植物基因组中的连锁不平衡
植物基因组领域重要文章

植物基因组领域重要文章花了一个周末加一个半天,根据记忆整理了我认为重要的植物基因组领域的一些重要或者说有趣的文章,大家如果都读过,而且能讲个一二三,那恭喜你三把斧头至少已经有一把在手了. 需要说明的,这仅是一家之言,而且整理之时并没有去读文章,对文章的推荐仅是凭之前读过的印象,所以错误在所难免.请大家辨证参考.当然,我也无法保证这真的是经典,呵呵. 一共分为13个领域,推荐了140篇文章,就当寒假作业好了,呵呵.欢迎指正补充.(注:因为是基因组,所以暂没有把功能基因组的内容包括进来)植物基因组学研究我感兴趣的几个领域1.基因组的结构和变异2.分子标记连锁图谱构建和基因定位3.QTL定位的原理和方法4.QTL精细定位5.基因和QTL的克隆5.1插入突变方法5.2图位克隆的方法(含比较图位克隆)5.3候选基因法6.资源评估和利用7.分子标记辅助选择(含分子设计育种)8.转基因8.1转基因体系和实证研究8.2转基因的生态学安全研究9.比较基因组9.1标记水平的比较研究9.2序列水平的比较研究9.3性状水平的比较研究9.4功能比较研究10.杂种优势研究10.1遗传学解释10.2分子生物学解释11.分子进化(主要是玉米进化)12.基于连锁不平衡的关联分析12.1实证研究12.2方法学研究13.基因组研究中的一些新技术运用13.1DNA芯片技术13.2 DNA shuffling13.3 Gene Trap13.4 Gene therapy in plants13.5 TILLING 技术1.植物基因组的结构和变异在越来越多的植物基因组被测完后,该研究的重要性逐渐显现,该方面的文章可以说是汗牛充栋.在玉米方面该领域的大牛是Buckler,ES; Messing, J, Dooner HK, Doebley J ; Gaut, BS.1. Buckler, E. S., Gaut, B. S. and McMullen, M. D. (2006)Molecular and functional diversity of maize. Curr. Opin. PlantBiol. 9, 172-176这是关于玉米基因组结构的REVIEW文章,先了解大概,在细读研究文章.其任何2个玉米自交系之间的遗传变异大于人和大猩猩之间的差异的经典论断充分说明玉米变异的广泛性.最近因为人类基因组研究的进展而似乎可以改写.2.Messing J, Dooner HK. Organization and variability of themaize genome. Curr Opin Plant Biol. 2006 Apr;9(2):157-63两位大牛的联合REVIEW, 值得一读.3.Goff S A, Ricke D, Lan T H, Presting G, Wang R, Dunn M,Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D,Martin C, Katagiri F, Lange B M, Moughamer T, Xia Y, Budworth P,Zhong J, Miguel T, et al. A Draft Sequence of the Rice Genome Oryzasativa L. ssp. japonica. Science, 2002, 296: 92-100大家或许都知道这篇文章,但我相信看完的不多,尽管全基因组测序的文章许多,强烈建议大家读这篇,讨论写的太好了.同期中国测序的文章就相形见拙许多,当然之后水稻精细图谱的公布,这篇文章也可以读读. 4. International Rice Genome Sequencing Project. The map-basedsequence genome. nature, 2005, 436: 793-8005.Fu H H, Dooner H K. Intraspecific violation of geneticcolinearity and its implications in maize. Proc Natl Acad Sci USA,2002, 99: 9573-9578改文章给我的启示许多,基因的存在和缺失也是等位基因的一种形式就是其一,尽管后来该文章的结论不断被修正.6.Song R, Messing J: Gene expression of a gene family in maizebased on noncolinear haplotypes. Proc Natl Acad Sci USA 2003,100:9055-9060.宋任涛代表作之一, 与Fu的文章有异曲同工之妙,给杂种优势提供了新的解释.7.Brunner S, Fengler K, Morgante M, Tingey S, Rafalski A:Evolution of DNA sequence non-homologies among maize inbreds. PlantCell 2005, 17:343-360.5,6工作的基础上提供了更多的数据8. Lai J, Li Y, Messing J, Dooner HK: Gene movement by Helitrontransposons contributes to the haplotype variability of maize. ProcNatl Acad Sci USA 2005, 102:9068-9073.赖锦盛的代表工作之一,为玉米基因组的扩张提供了全面的解释.9. Lai J, Ma J, Swigonova Z, Ramakrishna W, Linton E, Llaca V,Tanyolac B, Park YJ, Jeong OY, Bennetzen JL et al.: Gene loss andmovement in the maize genome. Genome Res 2004, 14:1924-1931部分阐述了玉米基因组的结构的成因,更多的是插入而不是缺失.10. Morgante M, Brunner S, Pea G, Fengler K, Zuccolo A, RafalskiA:Gene duplication and exon shuffling by helitron-like transposonsgenerate intraspecies diversity in maize.Nat Genet 2005,37:997-1002与8讲的同一个故事.11.Tenaillon MI, Sawkins MC, Long AD, Gaut RL, Doebley JF, GautBS: Patterns of DNA sequence polymorphism along chromosome 1 ofmaize (Zea mays ssp. mays L.). Proc Natl Acad Sci USA 2001,8:9161-9166该数据表明,在玉米基因组大约只保留了其祖先大刍草60%的遗传变异.12.Messing J, Bharti AK, Karlowski WM, Gundlach H, Kim HR, Yu Y,Wei F, Fuks G, Soderlund CA, Mayer KF et al.: Sequence compositionand genome organization of maize. Proc Natl Acad Sci USA 2004,101:14349-14354玉米有59000个基因的预测就出自此文.13. Bruggmann R, Bharti AK, Gundlach H, Lai J, Young S,Pontaroli AC, Wei F, Haberer G, Fuks G, Du C, Raymond C, Estep MC,Liu R, Bennetzen JL, Chan AP, Rabinowicz PD, Quackenbush J,Barbazuk WB, Wing RA, Birren B, Nusbaum C, Rounsley S, Mayer KF,Messing J. Uneven chromosome contraction andexpansion in the maize genome. Genome Res. 2006Oct;16(10):1241-5114.Emrich SJ, Li L, Wen TJ, Yandeau-Nelson MD, Fu Y, Guo L, ChouHH, Aluru S, Ashlock DA, Schnable PS. Nearly Identical Paralogs:Implications for Maize (Zea mays L.) Genome Evolution.Genetics.2007 Jan;175(1):429-39Schnable 提出的NIP概念给我们以后的关联分析和其他一系列研究提出了新的挑战,尽管在玉米基因组的频率只有1%.15. Fu Y, Emrich SJ, Guo L, Wen TJ, Ashlock DA, Aluru S,Schnable PS.Quality assessment of maize assembled genomic islands (MAGIs) andlarge-scale experimental verification of predicted genes. Proc NatlAcad Sci U S A. 2005 23;102(34):12282-7.看看什么是MAGI,也是Schnable的贡献,其超大的课题组(在美国而言)和永不疲倦的精力让他文章如麻,而且牛文不断。
孟德尔随机化连锁不平衡参数设置

