Gliclazide_21187-98-4_DataSheet_MedChemExpress
Healthy Blue SC会员手册说明书
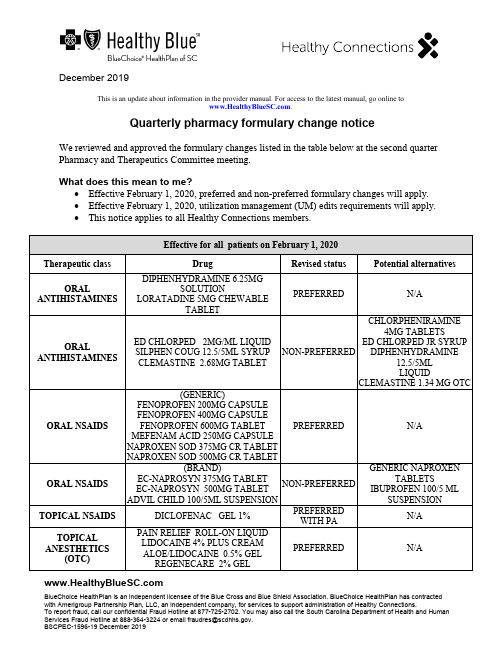
December 2019BlueChoice HealthPlan is an independent licensee of the Blue Cross and Blue Shield Association. BlueChoice HealthPlan has contracted with Amerigroup Partnership Plan, LLC, an independent company, for services to support administration of Healthy Connections.To report fraud, call our confidential Fraud Hotline at 877-725-2702. You may also call the South Carolina Department of Health and Human ************************************************************.BSCPEC-1596-19 December 2019This is an update about information in the provider manual. For access to the latest manual, go online to .Quarterly pharmacy formulary change noticeWe reviewed and approved the formulary changes listed in the table below at the second quarter Pharmacy and Therapeutics Committee meeting.What does this mean to me?• Effective February 1, 2020, preferred and non-preferred formulary changes will apply. • Effective February 1, 2020, utilization management (UM) edits requirements will apply. • This notice applies to all Healthy Connections members.Effective for all patients on February 1, 2020Therapeutic class DrugRevised status Potential alternativesORALANTIHISTAMINESDIPHENHYDRAMINE 6.25MGSOLUTIONLORATADINE 5MG CHEWABLETABLETPREFERREDN/AORALANTIHISTAMINESED CHLORPED 2MG/ML LIQUID SILPHEN COUG 12.5/5ML SYRUP CLEMASTINE 2.68MG TABLET NON-PREFERRED CHLORPHENIRAMINE4MG TABLETSED CHLORPED JR SYRUPDIPHENHYDRAMINE12.5/5MLLIQUIDCLEMASTINE 1.34 MG OTCORAL NSAIDS(GENERIC)FENOPROFEN 200MG CAPSULE FENOPROFEN 400MG CAPSULE FENOPROFEN 600MG TABLET MEFENAM ACID 250MG CAPSULE NAPROXEN SOD 375MG CR TABLET NAPROXEN SOD 500MG CR TABLETPREFERRED N/A ORAL NSAIDS(BRAND) EC-NAPROSYN 375MG TABLET EC-NAPROSYN 500MG TABLET ADVIL CHILD 100/5ML SUSPENSION NON-PREFERRED GENERIC NAPROXENTABLETSIBUPROFEN 100/5 ML SUSPENSIONTOPICAL NSAIDSDICLOFENAC GEL 1% PREFERREDWITH PAN/A TOPICALANESTHETICS(OTC)PAIN RELIEF ROLL-ON LIQUIDLIDOCAINE 4% PLUS CREAMALOE/LIDOCAINE 0.5% GELREGENECARE 2% GELPREFERRED N/ALIDODOSE 3% GELREGENECARE SPRAYALOCANE 4% GELAFTERBURN 2.5% GELXOLIDO 2% CREAM BURN RELIEF 0.5% AEROSAL ASPERCREME 4% SPRAYLIDOCAINE 3% CREAMLIDOCAINE 4% CREAMLIDOCAINE 5% CREAMAFTERSUN 0.5% GELLIDOCAINE 4% PADTOPICAL ANESTHETICS(RX)LIDOCAINE 3% CREAMLIDOCAINE 5% OINTMENT NON-PREFERREDOTC LIDOCAINEPRODUCTSRX LIDOCAINE5% PATCH(PA REQUIRED)MISCELLANEOUS ANTICONVULSANTSPREGABALIN 25MG CAPSULEPREGABALIN 50MG CAPSULEPREGABALIN 75MG CAPSULEPREGABALIN 100MG CAPSULEPREGABALIN 150MG CAPSULEPREGABALIN 200MG CAPSULEPREGABALIN 225MG CAPSULEPREGABALIN 300MG CAPSULEPREGABALIN SOL 20MG/MLPREFERREDWITH NO PRIORAUTHORIZATION(PA)N/AATOPICDERMATITIS PIMECROLIMUS 1% CREAMPREFERREDWITH STEPTHERAPY (ST)N/AFIBRATESFENOFIBRATE 130MG CAPSULEFENOFIBRATE 145MG TABLETFENOFIBRIC 35MG TABLETFENOFIBRIC 105MG TABLETFENOFIBRIC 135MG DR CAPSULENON-PREFERREDWITH STFENOFIBRATE134MG, 160MG, 200MG,43MG, 48MG,54 MG,67 MGFENOFIBRIC ACID 45 MGALCOHOL SWABS (MANUFACTURERS) GLOBAL DIABETICRITE AID NON-PREFERREDMANUFACTURERSBD DIABETESDYNAREXHEALTH MARTULTIMEDALCOHOL SWABS (MANUFACTURERS) BD DIABETESDYNAREXHEALTH MARTULTIMEDPREFERRED N/AIRON SUPPLEMENTS (GENERIC OTC)IRON 45MG TABLETSLOW-RELEASE FE 45MG TABLETHEMAX TABLETGENTLE IRON 28MG CAPSULEHIGH POTENCY FE 27MG TABLETNU-IRON 150 150MG CAPSULEABATRON AF TABLETSLOW IRON 50MG TABLETPREFERRED N/AFERGON 27MG TABLETIRON SUPPLEMENTS(BRAND OTC)FOLITAB 500 TABLET IRON 28MG TABLETFERROUS GLUC 324MG TABLETEZFE 200MG CAPSULEFERROUS GLUC TAB 324MGFERROUS SULF 324MG EC TABLETFERRETTS 325MG TABLETFERREX 150MG CAPSULEFERREX 28 MIS FERREX 150 PLUS CAPSULE FERREX 150 FORTE PL CAPSULECHEWABLE IRONPEDIATRIC IRON CHEWABLEFERROUS SUL 220/5ML LIQUIDFERROUS SULF 300/5ML SYRUPFEOSOL 200MG TABLETSLOW RELEASE FE 143MG CRTABLETNON- PREFERRED OTC GENERIC IRONSUPPLEMENTSRX PRODUCTS:HEMATOGEN FA CAPSULEHEMETAB TABLETMULTIGEN TABLETMULTIGEN PLS TABLETMULTIGEN FOLICTABLETFERRAPLUS 90 TABLETTARON FORTE CAPSULEFOLIVANE-F CAPSULEFOLIVANE-PLS CAPSULECENTRATEX CAPSULEIRON SUPPLEMENTS(PRESCRIPTIONSTRENGTH)IFEREX 150 FORTE CAPSULE HEMATOGEN CAPSULE HEMATOGEN FORTE CAPSULE TRICON CAPSULE MYFERON 150 FORTE CAPSULE FERROCITE PLUS TABLET FEROCON CAPSULE PUREVIT DUA FE PLUS CAPSULE HEMATINIC PL VIT/MIN TABLET HEMATINIC/FA TABLET POLY-IRON 150 FORT CAPSULE CORVITA 150 TABLET TRIGELS-F FORTE CAPSULE TL ICON CAPSULE SE-TAN PLUS CAPSULE NON- PREFERRED OTC GENERIC IRON SUPPLEMENTSRX PRODUCTS:HEMATOGEN FA CAPSULE HEMETAB TABLET MULTIGEN TABLET MULTIGEN PLS TABLET MULTIGEN FOLIC TABLET FERRAPLUS 90 TABLETTARON FORTE CAPSULE FOLIVANE-F CAPSULEFOLIVANE-PLS CAPSULE CENTRATEX CAPSULEUM edits — effective for all members no later than February 1, 2020 No changes in preferred/non-preferred status revision or addition to UM edit onlyANDROGENS*JATENZO CAPSULE ADD ST WITH QUANTITY LIMITS (QL)58 MG AND 198 MG QL: 4 PER DAY 237 MG QL: 2 PER DAY ANTICONVULSANTSNAYZILAM SPRAY 5MG ADD PA WITH QLQL: 50 MG PER 30 DAYS ANTICONVULSANTSOXTELLAR XR 150 MGOXTELLAR XR 600 MGREVISED QL LIMIT:150 MG: 3 TABLETS PER DAY 600 MG: 4 TABLETS PER DAYANTINEOPLASTICAGENTSPIQRAY 200 MG TABLETSPIQRAY 250 MG TABLETSPIQRAY 300 MG TABLETSADD PA WITH QL QL: 1 CARTON PER 28 DAYS ANTINEOPLASTICAGENTSXPOVIO PAK 60MGXPOVIO PAK 80MGXPOVIO PAK 100MGADD QL 1 CARTON PER 28 DAYSANTINEOPLASTICAGENTSNUBEQA 300MG TABLET ADD QL 4 TABLETS PER DAY ANTINEOPLASTICAGENTS TURALIO CAP 200MG ADD QL 4 TABLETS PER DAY ANTINEOPLASTICAGENTS PIQRAY 200MG TAB DOSE PIQRAY 300MG TAB DOSE PIQRAY 250MG TAB DOSE REVISE QL1 CARTON PER 28 DAYS CHOLESTEROLAGENTS EZALLOR SPRINKLE 5 MG CAP EZALLOR SPRINKLE 10 MG CAP EZALLOR SPRINKLE 20 MG CAP EZALLOR SPRINKLE 40 MG CAP ADD PA AND QLQL: 1 TABLET PER DAY COPD AGENTS DUAKLIR 400/12 INHALER ADD ST AND QLQL: 1 INHALER PER 30 DAYSCYSTIC FIBROSISAGENTSKALYDECO PAK 25MG ADD QL2 PACKETS PER DAYCYSTIC FIBROSISAGENTSORKAMBI GRANULES ADD QL2 PACKETS PER DAY HIVDOVATO TABLET EDURANT 25 MG TABLET DELSTRIGO TABLET COMPLERA TABLET ODEFSEY TABLET JULUCA TABLET ADD PA FOR NEW STARTS AND ADD QLQL: 1 PER DAY HIVINTELENCE TABLET ADD PA FOR NEW STARTS AND ADD QLQL:200 MG- 2 TABLETS PER DAY 400 MG- 4 TABLETS PER DAY 25 MG – 16 TABLETS PER DAYHIVATRIPLA TABLET BIKTARVY TABLET CIMDUO TABLET DESCOVY TABLETEMTRIVA 200 MG CAPSULE EPIVIR 300 MG TABLET EPZICOM TABLET EVOTAZ TABLET GENVOYA TABLET PIFELTRO 100 MG TABLET PREZCOBIX TABLET PREZISTA 800 MG TABLET REYATAZ 300 MG CAPSULESTRIBILD TABLET SUSTIVA 600 MG TABLETSYMFI TABLET SYMFI LO TABLET SYMTUZA TABLET TRIUMEQ TABLET TRUVADA TABLET TYBOST 150 MG TABLET VIDEX EC 400 MG CAPSULE VIDEX EC 250 MG CAPSULE VIRAMUNE XR 400 MG TABLETADD QL 1 PER DAYTEMIXYS TABLETHIVREYATAZ 200 MG CAPSULE REYATAZ 150 MG CAPSULE VIDEX EC 200 MG CAPSULE ZERIT 40 MG CAPSULE ZERIT 30 MG CAPSULE COMBIVIR TABLET DUTREBIS TABLET EPIVIR 150 MG TABLET ISENTRESS HD 600 MG TABLET PREZISTA 600 MG TABLET RETROVIR 300 MG TABLET SELZENTRY 75 MG TABLET TIVICAY 10 MG, 25 MG AND 50 MGTABLETTRIZIVIR TABLETVIRAMUNE 200 MG TABLET ZIAGEN 300 MG TABLET ADD QL 2 PER DAYHIV ISENTRESS 100 MG GRANULE PACKET FOR SUSPENSION ADD QL2 PACKETS PER DAYHIVVIDEX EC 125 MG CAPSULE VIRAMUNE XR 100MG TABLET ADD QL 3 PER DAYHIVAPTIVUS 250 MG CAPSULE INVIRASE 500 MG TABLET ISENTRESS 400 MG TABLET KALETRA 200 MG-50 MG TABLETLEXIVA 700 MG TABLET SELZENTRY 300 MG TABLET SELZENTRY 150 MG TABLET SUSTIVA 200 MG CAPSULE VIRACEPT 625 MG TABLET ZERIT 20 MG CAPSULE ZERIT 15 MG CAPSULEADD QL 4 PER DAYHIVREYATAZ 50 MG POWDER FORSUSPENSIONADD QL5 PACKETS PER DAYHIVCRIXIVAN 400 MG CAPSULE PREZISTA 150 MG TABLET RESCRIPTOR 200 MG TABLET RETROVIR 100 MG CAPSULE ISENTRESS 100 MG CHEWABLEADD QL 6 PER DAY HIV SELZENTRY 25 MG TABLET ADD QL 8 PER DAY HIV TROGARZO 150MG/ML VIAL ADD QL8 VIALS PER 28 DAYSHIVINVIRASE 200 MG CAPSULE KALETRA 100 MG-25 MG TABLETPREZISTA 75 MG TABLET VIRACEPT 250 MG TABLET ADD QL 10 PER DAY HIVCRIXIVAN 200 MG CAPSULE NORVIR 100 MG TABLET NORVIR 100 MG CAPSULEADD QL 12 PER DAYNORVIR 100 MG ORAL POWDERPACKETRESCRIPTOR 100 MG TABLET SUSTIVA 50 MG CAPSULEHIV APTIVUS 100 MG/ML SOLUTION ADD QL 13 ML PER DAYHIV PREZISTA 100 MG/ML SUSPENSION ADD QL 14 ML PER DAY HIV KALETRA 400 MG-100 MG/5 MLORAL SOLUTIONNORVIR 80 MG/ML ORAL SOLUTION ADD QL 16 ML PER DAY HIV ISENTRESS 25 MG CHEWABLE ADD QL24 TABLETS PER DAYHIV EMTRIVA 10 MG/ML SOLUTION ADD QL 29 ML PER DAYHIVEPIVIR 10 MG/ML ORAL SOLUTION ZIAGEN 20 MG/ML SOLUTION ADD QL 32 ML PER DAY HIVVIDEX 4 GM PEDIATRIC ORALSOLUTIONVIDEX 2 GM PEDIATRIC ORALSOLUTIONVIRAMUNE 50 MG/5 MLSUSPENSION ADD QL 40 ML PER DAY HIV VIRACEPT 50 MG/G POWDERADD QL 53 GM PER DAYHIV FUZEON 90 MG VIAL ADD QL60 VIALS PER 30 DAYSHIV LEXIVA 50 MG/ML SUSPENSION ADD QL 60 ML PER DAYHIV SELZENTRY 20 MG/ML ORALSOLUTION ADD QL 62 ML PER DAYHIV RETROVIR 10 MG/ML SYRUP ADD QL 64 ML PER DAYHIVZERIT 1 MG/ML SOLUTION ADD QL 80 ML PER DAY IRRITABLE BOWEL SYNDROME (IBS)AGENTSZELNORM 6MG TABLET ADD PA AND QL QL 2 TABLETS PER DAY LAMBERT-EATON MYASTHENIC SYNDROME AGENTSRUZURGI 10MG TABLET ADD PA AND QL QL 10 TABLETS PER DAYNARCOTIC ANTAGONISTS SUBLOCADE 100/0.5 INJECTION SUBLOCADE 300/1.5 INJECTION REMOVE PANARCOTIC ANTAGONISTS VIVITROL 380MG INEJCTION REMOVE PA AND ADD QL QL 1 VIAL PER 28 DAYSNARCOTIC ANTAGONISTS ZUBSOLV 2.9-0.71 SUB REVISE QL QL 5 PER DAY ORAL DIABETICAGENTS*QTERNMET XR TABLETADD ST AND QLQL:5 MG/5 MG/1000 MG, 10 MG/5 MG/1000 MG:1 TABLET PER DAY2.5 MG/2.5 MG/1000 MG, 5 MG/2.5 MG/10000MG: 2 TABLETS PER DAYORAL DIABETICAGENTS QTERN 5-5MG TABLET ADD QL1 TABLET 28 DAYSINJECTABLE DIABETIC AGENTSOZEMPIC 2/1.5ML INJECTION ADD QL 1 PER 28 DAYSPRENATAL VITAMINS DUET DHADUET DHA BALANCEDNESTABS ABC NESTABS DHA OBTREX DHA SELECT-OB+DHATHERANATAL COMPLETEVITAFOL FE+ VITAFOL-OB+DHABAL-CARE DHA ESSENTIAL ADD QL 2 PER DAYPRENATAL VITAMINS CITRANATAL B-CALMADD QL 3 PER DAYTOPICAL ANTIPRURITICS DOXEPIN HCL 5% CREAM,ZONALON 5% CREAM, PRUDOXIN5% CREAM ADD PA AND QLQL 1 TUBE PER FILL; 1 FILL PER 3 MONTHSTOPICAL ANESTHETIC COMBINATIONSLIDOCAINE/PRILOCAINE CREAMREVISE QL30 GM PER 30 DAYS* Clinical edits will be put in place as these new drugs to come market.What action do I need to take?Please review these changes and work with your Healthy Connections members to transition them to formulary alternatives. If you determine preferred formulary alternatives are notclinically appropriate for specific members, you will need to obtain prior authorization (PA) to continue coverage beyond the applicable effective date.What if I need assistance?We recognize the unique aspects of member cases. If your Healthy Connections member cannot be converted to a formulary alternative for medical reasons, call our Pharmacy department at 866-902-1689 and follow the voice prompts for pharmacy PA.You can find the Preferred Drug List on our website at > Providers > Pharmacy Information. If you need assistance with any other item, contact the Customer Care Center at 866-757-8286.。
比伐卢定

对于未接受 PTCA的不稳定心绞痛患者和其它冠状动脉疾病患者,尚无试验资料。在实施 PTCA之前,本品常规剂量为首剂 1.0 mg‘kg~ ,合并 2.5 mg?kg ?h 静脉滴注 4 h,如果需要,可按 0.2 rng ?kg ?h一 维持至 20 h。用 5%葡萄糖或 0.9%氯化钠注射液溶解后使用。本品不得用于肌内注射。 肾功能不全的患者 ,需根据表 1值酌情减量。
【不良反应】
常见的是出血,多见于动脉穿刺部位 ,也可能发生在身体其它部位。用药中,若血压或血容量突然下降,或有其它不明症状出现时,都应立刻停药并高度警惕出血的发生。其它 尚有背痛 、头痛 、低血压等。
【禁忌症】
本品禁用于大 出血活动期以及对药物过敏者。
【产品规格】
250mg/瓶;pa值:制备后溶液为5~6
注射用比伐卢定说明书
【批准文号】
【中文名称】
比伐卢定
【产品英文名称】
Bivalirudin
【生产企业】
【功效主治】
本品主要用于预防血管成型介入治疗不稳定性心绞痛,前后的缺血性并发症。
【化学成分】
抗凝成分是水蛭素衍生物(片断),系一合成 20肽,相对分子质量 2 180。结构式为 Phe—Pro——Arg——Pro ——Gly——Gly——Gly——Gly——Ash——Gly——Asp—— Phe—Glu—Glu—Ile—Pro—Glu—G1u—Tvr—Leu。
本品为凝血酶(thrombin)直接 的、特异的、可逆性抑制剂。无论凝血酶处于血循环中还是与血栓结合,本品均可与其催化位点和阴离子结合位点(又称底物识别位点)发生特异性结合,从而直接抑制凝血酶的活性…1,其作用与肝素不同,它不依赖于抗凝血酶1V(NI1_Ⅳ)、肝素辅因子 Ⅱ等。凝血酶是凝血反应中起核心作用的丝氨酸蛋白酶:它水解纤维蛋白原生成纤维蛋白单体 ;激活凝血因子 XⅢ;促进纤维蛋白交联形成稳定血栓的共价结构。同时,凝血酶激活凝血因子V,Ⅷ;激活血小板,促进血小板聚集和颗粒释放。因凝血酶可水解本品多肽顺序中Arg3和 Pro4之间的肽键 ,使本品失活,所以本品对凝血酶的抑制作用是可逆而短暂的。在体外实验中,本品以浓度依赖方式延长健康人血浆的活化部分凝 血酶时间 (activated partial thromboplastin time,af T)、凝血酶 时 间 (thrombin time,1vr)、凝血 酶原 时 间(prothrombin time,PT)。本品与游离型凝血酶或血栓型凝血酶的结合不受血小板释放物质影响。健康志愿者试验显示 :本品以剂量和浓度依赖方式延长活化凝血 时 间(activated clotting time,ACT),af耵 ,Prr和1vr。因血管梗塞≥70%而接受经皮穿刺腔 内冠状动脉成型术(PTCA)治疗的冠心病患者 ,给予本品后也可获得 与健康人相同的抗凝效果。在静脉注射本品后即产生抗凝作用,患
脱氢抗坏血酸(DHA)检测试剂盒(菲咯啉微板法)

脱氢抗坏血酸(DHA)检测试剂盒(菲咯啉微DHA)检测试剂盒(菲咯啉微板法)检测原理是利用还原剂将脱 氢抗坏血酸还原成还原型抗坏血酸,在酸性条件下,维生素 C(抗坏血酸)把三价铁离子还原 成亚铁离子,后者与菲咯啉形成稳定的红色螯合物,以酶标仪 534nm 处检测吸光度,在一 定浓度范围吸光度与抗坏血酸含量呈线性关系,获得抗坏血酸含量。该试剂盒主要用于植 物组织中的维生素 C(抗坏血酸)的检测,计算出总抗坏血酸含量,从中减去样品中原有的还 原型抗坏血酸含量,即得脱氢抗坏血酸含量,本试剂盒仅用于科研领域,不宜用于临床诊 断或其他用途。
组成:
编号 名称 试剂(A): 抗坏血酸标准(250μg/ml) 试剂(B): 组织匀浆液(5×) 试剂(C): DHA 还原液 试剂(D): NaOH 溶液 试剂(E): 酸性缓冲液 试剂(F): AsA Assay buffer 试剂(G): 菲咯啉显色液 使用说明书
TC2040 100T
Storage
DHA 含量(mg/100g)=(m0×VT×100)/(m1×VS×1000)
北京雷根生物技术有限公司
注意事项:
1、 上述低温试剂避免反复冻融,以免失效或效率下降。 2、 组织匀浆液(5×)久置或低温保存,容易产生乳白色浑浊。如果白色浑浊不明显,可以
直接使用,不影响效果;如果白色浑浊较多,应弃用。 3、 待测样本如不能及时测定,应置于 2~8℃保存,3 天内稳定。 4、 如果样品浓度过高,应用蒸馏水稀释后重测,结果乘以稀释倍数。
3、 制备 DHA 待测液:取 AsA 提取液,加入 DHA 还原液,用 NaOH 溶液调节 pH 至 7-8, 室温下静置,使脱氢抗坏血酸还原成抗坏血酸,再加入组织匀浆液(5×)和蒸馏水即为 DHA 待测液。
GLP-1类似物药物进展

