㊁亮蓝㊁日落黄的含量检测㊀高效液相色谱法:SN/ T1743 2006[S].北京:中国标准出版社,2006.State Administration of Quality Supervision,Inspection and Quarantine of the People s Republic of China.Determination of allure red AC,carmosine,brillint blue FCF,sunset yellow FCF in food:High performance liquid chromatogarphic method:SN/T 1743 2006[S].Beijing:China Standards Press,2006.[14]陈洁.高效液相色谱法测定市售自制饮料中多种色素㊁防腐剂及甜味剂[J].中国食品添加剂,2021,32(1):92G95.CHEN J.Simultaneous determination of various pigments, preservatives and sweeteners in vendorGmade beverages by HPLC [J].China Food Additives,2021,32(1):92G95.[15]吕小丽,朱春燕,陈林,等.HPLC法测定果汁和葡萄酒中的三种红色合成着色剂[J].食品工业,2020,41(6):299G302.LU X L,ZHU C Y,CHEN L,et al.Determination of three red synthetic pigments in juice and wine by HPLC[J].Food Industry, 2020,41(6):299G302.49安全与检测S A F E T Y&I N S P E C T I O N总第269期|2024年3月|。
同位素内标校正-概述说明以及解释

同位素内标校正-概述说明以及解释1.引言1.1 概述同位素内标校正是一种常用的分析方法,广泛应用于各个领域的科学研究中。
它是通过引入一种已知同位素比例的内标物质,来准确测量目标物质的浓度或比例的方法。
同位素内标校正可以消除由于样品制备和测量过程中引入的误差,提高分析结果的准确性和可靠性。
同位素内标校正的原理基于同一元素的不同同位素存在着自然丰度差异的事实。
在实际应用中,通常选择一个与目标物质具有相似化学性质的同位素作为内标。
通过在样品中添加已知比例的内标物质,可以将内标物质与目标物质在样品制备和分析过程中的损失、回收等因素同时考虑,从而准确计算出目标物质的浓度或比例。
同位素内标校正方法可以大幅度降低测量误差,提高分析结果的准确性和可靠性。
同位素内标校正在各个领域有着广泛的应用。
在环境科学中,同位素内标校正可用于确定大气和水体中有机物的来源和迁移途径。
在地质学和考古学中,它可以用于确定地球历史上的气候变化和生物演化过程。
在医学和生物学研究中,同位素内标校正可以用来研究生物代谢过程和药物代谢途径。
在食品安全领域,它可以用于检测食品中的重金属、农药和其他有害物质的含量。
同位素内标校正在各个领域中都发挥着重要的作用。
然而,同位素内标校正也存在一些限制。
首先,内标物质的选择要考虑到其与目标物质的化学性质相似,以确保在样品处理和分析过程中能够保持相似的行为。
其次,同位素内标校正的方法需要严格控制样品制备和测量过程中的各种误差来源,包括内标物质的纯度、稳定性和添加量等。
此外,同位素内标校正方法的应用还需要仪器设备的支持和专业的操作技术,增加了实验的复杂性和成本。
未来,同位素内标校正方法在科学研究中仍具有广阔的发展前景。
随着现代分析技术的不断发展,应用于同位素内标校正的新方法和新技术将会不断涌现,提高分析的准确性和灵敏度。
同时,同位素内标校正方法的应用范围也将进一步扩展,逐渐应用于更多的领域和问题的研究中。
1.2 文章结构文章结构是指文章的组织框架和内容安排,它直接关系到文章的逻辑性和可读性。
同位素内标稀释高效液相色谱-质谱法同时测定水中多种痕量抗生素

黄秋鑫1∗∗ 陈 琼1 雷 敏1 韦高玲2 丑天姝1
(1. 工业和信息化部电子第五研究所,中国赛宝环境评估与监测中心, 广州, 510610; 2. 广东省生态环境与土壤研究所,广东省农业环境综合治理重点实验室, 广州, 510650)
(1. CEPREI Environmental Assessment and Monitoring Center, The 5th Electronics Research Institute of the Ministry of Industry and Information Technology, Guangzhou, 510610, China; 2. Guangdong Key Laboratory of Agricultural Environment Pollution Integrated Control, Guangdong Institute of Eco⁃Environmental and Soil Sciences, Guangzhou, 510650, China)