孟德尔随机化连锁不平衡参数设置1. 背景介绍孟德尔遗传定律是基因遗传规律的首创性发现,其对遗传学和生物学科学的发展具有重要意义。
孟德尔遗传原理将连锁不平衡引入了遗传学的研究中,而随机化连锁不平衡参数设置则是在孟德尔遗传原理的基础上进一步推导和研究的结果。
2. 随机化连锁不平衡参数设置的定义随机化连锁不平衡参数设置是指在遗传连锁不平衡的情况下,通过统计学的方法来设置参数,以准确描述遗传连锁不平衡的程度和影响。
3. 随机化连锁不平衡参数设置的重要性在遗传学研究中,连锁不平衡是遗传连锁的一种情况,其对基因型和表现型的分离和组合产生了影响。
了解和设置连锁不平衡的参数,对于理解基因的遗传规律和特征具有重要意义。
随机化连锁不平衡参数设置可以帮助研究者更准确地分析遗传数据,推断基因的亲缘关系和遗传规律。
4. 随机化连锁不平衡参数设置的方法与步骤a. 收集遗传数据:首先需要收集一定数量的遗传数据,包括基因型和表现型的数据。
b. 分析数据:利用统计学的方法对收集的遗传数据进行分析,计算连锁不平衡的参数。
c. 设置参数:根据分析得到的结果,设置随机化连锁不平衡的参数。
d. 验证参数:通过实验或模拟验证设置的参数是否符合实际情况,对参数进行修正和完善。
5. 随机化连锁不平衡参数设置的应用随机化连锁不平衡参数设置在遗传学和生物学领域有着广泛的应用。
它可以用于遗传资源的保护和开发,遗传疾病的研究与诊断,作物育种和改良等方面。
通过合理设置连锁不平衡的参数,可以更好地理解遗传现象,指导实践工作。
6. 随机化连锁不平衡参数设置的挑战与展望在应用随机化连锁不平衡参数设置的过程中,仍然存在着一些挑战。
采集遗传数据的难度和成本,统计分析方法的完善和改进等。
但随着科学技术的不断发展,这些问题将得到解决,随机化连锁不平衡参数设置的研究也将取得更多的突破。
7. 结语随机化连锁不平衡参数设置作为孟德尔遗传原理的延伸和发展,为遗传学和生物学研究提供了重要的理论和方法支持。
关联分析

1
1
F
2
1
…
2
2
1
M
2
2
…
1
3
0
F
1
2
…
2
4
1
F
1
1
…
2
5
0
M
0
-9
…
1
sample id case/control
genotypes
三、关联检验
❖2、关联检验的模型
假定: 某个SNP位点有两个基等位A、a, 形成三个基因型:AA、Aa、aa。
开始检测之前A、a地位相同,我们假定A为 minor allele,对两个等位加以区别。
1、理解连锁与连锁不平衡
连锁,是位于同一条染色体上的基因(或位点)连在一起的伴同遗传的现象 与连锁相对应的概念是交换。 连锁不平衡,是不同座位上等位基因连锁状态的描述,指这些等位基因在 同一条染色体上出现的频率大于随机组合的预期值 与连锁不平衡相对应的概念是连锁平衡。
二、连锁与连锁不平衡的关系
3、连锁分析与关联分析简介
❖2、关联检验的模型
(1)Genotypic Model
❖ Hypothesis: all 3 different genotypes have different effects
AA vs. Aa vs. aa
三、关联检验
Genotypic Model的卡方检验: Null Hypothesis: Independence
注:连锁分析与关联分析的区别: 连锁分析中,连锁描述两个位点的位置关系,
可通过重组率来度量,需要重组的数据,因此需 要家系资料。
关联分析的基础—连锁不平衡,描述的是群体 中两个位点上的等位基因的关联性,需要群体数 据。
基于关联或连锁不平衡的分析方法

基于关联或连锁不平衡的分析方法中山大学公共卫生学院医学统计与流行病学系李彩霞博士licx@(020)87330673-83(引用时请注明资料来源以及作者信息)如果两个基因座上的等位基因是随机关联的,即不独立,这种情况就叫做等位基因关联(allelic association)或者连锁不平衡(linkage disequilibrium,LD)。
关联通常反映了分子标记与性状功能突变之间在统计学上的非独立性(连锁不平衡),但并不一定意味着因果关系。
如果一个群体在初始状态下连锁不平衡(δ0≠0),在随机婚配条件下,在n代以后,有δn=(1-θ)nδ0。
因此连锁不平衡状态随着代数增加逐渐演变为平衡状态。
当连锁很弱,即重组率θ很大(接近1/2)时,连锁不平衡参数将随着代数的增加而迅速减小。
如果两个基因座紧密连锁,重组率θ很小(接近0),则不平衡状态将持续很多代。
连锁分析考察重组,因此,考察连锁必须有家庭数据,而由等位基因关联性(或连锁不平衡性)可以由一般的群体数据观察到,有的连锁不平衡现象可能是因为群体混杂造成的,但过大的连锁不平衡通常被视为紧密连锁的证据。
传统的连锁分析的结果通常是将基因定位在较大(例如~30cM)的基因组区域,而连锁不平衡被视为一种精细定位的方法。
Ott(1999)指出,对于那些远系繁殖的大群体,连锁不平衡通常只能延伸到0.3cM。
群体关联分析传统的病例-对照研究是基于群体而非家系的疾病关联分析,它通过随机选择病例和对照,然后比较其在标记等位基因和基因型频率上的差异来说明位点与疾病的关联性。
其缺点是:阳性结果可能由混杂因素造成,如不同分层人群(stratified populations)混杂在一起造成的虚假联系。
为了克服不同分层人群混杂的影响,相应产生了基于家庭的病例-对照研究方法。
单倍型相对风险分析(HRR,haplotype relative risk)单倍型相对风险分析是基于家系的病例-对照研究方法。
生物统计学中的遗传分析方法

生物统计学中的遗传分析方法生物统计学是指运用统计学的原理、方法以及计算机技术对生物学研究和实验数据进行处理、分析和解释的学科。
遗传分析是其中一个重要的研究方向,它涉及到人类和动植物遗传特征的研究、生物信息学和生物医学的应用。
在遗传分析中,统计学方法是必不可少的,下文将介绍几种常用的遗传分析方法。
1. 连锁分析连锁分析是研究基因在染色体上位置的分析方法。
在连锁分析中,首先需要用多态性标记(如SNP、STR、VNTR等)来确定人群中特定基因的可变位点,然后根据不同基因座的连锁关系,分析它们是否同时传递或存在重组。
连锁分析常用于家系研究和遗传性疾病的基因定位。
2. 关联分析关联分析是研究基因和表型之间关系的方法。
在关联分析中,通过对一定数量的个体进行基因型和表型的测量,研究同一区域内的不同基因和表型之间的连锁不平衡关系。
这种方法常用于遗传性疾病的研究和基因组关联分析。
3. 追溯分析追溯分析是一种通过调查家系史和分析现有家庭成员的基因数据,确定疾病的遗传性质和模式的方法。
在追溯分析中,需要掌握家系中各成员的基因型、表型和家庭史等信息,通过分析这些信息,可以确定疾病的遗传方式。
4. 协同分析协同分析是一种将多种遗传因素综合起来研究其对人类疾病或表型的影响的方法。
在协同分析中,需要综合考虑多种因素,如基因型、环境因素、年龄和性别等。
5. 基因表达分析基因表达分析是一种研究基因的转录和表达水平的方法。
在基因表达分析中,通过对特定基因的mRNA表达量进行测量,分析其表达变化的规律和机制,从而探究基因与表型之间的关联。
以上是几种生物统计学中常用的遗传分析方法,这些方法均是基于统计学的原理和方法开展研究的。
随着技术的不断进步,这些方法也在不断优化和完善,将对人类和动植物的遗传和表型研究起到越来越重要的作用。
基因的连锁和互换规律

