过渡金属配合物的电子光谱
两种过渡金属配合物的合成和光谱表征

为 40 持 续 搅 拌 lh的 时 间 。将 沉 淀 物 反 复 水 洗 离 心 5次 , 1 0℃ 下 真 空 干 燥 2h, 得 到 黑 色 的 固 ., 在 5 即
体 样 0 3 , 率 9 % 。 素 分 析 结 果 : :23 % , 382 , 80 % , 照 化 学 式 c。 2e 2 计 算 c .6g产 0 元 C6 .2 H:. % N:.5 按 8 F N O2 H。 :
应 。 P 2 0 HN 型 自动 元 素 分 析 仪 , 国 Nioe T l AV AR 3 0F — I 红 外 光 谱 仪 ( r 片 ) E一 4 0 C 美 c lt — R AT 6 - I. F F I KB 压 , 扫 描 范 围 40 0—4 0c 。荧 光 光 谱 仪 : - 5 0 Hi c i 司 。 0 0 m~ F 40 , t h公 a
维普资讯
Z0 年 儿 月 07
安 庆师 范学院 学报 (自然科 学 I 皈)
J u l f n igT a h r olg ( aua S in e E i n) o ma qn e c esC l e N trl ce c dt oA e i o
12 配 合 物 的 合 成 【 - 甸 1 2 1 F Q2 合 成 -. e 的
称 取 03 38g的 F S H: .2 e O . O溶 于 5 蒸 馏 水 中 , 力 搅 拌 并 加 热 至 6 7 0 ml 磁 0℃ ; 化 学 计 量 比 l2称 按 :
取 HQO3 44g 加 入 适 量 尢 水 乙 醇 , 拌 使 其 充 分 溶 解 ; 两 者 混 合 均 匀 后 用 l:l .2 , 搅 将 0的 氨 水 调 解 p 值 H
无机化学 配合物

ML= 0 MS =0
(2S+1)(2L+1)=1
能量相同的微状态归为一组,得到自由离子的5个光谱项:
L=4, ML= 4, 3, 2, 1 0,
L=3, ML= 3, 2, 1 0,
S=0
S=1
MS= 0
MS= 1, 0
1G
3F 1D 3P 1S
L=2, ML= 2, 1 0,
ML=4, 3, 2, 1, 0
MS =0
(2S+1)(2L+1)=9
ML= 3, 2, 1, 0 MS = 1, 0 (2S+1)(2L+1)=21
ML=2, 1, 0 MS =0 (2S+1)(2L+1)=5
ML=1, 0 MS = 1, 0 (2S+1)(2L+1)=9
二. O大小的表征―电子光谱(或电子光谱,紫 外可见光谱) 1.单电子的近似的配合物光谱
定性判断:
ligand
excitation
显色
吸收颜色
O
Cu(NH3)42+
Cu(OH2)42+
强场
弱场
紫色
蓝色
黄色
橙色
大
小
Cr(NH3)63+
Cr(OH2)63+
强场
弱场
橙色
紫色
蓝色
黄色
大
小
ground
只考虑配位场作用,不考虑d电子之间的相互作用
根据这两点,可推出d2组态的5个谱项的能量顺序为:
3F
3P 1G 1D 1S ,
其中3F为 基谱项(最大S, 最大L)
但实际观察的d2组态(Ti2+)光谱项的能量顺序则为:
过渡金属配合物的发光材料课件

过渡金属配合物的发光材料课件
Thank you !
过渡金属配合物的发光材料课件
过渡金属配合物的发光材料课件
(四) 常用光学材料:
自从1828年W.Nicol发明 偏光显微镜以后,人们就系 统地研究天然矿物晶体的光 学性质,而偏光显微镜的心 脏就是由方解石制成的Nicol 棱镜。
过渡金属配合物的发光材料课件
几种常见的发光材料
过渡金属配合物的发光材料课件
过渡金属配合物的发光材料课件
过渡金属配合物的发光材料课件
• 2.电荷转移跃迁
电荷转移跃迁:是指络合物 中配位体和金属离子之间, 一方的电子向主要属于另一 方的轨道的跃迁,所产生的 吸收光谱称为荷移光谱。
过渡金属配合物的发光材料课件
• 3.金属离子影响下配体的 → * 跃迁
显色剂大多含有生色团和助色团,与金属离子配 位时,其共轭结构发生变化导致吸收光谱发生红移 或蓝移。
R=
图2 含锌-三联吡啶发色团的高分子聚合链
过渡金属配合物的发光材料课件
目前应用于有机EL 金属配合物的发光材料大多 配位数均为偶数,四配位的8-羟基喹啉锌配合物 (Znq2)的荧光来自受金属微扰的配体。
过渡金属配合物的发光材料课件
采用新的方法合成了三配位的锌配合物: 8-羟基喹啉对甲基苯酚合锌配合物(ZnqP, 见图3), 它的发光性质,应用于有机电致发光器件。
例:茜素磺酸钠 弱酸性-黄色- λmax=420nm 弱碱性-紫红色- λmax=560nm
过渡金属配合物的发光材料课件
激发态
h
激发
发射
一些含杂环配体的过渡金属配合物的微波合成、光谱、热化学性质和生物活性