精心整理2019年9月GLP-1类似物药物进展胰高血糖素样肽(glucagon-like peptide ,GLP )是小肠表皮细胞在食物刺激情况下分泌的单肽类肠促胰岛素,包括GLP-1、GLP-2两种类型。
其中GLP-2具有促进小肠生长,抑制细胞凋亡,促进胃排空,增加食欲的药理作用,临床上可用于治疗小肠短小综合症;而GLP-1具有促进胰岛素分泌,保护胰岛β细胞,抑制胰高血糖素分泌,抑制胃排空,降低食欲的药理作用,临床可用于二型糖尿病和肥胖症的治疗。
人体内具有生物活性的GLP-1主要是GLP-1(7-36)酰胺和GLP-1(7-37),天然GLP-1可被二肽基肽酶Ⅳ(dipeptidyl peptidase-Ⅳ, DPP-Ⅳ)迅速水解失活(半衰期小于5 min ),不具有临床使用价值,因此对GLP-1结构修饰,掩盖DPP-Ⅳ的结合位点,延长半衰期并保证疗效是该类药物研发的主要方向。
一、1. 合研发,,与GLP-1然GLP-1Amylin 批准。
Bydureon 次。
另外,以色列的2型糖尿病。
2. 延长生降低用剂量为3 mg ,高于其用于II 型糖尿病的剂量1.8 mg 。
临床数据显示,连续54周服用利拉鲁肽可减重约6.4 kg (6%)。
3. 利司那肽(Lixisenatide )利司那肽(商品名Lyxumia )由法国Sanofi Aventis 和Zealand 公司共同开发,于2013年相继获欧洲和日本批准。
利司那肽在艾塞那肽结构上去掉38位的Pro ,并在39位的Ser 接了6个Lys ,经过修饰,半衰期相对艾塞那肽有所延长,最高可达6.5小时,可每日一次皮下注射。
在中国仓鼠卵巢癌细胞系中,利斯那肽对GLP-1受体亲和力较天然GLP-1高出约4倍。
与艾塞那肽相比,在保持疗效的情况下延长半衰期,降低不良反应发生率。
4. 阿必鲁肽(Albiglutide )精心整理2019年9月阿必鲁肽(商品名Eperzan )是由GlaxoSmithKline 研发的每周一次皮下注射的长效GLP-1类似物,2014年首先在欧洲上市。
恩格列净联合西格列汀治疗老年2_型糖尿病患者的临床疗效分析

·药物与临床·糖尿病新世界 2023年3月DOI:10.16658/ki.1672-4062.2023.05.059恩格列净联合西格列汀治疗老年2型糖尿病患者的临床疗效分析臧道军,龚红燕江苏省常州市德安医院老年内科,江苏常州213000[摘要]目的探讨老年2型糖尿病患者使用恩格列净+西格列汀治疗的临床效果。
方法选取2020年1月—2021年12月常州市德安医院接诊的100例老年2型糖尿病患者作为研究对象,根据不同用药方式分为对照组与研究组,各50例,对照组接受西格列汀治疗,研究组接受恩格列净+西格列汀治疗,就两组患者血糖指标、炎性指标、胱抑素C(Cys-C)、血尿素氮(BUN)、血同型半胱氨酸(Hcy)指标进行比较。
结果治疗前两组血糖指标相比,差异无统计学意义(P>0.05),治疗后,研究组HbA1c、FPG及2 hPG明显低于对照组,差异有统计学意义(P<0.05);治疗前两组炎性指标比较,差异无统计学意义(P>0.05),治疗后,研究组IL-4、IL-6及TNF-α明显低于对照组,差异有统计学意义(P<0.05);治疗前两组Cys-C、BUN及Hcy相比,差异无统计学意义(P>0.05),治疗后,研究组患者Cys-C、BUN及Hcy明显低于对照组,差异有统计学意义(P<0.05)。
结论对于老年2型糖尿病患者开展恩格列净+西格列汀治疗能有效改善血糖指标,降低Hcy,提升肾功能,治疗效果显著。
[关键词] 老年人群;恩格列净;西格列汀;2型糖尿病[中图分类号] R4 [文献标识码] A [文章编号] 1672-4062(2023)03(a)-0059-04Clinical Efficacy Analysis of Empagliflozin Combined with Sitagliptin in the Treatment of Elderly Patients with Type 2 Diabetes MellitusZANG Daojun, GONG HongyanDepartment of Geriatric Medicine, Changzhou De'an Hospital, Changzhou, Jiangsu Province, 213000 China[Abstract] Objective To investigate the clinical effect of treatment with empagliflozin + sitagliptin in elderly patients with type 2 diabetes mellitus.Methods A total of 100 elderly patients with type 2 diabetes mellitus admitted to Chang⁃zhou De'an Hospital from January 2020 to December 2021 were selected as study subjects. The cases were divided into control group and study group according to different medication administration, fifty cases in each. The control group received sitagliptin treatment and the study group received empagliflozin + sitagliptin treatment. The blood glu⁃cose index, inflammatory index, cystatin C (Cys-C), blood urea nitrogen (BUN), and blood homocysteine (Hcy) index were compared between the two groups.Results There was no statistically significant difference in blood glucose in⁃dexes between the two groups before treatment (P>0.05). After treatment, HbA1c, FPG and 2 hPG of the study group were significantly lower than those in the control group, the difference was statistically significant (P<0.05). There was no statistically significant difference in inflammatory indexes between the two groups before treatment (P>0.05). After treatment, IL-4, IL-6 and TNF-α in the study group were significantly lower than those in the control group, the dif⁃ference was statistically significant (P<0.05). There was no statistically significant difference in the Cys-C, BUN and Hcy between the two groups before treatment (P>0.05). After treatment, the Cys-C, BUN and Hcy of the study group were significantly lower than those in the control group, the difference was statistically significant (P<0.05).Conclusion For elderly patients with type 2 diabetes mellitus, treatment with empagliflozin + sitagliptin can effec⁃tively improve blood glucose index, reduce Hcy and enhance renal function, with significant therapeutic effects.[作者简介]臧道军(1974-),男,本科,副主任医师,研究方向为老年内科。
阿法替尼中文说明书

【药物名】Afatinib(阿法替尼)【商品名】Gilotrif【美国上市时间】非小细胞肺癌,2013年【类别】激酶抑制剂【分子式】C32H33ClFN5O11【靶点】EGFR 【生产公司】Boehringer Ingelheim Pharmaceuticals 勃林格殷格翰公司【购买地】美国【剂型和规格】口服片剂,规格有:40mg/片、30mg/片、20mg/片。
40毫克药片:浅蓝色,薄膜包衣,圆形双凸面,斜角边。
一面有“T40”字样,另一面标有勃林格殷格翰的标志,国家药品验证号NDC: 0597-0138-30。
30毫克药片:深蓝色,薄膜包衣,圆形双凸面,斜角边。
一面有“T30”字样,另一面标有勃林格殷格翰的标志,国家药品验证号NDC: 0597-0137-30。
20毫克药片:白色到浅黄色,薄膜包衣,圆形双凸面,斜角边。
一面有“T20”字样,另一面标有勃林格殷格翰的标志,国家药品验证号NDC: 0597-0141-30。
【适应症和用法】EGFR突变阳性,转移性非小细胞肺癌。
使用限制:目前没有数据支持阿法替尼能够用于治疗肿瘤含有其他EGFR突变的病人; 铂化疗后的转移性非小细胞肺鳞癌。
【用法用量】病人的选择:根据病人肿瘤切片中EGFR19号外显子缺失或21号外显子替换突变的样式。
推荐剂量:口服40毫克/次/天,直到出现耐受性或者疾病的进展。
患者有严重的肾损伤(肾小球滤过率为15到29 毫升/分钟/1.73 m2):推荐剂量为:口服30毫克/次/天,一天口服一次。
用药时间:饭前1小时或餐后2小时。
在错过一剂量用药的十二个小时内不要进行下次用药。
出现副反应时的剂量调整:出现任何如下副反应,请立即停止用药:•3级或者更高级别的副作用•2级或更高级别腹泻;也可以在服用抑制腹泻药物的同时,持续坚持2天或两天以上•持续超过7天或难以忍受的皮肤反应•2级或者更高级的肾损伤当副作用降为1级或者回到基准线水平或者患者恢复正常状态时,恢复给药;但是剂量需要减少,如比原剂量减少10毫克/次/天。
达格列净联合二甲双胍治疗2_型糖尿病的效果观察

·药物与临床·糖尿病新世界 2023年9月DOI:10.16658/ki.1672-4062.2023.18.085达格列净联合二甲双胍治疗2型糖尿病的效果观察卢红艳1,21.广宁县人民医院呼吸科,广东肇庆526300;2.广宁县人民医院内分泌科,广东肇庆526300[摘要]目的探讨对2型糖尿病患者采用达格列净+二甲双胍药物完成治疗后获得临床效果。
方法选取2020年10月—2021年10月广宁县人民医院100例2型糖尿病患者,按照随机数字表法分为常规组和研究组,各50例。
常规组采用格列齐特缓释片+二甲双胍缓释片治疗,研究组采用达格列净+二甲双胍缓释片治疗。
比较两组患者治疗结果。
结果研究组治疗总有效率(98.00%)高于常规组(84.00%),差异有统计学意义(χ2= 5.983,P<0.05)。
治疗后,研究组三酰甘油、低密度脂蛋白、总胆固醇以及体质指数均低于常规组,差异有统计学意义(P<0.05)。
治疗后,研究组空腹血糖、糖化血红蛋白、餐后2 h血糖水平均低于常规组,差异有统计学意义(P<0.05)。
结论达格列净+二甲双胍缓释片联合应用,可显著提高患者治疗效果,显著改善血脂水平以及体质量,有效降低血糖水平,促进2型糖尿病患者总体预后水平改善。
[关键词] 2型糖尿病;达格列净;二甲双胍缓释片;格列齐特缓释片;治疗总有效率;血脂指标;体质指数;血糖指标[中图分类号] R47 [文献标识码] A [文章编号] 1672-4062(2023)09(b)-0085-04Efficacy of Dapagliflozin Combined with Metformin in the Treatment of Type 2 Diabetes MellitusLU Hongyan1,21. Department of Respiratory, Guangning County People's Hospital, Zhaoqing, Guangdong Province, 526300 China;2. Department of Endocrinology, Guangning County People's Hospital, Zhaoqing, Guangdong Province, 526300 China[Abstract] Objective To investigate the clinical effect of dapagliflozin plus metformin in patients with type 2 diabe‐tes. Methods 100 patients with type 2 diabetes in Guangning County People's Hospital from October 2020 to October 2021 were selected and divided into routine group and study group according to random number table, with 50 casesin each group. The routine group was treated with gliclazide sustained-release tablets and metformin sustained-release tablets, while the study group was treated with daggliflozin and metformin sustained-release tablets. Compared the treatment results of two groups of patients. Results The total effective rate of the study group (98.00%) was higher than that of the routine group (84.00%), the difference was statistically significant (χ2=5.983, P<0.05). After treat‐ment, the levels of triglycerides, low-density lipoprotein, total cholesterol, and body mass index in the study group were lower than those in the routine group, the difference was statistically significant (P<0.05). After treatment, the fasting blood glucose, glycated hemoglobin, and 2-hour postprandial blood glucose levels in the study group were sig‐nificantly lower than those in the routine group, the difference was statistically significant (P<0.05). Conclusion The combined application of dapagliflozin and metformin sustained-release tablets can significantly improve the therapeu‐tic effect of patients, significantly improve the level of blood lipid and body mass, effectively reduce the level of blood glucose, and promote the improvement of the overall prognosis of patients with type 2 diabetes.[Key words] Type 2 diabetes; Dapagliflozin; Metformin sustained-release tablets; Gliclazide sustained release tab‐lets; Total effective rate of treatment; Blood lipid index; Body mass index; Blood glucose index[作者简介]卢红艳(1982-),女,本科,副主任医师,研究方向为内分泌。
抗抑郁药---盐酸维拉佐酮

2014.10.20
前言 • MDD治疗的主要药物种类
• 第一代经典抗抑郁药:主要包括单胺氧化酶抑制剂(maoi)和三环类 抗抑郁药(tca)。
• 第二代新型抗抑郁药:以选择性五羟色胺(5-ht)再摄取抑剂为主。 • 盐酸维拉佐酮(vilazodone hydrochloride)是首个吲哚烷基胺类新 型抗抑郁药。
2014.10.20
药物背景 • 安全性
• 盐酸维拉佐酮在8个临床试验共计2 177例MDD患者的临床研究中显 示具有良好的耐受性和安全性。在与安慰剂,对照的Ⅲ临床研究中, 因不良反应导致盐酸维拉佐酮治疗组中止治疗的患者占7.1%,安慰 剂对照组为3.2%,治疗组导致停药的常见不良反应主耍为恶心(1. 3%)和腹泻(1.2%) 。 • 由于抗抑郁药可增加儿童、青少年和l 8~24岁年轻人服药初期自杀想 法和自杀行为的风险,因此以上患者要慎用。 • 维拉唑酮在治疗终止时,尤其突然终止时,会出现戒断症状(烦躁不 安、易怒、眩晕、感觉障碍、意识模糊等症状)。
2014.10.20
报告框架
• 1.药物背景
• 2.专利介绍 • 3.合成路线
Viibryd(盐酸维拉佐酮)
2014.10.20
药物背景
• • • • • • • • • • • • • • • • 通用名称:维拉佐酮(Vilazodone) 商品名:Viibryd 原研公司:德国Merck KGaA 基本专利: DE19934333254 优先权:1993 年9 月30 日 相关中国专利: CN94116585 类别:抑郁症治疗药 化合物类型:新分子实体 5-[4-[4-( 5-cyano-1H-indol-3-yl) butyl]-1(New molecular entity) piperazinyl]-2-benzofurancarboxamide 分子式:C26H27N5O2 ·HCl hydrochloride 相对分子质量:477.99 CAS 号:163521-08-2 适应症:重度抑郁症(MDD) 化学名: 5-[4-[4-( 5-氰基-1H-吲哚-3-基) 丁基]-1-哌嗪基]-2-苯并呋喃草酰胺盐酸 盐; 获批单位:Trovis 制药有限责任公司 批准日期:2011 年1 月21 日
依替巴肽注射液质量标准

依替巴肽注射液质量标准
依替巴肽注射液是一种治疗糖尿病的药物。
其通用名为exenatide,属于胰高血糖素样多肽-1(GLP-1)受体激动剂类药物。
该药物能够促进胰岛素释放,降低血糖水平,是一种重要的口服降糖药物。
根据中国药典2020年版,依替巴肽注射液的质量标准如下:
1. 外观:为无色透明液体。
2. 相对分子质量:约为4186。
3. 含量测定:用高效液相色谱法测定,依替巴肽的含量应符合规定。
4. PH值:该注射液的PH值在4.0~6.0之间。
5. 有关杂质:除依替巴肽外,注射液中不得检出重金属、铅、砷等有害物质。
6. 有关菌落:依替巴肽注射液的总菌落数不得超过100 CFU/ml,霉菌和酵母菌的总数不得超过10 CFU/ml。
7. 稳定性:依替巴肽注射液应储存在阴凉干燥处,避免阳光直射。
在储存期间,
药物的含量不应发生明显变化。
总之,依替巴肽注射液的质量标准非常严格,药厂必须遵循这些标准,确保药物的质量和安全性。
对于患有糖尿病的患者来说,依替巴肽注射液的质量标准尤为重要,他们需要使用高质量的药物来控制血糖水平,保持健康。
替卡格雷FDA说明书

______________________________________________________________________________________________________________________________________________________________HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to useBRILINTA safely and effectively. See full prescribing information for BRILINTA.BRILINTA ™(ticagrelor ) tablets, for oral useInitial U.S. Approval: 2011WARNING: BLEEDING RISK • BRILINTA, like other antiplatelet agents, can cause significant, sometimes fatal, bleeding (5.1, 6.1). • Do not use BRILINTA in patients with active pathological bleeding or a history of intracranial hemorrhage (4.1, 4.2). •Do not start BRILINTA in patients planned to undergo urgent coronary artery bypass graft surgery (CABG). When possible,discontinue BRILINTA at least 5 days prior to any surgery (5.1). •Suspect bleeding in any patient who is hypotensive and has recentlyundergone coronary angiography, percutaneous coronary intervention (PCI), CABG, or other surgical procedures in the setting of BRILINTA (5.1).•If possible, manage bleeding without discontinuing BRILINTA. Stopping BRILINTA increases the risk of subsequent cardiovascular events (5.5). WARNING: ASPIRIN DOSE AND BRILINTA EFFECTIVENESS •Maintenance doses of aspirin above 100 mg reduce the effectiveness of BRILINTA and should be avoided. After any initial dose, use with aspirin 75-100 mg per day (5.2, 14). -----------INDICATIONS AND USAGE ------------BRILINTA is a P2Y 12 platelet inhibitor indicated to reduce the rate of thrombotic cardiovascular events in patients with acute coronary syndrome (ACS) (unstable angina, non-ST elevation myocardial infarction, or ST elevation myocardial infarction). BRILINTA has been shown to reduce the rate of a combined endpoint of cardiovascular death, myocardial infarction, or stroke compared to clopidogrel. The difference between treatments was driven by CV death and MI with no difference in stroke. In patients treated with PCI, it also reduces the rate of stent thrombosis. (1)BRILINTA has been studied in ACS in combination with aspirin. Maintenance doses of aspirin above 100 mg decreased the effectiveness of BRILINTA. Avoid maintenance doses of aspirin above 100 mg daily. (1, 5.2, 14). ----------DOSAGE AND ADMINISTRATION -------•Initiate treatment with 180 mg (two 90 mg tablets) oral loading dose. (2) • Continue treatment with 90 mg twice daily. (2)•After the initial loading dose of aspirin (usually 325 mg), use BRILINTA with a daily maintenance dose of aspirin of 75-100 mg. (2) --------DOSAGE FORMS AND STRENGTHS ---------• 90 mg tablets (3) -------------CONTRAINDICATIONS --------------• History of intracranial hemorrhage (4.1) • Active pathological bleeding (4.2) •Severe hepatic impairment (4.3) -----------WARNINGS AND PRECAUTIONS --------• Like other antiplatelet agents, BRILINTA increases the risk of bleeding. (5.1)• In PLATO, use of BRILINTA with maintenance doses of aspirin above 100 mg decreased the effectiveness of BRILINTA. (5.2, 14) • Moderate Hepatic Impairment: Consider the risks and benefits of treatment, noting the probable increase in exposure to ticagrelor. (5.3) • Dyspnea: Dyspnea was reported more frequently with BRILINTA than with clopidogrel. Dyspnea resulting from BRILINTA is self-limiting. Rule out other causes. (5.4) • Discontinuation of BRILINTA: Premature discontinuation increases the risk of myocardial infarction, stent thrombosis, and death. (5.5) ---------------ADVERSE REACTIONS -------------Most common adverse reactions are bleeding 12% and dyspnea 14%. (5.1,5.4, 6.1) To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or /medwatch---------------DRUG INTERACTIONS -------------• Avoid use with strong CYP3A inhibitors or CYP3A inducers. (7.1, 7.2). • Patients receiving more than 40 mg per day of simvastatin or lovastatin may be at increased risk of statin-related adverse effects. (7.3)• Monitor digoxin levels with initiation of or any change in BRILINTA. (7.4)See 17 For PATIENT COUNSELING INFORMATION and Medication Guide.Revised: 07/2011FULL PRESCRIBING INFORMATION: CONTENTS*WARNING: BLEEDING RISKWARNING: ASPIRIN DOSE AND BRILINTA EFFECTIVENESS 1 INDICATIONS AND USAGE 1.1 Acute Coronary Syndromes 2 DOSAGE AND ADMINISTRATION 3 DOSAGE FORMS AND STRENGTHS 4 CONTRAINDICATIONS 4.1 History of Intracranial Hemorrhage 4.2 Active Bleeding 4.3 Severe Hepatic Impairment 5 WARNINGS AND PRECAUTIONS 5.1 General Risk of Bleeding 5.2 Concomitant Aspirin Maintenance Dose 5.3 Moderate Hepatic Impairment 5.4 Dyspnea 5.5 Discontinuation of BRILINTA 5.6 Strong Inhibitors of Cytochrome CYP3A 5.7 Cytochrome CYP3A Potent Inducers 6 ADVERSE REACTIONS 6.1 Clinical Trials Experience 7 DRUG INTERACTIONS 7.1 CYP3A inhibitors 7.2 CYP3A inducers 7.3 Simvastatin, lovastatin 7.4 Digoxin 7.5 Other Concomitant Therapy 8 USE IN SPECIFIC POPULATIONS 8.1 Pregnancy 8.3 Nursing Mothers 8.4 Pediatric Use 8.5 Geriatric Use 8.6 Hepatic Impairment 8.7 Renal Impairment 10 OVERDOSAGE 11 DESCRIPTION 12 CLINICAL PHARMACOLOGY 12.1 Mechanism of Action 12.2 Pharmacodynamics 12.3 Pharmacokinetics 13 NONCLINICAL TOXICOLOGY 13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility 14 CLINICAL STUDIES 16 HOW SUPPLIED/STORAGE AND HANDLING 17 PATIENT COUNSELING INFORMATION 17.1 Benefits and Risks 17.2 Bleeding 17.3 Other Signs and Symptoms Requiring Medical Attention 17.4 Invasive Procedures 17.5 Concomitant Medications *Sections or subsections omitted from the Full Prescribing Information are not listed .FULL PRESCRIBING INFORMATIONWARNING: BLEEDING RISK• BRILINTA, like other antiplatelet agents, can cause significant, sometimes fatal, bleeding (5.1, 6.1).