黑身残翅
(bbvv)
测交 后代
(BbVv) 42% (bbvv)42% (Bbvv) 8%
雌 果 蝇 的 连 锁 和 交 换 遗 传
(bbVv) 8%
灰身长翅 黑身残翅 灰身残翅 黑身长翅
p
配 子
B b V v 灰身长翅(父本)
b b x v v 黑身残翅(母本) b v
b b v v 黑身残翅
未交换精子 精原细胞数 精子数 Ab 80个未交换 80*4=320 20个交换 100 20*4=80 400
160
交换精子 AB ab
Ab
160
20
180
20
180 2A % A% A/2%
20
20
20
20
精原细胞的交换值为20% 交换值为10%
一种交换配子为5%
基因连锁和互换规律在实践上的应用 1、如果不利的性状和有利的性状连锁在一起,那
B V
b v
子 代
B b V v 灰身长翅
雄 果 蝇 的 连 锁 遗 传 解 释
1
:
1
基因连锁和交换的原因
灰身长翅果蝇的灰身基因和长翅基因位 于 同一染色体 上,以 ( B V )表示。 黑身残翅果蝇的黑身基因和残翅基因位 于 同一染色体 上,以 ( b v )表示。 经过杂交,F1是灰身长翅,其基因型是 B V ( )。 这样的雄果蝇,位于同一染色体上的两 个基因(B和V、b和v) 不分离 ,而是连 在一起随着生殖细胞传递下去。
• 美国的遗传学家摩尔根和他的同事用果 蝇做实验材料,进行了大量的遗传学研究, 终于解开了人们心中的疑团,这不仅证实 了孟德尔的遗传规律的正确性,并且丰富 发展了关于两对(或两对以上)基因的遗传 理论,提出了遗传的第三个规律----基因的 连锁互换规律
【word】人类基因组中的连锁不平衡方式