生 成 物 与 C ,o I, i ) C () 成 了 一 些 席 夫 碱 配 合 物 。用 元 素 分பைடு நூலகம்析 ,1 R, 原 子 轰 击 质 谱 , 尔 电 导 率 , C ()N( 和 u 合 Il Ⅱ Ⅱ F1 快 摩 电子 光 谱 , . H
N MR.S 磁 化 率 , 分 析 , E R, 热 电导 率 和 X D等 对 这 些 化 合 物进 行 了表 征 。这些 配合 物 在 空 气 中稳 定 并 有颜 色 。分 析 数 据表 明所 R 有 配合 物 金 属 与 配体 的 比均 为 1 . 位 数 为 4或 6 原 子 轰击 质谱 和热 分 析 数 据 说 明 了 这 些 配合 物 的 降解 方 式 。 R 图 给 出 了 :配 2 X D 这 些 配 合 物 的结 晶情 况 。席 夫 碱 的 上 述 金 属 配 合 物 对 革 兰 氏 阳性 菌 : 黄 色 葡 萄 球 菌 和 革 兰 氏 阴性 菌 : 肠 埃 希 菌 ( 肠杆 菌) 金 大 大
Ji a n r Mi r anR j daK e s aA P h
(e ate tfC e ir, y tei Iognc& C o ia o h mir L b r oi . D p r n o h ms y S nht nro i m t c or n t nC e s y a o tr s d i t a e
第 2 卷 第 8期 8
2 2年 8 月 01
无
机
化
学
学
报
Vo .8 No. 12 8
CHI S O NE E J URNAL OFI RGANI NO C CHEMI T S RY
1 8.69 6 719
一
10.11配合物结构测定1

如果能够直接观测到 M-N 和 M-O(M 表示金属 ,N、 、 O 表示配位原子 ) 等与配位键密切相关的红外振 动吸收带的话, 将是配合物形成的最有力证据, 动吸收带的话 将是配合物形成的最有力证据 但 是遗憾的是这些 M-N 和 M-O 等关键的红外振动 吸收带一般出现在远红外 (far infrared) 区 , 超出 了普通红外光谱的检测范围。 了普通红外光谱的检测范围。
若配合物 中的△ 与 中的△ν与 游离时的 △ν差不 差不 多 , 则是 双单齿形 式配位。 式配位。
多数抗衡阴离子在红外光谱中有强的特征吸收带: 多数抗衡阴离子在红外光谱中有强的特征吸收带: ClO4- : 约 1100cm-1; NO3- : 1350~1400cm-1; NO2- :约1250cm-1 ; 约 BF4- : 约 1050cm-1; PF6- : 约 840 及约 558cm-1等, 因此对于含有抗衡阴离子的配合物, 因此对于含有抗衡阴离子的配合物 可以从抗衡阴离子红 外吸收带的位置、分裂情况等来推测配合物的形成。 外吸收带的位置、分裂情况等来推测配合物的形成。需要 注意的是当这些抗衡阴离子与金属离子间有配位作用存在 其振动吸收会发生较大的变化。 时, 其振动吸收会发生较大的变化。
吸收带, 配体场吸收带就是人们常说的 d-d 吸收带 是电 子从一个 d 轨道跃迁到另一个 d 轨道时所产生的 吸收, 并可以从配合物的化学键理论得到解释。 吸收 并可以从配合物的化学键理论得到解释。 d-d 吸收带中又分为自旋允许吸收带 (spinallowed absorption band) 和自旋禁止吸收带 (sin-forbidden absorption band)2 种, 而且前者 强度较强, 后者强度较弱。 强度较强 后者强度较弱。
过渡金属配合物的电子光谱

紫 紫 蓝 480 红 红 绿蓝 490 650 蓝绿 橙 黄 绿 500 598 黄 绿
580
560
二 配体内部的电子光谱
配位体如水和有机分子等在紫外区经常出现吸收谱带。形成配合物后, 这 些谱带仍保留在配合物光谱中, 但从原来的位置稍微有一点移动。
配位体内部的光谱包括以下三种类型: ① n→* 处于非键轨道的孤对电子到最低未占据的空轨道 ζ*反键轨道的跃迁。水、醇、胺、卤化物等配体常发生这类跃 迁。 ② n→* 处于非键轨道的孤对电子到最低未占据空轨道* 反键分子轨道的跃迁, 常出现在含羰基的醛和酮类分子中。 ③ →* 处于最高占据轨道分子轨道的电子向最低未占 据的空轨道*反键分子轨道跃迁, 这类跃迁经常出现在含双键、 叁键的有机分子中。
Td d2 、 d 7
相 反 相 相反
Oh d 3 、 d8
相 反
同
相 同
Td d3 、 d8
相反
Oh d2 、 d7
由d2、d8、d3、d7组态 的Orgel图可以发现: ① F谱项在配位场中 分裂为T1、T2和A2, 而P谱 项不分裂但变成T1, 基态F 谱项与P谱项能量差为 15B'。 ② 相同类型的线 , 如 T1(P) 和 T1(F) ( 图的左边 ) 是禁止相交的 , 他们发生 弯曲, 互相回避, 其弯曲的 程度以C表示, 称为弯曲系 数。
ⅱ d-d跃迁吸收峰的半宽度
① 由于振动将使得配体-金属之间的键长不停地 变化, 从而分裂能将随键长的增加而减小。而分裂能的 变化将导致配位场谱项之间的能量间隔发生变化, 并维 持在一定的范围。
② Janh-Taller效应导致轨道能级进一步分裂, 这 种分裂常使吸收峰谱带加宽。
3.1-配合物的UV-vis光谱