• Do not use BRILINTA in patients with active pathological bleeding or a history of intracranial hemorrhage (4.1, 4.2).• Do not start BRILINTA in patients planned to undergo urgent coronary artery bypass graft surgery (CABG). When possible, discontinue BRILINTA at least 5days prior to any surgery (5.1).• Suspect bleeding in any patient who is hypotensive and has recently undergone coronary angiography, percutaneous coronary intervention (PCI), CABG, orother surgical procedures in the setting of BRILINTA (5.1).• If possible, manage bleeding without discontinuing BRILINTA. Stopping BRILINTA increases the risk of subsequent cardiovascular events (5.5). WARNING: ASPIRIN DOSE AND BRILINTA EFFECTIVENESS• Maintenance doses of aspirin above 100 mg reduce the effectiveness of BRILINTA and should be avoided. After any initial dose, use with aspirin 75-100 mg per day(5. 2, 14).1 INDICATIONS AND USAGEAcute Coronary SyndromesBRILINTA is a P2Y12 platelet inhibitor indicated to reduce the rate of thrombotic cardiovascular events in patients with acute coronary syndrome (ACS) (unstable angina, non-ST elevation myocardial infarction, or ST elevation myocardial infarction).BRILINTA has been shown to reduce the rate of a combined endpoint of cardiovascular death, myocardial infarction or stroke compared to clopidogrel. The difference between treatments was driven by CV death and MI with no difference in stroke. In patients treated with PCI, it also reduces the rate of stent thrombosis [see Clinical Studies (14)].BRILINTA has been studied in ACS in combination with aspirin. Maintenance doses of aspirin above 100 mg decreased the effectiveness of BRILINTA. Avoid maintenancedoses of aspirin above 100 mg daily [see Warnings and Precautions (5.2) and ClinicalStudies (14)].2 DOSAGE AND ADMINISTRATIONInitiate BRILINTA treatment with a 180 mg (two 90 mg tablets) loading dose andcontinue treatment with 90 mg twice dailyAfter the initial loading dose of aspirin (usually 325 mg), use BRILINTA with a dailymaintenance dose of aspirin of 75-100 mg.ACS patients who have received a loading dose of clopidogrel may be started onBRILINTA.BRILINTA can be administered with or without food.A patient who misses a dose of BRILINTA should take one 90 mg tablet (their nextdose) at its scheduled time.3 DOSAGE FORMS AND STRENGTHSBRILINTA (ticagrelor) 90 mg is supplied as a round, biconvex, yellow, film-coatedtablet marked with a “90” above “T” on one side.4 CONTRAINDICATIONS4.1 History of Intracranial HemorrhageBRILINTA is contraindicated in patients with a history of intracranial hemorrhage (ICH) because of a high risk of recurrent ICH in this population [see Clinical Studies(14)].Bleeding4.2 A ctiveBRILINTA is contraindicated in patients with active pathological bleeding such as peptic ulcer or intracranial hemorrhage [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].4.3 Severe Hepatic ImpairmentBRILINTA is contraindicated in patients with severe hepatic impairment because of a probable increase in exposure, and it has not been studied in these patients. Severe hepatic impairment increases the risk of bleeding because of reduced synthesis of coagulation proteins [see Clinical Pharmacology (12.3)].5 WARNINGS AND PRECAUTIONS5.1 General Risk of BleedingDrugs that inhibit platelet function including BRILINTA increase the risk of bleeding.BRILINTA increased the overall risk of bleeding (Major + Minor) to a somewhat greater extent than did clopidogrel. The increase was seen for non-CABG-related bleeding, but not for CABG-related bleeding. Fatal and life-threatening bleeding rates were not increased [see Adverse Reactions (6.1)].In general, risk factors for bleeding include older age, a history of bleeding disorders, performance of percutaneous invasive procedures, and concomitant use of medications that increase the risk of bleeding (e.g., anticoagulant and fibrinolytic therapy, higher doses of aspirin, and chronic nonsteroidal anti-inflammatory drugs [NSAIDS]).When possible, discontinue BRILINTA five days prior to surgery. Suspect bleeding in any patient who is hypotensive and has recently undergone coronary angiography, PCI, CABG, or other surgical procedures, even if the patient does not have any signs of bleeding.If possible, manage bleeding without discontinuing BRILINTA. Stopping BRILINTA increases the risk of subsequent cardiovascular events [see Warnings and Precautions(5.5) and Adverse Reactions (6.1)].5.2 Concomitant Aspirin Maintenance DoseIn PLATO, use of BRILINTA with maintenance doses of aspirin above 100 mg decreased the effectiveness of BRILINTA. Therefore, after the initial loading dose of aspirin (usually 325 mg), use BRILINTA with a maintenance dose of aspirin of 75-100 mg [see Dosage and Administration (2) and Clinical Studies (14)].5.3 Moderate Hepatic ImpairmentBRILINTA has not been studied in patients with moderate hepatic impairment.Consider the risks and benefits of treatment, noting the probable increase in exposure to ticagrelor.5.4 DyspneaDyspnea was reported in 14% of patients treated with BRILINTA and in 8% of patients taking clopidogrel. Dyspnea was usually mild to moderate in intensity and often resolved during continued treatment. If a patient develops new, prolonged, or worsened dyspnea during treatment with BRILINTA, exclude underlying diseases that may require treatment. If dyspnea is determined to be related to BRILINTA, no specific treatment is required; continue BRILINTA without interruption.In a substudy, 199 patients from PLATO underwent pulmonary function testing irrespective of whether they reported dyspnea. There was no significant difference between treatment groups for FEV1. There was no indication of an adverse effect on pulmonary function assessed after one month or after at least 6 months of chronic treatment.BRILINTAof5.5 DiscontinuationAvoid interruption of BRILINTA treatment. If BRILINTA must be temporarily discontinued (e.g., to treat bleeding or for elective surgery), restart it as soon as possible. Discontinuation of BRILINTA will increase the risk of myocardial infarction, stent thrombosis, and death.of Cytochrome CYP3A5.6 StrongInhibitorsTicagrelor is metabolized by CYP3A4/5. Avoid use with strong CYP3A inhibitors, such as atazanavir, clarithromycin, indinavir, itraconazole, ketoconazole, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin and voriconazole [see Drug Interactions(7.1) and Clinical Pharmacology (12.3)].5.7 Cytochrome CYP3A Potent InducersAvoid use with potent CYP3A inducers, such as rifampin, dexamethasone, phenytoin, carbamazepine, and phenobarbital [see Drug Interactions (7.2), and Clinical Pharmacology (12.3)].REACTIONS6 ADVERSE6.1 Clinical Trials ExperienceThe following adverse reactions are also discussed elsewhere in the labeling:•Dyspnea [see Warnings and Precautions (5.4)]Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.BRILINTA has been evaluated for safety in more than 10000 patients, including more than 3000 patients treated for more than 1 year.BleedingPLATO used the following bleeding severity categorization:• Major bleed – fatal/life-threatening. Any one of the following: fatal;intracranial; intrapericardial bleed with cardiac tamponade; hypovolemic shockor severe hypotension due to bleeding and requiring pressors or surgery;clinically overt or apparent bleeding associated with a decrease in hemoglobin(Hb) of more than 5 g/dL; transfusion of 4 or more units (whole blood or packedred blood cells (PRBCs)) for bleeding.• Major bleed – other. Any one of the following: significantly disabling (e.g., intraocular with permanent vision loss); clinically overt or apparent bleedingassociated with a decrease in Hb of 3 g/dL; transfusion of 2-3 units (wholeblood or PRBCs) for bleeding.• Minor bleed. Requires medical intervention to stop or treat bleeding (e.g., epistaxis requiring visit to medical facility for packing).• Minimal bleed. All others (e.g., bruising, bleeding gums, oozing from injection sites, etc.) not requiring intervention or treatment.Figure 1 shows major bleeding events over time. Many events are early, at a time of coronary angiography, PCI, CABG, and other procedures, but the risk persists during later use of antiplatelet therapy.Figure 1 - Kaplan-Meier estimate of time to first PLATO-defined ‘Total Major’ bleeding eventAnnualized rates of bleeding are summarized in Table 1 below. About half of the bleeding events were in the first 30 days.Table 1 - Non-CABG related bleeds (KM%)BRILINTA N=9235 Clopidogrel N=9186Total (Major + Minor) 8.7 7.0Major 4.5 3.8Fatal/Life-threatening 2.1 1.9Fatal 0.2 0.2Intracranial (Fatal/Life-threatening) 0.3 0.2As shown in Table 1, BRILINTA was associated with a somewhat greater risk of non-CABG bleeding than was clopidogrel. No baseline demographic factor altered the relative risk of bleeding with BRILINTA compared to clopidogrel.In PLATO, 1584 patients underwent CABG surgery. The percentages of those patients who bled are shown in Table 2. Rates were very high but similar for BRILINTA and clopidogrel.Table 2 –CABG bleeds (KM%)Patients with CABGBRILINTA N=770 Clopidogrel N=814Total Major 85.8 86.9Fatal/Life-threatening 48.1 47.9Fatal 0.9 1.1Although the platelet inhibition effect of BRILINTA has a faster offset than clopidogrel in in vitro tests and BRILINTA is a reversibly binding P2Y12 inhibitor, PLATO did not show an advantage of BRILINTA compared to clopidogrel for CABG-related bleeding. When antiplatelet therapy was stopped 5 days before CABG, major bleeding occurred in 75% of BRILINTA treated patients and 79% on clopidogrel.No data exist with BRILINTA regarding a hemostatic benefit of platelet transfusions. Drug DiscontinuationIn PLATO, the rate of study drug discontinuation attributed to adverse reactions was 7.4% for BRILINTA and 5.4% for clopidogrel. Bleeding caused permanent discontinuation of study drug in 2.3% of BRILINTA patients and 1.0% of clopidogrel patients. Dyspnea led to study drug discontinuation in 0.9% of BRILINTA and 0.1% of clopidogrel patients.Common Adverse EventsA variety of non-hemorrhagic adverse events occurred in PLATO at rates of 3% or more. These are shown in Table 3. In the absence of a placebo control, whether these are drug related cannot be determined in most cases, except where they are more common on BRILINTA or clearly related to the drug’s pharmacologic effect (dyspnea).Table 3 – Percentage of patients reporting non-hemorrhagic adverse events at least 3% or more in either group BRILINTA N=9235 ClopidogrelN=9186Dyspnea a 13.8 7.8Headache 6.5 5.8 Cough 4.9 4.6 Dizziness 4.5 3.9 Nausea 4.3 3.8 Atrial fibrillation 4.2 4.6Hypertension 3.8 4.0 Non-cardiac chest pain 3.7 3.3Diarrhea 3.7 3.3 Back pain 3.6 3.3Hypotension 3.2 3.3 Fatigue 3.2 3.2 Chest pain 3.1 3.5Includes: dyspnea, dyspnea exertional, dyspnea at rest, nocturnal dyspnea, dyspnea paroxysmal nocturnal Bradycardia In clinical studies BRILINTA has been shown to increase the occurrence of Holterdetected bradyarrhythmias (including ventricular pauses). PLATO excluded patients at increased risk of bradycardic events (e.g., patients who have sick sinus syndrome, 2nd or 3rd degree AV block, or bradycardic-related syncope and not protected with a pacemaker). In PLATO, syncope, pre-syncope and loss of consciousness were reported by 1.7% and 1.5% of BRILINTA and clopidogrel