人类基因组中的连锁不平衡方式第25卷第3期2005年6月国外医学?生理,病理科学与临床分册ForeignMedicalSciences?SectionofPathophysiologyandClinicalMedicine V0I.25NO.3June.20D5人类基因组中的连锁不平衡方式梁云综述周韧审校(浙江大学病理学与法医学研究所,浙江杭州310006)摘要:连锁不平衡(1inkagedisequilibrium,LD)伴随突变的多态性出现,由于位点间重组,LD程度逐渐下降.对于一个特定群体而言影响LD的因素很多,一系列人口历史因素起着重要的作用.LD程度的度量,目前常用的两种配对检验方法为D和r2.在染色体的部分区域存在一系列重组热点分割的单倍型块.目前LD主要应用于关联研究中以定位复杂的疾病基因.关键词:连锁不平衡;单倍型块;关联分析中图分类号:Q75文献标识码:A文章编号:1001?1773(2005)03-0247-04 随着人类全基因组高精度序列图的完成,越来越多的人开始把目光转向多基因疾病的基因定位, 克隆,诊断和治疗.过去对符合孟德尔遗传规律的单基因病的研究主要采用连锁分析,其数据来源于对受累家系的分析,而多基因病是受多个微效基因与某些环境因素共同影响所致.Risch等¨认为,在多基因疾病中若应用连锁分析定位微效基因,其所需要的家系数目将大得惊人,故提出了关联分析(testofassociation)的方法,其中基于连锁不平衡(1inkagedisequilibrium,LD)的关联分析在定位复杂疾病基因上显示出强大的功能.LD是基因定位的基础,因此有必要深入了解LD的本质,结构及不同人种在基因组不同部位的延伸程度.1LD的本质LD是相邻基因座位上等位基因的非随机性相关,当位于某一基因座位上的特定等位基因与同一条染色体另一基因座位上的某等位基因同时出现的几率大于人群中因随机分布而使两位点同时出现的几率时,就称这两个位点处于LD状态.LD的概念最初是用一等式来描述和衡量的:假设存在相邻基因座位1和2,座位1的等位基因为A,a,其频率为P,P;座位2的等位基因为B,b,其频率为P,P.等位基因A和B在同一染色体上同时出现的频率,即它们组成的单倍型频率为P则连锁不平衡值D =PAB—PA×PB.LD的产生可以简单地理解为由突变产生的多态性形成的.在染色体某一特定等位基因附近有新的突变产生时则LD出现,之后由于重组的发生,两位点间LD程度逐渐降低.理论上说,人群中LD强度将随着时间和重组距离而减弱,但实际上对于较短距离的LD,随机因素可能起着更重要的作用.另外,虽然也有数据显示随着距离的增加LD有减弱的趋势,但靠得很近的标记并非总呈现出LD_2;相反,亦有数据显示对于距离相当远的标记也存在LD[.2影响LD的因素突变和重组对LD起着极其重要的作用,但其他因素也影响了LD的程度和分布,人口历史因素就是其中最重要的方面.2.1遗传漂变这种现象指群体中世代间基因频率的随机变化最终会导致一个等位基因的固定或丢失,成为某个等位基因的纯合子.这种由于群体较小和偶然事件而造成的基因频率随机波动,在任何一个隔离的人类自然群体中,只要与其他群体间没有基因的交流都会发生这样的过程.一般认为在一个小而稳定(人数不增加)的群体中遗传漂变会使LD程度增加,单倍型种类减少.遗传漂变的另一种形式是”奠基者效应”,即一个小群体从一个大群体中分离出去并在此基础上逐渐发展起来,这是一种剧烈的漂变.2.2人口增长与群体结构快速的人口增长会降低遗传漂变,从而减弱了LD的强度.群体结构收稿日期:2004-12-06修回日期:2005-04.21作者简介:梁云(1977-),男,山西临汾人,硕士研究生,主要从事肿瘤病理研究.基金项目:浙江省自然科学基金(M303818),浙江省卫生厅科研基金(2002ZX010)247第3期国外医学?生理,病理科学与临床分册第25卷的很多方面都会影响LD,群体的增长可引起LD程度的降低,在长期增长的群体中此现象更为明显.相反,群体的再分则会增加LD的程度.2.3重组率的变化在基因组的不同位置重组率不同.由于重组是打断LD的主要原因,因此LD 程度与重组率呈反比.目前已知重组在很大程度上局限于热点区域.因此LD主要发生在非重组区,在重组热点区断裂.’2.4突变率的变化一些单核苷酸多态性位点(singlenucleotidepolymorphism,SNP)尤其是位于CpG岛上的SNP有很高的突变率,与邻近位点的标记几乎不表现出LD.2.5基因转换基因转换是指染色体的部分片段在减数分裂的过程中转移到另一片段的过程.类似重组或突变,基因转换也可打断LD.现已证明人类基因转换的发生率较高,并且对紧密相邻标记的LD影响较大.从以上可以看出,人口的历史因素对LD的影响是复杂的.当人类最初”走出非洲”后,世界上不同地域的群体经历了不同程度的迁移,混合或遗传漂变.大量的实验数据也证实了对于不同人群其LD程度不同.欧洲人群较之非洲人群其核苷酸多样性程度较低,而LD程度较高J.一般认为这是由于欧洲人群在其发展历史中经历了”奠基者效应”,从而形成强的LD效应.另外,LD程度在基因组的不同区域也不相同.曾有报道,在相对距离超过100kb的标记间仍有LD,相反亦有相当多的研究显示在很短距离的标记间只存在弱的LD.Abe. casis等认为物理距离对于LD变化的影响不超过50%,遗传漂变等人口历史因素可能起着更重要的作用.3LD的度量对于LD的度量已有多种不同的方法,其中大多数都是应用于双等位基因的配对检验.目前常用的两种配对检验方法为连锁不平衡系数(coefficient oflinkagedisequilibrium,D)和r.它们的取值范围都是从0(连锁平衡)到l(完全连锁不平衡).但它们的意义不同.3.1连锁不平衡系数D当两个基因座位上的等位基因频率确定后,D等于D除以D的最大可能值Dmax.连锁不平衡系数D的意义如下:D=1称为完全连锁不平衡(completeLD),说明两个位点间没有发生重组,在这种情况下由这两个位点构成248的4种单倍型在所选的样本中至多只能出现3种. 如果D<l则说明这两个位点间发生过重组(新发生的突变也会引起D<l,但对于SNP来说突变的概率较重组要小的多),这种情况下4种单倍型均可出现,但这时D值相对大小的意义就很模糊了(如D=0.3或D0.7,二者的区别就很模糊),因此如果D的计算结果接近于l,则提示两位点间历史上发生重组的可能性很小,但如果D处于中间值则不可用该数值来比较两位点LD程度的差别.而且,在小样本中D值会显着增加,这对于有少见等位基因(两个等位基因中频率较低的一个频率<5%)的SNP来说尤其明显,因此这时即使两个标记已达连锁平衡亦可能出现较高的D值.3.2rr代表两位点在统计学上的关系,在某种程度上可看作D的补充.r等于D除以两位点上4种等位基因频率的积.r等于l说明两位点没有被重组分开,且等位基因频率相同.在这种情况下由两位点构成的4种单倍型在所选样本中只出现2种(即AB,ab).r的数值表示一个位点可反映另一位点信息量的程度,r等于l称为完美连锁不平衡(perfectLD),这时只观察一个标记即可提供另一标记的全部信息.相比较而言D等于l时两位点等位基因频率并不需要相同,D等于1只是反映最近一次突变发生后突变位点与临近多态性位点的关系.另外,r在小样本中亦不会显着增加.目前r主要用于关联分析中¨….以上两种方法都是针对配对位点的LD强度测量.但有时需要知道包含有多个多态性位点的某区段的LD强度,或是不同群体间LD的差异,这就需要用到P测量法.对于估计P值大小计算方法的研究是现在的一个热点¨,在此不再详述.4单倍型块的概述2001年Daly等¨发现位于染色体5q3l的一个长约500kb区段上的单倍型结构可以分割成一系列彼此分离的单倍型块(haplotypeblock),其大小为3~92kh.这些单倍型块被重组热点所分割,在单倍型块内重组率很低,对应于某一区段的单倍型块,只有少数几种单倍型(平均2~4种).Jeff-reys等发现,MHC.I1类分子基因重组局限于一狭窄的热点范围内.基于以上研究结果,遗传学家提出了基因组可以分割为一系列由重组热点分割的高LD区域.根据这一假说,如果单倍型块样的结构普遍存在,那么在关联研究中所需的SNP数量将大大第3期梁云,等:人类基因组中的连锁不平衡方式第25卷减少.此后一些大范围的序列研究也证实,基因组可以分割为一系列高D或低单倍型种类的区域¨. 在已发现的单倍型块中,最长的单倍型块位于欧洲人群的22号染色体,该单倍型块延伸约804kb¨.当然.这只是特例,绝大多数报导的单倍型块长度在5~20kb之间.4.1单倍型块的定义染色体在一代代的传递中同源片段发生重组,多代之后祖先染色体片段的原有排布已被打乱.那些没有被重组打破的区域相互间被重组区域隔开,这些区域就是单倍型块.定义单倍型块的方法很多,但目前大多数学者更倾向于利用配对连锁不平衡系数D以确认重组热点,因为测量重组似乎更符合单倍型块的本质,而且利用配对连锁不平衡系数D更适合用于双等位基因.在此基础上,Reich等提出了以LD半衰期作为单倍型块的界限,所谓LD半衰期(LDhalf-life)是指平均D降低至0.5以下.Reich等用配对D在尼13利亚(约鲁巴人)人群中分析19个平均长度为160kb 区段上的272个高频SNP,根据LD半衰期作为单倍型块界限的原则发现了长为6~155kb不等的单倍型块.Gabriel等则定义单倍型块为有重组迹象的SNP对小于5%的区段.估计一对SNP在历史上是否发生过重组可通过D估计,但D会因样本量小而上升,故需使用D的可信区间(而非对某一点的估计)来计算重组,并定义Dl的可信区间上游小于9.0为重组依据.4.2标签SNP引入单倍型块概念在于只用少量的标签SNP(haplotypetagsSNP,htSNP)就能代表某一区段绝大多数的常见单倍型种类.htSNP是指位于染色体某一区段的单倍型结构能够用少量关键的标记代表,而无需找出该单倍型上的所有标记,用少量的几个标记代表的单倍型结构即可与其他单倍型相区别.Daly等..在250个欧洲人中应用103个常见SNP(频率大于5%,平均密度1SNP/5kb)寻找染色体5q31上长约500kb范围内的单倍型结构, 该研究显示只需少量的htSNP即可确定大小从10~100kb的单倍型块,并且2~4个单倍型就能够代表该区段中90%以上的单倍型种类.4.3单倍型块的特点到目前为止,对单倍型块的理解可归纳为以下几点:①并非染色体的所有区段都以单倍型块的方式存在,Gabriel等对欧裔美国人,非裔美国人,东亚人和非洲人等4种人群的研究显示,只有不到一半的序列以单倍型块的方式存在,并且对于不同人群单倍型块的大小亦不相同,在非洲和非裔美国人中单倍型块约22kb左右,在欧洲和亚洲人群中单倍型块约44kb左右.②对于已明确的单倍型块只需少量的多态标记即可得到绝大多数的单倍型种类,而要想确定一个区段是否为单倍型块则需要在一个足够大的样本中确定高密度的多态性标记,因为在一个只有较少标记的区段中可能会探测不到一些小的单倍型块.③目前绝大多数学者认为单倍型块模型以一种简单而有效的途径代表了LD的主要特点¨.4.4单倍型块的应用由于只用少量的htSNP就能代表一个单倍型块内的绝大多数单倍型种类, 故htSNP是寻找致病基因的一条捷径.如果某个单倍型块在特定疾病人群中更为普遍,那么在此单倍型块内可找到疾病相关的基因.通过这种方法可以发现心脏病,糖尿病等多基因疾病的致病基因,并可找到不同个体对相同药物具有不同反应的原因.5连锁不平衡和关联研究将LD应用到大规模的关联研究中,可定位复杂的疾病基因.在关联研究中,如果某一因素可增加某种疾病的发生风险,而该因素在疾病人群中的频率比在正常人群中高,就可认为该因素与疾病相关联.在关联分析中,主要关注基于LD的间接关联分析,其基本原理为:如果某致病基因座与遗传标记(多态性的等位基因)存在强的LD,那么就可以通过比较遗传标记在患者与正常个体间的差异,最终得到该致病基因座在疾病发生中的相对危险度.一般认为,影响关联分析效力的因素有以下几点:①所研究疾病位点的危险度;②疾病位点等位基因的频率;③标记位点等位基因的频率;④两者之间的LD强度.必须指出,在群体关联分析的实验设计中,各个环节都有可能影响数据分析的结果和准确性,其中以所研究群体的选择最为重要.在社会人群中,由于奠基者效应,种族差异以及性别比例和年龄结构不同等诸多因素,使得用于群体关联分析的标本在病例组和对照组不能保持匹配,出现所谓的”群体分层”,造成病例组和对照组多态性位点差异具有显着性,从而得出该位点与疾病相关的假阳性结果.因此,在实验设计时应尽可能选择相对同源的群体.参考文献01RischN,MerikangasK.Thefutu~ofgeneticstudiesofcomplexhu. mandiseases[J].Science,1996,273(5281):1516.1517.249第3期国外医学?生理,病理科学与临床分册第25卷02ArdlieK,”I卜Co~emSN,EberleMA,eta1.Lowerthanexpected linkagedisequilibriumbetweentighdylinkedma~ersinhumanssug- gestsaroleforgeneconversion[J].AmJHumGenet,2001,69(3):582-589.03StephensJC,SchneiderJA,TanguayDA,eta1.Haplotypevariation andlinkagedisequilibriumin313humangenes[J].Sc/ence,2001,293(5529):489-493.04FrisseL,HudsonRR,BartoszewiczA,eta1.Geneconversionand differentpopulationhistoriesmayexplmnthecontrastbetweenpoly? morphismandlinkagedisequilibriumlevels[J].AmJHumGenet, 2001,69(4):8313.05G~dardKA,HopkinsPJ,HallJM,eta1.Linkagedisequilibrium andallele—frequencydistributionsfor114sin~e—nucleotidepolymor- phismsinfivepopulations[J].AmJHumGenet,2000,66(1):2l6-234.06NakaiimaT,JordeLB,IshigamiT,cta1.Nucleotidediversityand haplotypestructureofthehumanangiotensinogengeneintwopopula- tions[J].AmJHumGenet,2002,70(1):108.123.07AbecasisGR,NoguchiE,HeinzmannA,eta1.Extentanddistribu. tionoflinkagedisequilibriuminthreegenomicregions[J].AmJ HumGenet,2001,68(1):191.197.08PritchardJK,PrzeworskiM.Linkagedisequilibriuminhumans: modelsanddata[J].AmJHumGenet,2001,69(1):1.14.09ArdlieKG,KruglyakL.SeielstadM.Patternsoflinkagedisequilib- riuminthehumangenome[J].NatRevGenet,2002,3(4):299.3o9.10WeissKM,ClarkAG.Linkagedisequilibriumandthemappingof complexhumantraits[J].TrendsGenet,2002,l8(1):19-24.121415McV eanG,AwedaliaP,FearnheadP.Acoalesc ent’basedmethed fordetectingandestimatingrecombinationfromgenesequences[J]. Genet/cs,2002,160(3):1231?1241.DalyMJ,RiouxJD,SchaffnerSF,eta1.High?resolutionhaplotype structureinthehumangenome[J].NatGenet,2001,29(2):229. 232.JeffreysAJ,KanppiL,NeumannR.Intenselypunctatemeioticre? combinationintheclassIIregionofthemajorhistocompatibilitycom? plex[J].NatGenet,2001,29(2):217-222.PhillipsMS,LawrenceR,SachidanandamR,eta1.Chromosome. widedistributionofhaplotypeblocksandtheroleofrecombinationhotspots[J].NatGenet,2003,33(3):382-387.DawsonE,AbeeasisGR,BumpsteadS,eta1.Afirst—generation linkagedisequilibriummapofhumanchromosome22[J].Nature, 2002,418(6897):544.54816ReichDE,CargillM,BolkS,eta1.Linkagedisequilibriuminthe humangenome[J].Nature,2001,411(6834):199-204.17GabrielSB,SchaffnerSF,NguyenH,eta1.Thestructureofhaplo. typeblocksinthehumangenome[J].Sc/ence,2002,296(5576):2225-2229.18CarlsonCS,EbedeMA.RiederMJ.eta1.AdditionalSNPsand linkage?disequilibriumanalysesarenecessaryforwhole.genomeas$o- ciationstudiesinhumans[J].NatGenet,2003,33(4):518-521.19WallJD,PritchardJK.Hapbtypeblocksandlinkagedisequilibrium inthehumangenome[J].NatRevGenet,2003,4(8):587397.20ZondervanKT.CardonLR.Thecomplexinterplayamongfactors thatinfluenceallelicassociation[J].NatRevGenet,2004,5(2):89.1o0250。
遗传学名词解释