过度金属配合物的紫外-可见吸收光谱主要是由于配体与金属离子间 的结合而引起的电子跃迁,因此也称为电子光谱(electronic spectrum)
在紫外-可见吸收光谱中,根据吸收带来源的 不同划分:
紫外-可见吸收光谱
配位场吸收带Leabharlann 电荷跃迁吸收带配体内的电子跃迁吸收带
定性定量分析
金属向配体的跃迁 MLCT
n
等跃迁,研究配体间的作用方式和关系。波长范围大多在近紫外-可见光区。
生色团:分子结构的某些基团吸收某种波长的光,而不吸收另 外波长的光,从而使人觉得好像这一物质“发出颜色”似的, 因此把这些基团称为“发色基团/发色团”。例如>C=C<, >C=O,-N=N-,-C C-,-C N-等。 助色团:分子中本身不吸收辐射而能使分子中生色基团的吸收峰 向长波长移动并增强其强度的基团,如羟基、胺基和卤素等。助 色团可以分为两类: 酸性助色团: -COOH, -OH, -SO3H 碱性助色团:-NHR,-NR2,-NH2
包括d-d跃迁和f-f跃迁,对于过渡金属配合物而言也称为d-d跃 迁吸收带,其位置变化和裂分可跟踪考察配合物的反应和形式, 波长范围大多在可见光区。下面为d-d跃迁。
中心离子所带电量越大,周期数越高,分裂能越大。不同的配位场, 平面四方>八面体>四面体。
配体向金属的跃迁 LMCT 电荷跃迁吸收带
吸收的光与透过的光互为补色。
定性分析
定量分析
MCM_41介孔分子筛组装过渡金属配合物的电子光谱解读

第24卷,第3期光谱学与光谱分析2004年3月SpectroscopyandSpectralAnalysisVol124,No13,pp2812284March,2004MCM241介孔分子筛组装过渡金属配合物的电子光谱霍涌前,李,王伟,弓亚琼,张逢星3西北大学化学系,陕西省物理无机化学研究所,陕西西安710069摘要通过γ2氨丙基修饰纳米孔材料MCM241,将其与过渡金属Mn,Cu,Co,Fe 的N,N2双水杨醛缩乙二胺席夫碱配合物进行化学组装。
用IR和Vis2UV等谱学技术对其进行了初步的表征。
结果表明,组装产物的IR光谱,呈现出氨基和Schiff碱相应基团的特征吸收,其中氨基较游离态表现出明显的红移,但Schiff碱相应基团的IR吸收较组装前的配合物变化不大。
主题词Schiff碱配合物;纳米孔材料;化学组装;N,N2双水杨醛缩乙二胺中图分类号:O614文献标识码:A文章编号:100020593(2004)0320281204 1992年Mobil的科学家Kresge[1,2]对M41S(MCM241,MCM248,MCM222,MCM250,MCM249)系列硅基纳米孔分子筛的合成揭开了分子筛科学的新纪元。
基于纳米孔材料大的孔径、比表面积和壁厚以及较高的化学和热力学稳定性[3],近年来以纳米孔分子筛作主体、功能分子如配合物作客体进行主2客体组装等纳米孔材料化学修饰工作得到了深入发展[4]。
其中,基于MCM241材料表面有许多端羟基,通过这些端羟基对其进行化学修饰,譬如与三乙氧基氯丙基硅烷缩合反应修饰上氯丙基[5,6],与三乙氧基氨丙基硅烷缩合反应修饰上氨丙基[7],然后再与各种金属配合物组装,用以扩展MCM241纳米孔材料原本质子酸碱功能位以外的许多新功能位,例如引入路易斯酸碱位、氧化还原活性位以及某些光学、电化学等特殊功能活性位,使得这种化学修饰后的纳米孔材料可以用于吸附、分离、催化、主客体化学、有机合成等方面。
过渡元素的配合物的成键理论过渡金属化合物的电子光谱过渡元素

一般地说, 只有惰性配位化合物才表现出异构现象, 因为不 安定的配位化合物常常会发生分子内重排, 最后得到一种最稳定 的异构体。
配合物的立体异构
立体异构可分为几何异构和光学异构两种
1 几何异构 在配合物中, 配体可以占据中心原子周围的不同位置。所研
1 电离异构 名词用于描述在溶液中产生不同离子的异构体, 一个经典
的例子是,[Co(NH3)5Br]SO4紫红色和[Co(NH3)5SO4]Br(红色), 它们在溶液中分别能产生SO42-和Br-。
2 溶剂合异构 当溶剂分子取代配位基团而进入配离子的内界所产生的溶
剂合异构现象。与电离异构极为相似, 最熟悉的例子是: [Cr(H2O)6]Cl3 [Cr(H2O)5Cl]Cl2·H2O [Cr(H2O)4Cl2]Cl·2H2O 它们各含有6、5、4个配位水分子, 这些异构体在物理和化
◆并非化学式为MX3都是三配位的。如, CrCl3为层状结 构, 是六配位的;而CuCl3是链状的, 为四配位, 其中含有氯桥 键, AuCl3也是四配位的, 确切的分子式为Au2Cl6。
3 四配位化合物
四配位是常见的配位, 包括 平面正方形和四面体 两种构型。
一般非过渡元素的四配位化合物都是四面体构型。这是因 为采取四面体空间排列, 配体间能尽量远离, 静电排斥作用最小 能量最低。但当除了用于成键的四对电子外, 还多余两对电子时 , 也能形成平面正方形构型, 此时, 两对电子分别位于平面的上下 方, 如XeF4就是这样。
十一配位的化合物极少, 理
论上计算表明, 配位数为十一的 配合物很难具有某个理想的配 位多面体。可能为单帽五角棱 柱体或单帽五角反棱柱体, 常见 于大环配位体和体积很小的双 齿硝酸根组成的络合物中。
第五章 配位场理论和配合物的电子光谱—(1)