patients, respectively.In a Holter substudy of about 3000 patients in PLATO, more patients had ventricular pauses with BRILINTA (6.0%) than with clopidogrel (3.5%) in the acute phase; rates were 2.2% and 1.6% respectively after 1 month.GynecomastiaIn PLATO, gynecomastia was reported by 0.23% of men on BRILINTA and 0.05% on clopidogrel.Other sex-hormonal adverse reactions, including sex organ malignancies, did not differ between the two treatment groups in PLATO.Lab abnormalitiesSerum Uric Acid:Serum uric acid levels increased approximately 0.6 mg/dL from baseline on BRILINTA and approximately 0.2 mg/dL on clopidogrel in PLATO. The differencedisappeared within 30 days of discontinuing treatment. Reports of gout did not differbetween treatment groups in PLATO (0.6% in each group). Serum Creatinine:In PLATO, a >50% increase in serum creatinine levels was observed in 7.4% of patients receiving BRILINTA compared to 5.9% of patients receiving clopidogrel. The increases typically did not progress with ongoing treatment and often decreased with continued therapy. Evidence of reversibility upon discontinuation was observed even in those with the greatest on treatment increases. Treatment groups in PLATO did not differ for renal-related serious adverse events such as acute renal failure, chronic renal failure, toxic nephropathy, or oliguria.INTERACTIONS7 DRUGEffects of other drugsTicagrelor is predominantly metabolized by CYP3A4 and to a lesser extent by CYP3A5.inhibitors7.1 CYP3AAvoid use of strong inhibitors of CYP3A (e.g., ketoconazole, itraconazole, voriconazole, clarithromycin, nefazodone, ritonavir, saquinavir, nelfinavir, indinavir, atazanavir and telithromycin) [see Warnings and Precautions (5.6) and Clinical Pharmacology (12.3)].7.2 CYP3A inducersAvoid use with potent inducers of CYP3A (e.g., rifampin, dexamethasone, phenytoin, carbamazepine and phenobarbital) [see Warnings and Precautions (5.7) and Clinical Pharmacology (12.3)].7.3 AspirinUse of BRILINTA with aspirin maintenance doses above 100 mg reduced the effectiveness of BRILINTA [see Warnings and Precautions (5.2) and Clinical Studies(14)].Effect of BRILINTA on other drugsTicagrelor is an inhibitor of CYP3A4/5 and the P-glycoprotein transporter.lovastatin7.4 Simvastatin,BRILINTA will result in higher serum concentrations of simvastatin and lovastatin because these drugs are metabolized by CYP3A4. Avoid simvastatin and lovastatin doses greater than 40 mg [see Clinical Pharmacology (12.3)].7.5 D igoxinDigoxin: Because of inhibition of the P-glycoprotein transporter, monitor digoxin levels with initiation of or any change in BRILINTA therapy [see Clinical Pharmacology(12.3)].7.6 Other Concomitant TherapyBRILINTA can be administered with unfractionated or low-molecular-weight heparin, GPIIb/IIIa inhibitors, proton pump inhibitors, beta-blockers, angiotensin converting enzyme inhibitors, and angiotensin receptor blockers.8 USE IN SPECIFIC POPULATIONS8.1 PregnancyPregnancy Category C:There are no adequate and well-controlled studies of BRILINTA use in pregnant women. In animal studies, ticagrelor caused structural abnormalities at maternal doses about 5 to 7 times the maximum recommended human dose (MRHD) based on body surface area. BRILINTA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.In reproductive toxicology studies, pregnant rats received ticagrelor during organogenesis at doses from 20 to 300 mg/kg/day. The lowest dose was approximately the same as the MRHD of 90 mg twice daily for a 60 kg human on a mg/m2 basis.Adverse outcomes in offspring occurred at doses of 300 mg/kg/day (16.5 times the MRHD on a mg/m2 basis) and included supernumerary liver lobe and ribs, incomplete ossification of sternebrae, displaced articulation of pelvis, and misshapen/misaligned sternebrae. When pregnant rabbits received ticagrelor during organogenesis at doses from 21 to 63 mg/kg/day, fetuses exposed to the highest maternal dose of 63 mg/kg/day(6.8 times the MRHD on a mg/m2 basis) had delayed gall bladder development andincomplete ossification of the hyoid, pubis and sternebrae occurred.In a prenatal/postnatal study, pregnant rats received ticagrelor at doses of 10 to 180 mg/kg/day during late gestation and lactation. Pup death and effects on pup growth were observed at 180 mg/kg/day (approximately 10 times the MRHD on a mg/m2 basis). Relatively minor effects such as delays in pinna unfolding and eye opening occurred at doses of 10 and 60 mg/kg (approximately one-half and 3.2 times the MRHD on a mg/m2 basis).Mothers8.3 NursingIt is not known whether ticagrelor or its active metabolites are excreted in human milk.Ticagrelor is excreted in rat milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from BRILINTA, a decision should be made whether to discontinue nursing or to discontinue drug, taking into account the importance of the drug to the mother.Use8.4 PediatricThe safety and effectiveness of BRILINTA in pediatric patients have not been established.8.5 Geriatric UseIn PLATO, 43% of patients were ≥65 years of age and 15% were ≥75 years of age.The relative risk of bleeding was similar in both treatment and age groups.No overall differences in safety or effectiveness were observed between these patients and younger patients. While this clinical experience has not identified differences in responses between the elderly and younger patients, greater sensitivity of some older individuals cannot be ruled out.8.6 HepaticImpairmentBRILINTA has not been studied in the patients with moderate or severe hepatic impairment. Ticagrelor is metabolized by the liver and impaired hepatic function can increase risks for bleeding and other adverse events. Hence, BRILINTA is contraindicated for use in patients with severe hepatic impairment and its use should be considered carefully in patients with moderate hepatic impairment. No dosage adjustment is needed in patients with mild hepatic impairment [see Contraindications(4), Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].8.7 RenalImpairmentNo dosage adjustment is needed in patients with renal impairment. Patients receiving dialysis have not been studied [see Clinical Pharmacology (12.3)].10 OVERDOSAGEThere is currently no known treatment to reverse the effects of BRILINTA, and ticagrelor is not expected to be dialyzable. Treatment of overdose should follow local standard medical practice. Bleeding is the expected pharmacologic effect of overdosing. If bleeding occurs, appropriate supportive measures should be taken.Other effects of overdose may include gastrointestinal effects (nausea, vomiting, diarrhea) or ventricular pauses. Monitor the ECG.11 DESCRIPTIONBRILINTA contains ticagrelor, a cyclopentyltriazolopyrimidine, inhibitor of platelet activation and aggregation mediated by the P2Y12 ADP-receptor. Chemically it is (1S,2S,3R,5S)-3-[7-{[(1R,2S)-2-(3,4-difluorophenyl)cyclopropyl]amino}-5(propylthio)-3H-[1,2,3]-triazolo[4,5-d]pyrimidin-3-yl]-5-(2hydroxyethoxy)cyclopentane-1,2-diol. The empirical formula of ticagrelor is C23H28F2N6O4S and its molecular weight is 522.57. The chemical structure of ticagrelor is:Ticagrelor is a crystalline powder with an aqueous solubility of approximately10 µg/mL at room temperature.BRILINTA tablets for oral administration contain 90 mg of ticagrelor and the following ingredients: mannitol, dibasic calcium phosphate, sodium starch glycolate, hydroxypropyl cellulose, magnesium stearate, hydroxypropyl methylcellulose, titanium dioxide, talc, polyethylene glycol 400, and ferric oxide yellow.PHARMACOLOGY12 CLINICAL12.1 Mechanism of ActionTicagrelor and its major metabolite reversibly interact with the platelet P2Y12 ADP-receptor to prevent signal transduction and platelet activation. Ticagrelor and its active metabolite are approximately equipotent.12.2 PharmacodynamicsThe inhibition of platelet aggregation (IPA) by ticagrelor and clopidogrel was compared in a 6 week study examining both acute and chronic platelet inhibition effects in response to 20 μM ADP as the platelet aggregation agonist.The onset of IPA was evaluated on Day 1 of the study following loading doses of 180 mg ticagrelor or 600 mg clopidogrel. As shown in Figure 2, IPA was higher in the ticagrelor group at all time points. The maximum IPA effect of ticagrelor was reached at around 2 hours, and was maintained for at least 8 hours.The offset of IPA was examined after 6 weeks on ticagrelor 90 mg twice daily or clopidogrel 75 mg daily, again in response to 20 µM ADP.As shown in Figure 3, mean maximum IPA following the last dose of ticagrelor was 88% and 62% for clopidogrel. The insert in figure 3 shows that after 24 hours, IPA in the ticagrelor group (58%) was similar to IPA in clopidogrel group (52%), indicating that patients who miss a dose of ticagrelor would still maintain IPA similar to the trough IPA of patients treated with clopidogrel. After 5 days, IPA in the ticagrelor group was similar to IPA in the placebo group. It is not known how either bleeding risk or thrombotic risk track with IPA, for either ticagrelor or clopidogrel.Figure 2 - Mean inhibition of platelet aggregation (±SE) following single oral doses of placebo, 180 mg ticagrelor, or 600 mg clopidogrelFigure 3 - Mean inhibition of platelet aggregation (IPA) following 6 weeks on placebo, ticagrelor 90 mg twice daily, or clopidogrel 75 mg daily•Ticagrelor ▲Clopidogrel ■PlaceboTransitioning from clopidogrel to BRILINTA resulted in an absolute IPA increase of26.4% and from BRILINTA to clopidogrel resulted in an absolute IPA decrease of24.5%. Patients can be transitioned from clopidogrel to BRILINTA withoutinterruption of antiplatelet effect [see Dosage and Administration (2)].12.3 P harmacokineticsTicagrelor demonstrates dose proportional pharmacokinetics, which are similar in patients and healthy volunteers.AbsorptionAbsorption of ticagrelor occurs with a median t max of 1.5 h (range 1.0–4.0). The formation of the major circulating metabolite AR-C124910XX (active) from ticagrelor occurs with a median t max of 2.5 h (range 1.5-5.0).The mean absolute bioavailability of ticagrelor is about 36%, (range 30%-42%).Ingestion of a high-fat meal had no effect on ticagrelor C max, but resulted in a 21% increase in AUC. The C max of its major metabolite was decreased by 22% with no change in AUC. BRILINTA can be taken with or without food.。
[VIP专享]孚来迪说明书
![[VIP专享]孚来迪说明书](https://img.taocdn.com/s3/m/2749618889eb172ded63b7b6.png)
孚来迪(瑞格列奈片)【药品名称】通用名称:瑞格列奈片商品名称:孚来迪英文名称:Repaglinide Tablets汉语拼音:Ruigelienai Pian【成份】本品主要成份为瑞格列奈。
化学名称:(S)-2-乙氧基-4-[2-[[甲基-1-[2-(1-哌啶基)苯基]丁基]氨基]-2-氧代乙基]苯甲酸。
化学结构式:分子式:C27H36N2O4分子量:452.59【性状】本品为白色或类白色片。
【适应症】用于饮食控制、降低体重及运动锻炼不能有效控制高血糖的2型糖尿病(非胰岛素依赖型)患者。
瑞格列奈片可与二甲双胍合用。
与各自单独使用相比,二者合用对控制血糖有协同作用。
【规格】0.5mg。
【用法用量】瑞格列奈片应在主餐前服用(即餐前服用)。
在口服瑞格列奈片30分钟内即出现促胰岛素分泌反应。
通常在餐前15分钟内服用本药。
服药时间也可掌握在餐前0~30分钟内。
请遵医嘱服用瑞格列奈片。
剂量因人而异,以个人血糖而定。
推荐起始剂量为0.5 mg(1片),以后如需要可每周或每两周作调整。
接受其它口服降血糖药治疗的病人可直接转用瑞格列奈片治疗。
其推荐起始剂量为1mg(2片)。
最大的推荐单次剂量为4mg(8片),进餐时服用。
但最大日剂量不应超过16mg(32片)。
对于衰弱和营养不良的患者,应谨慎调整剂量。
如果与二甲双胍合用,应减少瑞格列奈片的剂量。
尽管瑞格列奈主要由胆汁排泄,但肾功能不全的患者仍应慎用。
【不良反应】同其他口服降糖药一样,服用瑞格列奈可能引起血糖变化,如高血糖和低血糖。
同每种糖尿病治疗一样,这些反应的出现依赖于个体因素,如饮食习惯、剂量、运动和应激反应。
瑞格列奈及其它降血糖药物的临床应用显示,服用瑞格列奈可能发生以下不良反应:根据不良反应的发生率分别定义如下:罕见不良反应:发生率>1/10000,<1/1000。
非常罕见不良反应:发生率<1/10000。
1.免疫系统失调过敏反应可发生皮肤过敏反应,如瘙痒、发红、荨麻疹。
阿格列汀FDA说明书(英文)