遗传学名词解释●law of segregation(分离定律):一个遗传性状的两个等位基因在配子形成过程中是分离的,最终形成不同的配子●law of independent assortment(自由组合定律):应当具有两对(或更多对)相对性状的亲本进行杂交,在子一代产生配子时,在等位基因分离的同时,非同源染色体上的非等位基因表现为自由组合。
●The Law of Dominance(显性定律):在杂合子中,一个等位基因可以隐藏另一个等位基因的存在。
●allele(等位基因):是指位于一对同源染色体相同位置上控制同一性状不同形态的基因。
●test cross(测交):是一种特殊形式的杂交,是杂交子一代个体(F1)再与其隐性或双隐性亲本的交配,是用以测验子一代个体基因型的一种回交。
●monohybrid(单因子杂种):指只有1对等位基因不同的两个(同质的)亲本所形成的杂种。
●dihybrid(双基因杂种):二对等位基因不同的两亲间的杂种。
●Complete dominance(完全显性):发生在杂合子和显性纯合子表型相同的情况下。
●incomplete dominance(不完全显性):f1杂种的表型介于两个亲本的表型之间。
●codominance(共显性):两个显性等位基因以不同的方式影响表型。
●multiple allele(复等位基因):一个基因有两个以上的等位基因。
●allele frequency(等位基因频率):基因的每个等位基因占基因拷贝总数的一个百分比,这个百分比称为等位基因频率。
●monomorphic genes(单型的基因):这种基因只有一种常见的野生型等位基因。
●polymorphic genes(多态性基因):有些基因有一个以上的等位基因。
●Pleiotropy(多效性):一个基因可能导致几个特征。
●Recessive epistasis(隐性上位)隐性等位基因需要隐藏另一个基因的作用,这种掩蔽现象称为隐性上位。
基因的连锁和互换规律

b V
b v
灰身长翅(母本)
x
bb vv
雌 果Βιβλιοθήκη 黑身残翅 (父本) 蝇的
连
配子 B
b
B
b
b
锁
V
v
v
V
v
和
互
换
测交 B b b b B b b b 遗
后代 V v v v v
vV v 传
表现型 灰身长翅 黑身残翅 灰身残翅 黑身长翅 解
42% 42%
8%
8%
释
具有连锁关系的两个基因,其连锁
关系是可以改变的。在减数分裂
交换值:测交后代 重组型/后代总数
=F1 重组型配子/ F1 配子总数 =发生互换的初级性母细胞所占比例/2 =F1 任意一种重组型配子比例×2
精原细胞数
精子数
未交换精子 Ab Ab
交换精子 AB ab
80个未交换 80*4=320 160 160
20个交换 20*4=80 20 20 20 20
基因的连锁和交换现象 雌 果
蝇
P 灰身长翅 ×
(BBVV)
黑身残翅
(bbvv)
的 连
锁
F1测交
雌×
雄
和
灰身长翅 黑身残翅
(BbVv)
(bbvv)
交 换
测交 后代
遗 传
灰身长翅 黑身残翅 灰身残翅 黑身长翅
(BbVv) 42% (bbvv)42% (Bbvv) 8% (bbVv) 8%
P
B V
B v
(一)、 连锁遗传现象
1906年 W. Bateson & Pannett在香豌 豆两对相对性状杂交试验中首先发现。
翻译植物连锁不平衡和联分析目前的现状和未来的发展教材