的光谱项。(用谱学方法得到)
例如:
电子组态d 1, l = 2, ml = 2, 1, 0,s = 1/2, 电子的自旋取向ms可分 别为 1/2, 因此共有10种排列方式, 即10种微态(microstate).
自由离子光谱项(term) (多电子作用)
一. d 轨道在配位场中的能级分裂
eg
eg
6Dq
4Dq
t 2g
t 2g
d5 , High spin(弱场) 八面体场
O=10Dq
t2
4Dq
eg
6Dq
e
t 2g
四面体场 T=
4/9 O
d5 , low spin(强场)
影响分裂能的因素: 10Dq=fligand gion 1. 配体, 配位场的强度, 光谱化学系列 I Br S2 SCN Cl NO3 F OH ox2 H2O NCS CH3CN NH3 en dipy phen NO2 PR3 CN CO 2. 金属离子Mn+, n越大, 分裂能越大
②在可见光区有吸收, 但强度不大。但在紫外区, 常有强度 很大的配位体内部吸收带。
过渡金属配合物电子运动所吸收的辐射能量一般处于可见区 或紫外区, 所以这种电子光谱通常也称为可见光谱及紫外光谱。 当吸收的辐射落在可见区时, 物质就显示出颜色。物质所 显示的颜色是它吸收最少的 那一部分可见光的颜色, 或 者说是它的吸收色的补色。 表10和下图给列出可见 光的吸收与物质颜色之间的 对应关系。
MS= 1, 0
1G
3F 1D 3P 1S
L=2, ML= 2, 1 0,
L=1, ML= 1, 0, L=0, ML= 0,
第4章(4)过渡金属配合物的电子光谱
二、配合物电子光谱所包含的成份(参见过渡金属配合物.ppt)电荷迁移光谱(荷移光谱)由于电子在金属与配体间迁移产生的光谱。
—轨道角量子数注*矢量用黑体字母表示。
*角动量:就是质量乘以角速度(单位角度/秒)。
自旋角动量:角动量是由物体自旋产生的,而不是外力给它的。
轨道角动量:角动量是由轨道运动产生的2、电子间相互作用在多电子体系中,l i与l j主要是通过电性相互作用;而s i与l i或s j之间则主要通过磁性作用。
s i s jl i l j对轻元素(原子序数<30),电子间偶合强于电子内偶合,即:l i——l js i——s j的作用要大于s i——l i的作用。
此时电子间相互作用,可用L—S偶合方案处理:(参见L—S偶合方案.pdf)Σl L (总轨道角动量)Σs S (总自旋角动量)即可用L、S描述多电子体系的状态。
│S│=[S(S+1)]1/2(h/2π) │L│=[L(L+1)] 1/2(h/2π)S——总自旋角量子数L——总轨道角量子数如何求S、L见“物质结构”。
3、d n组态金属离子的谱项多电子体系的能量状态可用谱项符号表示:2S+1L L 0 1 2 3 4 5符号S P D F G H(2S+1)为谱项的自旋多重度。
如S=1/2,L=2时,为2D谱项。
组态谱项d1 d92Dd2 d83F,3P,1G,1D,1Sd3 d74F,4P,2H,2G,2F,2x2D,2Pd4d65D,3H,3G,2x3F,3D,2x3P,1I,2x1G,1F,2x1D,2x1Sd56S,4G,4F,4D,4P,2I,2H,2x2G,2x2G,2x2F,3x2D,2P,2S* d n体系,不考虑电子间作用时,只有一种能量状态。
考虑电子间作用后,产生多个能量状态(谱项)。
d1体系除外,因其只含一个电子。
4、基态谱项1)定义:能量最低的谱项称为基态谱项(基谱项)。
1)如何确定基谱项?A、同一电子组态的各谱项中,自旋多重度最大者能量最低。
第五节电子光谱

在弱配位场中的分裂及能量变化情况。可用于解释自
旋允许的电子跃迁光谱。
T-S图:
描述弱场和强场中各能量状态变化的情况。可用于 解释所有可能的电子跃迁.
⑴.Orgel图
a.d1,d4,d6和d9组态(单电子或拟单电子组态)
电子组态
d10 d1, d9 d2 , d8 d3 , d7 d4 , d6 d5
其中(1)和(2)是配合物显色的主要原因
3.过渡金属配合物电子光谱的特点
(1).通常是带状光谱(非线性光谱) 因为电子从基态能级向激发态能级跃迁时伴随有不 同的振动精细结构能级间的跃迁。 (2).配合物大多在可见区有吸收但强度不大(通常其摩
尔吸光系数A<102 ), 而在紫外区却常有强度很大的
配体内部吸收带(A=104 ~105)。
1.原子和自由离子的微观态与光谱项
①.电子组态: 指明每个轨道上的电子数目的符号。 如 p3, d4 ②.微观态: 某一给定的电子组态中, 电子对轨道的各种占据 方式叫做该组态的微观态。
如:d1电子组态的微观态 轨道角量子数l=2, 角量子在磁场的分量有2l+1个 取向, 即磁量子数m的取值有5个: ±2, ±1,0。 即一个d电子可排布在轨道角动量取向不同的5个轨道 上。由于自旋角动量在磁场方向上的分量有两个取向 (自旋量子数 ms=±1/2),所以,这一个d电子有十种 排布方式,即有10种微观态。
2.电子光谱类型
根据电子跃迁的机制,可将配合物的电子光谱
分为三种类型:
配位场光谱(由d-d跃迁产生)
电荷迁移光谱(简称荷移光谱) 配位体内的电子光谱(配位体光谱)
(1).配位场光谱
过渡金属配合物中, 由于金属离子的d轨道能级发 生分裂,当它吸收可见区或紫外区某一波段的光时, d电子便可从较低的能态跃迁+S1, L+S2, „„︱LS︱
第6章-过渡金属羰基配合物及原子簇合物