CENTER FOR DRUG EVALUATION ANDRESEARCHAPPLICATION NUMBER:022271Orig1s000LABELINGHIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use NESINA safely and effectively. See full prescribing information for NESINA.NESINA (alogliptin) tabletsInitial U.S. Approval: 2013----------------------------INDICATIONS AND USAGE---------------------------- NESINA is a dipeptidyl peptidase-4 (DPP-4) inh bitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. (1.1, 14)Limitation of Use: Not for treatment of type 1 diabetes or diabetic ketoacidosis. (1.2)------------------------DOSAGE AND ADMINISTRATION----------------------- ∙The recommended dose in patients with normal renal function or mild renal impairment is 25 mg once daily. (2.1)∙Can be taken with or without food. (2.1)∙Adjust dose if moderate or severe renal impairment or end-stage renal disease (ESRD). (2.2)Degree of Renal Impairment CreatinineClearance(mL/min) RecommendedDosingModerate ≥30 to <60 12.5 mg once dailySevere/ESRD <30 6.25 mg once daily----------------------DOSAGE FORMS AND STRENGTHS--------------------- Tablets: 25 mg, 12.5 mg and 6.25 mg (3)-----------------------------CONTRAINDICATIONS--------------------------- History of a serious hypersensitivity reaction to alogliptin-containing products, such as anaphylaxis, angioedema or severe cutaneous adverse reactions. (4) -----------------------WARNINGS AND PRECAUTIONS-------------------∙Acute pancreatitis: There have been postmarketing reports of acute pancreatitis. If pancreatitis is suspected, promptlydiscontinue NESINA. (5.1)∙Hypersensitivity: There have been postmarketing reports of serious hypersensitivity reactions in patients treated with NESINA such as anaphylaxis, angioedema and severe cutaneous adverse reactions. In such cases, promptly discontinue NESINA, assessfor other potential causes, institute appropriate monitoring andtreatment, and initiate alternative treatment for diabetes. (5.2)∙Hepatic effects: Postmarketing reports of hepatic failure, sometimes fatal. Causality cannot be excluded. If liver injury isdetected, promptly interrupt NESINA and assess patient forprobable cause, then treat cause if possible, to resolution orstabilization. Do not restart NESINA if liver injury is confirmedand no alternative etiology can be found. (5.3)∙Hypoglycemia: When an insulin secretagogue (e.g. sulfonylurea) or insulin is used in combination with NESINA, a lower dose of the insulin secretagogue or insulin may be required to minimize therisk of hypoglycemia. (5.4)∙Macrovascular outcomes: There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with NESINA or any other antidiabetic drug. (5.5)----------------------------ADVERSE REACTIONS---------------------------- Common adverse reactions (reported in ≥4% of patients treated with NESINA 25 mg and more frequently than in patients who received placebo) are: nasopharyngitis, headache, and upper respiratory tract infection. (6.1)To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals at 1-877-TAKEDA-7 (1-877-825-3327) or FDA at1-800-FDA-1088 or /medwatch.See 17 for PATIENT COUNSELING INFORMATION and Medication GuideRevised: 01/2013FULL PRESCRIBING INFORMATION: CONTENTS*1 INDICATIONSANDUSAGE1.1 Monotherapy and Combination Therapy1.2 Limitation of Use2 DOSAGEANDADMINISTRATION2.1 RecommendedDosing2.2 Patients with Renal Impairment3 DOSAGE FORMS AND STRENGTHS4 CONTRAINDICATIONS5 WARNINGSANDPRECAUTIONS5.1 Pancreatitis5.2 HypersensitivityReactions5.3 HepaticEffects5.4 Use with Medications Known to Cause Hypoglycemia5.5 MacrovascularOutcomes6 ADVERSEREACTIONS6.1 Clinical Studies Experience6.2 PostmarketingExperience7 DRUGINTERACTIONS8 USE IN SPECIFIC POPULATIONS8.1 Pregnancy8.3 NursingMothers8.4 PediatricUse8.5 GeriatricUse8.6 HepaticImpairment10 OVERDOSAGE11 DESCRIPTION12 CLINICAL PHARMACOLOGY12.1 Mechanism of Action12.2 Pharmacodynamics12.3 Pharmacokinetics13 NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility14 CLINICAL STUDIES14.1 Patients with Inadequate Glycemic Control on Diet andExercise14.2 Combination Therapy16 HOW SUPPLIED/STORAGE AND HANDLING17 PATIENT COUNSELING INFORMATION17.1 Instructions* Sections or subsections omitted from the full prescribing information are not listedFULL PRESCRIBING INFORMATION121 INDICATIONS AND USAGE31.1 Monotherapy and Combination TherapyNESINA is indicated as an adjunct to diet and exercise to improve glycemic control in45adults with type 2 diabetes mellitus in multiple clinical settings[see Clinical Studies6(14)].71.2 Limitation of Use8NESINA should not be used in patients with type 1 diabetes mellitus or for the treatment 9of diabetic ketoacidosis, as it would not be effective in these settings.2 DOSAGE AND ADMINISTRATION10112.1 Recommended Dosing12The recommended dose of NESINA is 25 mg once daily.13NESINA may be taken with or without food.142.2 Patients with Renal Impairment15No dose adjustment of NESINA is necessary for patients with mild renal impairment16(creatinine clearance [CrCl] ≥60 mL/min).17The dose of NESINA is 12.5 mg once daily for patients with moderate renal impairment 18(CrCl ≥30 to <60 mL/min).19The dose of NESINA is 6.25 mg once daily for patients with severe renal impairment20(CrCl ≥15 to <30 mL/min) or with end-stage renal disease (ESRD) (CrCl <15 mL/min or 21requiring hemodialysis). NESINA may be administered without regard to the timing of 22dialysis. NESINA has not been studied in patients undergoing peritoneal dialysis [see 23Clinical Pharmacology (12.3)].24Because there is a need for dose adjustment based upon renal function, assessment of 25renal function is recommended prior to initiation of NESINA therapy and periodically26thereafter.273 DOSAGE FORMS AND STRENGTHS28∙25 mg tablets are light red, oval, biconvex, film-coated, with “TAK ALG-25” printed 29on one side.30∙12.5 mg tablets are yellow, oval, biconvex, film-coated, with “TAK ALG-12.5”31printed on one side.32∙ 6.25 mg tablets are light pink, oval, biconvex, film-coated, with “TAK ALG-6.25”33printed on one side.344 CONTRAINDICATIONS35History of a serious hypersensitivity reaction to alogliptin-containing products, such as 36anaphylaxis, angioedema or severe cutaneous adverse reactions.375 WARNINGS AND PRECAUTIONS385.1 Pancreatitis39There have been postmarketing reports of acute pancreatitis in patients taking NESINA.40After initiation of NESINA, patients should be observed carefully for signs and41symptoms of pancreatitis. If pancreatitis is suspected, NESINA should promptly be42discontinued and appropriate management should be initiated. It is unknown whether 43patients with a history of pancreatitis are at increased risk for the development of44pancreatitis while using NESINA.455.2 HypersensitivityReactions46There have been postmarketing reports of serious hypersensitivity reactions in patients 47treated with NESINA. These reactions include anaphylaxis, angioedema, and severe 48cutaneous adverse reactions including Stevens-Johnson syndrome. If a serious49hypersensitivity reaction is suspected, discontinue NESINA, assess for other potential 50causes for the event, and institute alternative treatment for diabetes [see Adverse51Reactions (6.2)]. Use caution in a patient with a history of angioedema with another52DPP-4 inhibitor because it is unknown whether such patients will be predisposed to53angioedema with NESINA.545.3 HepaticEffects55There have been postmarketing reports of fatal and non-fatal hepatic failure in patients 56taking NESINA, although some of the reports contain insufficient information necessary 57to establish the probable cause [see Adverse Reactions (6.2)]. In randomized controlled 58studies, serum alanine aminotransferase (ALT) elevations greater than three times the 59upper limit of normal (ULN) were observed: 1.3% in alogliptin-treated patients and 1.5% 60in all comparator-treated patients.61Patients with type 2 diabetes may have fatty liver disease which may cause liver test62abnormalities, and they may also have other forms of liver disease, many of which can 63be treated or managed. Therefore, obtaining a liver test panel and assessing the patient 64before initiating NESINA therapy is recommended. In patients with abnormal liver tests, 65NESINA should be initiated with caution.Measure liver tests promptly in patients who report symptoms that may indicate liver6667injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine or68jaundice. In this clinical context, if the patient is found to have clinically significant liver 69enzyme elevations and if abnormal liver tests persist or worsen, NESINA should be70interrupted and investigation done to establish the probable cause. NESINA should not 71be restarted in these patients without another explanation for the liver testabnormalities.72735.4 Use with Medications Known to Cause Hypoglycemia74Insulin and insulin secretagogues, such as sulfonylureas, are known to causehypoglycemia. Therefore, a lower dose of insulin or insulin secretagogue may be7576required to minimize the risk of hypoglycemia when used in combination with NESINA.775.5 Macrovascular OutcomesThere have been no clinical studies establishing conclusive evidence of macrovascular7879risk reduction with NESINA or any other antidiabetic drug.6 ADVERSE80REACTIONS816.1 Clinical Studies ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction8283rates observed in the clinical trials of a drug cannot be directly compared to rates in the 84clinical trials of another drug and may not reflect the rates observed in clinical practice.85Approximately 8500 patients with type 2 diabetes have been treated with NESINA in 14 86randomized, double-blind, controlled clinical trials with approximately 2900 subjects87randomized to placebo and approximately 2200 to an active comparator. The mean88exposure to NESINA was 40 weeks with more than 2400 subjects treated for more than 89one year. Among these patients, 63% had a history of hypertension, 51% had a history 90of dyslipidemia, 25% had a history of myocardial infarction, 8% had a history of unstable 91angina, and 7% had a history of congestive heart failure. The mean duration of diabetes was 7 years, the mean body mass index (BMI) was 31 kg/m2 (51% of patients had a9293BMI ≥30 kg/m2), and the mean age was 57 years (24% of patients ≥65 years of age).94Two placebo-controlled monotherapy trials of 12 and 26 weeks of duration were95conducted in patients treated with NESINA 12.5 mg daily, NESINA 25 mg daily and96placebo. Four placebo-controlled add-on combination therapy trials of 26 weeks97duration were also conducted: with metformin, with a sulfonylurea, with athiazolidinedione, and with insulin.9899Five placebo-controlled trials of 16 weeks up through two years in duration wereconducted in combination with metformin, in combination with pioglitazone and with100101pioglitazone added to a background of metformin therapy.102Three active-controlled trials of 52 weeks in duration were conducted in patients treated 103with pioglitazone and metformin, in combination with metformin and as monotherapy 104compared to glipizide.105In a pooled analysis of these 14 controlled clinical trials, the overall incidence of adverse 106events was 66% in patients treated with NESINA 25 mg compared to 62% with placebo 107and 70% with active comparator. Overall discontinuation of therapy due to adverse108events was 4.7% with NESINA 25 mg compared to 4.5% with placebo or 6.2% with109active comparator.110Adverse reactions reported in ≥4% of patients treated with NESINA 25 mg and more 111frequently than in patients who received placebo are summarized in Table 1.112113Table 1. Adverse Reactions Reported in ≥4% Patients Treated with NESINA 25 mg and More Frequently Than in Patients Given Placebo in Pooled StudiesNumber of Patients (%)NESINA 25 mg Placebo ActiveComparatorN =5902 N=2926 N=2257Nasopharyngitis 257 (4.4) 89 (3.0) 113 (5.0)Headache 247 (4.2) 72 (2.5) 121 (5.4)Upper respiratory tract infection 247 (4.2) 61 (2.1) 113 (5.0)Pancreatitis114In the clinical trial program, pancreatitis was reported in 11 of 5902 (0.2%) patients 115receiving NESINA 25 mg daily compared to 5 of 5183 (˂0.1%) patients receiving all 116comparators.117Hypersensitivity Reactions118In a pooled analysis, the overall incidence of hypersensitivity reactions was 0.6% 119with NESINA 25 mg compared to 0.8% with all comparators. A single event of serum 120sickness was reported in a patient treated with NESINA 25 mg.121Hypoglycemia122Hypoglycemic events were documented based upon a blood glucose value and/or 123clinical signs and symptoms of hypoglycemia.124In the monotherapy study, the incidence of hypoglycemia was 1.5% in patients125treated with NESINA compared to 1.6% with placebo. The use of NESINA as add-on 126therapy to glyburide or insulin did not increase the incidence of hypoglycemia127compared to placebo. In a monotherapy study comparing NESINA to a sulfonylurea 128in elderly patients, the incidence of hypoglycemia was 5.4% with NESINA compared 129to 26% with glipizide (Table 2).130Page 6 of 30131 Table 2. Incidence and Rate of Hypoglycemia* in Placebo and Active-ControlledStudies when NESINA was Used as Add-on Therapy to Glyburide, Insulin,Metformin, Pioglitazone, or Compared to Glipizide Add-on to Glyburide(26 Weeks) NESINA 25 mg + Glyburide Placebo + GlyburideN =198 N=99 Overall (%) 19 (9.6)11 (11.1)Severe (%)†0 1 (1)Add-on to Insulin (+/- Metformin)(26 Weeks)NESINA 25 mg + Insulin (+/- Metformin) Placebo+ Insulin (+/- Metformin)N =129 N =129Overall (%) 35 (27)31 (24)Severe (%)†1 (0.8)2 (1.6)Add-on to Metformin(26 Weeks)NESINA 25 mg + MetforminPlacebo + MetforminN =207 N =104 Overall (%) 0 3 (2.9) Severe (%)†0 0Add-on to Pioglitazone (± Metformin or Sulfonylurea) (26 Weeks) NESINA 25 mg + PioglitazonePlacebo + PioglitazoneN =199 N =97Overall (%) 14 (7.0)5 (5.2)Severe (%)† 0 1 (1)Compared to Glipizide (52 Weeks)NESINA 25 mgGlipizideN =222 N =219 Overall (%) 12 (5.4)57 (26)Severe (%)† 0 3 (1.4)Add on to Metformin (26 Weeks) NESINA 25 mgMetformin 500 mgtwice dailyN =112 N =109Overall (%) 2 (1.8) 2 (1.8) Severe (%)†00Add on to Metformin Compared to Glipizide (52 Weeks)NESINA 25 mg+ MetforminGlipizide+ MetforminN =877N=869Overall (%) 12 (1.4) 207 (23.8)Severe (%)†0 4(0.5)*Adverse reactions of hypoglycemia were based on all reports of symptomatic andasymptomatic hypoglycemia; a concurrent glucose measurement was not required; intent-to-treat population.