植物连锁不平衡和关联分析目前的现状和未来的发展在过去的20年里,DNA分子标记已经广泛地应用到植物和动物的许多研究中。
一个主要的应用是构建分子遗传图谱来研究简单或复杂的性状。
然而这些研究主要是基于作图群体的连锁分析,这在许多植物系统中受到许多限制。
所以,已经提出了替代的策略,其中一个主要的策略是利用基于连锁不平衡的关联分析。
虽然这种策略已经用到分析人类复杂性状的遗传,例如不同疾病的遗传,但它在植物中的应用还刚开始。
在这篇综述中,我们首先区分连锁不平衡作图和关联作图,然后简单描述各种不同的衡量LD的方法及这种方法的应用,并列出了一系列影响LD的因素,讨论LD在植物研究中的一些内容。
我们也描述了LD在植物基因组学方面的不同应用,及在不同的植物基因组研究中LD相关研究目前的状态。
最后,简单讨论了植物LD研究未来的发展方向,列出了LD相关研究的一系列软件。
开发和检测动植物系统的DNA多态性标记是分子生物学和生物技术研究中的一个发展迅速的领域。
这是导致植物基因学研究20多年来进展迅速的主要原因,同时也使分子标记在植物遗传学研究中的应用变得越来越活跃。
产生DNA分子标记多态性两个主要原因是:突变和重组(突变产生新的等位基因,重组产生不同位点不同的等位基因组合)。
因此在许多研究中检测连锁和追踪DNA多态性的产生历史成为分子标记的中心任务,然而对连锁的研究需要设计适合的杂交,有时需要建立作图群体甚至近等基因系。
在有些情况下这是一个比较严重的限制因素,因为有的杂交不可能获得(例如树种),或者有时作图群体太小。
所以其它的策略已经被发展起来研究连锁和重组,以及群体的突变历史。
其中的一个策略是基于LD的关联分析。
近几年来,它越来越受到植物遗传学家们的重视。
这个策略有进行QTL作图以及鉴定引起表型变异的序列多态性的潜力。
它也可以鉴定单个基因或多个基因的单倍型(This also allows the identification of haplotype blocks and haplotypes representing different alleles of a gene.)可以根据特定区域的LD的存在来设计关联分析(In using this approach, an idea of the length of a region over which LD persists is also possible, so that one can plan and design studies for association analysis.)。
GWAS与QTL的分析内容与原理的比较
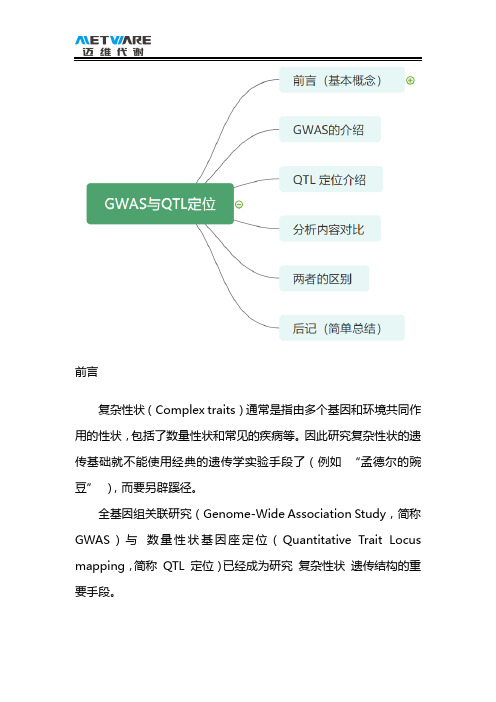
前言复杂性状(Complex traits)通常是指由多个基因和环境共同作用的性状,包括了数量性状和常见的疾病等。
因此研究复杂性状的遗传基础就不能使用经典的遗传学实验手段了(例如“孟德尔的豌豆”),而要另辟蹊径。
全基因组关联研究(Genome-Wide Association Study,简称GWAS)与数量性状基因座定位(Quantitative Trait Locus mapping,简称QTL 定位)已经成为研究复杂性状遗传结构的重要手段。
GWAS首先来聊聊GWAS,虽然才发展了十几年,已经在疾病研究和植物农艺性状遗传研究当中获得了广泛的应用,算是一个大热点领域。
此处需要祭出这张图:“Common Variants,common diseases”,这个假设就是GWAS研究的前提。
是的,没看错,人类对自身的探索了解和掌握永远是技术进步的一大动力,GWAS 研究开始也是为了研究人类疾病以及其他特征的。
GWAS 的核心是基于分子标记的连锁不平衡(LD),利用LD 来研究标记与性状之间关系。
名词解释:连锁不平衡连锁不平衡(linkage disequilibrium,简称LD),是指在指定群体内,不同位点等位基因间的非随机性关联,包括两个标记间/两个基因间/一个基因与一个标记间的非随机关联。
简单的说就是:两个不同位置的等位基因同时出现(连锁)的概率,高于随机出现的概率(不平衡)。
GWAS - 全基因组关联分析,顾名思义,是在全基因组范围内,检测多个个体的遗传变异多样性,获得群体中每个个体的基因型;然后与性状(即我们常说的表型)进行统计学关联分析,根据统计量(主要指P 值)筛选出候选变异位点和基因。
QTL 定位接下来,我们说说QTL 定位,与GWAS 相比,QTL定位可算历史悠久,已经发展了近一个世纪,是研究数量性状遗传基础的主要手段。
有趣的是,GWAS 实质是利用连锁不平衡定位,而QTL的实质,是确定分子标记与QTL 之间的连锁关系,基本原理是QTL 与连锁标记的共分离。
基于连锁不平衡的体细胞单核苷酸变异检测方法

基于连锁不平衡的体细胞单核苷酸变异检测方法基于连锁不平衡的体细胞单核苷酸变异检测方法随着基因组学的快速发展,越来越多的单核苷酸变异(Single Nucleotide Variations,SNVs)被发现与人类疾病的发生和发展密切相关。
SNVs是一种常见的遗传变异类型,它在基因组中的单个核苷酸位置发生变异,可能导致基因功能改变,进而影响个体的生理和病理状态。
因此,准确检测和鉴定SNVs对于疾病诊断和治疗具有重要意义。
然而,由于体细胞中存在连锁不平衡现象,即在同一染色体上的两个不同位点之间的遗传关系无法独立于其他位点进行分析,传统的SNV检测方法往往难以解决此问题。
为了克服这一困难,研究人员开发了一种基于连锁不平衡的体细胞SNV变异检测方法。
首先,为了确定SNV的存在和位置,人们通常采用高通量测序技术。
通过对基因组进行测序,可以生成大量的DNA片段序列信息。
然后,通过对这些序列进行比对,可以鉴定出SNVs的组成。
然而,在连锁不平衡情况下,仅仅依靠序列信息无法准确判断SNVs的存在与否,因为同一染色体上的多个SNVs之间可能存在复杂的遗传关系。
为了解决这一问题,研究人员提出了基于连锁不平衡的SNV变异检测方法。
该方法首先通过建立多个SNVs之间的连锁关系网络,来描述它们在同一染色体上的遗传关系。
基于该网络,可以计算出不同SNVs之间的连锁不平衡度,并据此推断SNVs的存在情况。
具体而言,该方法使用了碱基窗口平均方法对SNVs进行连锁分析。
首先,将染色体划分为多个重叠的碱基窗口,然后对每个窗口内的SNVs进行分析。
在每个窗口内,根据SNVs之间的连锁情况,构建SNV连锁网络。
接下来,通过计算连锁网络中的连锁不平衡度,可以确定SNVs的存在与否。
除了连锁不平衡度,该方法还考虑了突变频率和遗传距离等因素,以提高检测结果的准确性。
此外,为了进一步验证检测结果的可靠性,研究人员还引入了参照数据集和统计学方法。
植物基因组中的连锁不平衡