C6H6
Cr(C6H6)(CO)3 + 3CO
M(CO)6 + Py
M(CO)5Py + CO
M=Cr,Mo,W
2 与碱反应
Fe(CO)5 + 4NaOH
RX
Na+2[Fe(CO)4]2- + Na2CO3 + 2H2O
O
Na2[Fe(CO)4]
RCX
H RH
R'X RFe(CO)4
L(CO, PPh3)
O
RCHO
O2
RCOH
O2
O
X2
X2
RCX
R'R''NH O
H2O O R'OH
RCNR'R''
RCOR'
RCOOH
6.2.2过渡金属羰基簇合物的反应 1 置换反应
Mn2(CO)10 + PH3
hv
hv Fe2(CO)9 + bipy
Fe2(CO)9 + PH3
Mn2(CO)9PH3 + CO Fe2(CO)7(bipy) + 2CO Fe(CO)4PH3 + Fe(CO)5
第6章 过渡金属羰基配合物及 原子簇合物
CO与过渡金属组成的配合物称之为 过渡金属羰基配合物。含一个过渡金属 的单核羰基配合物及含两个以上过渡金 属并存在金属-金属键的多核配合物, 也称之为羰基簇合物。
表6-1 过渡金属羰基配合物
ⅣB ⅤB
ⅥB
ⅦB
ⅧB
ⅨB
Ti V(CO)6 Cr(CO)6 Mn2(CO)10 Fe(CO)5
本科配位化学

可以参照d轨道的对称性来理解D谱项:
22
可以参照d轨道的对称性来理解D谱项: 在八面体场中: d轨道分裂为eg和t2g, 同样D谱项也能分裂为Eg和T2g。 对d1和d6, 其能量关系是Eg>T2g; d4和d9与d1和d6的静电行为相反, 其能量关系是Eg<T2g 。
在四面体场中, 能级次序正好与八面体场相反。
23
这些概念可以用图形来表示, 以纵坐标代表谱项的能量, 横 坐标代表配位场分裂能,于是就得到了d1、d9、d4、d6组态在配 位场中的Orgel图, 其中d1与d4、d6与d9互为倒反, 八面体场和四
面体场互为倒反。
Td d1、d6
相反
Oh d1、d6
1
0 (13)
0
0 (14)
-1
0 (15)
5
把这15种可能的排布方式重新整理, 按每组的ML, Ms所包含 的微态数可以列成下面左上角的表。
MLMs +1 0 -1
+2
1
+1 1 2 1
0 1 31
-1
1 21
-2
1
根据这 个表, 我们 可以从中找 出相应的光 谱项。例如, 取出一组:
MLMs 0
其静电行为相同, 稳定化能均为0;
●而d6可认为是在d5上增加1个电子, 尤如从d0上增加1个电子 成d1一样, 因而d1和d6的静电行为应该相同;
●d4和d9, 可认为是在d5和d10状态上出现了一个空穴, 因而d4 和d9的静电行为也应相同。一个空穴相当于一个正电子, 其静电 行为正好与一个电子的静电行为相反, 电子最不稳定的地方, 正电 子就最稳定。因此可以预期d4与d6、d1与d9 、d1与d4、d6与d9的静 电行为都应该相反。
取代124-三氮唑过渡金属配合物的合成结构和性质

取代124-三氮唑过渡金属配合物的合成结构和性质三氮唑过渡金属配合物是一类重要的过渡金属配合物,在许多领域中具有广泛的应用。
其合成、结构和性质的研究对于进一步认识这类化合物的性质和应用具有重要意义。
本文将对三氮唑过渡金属配合物的合成方法、结构特点以及性质进行详细讨论。
三氮唑是一种含氮杂环化合物,具有良好的配位能力,能够与过渡金属形成稳定的配合物。
常用的合成方法有两种:一种是通过隔氧杂氮吡唑(PyrOz)和对苯二酚反应得到的,即用PyrOz与金属离子在适当的条件下反应生成配合物。
另一种是通过水热法合成,即将金属盐和三氮唑在高温高压的条件下反应得到配合物。
三氮唑过渡金属配合物的结构特点主要有以下几个方面:1.配合物中的过渡金属通常与三氮唑配体形成独特的配位模式,如配体可以通过N原子与金属形成配位键,形成五配位、六配位或更高配位数的配合物。
2.配合物的结构可以通过X射线衍射等实验方法确定。
通过结构分析可以得到配合物中金属离子的坐标及其配位键长度、角度等信息。
3.三氮唑配体通常可以使用不同类型的官能团进行修饰,从而改变配合物的性质。
例如,引入-cOOR基团可以使得配合物具有较好的溶解性和稳定性。
三氮唑过渡金属配合物的性质主要包括磁性、光谱性质以及催化性质等方面。
1.磁性:许多过渡金属配合物具有磁性。
根据配合物中金属离子的不同价态和配位形式,配合物的磁性可以是顺磁性或反磁性。
通过磁性测量可以了解配合物的电子结构和磁性行为。
2.光谱性质:三氮唑过渡金属配合物通常表现出特定的光谱性质,如紫外可见吸收光谱、红外光谱和核磁共振谱等。
这些光谱数据可以帮助我们了解配合物中金属离子和配体之间的相互作用及键的性质。
3.催化性质:一些三氮唑过渡金属配合物表现出很好的催化性能。
这类配合物通常通过调节配体结构和金属离子的配位环境来改变催化性能。
例如,三氮唑配体可以形成与金属中心配位的活性位点,从而促进催化反应的进行。
综上所述,三氮唑过渡金属配合物的合成、结构和性质的研究对于探索其应用的潜力具有重要意义。
课件过渡金属配合物的电子光谱