†Severe events of hypoglycemia were defined as those events requiring medical assistance orexhibiting depressed level or loss of consciousness or seizure.Vital Signs132No clinically meaningful changes in vital signs or in electrocardiograms were observed 133in patients treated with NESINA.134Laboratory Tests135No clinically meaningful changes in hematology, serum chemistry, or urinalysis were 136observed in patients treated with NESINA.1376.2 Postmarketing Experience138The following adverse reactions have been identified during the postmarketing use of 139NESINA outside the United States. Because these reactions are reported voluntarily 140from a population of uncertain size, it is not always possible to reliably estimate their 141frequency or establish a causal relationship to drug exposure.142Hypersensitivity reactions including anaphylaxis, angioedema, rash, urticaria, and143severe cutaneous adverse reactions including Stevens-Johnson syndrome; hepatic 144enzyme elevations; fulminant hepatic failure; and acute pancreatitis.1457 DRUG INTERACTIONS146NESINA is primarily renally excreted. Cytochrome (CYP) P450-related metabolism is 147negligible. No significant drug-drug interactions were observed with the CYP-substrates 148or inhibitors tested, or with renally excreted drugs [see Clinical Pharmacology (12.3)]. 1498 USE IN SPECIFIC POPULATIONS1508.1 Pregnancy151Pregnancy Category B152No adequate or well-controlled studies in pregnant women have been conducted with 153NESINA. Based on animal data, NESINA is not predicted to increase the risk of154developmental abnormalities. Because animal reproduction studies are not always155156predictive of human risk and exposure, NESINA, like other antidiabetic medications, 157should be used during pregnancy only if clearly needed.158Alogliptin administered to pregnant rabbits and rats during the period of organogenesis 159was not teratogenic at doses of up to 200 and 500 mg/kg, or 149-times and 180-times, 160respectively, the clinical dose based on plasma drug exposure (AUC).161Doses of alogliptin up to 250 mg/kg (approximately 95-times clinical exposure based on 162AUC) given to pregnant rats from gestation day 6 to lactation day 20 did not harm the 163developing embryo or adversely affect growth and development of offspring.164Placental transfer of alogliptin into the fetus was observed following oral dosing to165pregnant rats.8.3 Nursing Mothers166167Alogliptin is secreted in the milk of lactating rats in a 2:1 ratio to plasma. It is not known 168whether alogliptin is excreted in human milk. Because many drugs are excreted inhuman milk, caution should be exercised when NESINA is administered to a nursing 169170woman.1718.4 Pediatric Use172Safety and effectiveness of NESINA in pediatric patients have not been established. 1738.5 Geriatric Use174Of the total number of patients (N=8507) in clinical safety and efficacy studies treated 175with NESINA, 2064 (24.3%) patients were 65 years and older and 341 (4%) patients 176were 75 years and older. No overall differences in safety or effectiveness were177observed between patients 65 years and over and younger patients. While this clinical 178experience has not identified differences in responses between the elderly and younger 179patients, greater sensitivity of some older individuals cannot be ruled out.1808.6 Hepatic Impairment181No dose adjustments are required in patients with mild to moderate hepatic impairment 182(Child-Pugh Grade A and B) based on insignificant change in systemic exposures (e.g., 183AUC) compared to subjects with normal hepatic function in a pharmacokinetic study. 184NESINA has not been studied in patients with severe hepatic impairment (Child-Pugh 185Grade C). Use caution when administering NESINA to patients with liver disease [see 186Warnings and Precautions (5.3)].18710 OVERDOSAGE188The highest doses of NESINA administered in clinical trials were single doses of 800 189mg to healthy subjects and doses of 400 mg once daily for 14 days to patients with type 1902 diabetes (equivalent to 32 times and 16 times the maximum recommended clinical 191dose of 25 mg, respectively). No serious adverse events were observed at these doses. 192In the event of an overdose, it is reasonable to institute the necessary clinical monitoring 193and supportive therapy as dictated by the patient's clinical status. Per clinical judgment, it may be reasonable to initiate removal of unabsorbed material from the gastrointestinal 194195tract.Alogliptin is minimally dialyzable; over a 3-hour hemodialysis session, approximately 7% 196 of the drug was removed. Therefore, hemodialysis is unlikely to be beneficial in an 197 overdose situation. It is not known if NESINA is dialyzable by peritoneal dialysis. 198 11 DESCRIPTION199 NESINA tablets contain the active ingredient alogliptin, which is a selective, orally-200 bioavailable inhibitor of the enzymatic activity of dipeptidyl peptidase-4 (DPP-4). 201 Chemically, alogliptin is prepared as a benzoate salt, which is identified as 2-({6-[(3R )-3-202 aminopiperidin-1-yl]-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H )-203 yl}methyl)benzonitrile monobenzoate. It has a molecular formula of C 18H 21N 5O 2•C 7H 6O 2 204 and a molecular weight of 461.51 daltons. The structural formula is:205NN(Z)CNOO2HO 2CH 3C •206 Alogliptin benzoate is a white to off-white, crystalline powder containing one asymmetric 207 carbon in the aminopiperidine moiety. It is soluble in dimethylsulfoxide, sparingly soluble 208 in water and methanol, slightly soluble in ethanol, and very slightly soluble in octanol 209 and isopropyl acetate.210 Each NESINA tablet contains 34 mg, 17 mg, or 8.5 mg alogliptin benzoate which is 211 equivalent to 25 mg, 12.5 mg, or 6.25 mg, respectively, of alogliptin and the following 212 inactive ingredients: mannitol, microcrystalline cellulose, hydroxypropyl cellulose,213 croscarmellose sodium, and magnesium stearate. In addition, the film-coating contains 214 the following inactive ingredients: hypromellose, titanium dioxide, ferric oxide (red or 215 yellow), and polyethylene glycol, and is marked with printing ink (Gray F1). 216 12 CLINICAL PHARMACOLOGY217 12.1 Mechanism of Action218 Increased concentrations of the incretin hormones such as glucagon-like peptide-1 219 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are released into the 220 bloodstream from the small intestine in response to meals. These hormones cause 221 insulin release from the pancreatic beta cells in a glucose-dependent manner but are 222 inactivated by the DPP-4 enzyme within minutes. GLP-1 also lowers glucagon secretion 223 from pancreatic alpha cells, reducing hepatic glucose production. In patients with type 2 224 diabetes, concentrations of GLP-1 are reduced but the insulin response to GLP-1 is 225 preserved. Alogliptin is a DPP-4 inhibitor that slows the inactivation of the incretin 226 hormones, thereby increasing their bloodstream concentrations and reducing fasting 227 and postprandial glucose concentrations in a glucose-dependent manner in patients228with type 2 diabetes mellitus. Alogliptin selectively binds to and inhibits DPP-4 but not 229230DPP-8 or DPP-9 activity in vitro at concentrations approximating therapeutic exposures. 23112.2 Pharmacodynamics232Single-dose administration of NESINA to healthy subjects resulted in a peak inhibition of233DPP-4 within 2 to 3 hours after dosing. The peak inhibition of DPP-4 exceeded 93%234across doses of 12.5 mg to 800 mg. Inhibition of DPP-4 remained above 80% at 24hours for doses greater than or equal to 25 mg. Peak and total exposure over 24 hours 235236to active GLP-1 were 3- to 4-fold greater with NESINA (at doses of 25 to 200 mg) than237placebo. In a 16-week, double-blind, placebo-controlled study, NESINA 25 mgdemonstrated decreases in postprandial glucagon while increasing postprandial active 238239GLP-1 levels compared to placebo over an 8-hour period following a standardized240meal. It is unclear how these findings relate to changes in overall glycemic control in241patients with type 2 diabetes mellitus. In this study, NESINA 25 mg demonstrated242decreases in 2-hour postprandial glucose compared to placebo (-30 mg/dL versus 17243mg/dL, respectively).244Multiple-dose administration of alogliptin to patients with type 2 diabetes also resulted in245a peak inhibition of DPP-4 within 1 to 2 hours and exceeded 93% across all doses (25246mg, 100 mg, and 400 mg) after a single dose and after 14 days of once-daily dosing. At247these doses of NESINA, inhibition of DPP-4 remained above 81% at 24 hours after 14days of dosing.248249Cardiac Electrophysiology250In a randomized, placebo-controlled, 4-arm, parallel-group study, 257 subjects were251administered either alogliptin 50 mg, alogliptin 400 mg, moxifloxacin 400 mg, or placebo252once-daily for a total of 7 days. No increase in QTc was observed with either dose of253alogliptin. At the 400 mg dose, peak alogliptin plasma concentrations were 19-fold254higher than the peak concentrations following the maximum recommended clinical dose255of 25 mg.25612.3 Pharmacokinetics257The pharmacokinetics of NESINA has been studied in healthy subjects and in patients258with type 2 diabetes. After administration of single, oral doses up to 800 mg in healthysubjects, the peak plasma alogliptin concentration (median T max) occurred 1 to 2 hours 259260after dosing. At the maximum recommended clinical dose of 25 mg, NESINA waseliminated with a mean terminal half-life (T1/2) of approximately 21 hours.261After multiple-dose administration up to 400 mg for 14 days in patients with type 2262263diabetes, accumulation of alogliptin was minimal with an increase in total (i.e., AUC) and264peak (i.e., C max) alogliptin exposures of 34% and 9%, respectively. Total and peak265exposure to alogliptin increased proportionally across single doses and multiple doses266of alogliptin ranging from 25 mg to 400 mg. The inter-subject coefficient of variation for267alogliptin AUC was 17%. The pharmacokinetics of NESINA was also shown to besimilar in healthy subjects and in patients with type 2 diabetes.268269Absorption270The absolute bioavailability of NESINA is approximately 100%. Administration ofNESINA with a high-fat meal results in no significant change in total and peak exposure 271272to alogliptin. NESINA may therefore be administered with or without food.Distribution273274Following a single, 12.5 mg intravenous infusion of alogliptin to healthy subjects, the 275volume of distribution during the terminal phase was 417 L, indicating that the drug is 276well distributed into tissues.277Alogliptin is 20% bound to plasma proteins.278MetabolismAlogliptin does not undergo extensive metabolism and 60% to 71% of the dose is279280excreted as unchanged drug in the urine.281Two minor metabolites were detected following administration of an oral dose of282[14C] alogliptin, N-demethylated, M-I (<1% of the parent compound), and N-acetylated 283alogliptin, M-II (<6% of the parent compound). M-I is an active metabolite and is an 284inhibitor of DPP-4 similar to the parent molecule; M-II does not display any inhibitory 285activity towards DPP-4 or other DPP-related enzymes. In vitro data indicate that286CYP2D6 and CYP3A4 contribute to the limited metabolism of alogliptin.287Alogliptin exists predominantly as the (R)-enantiomer (>99%) and undergoes little or no 288chiral conversion in vivo to the (S)-enantiomer. The (S)-enantiomer is not detectable at 289the 25 mg dose.290ExcretionThe primary route of elimination of [14C] alogliptin-derived radioactivity occurs via renal 291excretion (76%) with 13% recovered in the feces, achieving a total recovery of 89% of 292293the administered radioactive dose. The renal clearance of alogliptin (9.6 L/hr) indicates some active renal tubular secretion and systemic clearance was 14.0 L/hr.294295Specific Populations296Renal ImpairmentA single-dose, open-label study was conducted to evaluate the pharmacokinetics of 297298alogliptin 50 mg in patients with chronic renal impairment compared with healthy299subjects.300In patients with mild renal impairment (creatinine clearance (CrCl) ≥60 to <90 mL/min), 301an approximate 1.2-fold increase in plasma AUC of alogliptin was observed. Because 302increases of this magnitude are not considered clinically relevant, dose adjustment for 303patients with mild renal impairment is not recommended.304In patients with moderate renal impairment (CrCl ≥30 to <60 mL/min), an approximate 3052-fold increase in plasma AUC of alogliptin was observed. To maintain similar systemic 306exposures of NESINA to those with normal renal function, the recommended dose is 30712.5 mg once daily in patients with moderate renal impairment.308In patients with severe renal impairment (CrCl ≥15 to <30 mL/min) and end-stage renal 309disease (CrCl <15 mL/min or requiring dialysis), an approximate 3- and 4-fold increase 310in plasma AUC of alogliptin were observed, respectively. Dialysis removed311approximately 7% of the drug during a 3-hour dialysis session. NESINA may beadministered without regard to the timing of the dialysis. To maintain similar systemic 312313exposures of NESINA to those with normal renal function, the recommended dose is。