连锁不平衡的理论基础11连锁不平衡的概念连锁不平衡linkageld亦被称disequilibrium为配子相不平衡gameticphasedisequilibrium配子不平衡gameticdisequilibrium位基因关联allelicassociation指的是一个群体内不同座位等位基因之间的非随机关联包括两个标记间或两个基因qtl间或一个基因qtl与一个标记座位间的非随机关联连锁不平衡与连锁是相关但完全不同的两个概念
有两个值得注意的问题是: 某些自交物种如大 麦[14]虽然是由同质个体组成, 但在群体水平却具有 很高的遗传多样性。另外, 自然进化进程和人为介 入可改变某物种的杂交类型。如栽培大豆的异交率 为 1%, 而其祖先的异交率高达 13%[15]。异交率的 改变将影响群体的 LD 水平。
2.2 群体特性
LD 作图利用的是自然群体中的自然变异, 即通 过分析自然群体中标记与紧密连锁 QTL 间的 LD 关 系来鉴定和定位 QTL, 而且可以鉴定由 QTL 所代表 的真正与被研究目的性状相关联的基因。LD 的一个 明显特性是群体依赖性。即使来自同一物种的不同 群体也可能有明显不同的 LD 特性。影响群体大小 的瓶颈效应将致使仅仅少数等位基因组合能够传递 到后代中, 而低频率多态性则丧失掉, 因此其 LD 水 平大大增加。但在没有其他减轻因素(比如群体亚结 构)的情况下, 这种影响是短期的[16]。选择的群体不 同, 其 LD 水平显著不同。多样性较高的群体包括更 多不同来源的研究个体, 因此其 LD 水平较低; 而当 所用群体来源有限时, 其 LD 将维持在一个较高水 平。如玉米中, 地方品种在 600 bp 范围内存在 LD 衰减[17]; 不同育种自交系在 2 000 bp 范围内存在 LD
基因完全连锁和不完全连锁

基因完全连锁和不完全连锁
基因的连锁是指两个或多个基因在遗传过程中常常共同遗传,且它们之间的遗传关系通常是稳定的。
基因的连锁可以分为完全连锁和不完全连锁两种类型。
完全连锁是指两个或多个基因在染色体上的位置非常接近,因此在遗传过程中通常一起遗传,难以分开。
这种情况下,这些基因往往会以一定的概率同时出现在后代中。
不完全连锁是指两个或多个基因在染色体上的位置并不非常接近,因此在遗传过程中可能会相互独立遗传。
在这种情况下,这些基因往往不会以固定的概率同时出现在后代中,而是可能出现在不同的后代中。
基因的连锁是遗传学中非常重要的概念,对于理解遗传过程和预测后代表现非常重要。
小豆遗传差异、群体结构和连锁不平衡水平的SSR分析

作物学报 ACTA AGRONOMICA SINICA 2014, 40(5): 788−797/ISSN 0496-3490; CODEN TSHPA9E-mail: xbzw@本研究由国家现代农业产业技术体系建设专项(CARS-09), 国家公益性行业(农业)科研专项经费(nyhyzx07-017), 作物种质资源保护(NB2013-2130135-25-09)和中国农业科学院科技创新工程资助。
*通讯作者(Corresponding author): 程须珍, E-mail: chengxz@, Tel: 010-********第一作者联系方式: E-mail: bai_peng02@Received(收稿日期): 2013-11-18; Accepted(接受日期): 2014-01-12; Published online(网络出版日期): 2014-03-24. URL: /kcms/detail/11.1809.S.20140324.1334.006.htmlDOI: 10.3724/SP.J.1006.2014.00788小豆遗传差异、群体结构和连锁不平衡水平的SSR 分析白 鹏 程须珍* 王丽侠 王素华 陈红霖中国农业科学院作物科学研究所, 北京100081摘 要: 利用57对小豆SSR 标记和31对绿豆SSR 标记, 用5份日本材料作对照, 对249份中国小豆种质进行遗传差异、群体结构和连锁不平衡(LD)分析。
结果表明, 共检测到630个等位变异, SSR 位点等位变异数在2~17之间, 遗传多样性指数范围为0.024~0.898, 平均为0.574。
15个不同地理来源群体间表现出显著的遗传多样性差异, 其中中国云南最高, 河北和天津最低。
聚类分析将254份材料划分为3个类群, 在一定程度上和地理生态环境相关。
LD 分析显示和其他作物相比, 小豆LD 衰减距离较短, 最大衰减距离为5.8 cM (R 2>0.1), 基因组LD 平均衰减距离小于1 cM (R 2>0.1, P <0.001)。
棉花品种资源群体结构与连锁不平衡分析