一 电子光谱
过渡金属配合物的电子光谱属于分子光谱, 它是分子中电子在不同能级 的分子轨道间跃迁而产生的光谱。
根据电子跃迁的机理, 可将过渡金属配合物的电子光谱分为 三种:
★d轨道能级之间的跃迁光谱, 即配位场光谱;
★配位体至金属离子或金属离子至配位体之间的电荷迁移光谱; ★配体内部的电子转移光谱。
4T2
4T1g(F)
g
15000
25000
cm-1
35000
使用左边部分进行计算, 4A2g→4T2g, 1=17400 cm-1, 相应于Oh场中的分裂能, 于是由 10 Dq=17400 cm-1, 得 Dq=1740 cm-1 由4A2g→4T1g(F), 2=24600 cm-1=18 Dq-C, 由此得 C=18×1740-24600=6720 c图 1 Orgel能级图
右 面 示 出 d1 、 d4、d6、d9组态在八 面体弱场和四面体 场中谱项分裂的 Orgel能级图。
为什么可以把d1、d4、d6、d9组态放到一张图中?
因为:
●d0、d5、d10 在八面体弱场和四面体场中都是球形对称的,
其静电行为相同, 稳定化能均为0;
表
物质就显示出颜色。物质所
显示的颜色是它吸收最少的
那一部分可见光的颜色, 或
者说是它的吸收色的补色。
右表和下图给列出可见
光的吸收与物质颜色之间的
对应关系。
380 435
780
紫紫 红
蓝
480
红
650
橙
绿蓝 蓝绿 490
598
黄黄 绿
绿 500
580 560
二 配体内部的电子光谱
配位体如水和有机分子等在紫外区经常出现吸收谱带。形成配合物后, 这 些谱带仍保留在配合物光谱中, 但从原来的位置稍微有一点移动。
第四章配合物的电子光谱

定性判断:
ligand 显色
Cu(NH3)42+ Cu(OH2)42+
强场 弱场
紫色 蓝色
Cr(NH3)63+ Cr(OH2)63+
强场 弱场
橙色 紫色
吸收颜色 O
黄色
大
橙色
小
蓝色
大
黄色
小
只考虑配位场作用,不考虑d电子之间的相互作用。 O的能级范围在紫外可见区域,d区元素的配合物有色.
一、d电子间相互作用 1. 单电子运动的描述 运动: 自旋运动 描述: 自旋角动量 s
特点: ①一般包含一个或多个吸收带; ② 强度比较弱, 因为d-d跃迁是光谱选律禁阻的跃迁; ③ 跃迁能量较小, 一般出现在可见区, 所以许多过渡金属
配合物都有颜色。
配合物离子的颜色 许多过渡金属配合物的颜色产生于 d 电子在晶体
场分裂而得的两组 d 轨道之间的跃迁, 即通常所谓的 d-d 跃迁 。
在无外场的情况下,这10种排列的能量是简并的, 用2D表示,D称为光谱项(term)。
自旋角量子数 s = 1/2
l:轨道角量子数
dn 体系,不考虑电子间作用时,只有一种能量状态。 考虑电子间作用后,产生多个能量状态(谱项)。d1体 系除外,因其只含一个电子。
光谱项的通式为:2S+1L
L为各个电子轨道角动量的矢量和
S 值越大,能量越低。
(2) 对于给定多重度(S相同),L大则电子间作用力小; L小, 电子间作用力大,能量高.
例: 3F的能量低于3P。 L越大,能量越低。
根据这两点,可推出 d2 组态的5个谱项的能量顺序为:
3F 3P 1G 1D 1S
其中 3F 为基谱项 (最大S, 最大L) 但实际观察的d2组态(Ti2+)光谱项的能量顺序则为:
配合物的电子光谱及磁学性质

配合物的电子光谱及磁学性质
配合物的电子光谱概述
原子光谱
第一节
光谱 分子光谱
原子光谱(包括离子光谱)主要由于原子外层电子能级变化而产生的光子发射或吸收,它是 线状光谱; 而过渡金属配合物的电子光谱则属于分子光谱。 分子光谱是分子中电子在不同能 级分子轨道间跃迁而产生的。 但在分子中除分子轨道能级外, 还有振动能级和转动能级且能 级差顺序为: ∆E e >> ∆E v >> ∆E r 因此电子跃迁的同时会伴随有振动和转动能级的跃迁, 由于这三种能量差相差很大, 吸收光谱的波长必然分布在很大的范围, 因此分子光谱是带状 光谱。 由于分子中的电子在发生跃迁时所吸收的辐射的能量一般在可见光区或紫外区, 因此 这种分子光谱常称为电子光谱或紫外-可见光谱(UV-Vis 谱) 。当吸收的辐射在可见光区时, 物质就显示出颜色,物质所显示的颜色是它最少吸收的那部分可见光的颜色-视色(吸收光 谱的补色)见表 4-1。 表 4-1 物质颜色与吸收光颜色的对应关系 λmax(nm) 吸收光 颜色 物质颜 色 400-425 紫 黄绿 425-450 深蓝 黄 450-480 蓝 橙 480-500 蓝绿 红 500-530 绿 洋红 530-560 黄绿 紫 560-600 黄 深蓝 600-640 橙 蓝 640-750 红 蓝绿
3
2
+ − − + 1 1 2 0 + + 1 0 + + - 1 1
0
1
+ − − + 1 0 1 0 + − − + - 1 1 - 1 1 + − 0 0
高等无机化学习题汇总