维格列汀与常见降糖药联合治疗2型糖尿病的研究进展

2020年12月 第17卷 第24期2型糖尿病(Type 2 Diabetes Mellitus,T2DM)以β细胞功能障碍[1-3]、较高的体质指数[4-6]、胰岛素抵抗指数[7-9]等为主要的表型特征。
在T2DM 的治疗指南中,建议将维格列汀等二肽基肽酶4抑制剂作为二线或三线药物[10]。
与其他降糖药相比,维格列汀等具有良好的药物耐受性和安全性。
那么,维格列汀与常见降糖药联合治疗T2DM的疗效与安全性如何呢?本文将综述相关研究成果。
1 维格列汀+二甲双胍联合治疗的相关研究1.1 维格列汀+二甲双胍联合用药与其他联合疗法的比较Peng等[11]以二甲双胍为基础,通过随机对照试验验证了格列美脲、吡格列酮、艾塞那肽、格列本脲、罗格列酮和维格列汀6种降血糖药物在T2DM中的不同疗效。
网络荟萃分析结果表明,这6种药物均能降低HbA1c水平。
与其他方案相比,艾塞那肽+二甲双胍联合治疗降低空腹血糖(Fasting Plasma Glucose,FPG)、空腹血浆胰岛素(Fasting Plasma Insulin,FPI)、总胆固醇(Total Cholesterol,TC)和稳态模型评估胰岛素抵抗指数(Homeostasis Model Assessment Insulin Resistance Index,HOMA-IR),且可提高高密度脂蛋白胆固醇(High-density Lipoprotein Cholesterol,HDL-C)水平;维格列汀+二甲双胍联合用药可降低FPI和低密度脂蛋白胆固醇(Low-density Lipoprotein Cholesterol,LDL-C)水平;格列本脲+二甲双胍联合用药可降低FPG水平,且提高HDL-C水平;格列美脲+二甲双胍联合用药可降低TC水平;罗格列酮+二甲双胍联合用药可降低LDL-C水平。
研究结果表明,艾塞那肽+二甲双胍和维格列汀+二甲双胍联合用药对T2DM有较好的疗效,二者均能改善胰岛素敏感性。
Tyrphostin_AG_879_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:TyrphostinAG879 is a tyrosine kinase inhibitor that inhibits TrKA phosphorylation, but not TrKB and TrKC.[1] also a ErbB2 kinase inhibitor, has at least 500–fold higher selectivity to ErbB2 (IC50 = 1 μmol/L) than EGFR (IC50 >500 μmol/L).target: TrKA [1], ErbB2 [2].IC 50: ErbB2 1 μmol/L [2].In vitro: TyrphostinAG879 significantly inhibit the A–type potassium currents in the cultured hippocampus neurons.[2]TyrphostinAG879 can reduce gephyrin puncta in GABAergic neurons and PSD–95 puncta in glutamatergic neurons where ErbB4expression was low. [3]In vivo: Treatment with TyrphostinAG879 in immunodepressed mice graft with leiomyosarcoma or promyelocytic leukemia cells result in dramatic reductions in tumor sizes. [1]References:[1]. Rende M et al. Role of nerve growth factor and its receptors in non–nervous cancer growth: efficacy of a tyrosine kinase inhibitor (AG879) andneutralizing antibodies antityrosine kinase receptor A and antinerve growth factor: an in–vitro and in–vivo study. Anticancer Drugs. 2006 Sep;17(8):929–41.[2]. Zhou Y et al. Blockade of EGFR and ErbB2 by the novel dual EGFR and ErbB2 tyrosine kinase inhibitor GW572016 sensitizes human colon carcinoma GEO cells to apoptosis. Cancer Res. 2006 Jan 1;66(1):404–11.[3]. Ting AK et al. Neuregulin 1 promotes excitatory synapse development and function in GABAergic interneurons. J Neurosci. 2011 Jan 5;31(1):15–25.Product Name:Tyrphostin AG 879Cat. No.:HY-20878CAS No.:148741-30-4Molecular Formula:C 18H 24N 2OS Molecular Weight:316.46Target:EGFR; EGFR; Trk Receptor Pathway:JAK/STAT Signaling; Protein Tyrosine Kinase/RTK; Protein Tyrosine Kinase/RTK Solubility:DMSO: ≥ 30 mg/mLCaution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@ Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
伊利替康 结构式 -回复

伊利替康结构式-回复伊利替康(Elidel)是一种局部用药治疗湿疹(atopic dermatitis)的药物。
它的主要成分是酯类化合物pimecrolimus。
伊利替康是一种非激素药物,被广泛使用于针对湿疹患者的长期治疗中,有效减轻患者的症状,并且具有较少的副作用。
本文将详细解析伊利替康的结构式,为读者提供更深入的了解。
首先我们来看一下伊利替康的结构式:酯类化合物pimecrolimus的化学名为(1S, 9S, 12S, 13E, 21R, 23R)-9, 21-dihydroxy-12-isopropyl-12-(2-methyl-1-oxobutoxy)-2,5,11,13,1 9-pentamethyl-8,21-dioxo-1H,20H-[1,4]oxazino[3,4]quinolizino[2,1 -a]isoquinoline-13-yl-2-methylpropanoate。
分析伊利替康的结构式,我们可以看到它由几个部分组成。
首先是氧杂吡咯(oxazino)的环结构,它是伊利替康的骨架。
吡咯环上连接着一个氮杂喹啉环(quinolizino)和一个异戊酸酯侧链。
在氮杂喹啉环上有一个吡美沙酮(pimecrolimus)基团。
伊利替康的结构中还有一些关键的官能团。
首先是两个羟基基团(hydroxyl group),它们分别连接在吡美沙酮基团的2号和21号碳原子上。
这些羟基基团在药效中扮演着重要的角色。
接下来是一个异丙基基团(isopropyl group),连接在吡美沙酮基团的12号碳原子上。
最后是一个异丁基基团(2-methyl-1-oxobutoxy),连接在吡美沙酮基团的12号羟基上。
伊利替康属于一类被称为calcineurin抑制剂(calcineurin inhibitors)的药物。
它通过抑制T细胞的活化和炎性细胞因子的释放,从而减轻过敏反应引起的炎症和瘙痒症状。
与传统的皮质类固醇类药物(如氢化可的松)相比,伊利替康作用于皮肤外层而不影响全身免疫系统,因此具有较少的副作用,尤其是在儿童和长期使用患者中。
T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects

DOI: 10.1126/scitranslmed.3002842, 95ra73 (2011);3 Sci Transl Med , et al.Michael Kalos and Can Establish Memory in Patients with Advanced Leukemia T Cells with Chimeric Antigen Receptors Have Potent Antitumor EffectsEditor's Summarythe potential for CAR-modified T cells to bring cancer therapy up to speed.treatment had complete remission of their leukemia. Although this is early in the clinical study, these results highlight scale with a second exposure to CLL cells. Indeed, two of the three CLL patients who underwent the CAR T cell CAR T cells persisted with a memory phenotype, which would allow them to respond more quickly and on a larger these CAR T cells expanded >1000-fold, persisted for more than 6 months, and eradicated CLL cells. Some of these allowing for much broader cellular targeting than is obtained with normal T cells. After transfer into three CLL patients,receptor could activate T cells in response to CD19 in the absence of major histocompatibility complex restriction, specific intracellular signaling domain. The resulting chimeric −specific costimulatory domain and a T cell −both a T cell bind in a restricted manner to the CD19 protein (which is found solely on normal B cells and plasma cells) attached to The CAR T cells used in this study expressed an antigen receptor that consists of antibody binding domains that as reflected by decreased numbers of B cells and plasma cells and the development of hypogammaglobulinemia.tumor cells after transfer into patients; they also mediated cancer remission. Innocent bystanders were also targeted, chronic lymphocytic leukemia (CLL) (a B cell cancer). The designer T cells not only expanded, persisted, and attacked modified T cells to express a chimeric antigen receptor (CAR) to yield so-called CAR T cells that specifically target . have genetically et al cells to the tumor and maintaining these cells in patients remains challenging. Now, Kalos harness the power of the immune system to fight cancers such as leukemia; however, targeting functional immune T to healthy tissues, such as infection or cancer, and then try to deter dangerous activity. Researchers have long sought As members of the body's police force, cells of the immune system vigilantly pursue bad actors that harmGo CAR-Ts in the Fast Lane/content/3/95/95ra73.full.html can be found at:and other services, including high-resolution figures,A complete electronic version of this article /content/suppl/2011/08/08/3.95.95ra73.DC1.htmlcan be found in the online version of this article at: Supplementary Material/about/permissions.dtl in whole or in part can be found at:article permission to reproduce this of this article or about obtaining reprints Information about obtaining last week in December, by the American Association for the Advancement of Science, 1200 New York Avenue (print ISSN 1946-6234; online ISSN 1946-6242) is published weekly, except the Science Translational Medicine o n F e b r u a r y 20, 2012s t m .s c i e n c e m a g .o r g D o w n l o a d e d f r o mL E U K E M I AT Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced LeukemiaMichael Kalos,1,2*Bruce L.Levine,1,2*David L.Porter,1,3Sharyn Katz,4Stephan A.Grupp,5,6 Adam Bagg,1,2Carl H.June1,2†Tumor immunotherapy with T lymphocytes,which can recognize and destroy malignant cells,has been limited by the ability to isolate and expand T cells restricted to tumor-associated antigens.Chimeric antigen receptors(CARs) composed of antibody binding domains connected to domains that activate T cells could overcome tolerance by allowing T cells to respond to cell surface antigens;however,to date,lymphocytes engineered to express CARs have demonstrated minimal in vivo expansion and antitumor effects in clinical trials.We report that CAR T cells that target CD19and contain a costimulatory domain from CD137and the T cell receptor z chain have potent non–cross-resistant clinical activity after infusion in three of three patients treated with advanced chronic lymphocytic leukemia(CLL).The engineered T cells expanded>1000-fold in vivo,trafficked to bone marrow,and continued to express functional CARs at high levels for at least6months.Evidence for on-target toxicity included B cell aplasia as well as decreased numbers of plasma cells and hypogammaglobulinemia.On average,each infused CAR-expressing T cell was calculated to eradicate at least1000CLL cells.Furthermore,a CD19-specific immune re-sponse was demonstrated in the blood and bone marrow,accompanied by complete remission,in two of three patients.Moreover,a portion of these cells persisted as memory CAR+T cells and retained anti-CD19effector functionality,indicating the potential of this major histocompatibility complex–independent approach for the ef-fective treatment of B cell malignancies.INTRODUCTIONUsing gene transfer technologies,T cells can be genetically modified to stably express antibody binding domains on their surface that con-fer novel antigen specificities that are major histocompatibility com-plex(MHC)–independent.Chimeric antigen receptors(CARs)are an application of this approach that combines an antigen recognition domain of a specific antibody with an intracellular domain of the CD3-z chain or Fc g RI protein into a single chimeric protein(1,2). Trials testing CARs are presently under way at a number of academic medical centers(3,4).In most cancers,tumor-specific antigens are not yet well defined,but in B cell malignancies,CD19is an attractive tumor target.Expression of CD19is restricted to normal and malig-nant B cells(5),and CD19is a widely accepted target to safely test CARs.Although CARs can trigger T cell activation in a manner sim-ilar to an endogenous T cell receptor,a major impediment to the clin-ical application of this technology to date has been the limited in vivo expansion of CAR+T cells,rapid disappearance of the cells after in-fusion,and disappointing clinical activity(4,6).CAR-mediated T cell responses may be further enhanced with ad-dition of costimulatory domains.In a preclinical model,we found that inclusion of the CD137(4-1BB)signaling domain significantly increased antitumor activity and in vivo persistence of CARs com-pared to inclusion of the CD3-z chain alone(7,8).To evaluate the safety and feasibility for adoptive transfer of T cells gene-modified to express such CARs,we initiated a pilot clinical trial using autologous T cells expressing an anti-CD19CAR including both CD3-z and the 4-1BB costimulatory domain(CART19cells)to target CD19+malig-nancies.To date,we have treated three patients under this protocol. Some of the findings from one of these patients are described in(9), which reports that this treatment results in tumor regression,CART19 cell persistence,and the unexpected occurrence of delayed tumor lysis syndrome.Here,we show that the CART19cells mediated potent clinical antitumor effects in all three patients treated.On average,each infused CAR T cell and/or their progeny eliminated more than 1000leukemia cells in vivo in patients with advanced chemotherapy-resistant chronic lymphocytic leukemia(CLL).CART19cells underwent robust in vivo T cell expansion,persisted at high levels for at least6 months in blood and bone marrow(BM),continued to express func-tional receptors on cells with a memory phenotype,and maintained anti-CD19effector function in vivo.RESULTSClinical protocolThree patients with advanced,chemotherapy-resistant CLL were enrolled in a pilot clinical trial for CART19cell therapy.Figure1presents a summary of the manufacturing process for the gene-modified T cells (A)and the clinical protocol design(B).All patients were extensively pretreated with various chemotherapy and biologic regimens(Table1). Two of the patients had p53-deficient CLL,a deletion that portends poor response to conventional therapy and rapid progression(10). Each of the patients had a large tumor burden after the preparative1Abramson Cancer Center,University of Pennsylvania,Philadelphia,PA19104,USA.2De-partment of Pathology and Laboratory Medicine,University of Pennsylvania,Philadelphia, PA19104,USA.3Department of Medicine,University of Pennsylvania,Philadelphia,PA 19104,USA.4Department of Radiology,University of Pennsylvania,Philadelphia,PA19104, USA.5Department of Pediatrics,University of Pennsylvania,Philadelphia,PA19104,USA. 6Division of Oncology,Children’s Hospital of Philadelphia,Philadelphia,PA19104,USA.*These authors contributed equally to this work.