磺胺类: 磺 胺 嘧 啶 ( SDZ, ≥ 99. 0%) 、 磺 胺 甲 噁 唑 ( SMX, ≥ 99. 0%) 、 磺 胺 二 甲 基 嘧 啶 ( SMZ, ≥99.0%) 、磺胺甲嘧啶( SMR,≥99.0%) 、磺胺吡啶( SPD,≥99.0%) ;甲氧苄胺嘧啶( TMP,≥98.0%) ;大 环内酯类:阿奇霉素( AZM,≥95.0%) 、克拉霉素( CTM,≥95.0%) 、红霉素( ETM,≥95.0%) 、罗红霉素 ( RTM,≥90. 0%) 、 螺旋霉素 ( SRM,95. 0%) ; 喹诺酮类: 环丙沙星 ( CPX, ≥98. 0%) 、 恩 诺 沙 星 ( EFX, ≥98.0%) 、洛美沙星( LEF,≥98.0%) 、马波沙星( MAB,≥98.0%) 、诺氟沙星( NFX,≥98.0%) 、氧氟沙星 ( OFX,≥98.0%) ;四环素类:土霉素( OTC,≥97.0%) 、四环素( TTC,≥97.0%) ;氯霉素( CAP,≥99.0%) 和黄连素( BEB,≥95.0%) .以上试剂均为固体粉末标样,购自 Sigma Aldrich 公司( 美国) .固体粉末氘代 环丙沙星( CPX⁃d8) 、 氘代磺胺二甲基嘧啶 ( SMZ⁃d4) 、 氘代甲 氧 苄 胺 嘧 啶 ( TMP⁃d9) 、 氘 代 恩 诺 沙 星 ( EFX⁃d5) 购自 C / N / D Isotopes( Pointe⁃Claire, 加拿大) ,纯度≥95.0%.
稳定性同位素稀释气相色谱-质谱法测定白酒中DBP、DEHP含量及不确定度评定

稳定性同位素稀释气相色谱-质谱法测定白酒中DBP、DEHP含量及不确定度评定李凤华;李蔚;曹艳平【摘要】采用稳定性同位素稀释气相色谱-质谱法对白酒中DBP、DEHP的含量进行测定,并对整个测试过程的不确定度来源进行系统分析。
样品提取、离心后取上层有机相进行测定,利用选择离子监测模式,以氘代同位素标记物作内标,以m/z 149和m/z153为定量离子对,对其定性定量测定,并对测试过程中引入的各个分量进行评定与合成。
结果表明,DBP、DEHP在0.2~2.0 mg/L浓度范围内线性关系良好,r≧0.999,定量限为0.05 mg/kg,不同加标水平中的平均回收率为DBP:91.2%~100.4%,RSD为2.24%~2.79%(n=5);DEHP:99.0%~110.6%,RSD为4.98%~5.44%(n=5)。
当DBP测定结果为0.572 mg/kg时,扩展不确定度为0.040 mg/kg,DEHP测定结果为0.251 mg/kg时,扩展不确定度为0.025 mg/kg,k=2,p=95%。
该方法准确可靠,可用于白酒中DBP、DEHP的含量测定。
并且建立的不确定度评定方法可用于气相色谱-质谱法测定白酒中DBP、DEHP浓度的不确定度评估。
标准溶液配制和标准曲线拟合、样品前处理以及分析仪器是主要的不确定度来源。
%In this study, DBP and DEHP content in Baijiu(liquor) was determined by stable isotope dilution GC-MS, and the uncertainties source in the determination process was analyzed systematically. In the operation process, after liquor sample extraction and centrifugation, the up-per-layer organic phase was determined by GC-MS with m/z149 and m/z153 as the quantitative ionpair in selective ions monitoring mode and deuterium isotope used as the internal standards, and each component introduced in the determinationprocess was evaluated and synthesized. The experimental results showed that, DBP and DEHP presented