第二章原子结构和分子结构一、判断题3.杂化轨道中含p成分越多,原子的电负性越大。
×4.根据VSEPR理论,在SiF62-中,中心原子的价层电子总数为10个。
×5.根据VSEPR理论,氧族原子提供的电子数为6。
×6.在SO3-中,中心原子的价层电子总数为12个。
×7.SnCl2几何构型为直线型。
×8.ICl4—几何构型为四面体。
×9.NH3和NO3-的几何构型均为平面三角型。
×10.H2O和XeF2的几何构型均为平面三角型。
×11.SO32-和NO3-的几何构型均为平面三角型。
×12.下列三种离子,其极化作用顺序为:Al3+ > Mg2+ > Na+ √13.下列三种离子,其极化作用顺序为:Pb2+ > Fe2+ > Mg2+√14.Ag+的极化作用大于K+的极化作用,因此Ag+的极化率小于K+的极化率。
×15.H+的极化能力很强。
√16.极化作用愈强,激发态和基态能量差愈小,化合物的颜色就愈深。
√17.温度升高,离子间的相互极化作用增强。
√18.半径相近、电子层构型相同时,阳离子正电荷越大,极化作用越强。
√19.其它条件相同或相近时,阴离子半径越大,变形性越大。
√20.无机阴离子团的变形性通常较大。
×二、选择题3、与元素的电离能和电子亲和能有关的电负性标度是(B):(A)鲍林标度(B)密立根标度(C)阿莱-罗周标度(D)埃伦标度4、下列基团中,电负性值最大的是(A):(A)CF3- (B)CCl3- (C)CBr3- (D)CI3-5、在以下化合物中,碳原子电负性最大的是(C):(A)CH4 (B)C2H4 (C)C2H2 (D)电负性相同7、XeO3离子的几何构型为(A)(A) 三角锥 (B) 四面体 (C) V型 (D) 平面三角形8、根据VSEPR理论,多重键对成键电子对的排斥作用最大的是(A)(A) 叁重键 (B) 双重键 (C) 单重键9、根据VSEPR理论,成键电子对(BP)和孤电子对(LP)之间相互排斥作用最大的是(A)(A) LP-LP (B) LP-BP (C) BP-BP10、ClO3-离子的几何构型为(A)(A) 三角锥 (B) 四面体 (C) V型 (D) 平面三角形11、ClF3的几何构型为(C):(A)平面三角型(B)三角锥型(C)T型(D)V型12、NF3的几何构型为(B):(A)平面三角型(B)三角锥型(C)T型(D)V型13、BrF3的几何构型为(C):(A)平面三角型(B)三角锥型(C)T型(D)V型14、下列分子中键角最大的是(A):(A)NH3(B)NBr3(C)NCl3(D)NF315、下列分子中键角最大的是(A)(A) CH4 (B) NH3 (C)H2O (D)H2S16、下列分子中键角最大的是(A)(A) NH3 (B) PH3 (C) AsH3 (D)SbH317、下列分子中键角最小的是(D):(A)PI3(B)PBr3(C)PCl3(D)PF318、若阳离子电荷相同,半径相近,则最外层电子层构型为(A)电子构型的阳离子的变形性最小。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第4章(4)过渡金属配合物的电子光谱
第一节概论
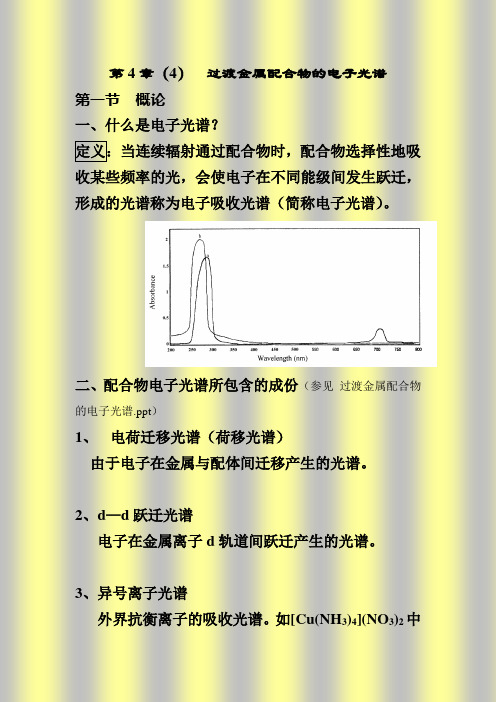
一、什么是电子光谱?
定义:当连续辐射通过配合物时,配合物选择性地吸收某些频率的光,会使电子在不同能级间发生跃迁,形成的光谱称为电子吸收光谱(简称电子光谱)。
二、配合物电子光谱所包含的成份(参见过渡金属配合物的电子光谱.ppt)
1、电荷迁移光谱(荷移光谱)
由于电子在金属与配体间迁移产生的光谱。
2、d—d跃迁光谱
电子在金属离子d轨道间跃迁产生的光谱。
3、异号离子光谱
外界抗衡离子的吸收光谱。
如[Cu(NH3)4](NO3)2中
二、d电子间相互作用(谱项与基谱项;又称光谱项、光谱支项)
原子光谱的光谱项符号是:2s+1L或2s+1L J 其构成方法为:(1)用字母表示总轨道角动量量子数L的值,对应规则是L=0,1,2,3,4,…→S,P,D,F,G,…;(2)用数字表示光谱项的多重性2S+1,其中S为原子的总自旋角动量量子数;(3)谱项的支项用右下标的J值加以区分;原子总角量子J 的取值为L + S ,L + S −1,……L −S )。
一个原子的一定的电子组态存在多个能级,相应就可以有多个原子光谱项;每个光谱项可有多个光谱支项,代表精细的能级;每个光谱支项还对应有2J +1个量子态,说明精细能级在外磁场中会进一步分裂。
1、单电子运动的描述
运动:自旋运动轨道运动
描述:自旋角动量s 轨道角动量l
│s│=[s(s+1)]1/2(h/2π) │l│=[l(l+1)] 1/2(h/2π)
自旋角量子数s=1/2
—轨道角量子数
注*矢量用黑体字母表示。
*角动量:就是质量乘以角速度(单位角度/秒)。
自旋角动量:角动量是由物体自旋产生的,而不是外力给它的。