棉花学报Cotton Science 2011,23(6):500~506棉花品种资源群体结构与连锁不平衡分析张友昌1,别墅1,易先达1,张成1,李成奇2,秦鸿德1*(1.湖北省农业科学科院经济作物研究所,武汉430064;2.河南科技学院生命科技学院,新乡453003)摘要:用基因组扫描的方法,利用棉花9个连锁群上的79个微卫星标记(Simple sequence repeat ,SSR),对收集的204份陆地棉品种(系)组成的品种资源群体进行群体结构和连锁不平衡(Linkage disequilibrium,LD)分析。
结果表明:本研究群体可划分为3个群体,其中两个群体分别由3个亚群体组成。
群体中,47%的标记位点之间可以观察到显著的LD(P ≤0.05)。
LD 在遗传距离小于120cM 范围内普遍存在。
在决定系数r 2≤0.05时,LD 在0.01单位的平均衰减距离为29.7cM ,在r 2≥0.05的条件下能观察到的LD 最大遗传距离为31.4cM ,在r 2≥0.1时能观察到LD 的最大遗传距离缩小到3.4cM 。
群体的连锁不平衡状况表明本研究群体可用于重要育种性状的关联分析。
关键词:陆地棉;数量性状位点;分子标记;连锁不平衡;群体结构中图分类号:S562.024文献标志码:A文章编号:1002-7807(2011)06-0500-07Population Structure and Linkage Disequilibrium Analysis of Germplasm Resources in Upland CottonZHANG You-chang 1,BIE Shu 1,YI Xian-da 1,ZHANG Cheng 1,LI Cheng-qi 2,QIN Hong-de 1*(1.Cash Crop Institute,Hubei Academy of Agricultural Sciences,Wuhan 430064,China ;2.Henan Institute of Technology,Xinxiang,Henan 453003,China )Abstract:Population structure and LD (Linkage disequilibrium )of 204upland cotton accessions were analyzed with 79SSR (Simple sequence repeat)markers located on nine chromosomes.Analysis of population genetic structure based on SSR data re-vealed that this population could be divided into three groups,two out of which were composed of three subgroups,respectively.47%of the SSR loci pairs showed LD at significant level of P ≤0.05.The maximum genetic distance of LD could be observed extended to 120cM.The LD average decay distance was 29.7cM at r 2≤0.05.Genome wide LD reduced to 3.4cM at r 2≥0.1,providing evidence of the potential for association mapping of important traits in cotton breeding program.Key words:upland cotton;quantitative trait locus;molecular marker;linkage disequilibrium;population structure收稿日期:2010-09-25作者简介:张友昌(1984-),男,硕士研究生;*通讯作者,qinhongde2002@基金项目:国家自然科学基金(30971823)目前对作物数量性状位点(Quantitative trait locus ,QTL )的定位主要用连锁分析的方法,即以标记基因型为依据,对由两自交亲本杂交形成的分离群体进行分组,通过比较不同基因型间目标性状的差异显著性,来推断影响该性状的基因与标记位点的连锁关系。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
连锁不平衡(linkage disequilibrium, LD)亦被称 为配子相不平衡(gametic phase disequilibrium)、配子 不 平 衡 (gametic disequilibrium) 或 等 位 基 因 关 联 (allelic association), 指的是一个群体内不同座位等 位基因之间的非随机关联, 包括两个标记间或两个 基因/QTL 间或一个基因/QTL 与一个标记座位间的 非随机关联[3]。
2.1 杂交类型
不同杂交类型植物间的 LD 水平存在很大差异。 在拟南芥[7, 8]、水稻[9]、大麦[10]和大豆[11]等自交物种 中, 个体绝大多数为纯合子, 虽然重组仍然发生但 不再对 LD 产生任何影响, 即其有效重组率较低, 因 此这些物种在很长的物理距离内(可达几百 kb)存在 LD[8, 12]。与自交物种相比, 异交物种如玉米中有效 重组率高, 重组导致连锁的位点彼此独立存在, 从 而削弱染色体内部的 LD, 因此异交物种中的 LD 迅 速衰减[13]。
DNA 测序技术和高通量 SNP 分析技术的发展及 2001 年 LD 作图(LD mapping) 在植物中的首次成功运 用, 引起了近年来科研工作者对 LD 研究的强烈兴趣。 本文将对 LD 的基础理论及其在植物基因组学研究中 的应用、目前研究热点及展望进行全面介绍。
1 连锁不平衡的理论基础
1.1 连锁不平衡的概念
另一方面, 揭示植物基因型和表型之间的内在 联系也是植物基因组学研究的重要内容。在过去的 几十年中, 人们分别从植物的形态学特征、生理生 化特性和分子机理等不同水平上进行了大量多层次 研究和不懈努力。随着人们认知的深入, 探索表型 和基因型之间内在联系的共识加速了植物生命现象 的深入研究。植物基因组中多态性位点间的 LD 是 联系结构基因组学和表型组学的一座桥梁。LD 为植 物基因组中新基因的发掘及揭示特定基因型和表型 之间的内在联系提供了一个全新的契机。
连锁不平衡与连锁是相关但完全不同的两个概 念。连锁不平衡指的是群体内等位基因之间的相关, 而连锁指的是位于同一条染色体上的基因联合传递 的现象。紧密连锁可导致较高的 LD 水平, 但这种 LD 纯粹是由突变产生的等位基因出现后紧密连锁 座位间所有重组事件的结果。
1.2 连锁不平衡的度量
所有 LD 统计的是实际观测到的单倍型频率与 随机分离时单倍型的期望频率之间的差异。LD 的计
r2 和 D'是两个座位间 LD 的度量。对于基因组 内某区域的 LD 分布状况, 通常用两种形象化的方 式来表示: LD 衰退图和 LD 矩阵。LD 衰退图是以位 点间的 LD 对遗传距离作图来表示一个区域内的 LD 分布情况, 这种表示方法也便于对不同物种中的 LD 水平进行比较。LD 矩阵是某基因内或某染色体上多 态性位点间 LD 的线性排列。
第 11 期
王荣焕等: 植物基因组中的连锁不平衡
1319
2 连锁不平衡的影响因素
通常, 随机匹配群体中, 在没有选择、突变或迁 移因素的影响时, 多态性位点处于连锁平衡状态[5], 相反, 连锁、选择和群体混合将增加 LD 的水平。群 体中的 LD 水平是许多遗传因素和非遗传因素综合 作用的结果。突变可导致新的多态性产生, 而重组 则可通过重新组合序列变异而削弱染色体内部的 LD, LD 的程度与重组率成反比[6]。植物中影响 LD 水平的因素主要包括:
关键词: 连锁不平衡; LD 结构; LD 作图
Linkage disequilibrium in plant genomes
WANG Rong-Huan, WANG Tian-Yu, LI Yu
Institute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing 100081, China
Dab = (πAB − πAπB )
r2 的计算公式为 D'的计算公式为
r2 = (Dab )2 πΑπaπΒπb
D′
=
(Dab )2
for
min(π Aπb ,π aπ B )
Dab
<
0
D'
=
( min(π
Dab )2 Aπ B ,π
aπ
b
ห้องสมุดไป่ตู้
)
for
Dab
>0
r2 和 D'反映了 LD 的不同方面。 r2 包括了重组 史和突变史, 而 D'仅包括重组史。D'能更准确地估 测重组差异, 但样本较小时发现低频率 4 种等位基 因组合的可能性大大减小, 因此 D'不适宜小样本研 究中的应用。r2 可以提供标记是否能与 QTL 相关的 信息, 因此 LD 作图中通常采用 r2 来表示群体的 LD 水平。
算依研究座位的性质和数目而异。 对于只有两个等位基因的座位如 SNP 和 AFLP,
通常用 r2 和 D'来估计两个座位之间的 LD 水平[4]。 假设有两个连锁的座位 A 和 B, 其等位基因分别为 A、a 和 B、b, 4 个等位基因的频率分别为 πA、πa、 πB、πb, 4 种单倍型 AB、aB、Ab 和 ab 的频率分别为 πAB、πaB、πAb 和 πab。那么, 实际观测到的单倍型频 率与期望单倍型频率之间的差异 D 的计算公式为:
Hi-Tech Research and Development Program of China (863 Program) (No. 2006AA10Z188) and the National Natural Science Foundation of China (No.30571133)] 作者简介: 王荣焕(1980−), 女, 河北衡水人, 在读博士, 研究方向:基于基因组学的作物种质资源研究。Tel: 010-62186652; E-mail: ronghuanwang@ 通 讯 作 者 : 黎 裕 (1966−), 男 , 四 川 仪 陇 人 , 研 究 员 , 博 士 , 研 究 方 向 : 玉 米 种 质 资 源 和 基 因 组 学 研 究 。 Tel: 010-62131196; E-mail: yuli@
1318
HEREDITAS (Beijing) 2007
第 29 卷
种质资源中挖掘优异基因, 尽快实现我国由种质资 源优势向基因资源优势的转变是新时期摆在我们面 前的一个迫切需要解决的问题。随着植物基因组学 的迅猛发展, 新的基因发掘方法不断涌现[1]。近年来, 连锁不平衡(Linkage disequilibrium, LD)作图方法已 被证明不仅是基因发掘, 而且也是等位基因发掘的 有效手段[2]。
Abstract: Linkage disequilibrium (LD) is one of the most recently focused interests in the field of plant genomics. LD
mapping is an effective approach to discovering novel genes and a bridge for connecting structural genomics to phenomics. LD mapping was first applied in plants in 2001. Since then, researches on the structure and extent of LD and LD mapping have been reported in a wide range of plant species. The basic theory of LD and its application in LD mapping, haplotype diversity analysis, htSNP identification and population genetics were reviewed in this paper. And advances of LD research in plants including influences of population structure, gene conversion, epistasis and G×E interactions, and future prospects were also presented. China has abundant germplasm resources, but gene discovery lags behind. Intensive researches on LD will certainly accelerate rapid development of plant genomics, especially the progress of gene discovery based on germplasm resources in China. Keywords: linkage disequilibrium (LD); LD structure; LD mapping
HEREDITAS (Beijing) 2007 年 11 月, 29(11): 1317―1323 ISSN 0253-9772
DOI: 10.1360/yc-007-1317
综述
植物基因组中的连锁不平衡