a good linear relationship within the range of 0.2~2.0 mg/L with r≧0.999, LOQ=0.05 mg/kg, the average recoveries of DBP between 91.2%to 100.4%and its RSD with the range of2.24%~2.79%(n=5), and the aver-age recoveries of DEHP between 99.0%to 110.6%and its RSD within the range of 4.98%~5.44%(n=5). As the determination results of DBP and DEHP were 0.572 mg/kg and 0.251mg/kg, their expanded uncertainties were 0.040 mg/kg and 0.025 mg/kg respectively(k=2,p=95%). Such method was suitable for the determination of DBP and DEHP content in Baijiu(liquor). The uncertainties source in the determination mainly came from the preparation of standard solution, standard curve fitting, pretreatment of liquor samples and the analytical instrument.【期刊名称】《酿酒科技》【年(卷),期】2014(000)010【总页数】5页(P111-115)【关键词】气相色谱-质谱(GC-MS);DBP;DEHP;同位素稀释;不确定度;白酒【作者】李凤华;李蔚;曹艳平【作者单位】山东省疾病预防控制中心,山东济南250014;山东省疾病预防控制中心,山东济南250014;山东省疾病预防控制中心,山东济南250014【正文语种】中文【中图分类】TS262.3;TS261.7;TS261.4;O657.63;R155.5邻苯二甲酸酯(Phthalic Acid Esters,PAEs)又称酞酸酯,由于其良好的性能而被广泛应用于塑料玩具、食品包装袋、纺织品、化妆品等各类产品的生产之中[1-2]。
常见定量f谱的内标

常见定量f谱的内标
常见的定量质谱(MS)分析中使用的内标包括氘代内标和稳定同位素内标。
氘代内标是指将分子中的氢原子替换为氘原子的化合物,因为氘原子比普通氢原子重,因此可以在质谱中形成明显的质量差异,用于定量分析。
另一种常见的内标是稳定同位素内标,例如^13C,^15N和^18O等同位素。
这些同位素在质谱中也会产生明显的质量差异,因此可以用于定量分析。
在定量质谱分析中,选择合适的内标对于准确测定目标化合物的含量非常重要。
内标应具有稳定的化学性质,易于与目标分析物区分开来,并且在质谱分析中能够产生明显的峰。
选择内标时需要考虑到其在样品制备和分析过程中可能引入的误差,并且内标的选择应该能够准确地反映目标分析物的含量变化。
总的来说,内标在定量质谱分析中起着至关重要的作用,合理选择和使用内标可以提高定量分析的准确性和可靠性。
同位素内标-液相色谱-串联质谱法测定动物源性食品中氯霉素残留

— —
最终 定容 体 积 , L; m
称样 量 , 。 g
破碎 , 尤其 是肠 衣 , 在 试 验 中 , 过超 声 的方 法 改 故 通 善提取 效果 , 入 同位 素 内标 , 偿 提 取 偏 差 , 去 加 补 免
小柱净 机试 剂
~
色谱 柱 : gl t cis X B—c8 ( 5 m × A i n l e D e E p 1 柱 10 m
4 6m 5 m) 柱 温 :5C; . m, ; 2 流速 :. / i ; o 0 5mL m n 进样
量 :5 I 流动相 : 2 L L; A相为 甲醇 , B相 为水 。
在龙虾 样 品 中取 出有代 表性 的样 品约 5 0 g 取 0 , 可食 部分用 粉碎 机 绞碎 ; 从所 取 肠 衣 样 品 中取 出有 代表 性 样 品 约 3 0 g 用 剪 刀 剪 碎 ( 于 0 5 c 。 0 , 小 . m) 将 粉 碎 的样 品混 合均 匀 , 均分 成 两份 , 别装入 洁净 分 容器 作为待 测样 , 密封 , 一1 ℃ 条件下 保存 。 在 8
表 1 氯 霉 素 的 定性 和 定量 条 件 药 物 氯 霉 素 定 性 离 子 对 ( z 定 量离 子 对 ( z 碰 撞 能 量/ m/ ) m/) V
3 0. /1 . 2 8 51 8 30 2
.