轨道角动量:角动量是由轨道运动产生的
2、电子间相互作用
在多电子体系中,l i与l j主要是通过电性相互作用;而s i与l i或s j之间则主要通过磁性作用。
s i s j
l i l j
对轻元素(原子序数<30),电子间偶合强于电子内偶合,即:
l i——l j
s i——s j的作用要大于s i——l i的作用。
此时电子间相互作用,可用L—S偶合方案处理:(参见L—S偶合方案.pdf)
Σl L (总轨道角动量)
Σs S (总自旋角动量)
即可用L、S描述多电子体系的状态。
│S│=[S(S+1)]1/2(h/2π) │L│=[L(L+1)] 1/2(h/2π)
S——总自旋角量子数L——总轨道角量子数
如何求S、L见“物质结构”。
3、d n组态金属离子的谱项
多电子体系的能量状态可用谱项符号表示:
2S+1L L 0 1 2 3 4 5
符号S P D F G H
(2S+1)为谱项的自旋多重度。
如S=1/2,L=2时,为2D谱项。
组态谱项
d1 d92D
d2 d83F,3P,1G,1D,1S
d3 d74F,4P,2H,2G,2F,2x2D,2P
d4d65D,3H,3G,2x3F,3D,2x3P,1I,2x1G,1F,2x1D,
2x1S
d56S,4G,4F,4D,4P,2I,2H,2x2G,2x2G,
2x2F,3x2D,2P,2S
* d n体系,不考虑电子间作用时,只有一种能量状态。
考虑电子间作用后,产生多个能量状态(谱项)。
d1体系除外,因其只含一个电子。
4、基态谱项
1)定义:能量最低的谱项称为基态谱项(基谱项)。
1)如何确定基谱项?
A、同一电子组态的各谱项中,自旋多重度最大者能量最低。
B、在自旋多重度最大的各谱项中,轨道角量子数最大者能量最低。
例:d2组态:3F(基)、3P、1G、1D、1S
B、如果忽略化学环境对电子自旋的影响,则一个
谱项被配体场分裂后产生的配体场谱项与原谱项具有相同的自旋多重度。
2)用群论方法讨论谱项的分裂
由于单电子轨道波函数和谱项波函数在空间的分布状况分别依赖于量子数l和L,若L=l,则该谱项波函数在空间的分布状况类似于该单电子轨道波函数。
因此谱项波函数分裂的结果与单电子轨道波函数的分裂结果相同。
* 下标g、u的用法:
A、单电子轨道波函数,如果配体场无对称中心,则不用这两个下标;若有对称中心,则l为偶数的轨道(s、d、g)用下标g,l为奇数的轨道(p、f)用下标u。
B、对于谱项波函数,如果配体场无对称中心,则不用这两个下标;若有对称中心,则用g或u,取决于产生这个谱项的电子组态中各个电子的单电子轨道波函数的本质。
我们感兴趣的来自d n组态的那些谱项,它们在中心对
称场中全部是g特征状态。
单电子轨道波函数的分裂
轨道O h T d D4h
s a1g a1a1g
p t1u t1 a2u+e u
d e g+t2g e+t2 a1g+b1g+b2g+e g
f a2u+t1u+t2u a2+t1+t2 a2u+b1u+b2u+2e u
g a1g+e g+t1g+t2g a1+e+t1+t2 2a1g+a2g+b1g+b2g+2e g
h e u+2t1u+t2u e+2t1+t2 a1u+2a2u+b1u+b2u+3e u
i a1g+a2g+e g+t1g+2t2g a1+a2+e+t1+2t2 2a1g+a2g+2b1g+2b2g+3e g
d2组态谱项波函数分裂的结果
谱项O h T d D4h
1S 1A1g1A11A1g
3P 3T1g3T13A2g+3E g
1D 1E g+1T2g1E+1T2
B、自旋多重度
通过降低对称性将多维表示过渡到一维表示,先求出低对称性时状态的自旋多重度。
由于对称性降低后,自旋多重度保持不变,从而可由低对称性的自旋多重度推出高对称性时状态的自旋多重度。
O h群的相关表
该表指出当对称性降低时,O h群的表示如何改变或分解为它的子群表示。
O h T d D4h C4v D3 C2h
A1g A1A1g A1A1 A g
A2g A2B1g B1A2 B g
E g E A1g+B1g A1+B1 E A g+B g
T1g T1A2g+E g A2+E A2+E A g+2B g
T2g T2B2g+E g B2+E A1+E 2A g+B g
A1u A2A1u A2A1 A u
2、破坏跃迁选律的机制
如果严格遵循上述跃迁选律,则八面体配合物不会出现d→d跃迁。
1)对于ΔS≠0的禁阻跃迁,可通过旋—轨偶合作用实现跃迁。
随着旋—轨偶合常数增大,其跃迁强度增大,不过总的来说强度还是很弱。
2)对于Laporte规则
中心对称配合物:可通过“电子—振动偶合”机制,即电子波函数与振动波函数的偶合,获得跃迁强度。
对于非中心对称配合物:可通过p-d轨道混合获得跃迁强度。
3、群表示理论对跃迁选律的解释
1)基本原理与选择定则
2)“电子—振动偶合”机制
3)电子—振动的偏振作用
五、d→d跃迁光谱。