5 0mL具 塞离 心管 中 , 确加 入 2 0 L氯 霉 素 一d 准 0
实验 步骤 操作 , 以峰面 积 比 ( 准峰 面积 与 内标 峰 标 面积 之 比 ) 为横 坐标 , 氯霉 素 浓 度 C n / L 为 纵坐 (g m ) 标 , 制标 准 曲线 , 绘 线性 回归方 程 为 C=0 2 57 . 4 X一 0 0 1 , = .9 4 标 准工 作 曲线线 性范 围 为0 0 . 0 r 0 9 9 , 3 .5
基质效应在生物样品质谱分析中的优化措施研究

基质效应在生物样品质谱分析中的优化措施研究吴文静【摘要】高效液相色谱-质谱联用法(LC - MS /MS )由于其高灵敏度和高选择性现阶段被广泛应用于食品检测、环境评估等方面的样品定量分析.然而,由于实际样品特别是复杂样品分析中基质效应的存在,样品分析进程以及检测结果的特异性、灵敏度和准确度都会受到影响.本文立足于实际的生物样品质谱分析,阐述了基质效应的产生原因、检测及评定方法,其优化措施包括四个方面,即样品前处理的优化、色谱条件的优化、质谱条件的优化以及同位素内标的选择.【期刊名称】《信阳农林学院学报》【年(卷),期】2017(027)004【总页数】5页(P115-118)【关键词】高效液相色谱一质谱联用基质效应生物样品分析【作者】吴文静【作者单位】安徽公安职业学院公安科学技术系,安徽合肥230031;【正文语种】中文【中图分类】O657.63高效液相色谱-质谱联用法(液相质谱,HPLC-MS/MS)是一种高灵敏度和高选择性的样品定量分析方法,由于其高效样品的选择性和准确的测定能力而被广泛推广应用于食品相关检测、环境风险评估、农药残留分析、药物组分以及代谢研究中[1-2]。
近几年,随着液质技术的快速发展,与检测相关的基质效应问题也开始被广泛关注。
基质效应作为质谱检测中存在的必然问题,对样品检测、分析方法和结果的特异性、灵敏度和准确度都有显著影响[3]。
目前,国外的学者已经开展了大量的与基质效应相关的工作和研究,但国内相关的研究还未能构成完整的研究体系。
本文结合国内外相关文献,对液质检测过程中基质效应的产生原因、相关作用以及目前常规的检测方法和消除或降低基质干扰的途径等问题进行阐述。
1 基质效应产生机制及影响基质效应指的是在样品检测过程中,除待测组分以外的其它物质对待测组分的分析进程产生的干扰,并影响检测结果的灵敏度和准确性。
基质效应的产生主要是源于样品中的待测组分与基质成分在离子化过程中的竞争。
《精准医疗(学)运用质谱技术对疾病的研究》

《精准医疗(学)运用质谱技术对疾病的研究》10.11东莞市松山湖中心人民医院 523000【摘要】目的:探讨计划的医学科技发展和医疗模式的影响及对我国的启示具有前沿性,超前意识的论述质谱技术、中国质谱技术现状。
懂质谱的人不懂临床,相反懂临床的人不懂质谱,有质谱知识和背景的人甚少。
笔者呼吁中国已到规范化开展质谱普及到地市级三甲医院,还未被院领导重视任重而道远。
国家已列入“十四五”计划,重点研发计划在《诊疗设备与生物医用》的计划中,促进我国高端诊疗装备和生物医用材料整体水平进入国际先进行列。
【关键词】质谱技术;疾病;组学;精准医学;测序;材料与方法什么是质谱技术质谱的核心精髓是换柱子技术,纯定量检测。
质谱属超定量分析每次要做定标。
检测方法学:质谱检测法;超高相液相色谱;液相色谱;质谱;气相色谱质谱;高效液相色谱串联质谱检测系统全自动多功能样本前处理系统支持SPE、SLE PLD PPT的自动化;独特双模式设计,支持96孔板和小柱模式;8个自动化移液通道;5个自动添加溶剂的溶剂池;耐溶剂触摸屏控制,操作界面直观易用;正压处理,实现精确的移液精度和准确度;适用项目维生素、新生儿遗传代谢疾病筛查、多种激素、多种脂肪酸、多种氨基酸等检测项目。
高效液相色谱串联质谱技术(LC-MS/MS),将高效液相色谱的高分离度、高效率与质谱的高灵敏度、高选择性完美结合,最终通过同位素内标及外标实现精准定量。
YS EXT9050 MD与YSEXT9900MD皆为新一代三重四极杆质谱仪,基于离子源、离子传输组件、质量分析器的多维创新,可针对基质中极低浓度的化合物进行定量分析。
具有超凡的定量能力,超快扫描,更高灵敏度,超宽动态范围,极具耐用性。
(下图为检测原理)临床应用维生素、新生儿遗传代谢疾病筛查、多种激素、多种脂肪酸、多种氨基酸、多种胆汁酸、治疗药物浓度等70余项检测项目。
新生儿遗传代谢疾病筛查系统串联质谱法检测原理液相色谱-串联质谱法(LC-MS/MS)用含稳定同位素内标的萃取液将样本中氨基酸、肉碱、琥珀酰丙酮提取出来,然后采用液相色谱-串联质谱仪进行检测,记录每种分析物的响应强度与对应稳定同位素内标的响应强度。
质谱介绍及质谱图的解析

质谱介绍及质谱图的解析质谱用于定量分析,其选择性、精度和准确度较高。
化合物通过直接进样或利用气相色谱和液相色谱分离纯化后再导入质谱。
质谱定量分析用外标法或内标法,后者精度高于前者。
定量分析中的内标可选用类似结构物质或同位素物质。
前者成本低,但精度和准确度以使用同位素物质为高。
使用同位素物质为内标时,要求在进样、分离和离子化过程中不会丢失同位素物质。
在使用FAB质谱和LC/MS(热喷雾和电喷雾)进行定量分析时,一般都需要用稳定的同位素内标。
分析物和内标离子的相对丰度采用选择离子监测(只监测分析物和内标的特定离子)的方式测定。
选择离子监测相对全范围扫描而言,由于离子流积分时间长而增加了选择性和灵敏度。
利用分析物和内标的色谱峰面积或峰高比得出校正曲线,然后计算样品中分析物的色谱峰面积或它的量。
解析未知样的质谱图,大致按以下程序进行。
(一)解析分子离子区(1)标出各峰的质荷比数,尤其注意高质荷比区的峰。
(2)识别分子离子峰。
首先在高质荷比区假定分子离子峰,判断该假定分子离子峰与相邻碎片离子峰关系是否合理,然后判断其是否符合氮律。
若二者均相符,可认为是分子离子峰。
(3)分析同位素峰簇的相对强度比及峰与峰间的Dm值,判断化合物是否含有CI、Br、S、Si等元素及F、P、I等无同位素的元素。
(4)推导分子式,计算不饱和度。
由高分辨质谱仪测得的精确分子量或由同位素峰簇的相对强度计算分子式。
若二者均难以实现时,则由分子离子峰丢失的碎片及主要碎片离子推导,或与其它方法配合。
(5)由分子离子峰的相对强度了解分子结构的信息。
分子离子峰的相对强度由分子的结构所决定,结构稳定性大,相对强度就大。
对于分子量约200的化合物,若分子离子峰为基峰或强蜂,谱图中碎片离子较少、表明该化合物是高稳定性分子,可能为芳烃或稠环化合物。
例如:萘分子离子峰m/z 128为基峰,蒽醌分子离子峰m/z 208也是基峰。
分子离子峰弱或不出现,化合物可能为多支链烃类、醇类、酸类等。
食品中高氯酸盐稳定同位素液相色谱质谱检测方法建立
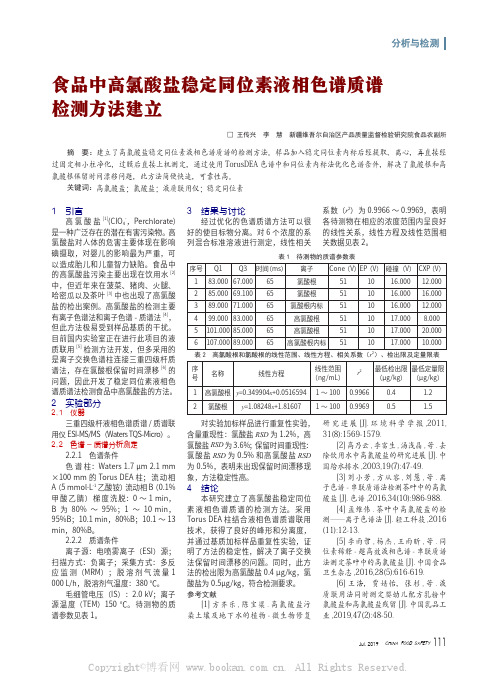
(ng/mL)
(μg/kg) (μg/kg)
1 高氯酸根 y=0.349904x+0.0516594 1 ~ 100 0.9966
0.4
1.2
2 氯酸根 y=1.08248x+1.81607 1 ~ 100 0.9969
0.5
1.5
对实验加标样品进行重复性实验, 含量重现性:氯酸盐 RSD 为 1.2%,高 氯酸盐 RSD 为 3.6%;保留时间重现性: 氯酸盐 RSD 为 0.5% 和高氯酸盐 RSD 为 0.5%,表明未出现保留时间漂移现 象,方法稳定性高。
[6] 王 浩, 贾 婧 怡, 张 杉 , 等 . 液 质联用法同时测定婴幼儿配方乳粉中 氯酸盐和高氯酸盐残留 [J]. 中国乳品工 业 ,2019,47(2):48-50.
111 Jul. 2019 CHINA FOOD SAFETY Copyright©博看网 . All Rights Reserved.
5 101.000 85.000 65
高氯酸根
51
10 17内标 51
10 17.000 10.000
表 2 高氯酸根和氯酸根的线性范围、线性方程、相关系数(r2)、检出限及定量限表
序 名称
号
线性方程
线性范围
r2 最低检出限 最低定量限
[1] 方 齐 乐 , 陈 宝 梁 . 高 氯 酸 盐 污 染土壤及地下水的植物 - 微生物修复
研 究 进 展 [J]. 环 境 科 学 学 报 ,2011, 31(8):1569-1579.
[2] 高乃云 , 李富生 , 汤浅晶 , 等 . 去 除饮用水中高氯酸盐的研究进展 [J]. 中 国给水排水 ,2003,19(7):47-49.
同位素内标

同位素内标1. 什么是同位素内标?同位素内标是一种分析化学中常用的技术,用于定量分析中的质量测量。
在同位素内标技术中,同位素被用作参考物质,通过与待测样品进行比较,可以准确测量出待测样品中目标物质的含量。
2. 同位素内标的原理同位素内标的原理基于同位素的特性。
同位素是指具有相同原子序数(即原子核中质子的个数相同),但质量数不同的原子核。
同位素之间的差异主要体现在质量上,因此同位素可以作为质量标记物质。
同位素内标的原理可以简单描述为以下几个步骤:2.1 同位素标记首先,选择一个稳定同位素作为内标。
稳定同位素是指具有相对较长半衰期的同位素,不会发生放射性衰变。
常用的稳定同位素有氢的氘同位素(D)、碳的13C同位素、氮的15N同位素等。
2.2 内标添加将已标记的同位素加入待测样品中。
内标的添加量应该适量,既要保证内标与待测物质的浓度比例适当,又不会对待测物质的测量结果产生显著影响。
2.3 分析测量使用适当的分析仪器对待测样品进行测量。
常用的分析技术包括质谱法、光谱法、电化学法等。
通过测量待测物质与内标的信号强度比值,可以计算出待测物质的含量。
3. 同位素内标的应用同位素内标技术在各个领域都有广泛的应用,以下列举几个常见的应用领域:3.1 环境科学同位素内标技术在环境科学中的应用较为广泛。
例如,通过添加氢的氘同位素作为内标,可以准确测量水中有机物的含量。
同样地,通过添加碳的^13C同位素作为内标,可以测量土壤中有机质的含量。
3.2 医学研究同位素内标技术在医学研究中也有重要的应用。
例如,通过添加氮的^15N同位素作为内标,可以测量体内蛋白质的新陈代谢情况。
同位素内标技术还可以用于药物代谢动力学研究,通过测量药物及其代谢产物中同位素的比例,可以了解药物在体内的代谢过程。
3.3 食品安全同位素内标技术在食品安全领域也有广泛的应用。
例如,通过添加氢的氘同位素作为内标,可以测量食品中残留农药的含量。
同位素内标技术还可以用于检测食品中的添加剂、污染物等。
兽药残留检测中稳定性同位素的应用研究

兽药残留检测中稳定性同位素的应用研究摘要:近些年伴随着各种兽药的广泛应用,出现了一系列重大食品安全事件,人们对于兽药的残留问题日益重视,受国际间贸易等因素的影响,极大的增加了兽药残留成分分析对象、测定难度和样本数量,面对对这一现象,对兽药残留分析迫切需要采用一种灵敏、快速且简便的方法,同时还可以对大批量样品兽药残留进行同时处理测定分析。
本次研究对稳定性同位素在兽药残留检测中的应用进行研究,阐明此种检测方法对我国食品安全监测发展发挥的重要作用。
关键词:兽药残留稳定性同位素检测应用伴随着人们生活水平的不断提升,对食品的安全要求也逐渐增高,目前食品安全尤其是动物食品安全要求并不容乐观,动物食品安全下降的主要原因是兽药残留因素[1]。
兽药残留是指在对畜禽用药之后残留或积蓄在其机体内或产物中能够的其他代谢物或中原型药物,同时还包括了和兽药相关的残留杂质。
伴随着人们对动物源食品的质量型转变,全世界开始普遍关注兽药残留问题,自20世纪60年代开始,污染物和视频添加剂联合专家委员会对兽药残留毒性进行相关评价,提供了人们认识兽药残留危害和控制的依据。
兽药可以有效防止动物疾病,对改善畜产品的质量、提升畜产品生产效率作用重大。
但受养殖人员缺乏科学知识和追求经济利益心理影响,当前畜牧业中普遍存在滥用兽药现象,造成兽药残留现象严重,不仅对人体健康产生了直接的危害,还严重影响了畜牧业的发展,造成生态环境破坏[2]。
1 兽药残留检测分析中稳定性同位素的应用兽药残留分析时存在了兽药残留代谢产物多样、兽药残留代谢产物不明确、干扰物质多、待测物质浓度低、样品基质复杂等多种现象特点,目前广泛采用的主流筛选法,如荧光分析法、液相色谱法、薄层色谱法、气相色谱法等技术方法均存在了一定的局限性,需要对检测阳性结构进行明确验证定量、定性分析,采用质谱可以同时提供定量信息和定性信息。
所以在发达国家均采用质谱法对兽药残留进行检测。
据统计资料显示,为了将前后处理、质谱检测器和视频复杂基质效应等因素造成的测定结果偏差影响避免,兽药残留检测方法在80%以上应用同位素内标,可以将出现的误差有效避免,极大的提升了检测方法稳定性[3]。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
稳定性同位素内标与质谱检测
稳定性同位素内标是质谱方法(稳定性同位素稀释法)独有的,没有别的临床检测方法用到同位素内标。
比如光吸收和免疫的方法,都无法分辨出被检测物和同位素内标的区别,因为它们的理化性质太接近了。
如果真的加进内标,那测出来的值肯定大大的偏高。
只有质谱才能把同位素内标和要检测的物质分得开,虽然它们的差别只有几个道尔顿。
现在的内标基本都是稳定同位素标记的,最常见的是D和13C。
同位素内标和被检测物是同一个物质,但是其中的几个氢原子被氘所取代,或者是12C换成了13C,理化性质基本不变。
同样是标记,13C就比D要好。
但是D要比13C便宜很多。
绝大多数D做的内标性能是很好的。
出问题的经常是一些疏水性比较差,保留时间比较短的物质。
出峰的时候跟很多其它物质一起出来,些许的偏差就能引起浓度测不准。
临床检测的数据要想测的准,离不开一条好的标准曲线。
通俗来讲,标准曲线就像一把尺子。
只有把尺子做准确了,才能把未知物品的长度测准确。
从科学上来讲,标准曲线就是检测物质的浓度和仪器读数的一种线形关系。
一般是浓度越高,读数越大。
标准品的浓度是已知的,高中低都有。
测完标准品以后,把它们的浓度和仪器测得的读数在x/y的坐标纸上一画,连一条线就成了。
测未知的病人样品时,浓度(x)是未知的,只有仪器的读数(y),通过这条曲线可以把y 换算成浓度。
标准品应该怎么做,怎么用,这里面有很多学问。
质谱是新鲜技术,大多的检测项目还买不到标准品,只能自己配。
做标准品需要有纯样品。
最好的纯样品应该是浓度和纯度都有保证书的,这样用起来放心。
如果是液体的溶液就更好了,省去自己称量和溶解的麻烦。
高浓度的纯样品要稀释到不同的低浓度才能使用。
用什么来稀释是下一个非常关键的步骤,这里面牵扯到基质效应。
因为基质效应这块儿瓦是质谱临床应用里比较难理解的一个概念。
目前同位素内标广泛应用于临床检测中:
激素检测
儿茶酚胺检测维生素检测
全谱氨基酸检测
全谱氨基酸
1-甲基-L-组氨酸(1MHis)L-组氨酸(His)
3-甲基-L-组氨酸(3MHis)δ-羟基赖氨酸(Hyl)L-α-氨基己二酸(Aad)羟基-L-脯氨酸(Hyp)
L-α-氨基正丁酸(Abu)L-异亮氨酸(Ile)
L-丙氨酸(Ala)L-亮氨酸(Leu)
L-氨肌肽(甲肌肽)(Ans)L-赖氨酸(Lys)
L-精氨酸(Arg)L-蛋氨酸,L-甲硫氨酸(Met)精氨基琥珀酸(Asa)L-正亮氨酸(Nle)
L-天门冬酰胺(Asn)L-正缬氨酸(Nva)
L-天门冬氨酸(Asp)L-鸟氨酸(Orn)
D,L-β-氨基异丁酸(bAib)O-磷酸乙醇胺(PEtN)
β-丙氨酸(bAla)L-苯基丙氨酸(Phe)
L-肌肽(Car)L-脯氨酸(Pro)
L-瓜氨酸(Cit)O-磷酸-L-丝氨酸(Pser)
胱硫醚,丙氨酸丁氨酸硫醚(Cth)肌氨酸(Sar)
L-胱氨酸(Cys)L-丝氨酸(Sar)
乙醇酸(Cys)牛磺酸(Tau)
γ-氨基正丁酸(GABA)L-苏氨酸(Thr)
L-谷氨酰胺(Gln)L-色氨酸(Trp)
L谷氨酸(Glu)L-酪氨酸(Tyr)
甘氨酸(Gly)L-缬氨酸(Val)
高瓜氨酸(Hcit)别异亮氨酸(Allo-lle)。