†To whom correspondence should be addressed.E-mail:cjune@ o n F e b r u a r y 2 0 , 2 0 1 2 s t m . s c i e n c e m a g . o r g D o w n l o a d e d f r o mchemotherapy,including extensive BM infiltration(40to95%)and lymphadenopathy;UPN02also had peripheral lymphocytosis.There was a low abundance of T cells in the apheresis products(2.29to4.46%) (table S1)as well as likely impaired T cell activation,as has been shown previously in CLL patients(11).Additional details of the cell manufac-turing and product characterization for the CART19cell preparation for each patient are shown in table S1.All patients were pretreated1to 4days before CART19cell infusions with lymphodepleting chemo-therapy(Table1).A split-dose cell infusion schedule was used to address potential safety concerns related to the evaluation of a previously untested CAR that incorporated the4-1BB costimulatory signaling domain. In vivo expansion,persistence,and BM trafficking of CART19cellsOur preclinical data in two animal models,including mice bearing xenografts of primary human precursor-B acute lymphoblastic leuke-mia(7,8),indicated that CAR+T cells that express a4-1BB signaling domain expanded after stimulation with anti-CD3/anti-CD28mono-clonal antibody–coated beads(12)and had improved persistence com-pared to CAR+T cells lacking4-1BB.We developed a quantitative polymerase chain reaction(qPCR)assay to enable quantitative tracking of CART19cells in blood and BM.CART19cells expanded and persisted in the blood of all patients for at least6months(Fig.2, A and B).Moreover,CART19cells expanded1000-to10,000-fold in the blood of patients UPN01and03during the first month after infusion,reaching peak frequencies of10to>95%of circulating white blood cells in UPN01and03(Fig.2C).The peak expansion levels coincided with onset of the clinical symptoms after infusion in UPN01 (day15)and UPN03(day23).Furthermore,after an initial decay,which can be modeled with first-order kinetics,the CART19cell numbers stabilized in all three patients from days90to180after infusion (Fig.2B).The CART19cells also trafficked to the BM in all patients, albeit in5-to10-fold fewer numbers than observed in blood(Fig.2D). CART19cells had a log-linear decay in the BM in UPN01and03, with a disappearance half-life of~35days.Induction of specific immune responses in the peripheral blood and BM compartments after CART19infusion Peripheral blood(PB)and BM serum samples from all patients were collected and batch-analyzed to quantitatively determine cytokine levels.A panel of30cytokines,chemokines,and other soluble factors were assessed for potential toxicities and to provide evidence of CART19cell function.The full data set for all of the cytokines measured in each of the three patients through the date of thisapheresisSeed in gas-permeable bags.Transduction w/αCD19-41BBζvectorVector washout.Culture in gas-permeable bagsCulture in WAV EbioreactorHarvest, wash, concentrateCryopreserve final product ininfusible cryomediaCD3/28-positive selection ofT cells with anti-CD3/anti-CD28 mAb-coated magneticbeadsDay 0Day 0-1Day 3Day 5Harvest day(10 ±2)ABManufacture/cryopreservationFig.1.Schematic representation of the gene transfer vector and trans-gene,gene-modified T cell manufacturing,and clinical protocol design.(A)T cell manufacturing.Autologous cells were obtained via leukapher-esis,and T cells were enriched by mononuclear cell elutriation,washed, and expanded by addition of anti-CD3/CD28–coated paramagnetic beads for positive selection and activation of T cells.Residual leukemic cells were depleted.The lentiviral vector was added at the time of cell activation and was washed out on day3after culture initiation.Cells were expanded on a rocking platform device(WAVE Bioreactor System) for8to12days.On the final day of culture,the beads were removed by passage over a magnetic field and the CART19cells were harvested and cryopreserved in infusible medium.mAb,monoclonal antibody.(B)Clin-ical protocol design.Patients were given lymphodepleting chemo-therapy as described,followed by CART19infusion#1by intravenous gravity flow drip over a period of15to20min.The infusion was given using a split-dose approach over3days(10,30,and60%)beginning1to5days after completion of chemotherapy.Endpoint assays were conducted on study week4.At the conclusion of active monitoring, subjects were transferred to a destination protocol for long-term follow-up as per FDA guidance.onFebruary2,212stm.sciencemag.orgmpublication is presented in tables S2to S5.Of the analytes tested, 11had a threefold or more change from baseline,including four cytokines[interleukin-6(IL-6),interferon-g(IFN-g),IL-8,and IL-10], five chemokines[macrophage inflammatory protein–1a(MIP-1a), MIP-1b,monocyte chemotactic peptide–1(MCP-1),CXC chemokine ligand9(CXCL9),and CXCL10],and the soluble receptors IL-1R a and IL-2R a;IFN-g had the largest relative change from baseline (Fig.3).The peak time of cytokine elevation in UPN01and03 correlated temporally with both the previously described clinical symptoms and the peak levels of CART19cells detected in the blood for each patient.Notably,cytokine modulations were transient,and levels reverted to baseline relatively rapidly despite continued func-Table1.Patient demographics and response.CR,complete response;PR,partial response;N/A,not available.Subject UPNAge/sexkaryotypePrevious therapiesCLL tumor burden at baselineTotaldoseof CART19(cells/kg)Response day+30(duration)BM(study day)‡Blood(studyday)‡Nodes/spleen(study day)‡0165/Mnormal Fludarabine×four cycles(2002)Hypercellular70%CLL N/A 6.2×1011to1.0×1012CLL cells(day−37)1.1×109(1.6×107/kg)CR(11+months)Rituximab/fludarabine×four cycles(2005)2.4×1012CLL cells(day−14)Alemtuzumab×12weeks(2006)1.7×1012CLL cells(day−1)Rituximab(two courses,2008to2009)R-CVP×two cycles(2009)Lenalidomide(2009)PCR×two cycles(5/18/2010to6/18/2010)Bendamustine×one cycle(7/31/10to8/1/10)pre-CART190277/M del(17)(p13)*Alemtuzumab×16weeks(6/2007)Hypercellular>95%CLL2.75×1011CLL cells(day−1)1.2×1012to2.0×1012CLL cells(day−24)5.8×108(1.0×107/kg)PR(7months)Alemtuzumab×18weeks(3/2009)3.2×1012CLL cells(day−47)Bendamustine/rituximab:7/1/2010(cycle1)7/28/2010(cycle2)8/26/2010(cycle3)pre-CART190364/M del(17)(p13)†R-Fludarabine×twocycles(2002)Hypercellular40%CLLN/A 3.3×1011to5.5×1011CLL cells(day−10)1.4×107(1.46×105/kg)CR(10+months)R-Fludarabine×four cycles(10/06to1/07)8.8×1011CLL cells(day−1)R-Bendamustine×one cycle(2/09)Bendamustine×three cycles(3/09to5/09)Alemtuzumab×11weeks(12/09to3/10)Pentostatin/cyclophosphamide(9/10/10)pre-CART19*UPN02karyotype[International System for Human Cytogenetic Nomenclature(ISCN)]:45,XY,del(1)(q25),+del(1)(p13),t(2;20)(p13;q11.2),t(3;5)(p13;q35),add(9)(p22),?del(13)(q14q34),-14,del(17) (p13)[cp24].†UPN03karyotype(ISCN):46,XY,del(17)(p12)[18]/44~46,idem,der(17)t(17;21)(p11.2;q11.2)[cp4]/40~45,XY,-17[cp3].‡See the Supplementary Material for methods of tumor burden determination.o n F e b r u a r y 2 0 , 2 0 1 2 s t m . s c i e n c e m a g . o r g D o w n l o a d e d f r o mtional persistence of CART19cells.Only modest changes in cytokine levels were noted in UPN 02,possibly as a result of corticosteroid treatment.We also noted a robust induction of cytokine secretion in the supernatants from BM aspirates of UPN 03(Fig.3D and table S5).Although a pretreatment marrow sample was not available,compared to the late time point (+176),we also observed elevated levels for a number of factors in the +28marrow sample for UPN 01including IL-6,IL-8,IL-2R,and CXCL9;in contrast,compared to the pretreatment marrow sample,no elevation in cytokines was de-tected in the +31day sample for UPN 02(table S5).One of the preclinical rationales for developing CAR +T cells with 4-1BB signaling domains was a projected reduced propensity to trigger IL-2and tumor necrosis factor –a (TNF-a )secretion compared to CAR +T cells with CD28signaling domains (7);indeed,elevated amounts of soluble IL-2and TNF-a were not detected in the serum of the patients.Lower levels of these cytokines may be related to sustained clinical ac-tivity:Previous studies have shown that CAR +T cells are potentially suppressed by regulatory T cells (13),which can be elicited by either CARs that secrete substantial amounts of IL-2or by the provision of exogenous IL-2after infusion.Moreover,the TNF-a is complicit in cy-tokine storm –related effects in patients,which are absent here.Prolonged receptor expression and establishment of a population of memory CART19cells in bloodA central question in CAR-mediated cancer immunotherapy is whether optimized cell manufacturing and costimulation domains will enhance the persistence of genetically modified T cells and permit the establishment of CAR +memory T cells in patients.Previous studies have not demonstrated robust expansion,prolonged persist-ence,or functional expression of CARs on T cells after infusion (14–17).The high persistence of CART19cells that we observed at late time points for UPN 03facilitated a more detailed phenotypic analysis ofpersisting cells.Flow cytometric analysis of samples from both blood and BM 169days after infusion revealed the presence of CAR19-expressing cells in UPN 03as well as an absence of B cells (fig.S1,Aand B).These CAR +cells persisted in allthree patients beyond 4months,as shown by qPCR (Fig.2).The in vivo frequency of CAR +cells by flow cytometry closely matched the values obtained from the PCR assay for the CAR19transgene.CAR expression was also detected on the surface of 5.7and 1.7%of T cells in the blood of patient UPN 01on days 71and286after infusion (fig.S2).We next used polychromatic flow cy-tometry to perform detailed studies and further characterize the expression,phe-notype,and function of CART19cells in UPN 03using an anti-CAR idiotype anti-body (MDA-647)and the gating strategy shown in fig.S3.We observed differencesin the expression of memory and activa-tion markers in both CD4+and CD8+T cells based on CAR19expression.In the CD4+compartment,at day 56,CART19cells were characterized by auniform lack of CCR7,a predominance of CD27+/CD28+/PD-1+cells distributed within both CD57+and CD57−compart-ments,and an essential absence of CD25and CD127expression,the latter two markers defining regulatory CD4+T cells (18)(Fig.4A).In contrast,CAR −cells at this time point were heterogeneous in CCR7,CD27,and PD-1expression;expressed CD127;and also contained a substantialCD25+/CD127−population.By day 169,although CD28expression remained uni-formly positive in all CART19CD4+cells,a fraction of the CART19CD4+cells had acquired a central memory phenotype,withA CB Day (after infusion)110100100010000100000Day (after infusion)T o t a l c e l l s i n c i r c u l a t i o nDay (after infusion)110100100010000D C o p i e s /µg g D N A% W B CDay (after infusion)C o p i e s /µg gD N AWBC and CART 19: Blood CART 19: Marrow 10101010101010Fig.2.Sustained in vivo expansion and persistence in blood and marrow of CART19cells.(A to D )qPCRanalysis was performed on DNA isolated from whole blood (A to C)or bone marrow (BM)(D)samples obtained from UPN 01,UPN 02,and UPN 03to detect and quantify CAR19sequences.The frequency of CART19cells is shown as average transgene copies (A),total calculated CART19cells in circulation (B),or as a fraction of circulating white blood cells (WBCs)(C).(A)Copies CAR19/microgram DNA is calculated as de-scribed in Materials and Methods.(B)The total number of lymphocytes (total normal and CLL cells)versus total CART19+cells in circulation is plotted for all three subjects using the absolute lymphocyte count from complete blood count values and assuming a 5.0-liter volume of peripheral blood.(C)%WBC is calculated as described in Materials and Methods.(D)Bulk qPCR analysis of marrow to quantify CART19sequences.The data from patient UPN 03in (A,C,and D)has been published in (9)and is reprinted here with permission.Each data point represents the average of triplicate measurements on 100to 200ng of genomic DNA,withmaximal percent coefficient of variation (CV)less than 1.56%.Pass/fail parameters for the assay included preestablished ranges for slope and efficiency of amplification,and amplification of a reference sample.The lower limit of quantification for the assay established by the standard curve range was two copies of transgene per microgram of genomic DNA;sample values below that number are considered estimates and presented if at least two of three replicates generated a C t value with percent CV for the values 15%.CART19cells were infused at days 0,1,and 2for UPN 01and 03and at days 0,1,2,and 11for UPN 02. o n F e b r u a r y 20, 2012s t m .s c i e n c e m a g .o r g D o w n l o a d e d f r o mCCR7expression,a higher percentage of CD27−cells,the appearance of a PD-1−subset,and acquisition of CD127expression.At day 169,CAR −cells remained reasonably consistent with their day 56counterparts,with the exception of a reduction in CD27expression and a decrease in the percentage of CD25+/CD127−cells.In the CD8+compartment,at day 56,CART19CD8+cells displayed primarily an effector memory phenotype (CCR7−,CD27−,CD28−),con-sistent with prolonged and robust exposure to antigen (Fig.4B).In con-trast,CAR −CD8+T cells consisted of mixtures of effector and central memory cells,with CCR7expression in a subset of cells,and substantial numbers of cells in the CD27+/CD28−and CD27+/CD28+fractions.Al-though a large percentage of both CART19and CAR −cell populations expressed CD57,a marker associated with memory T cells with high cytolytic potential (19),this molecule was uniformly coexpressed with PD-1in the CART19cells,a possible reflection of the extensive replicative history of these cells.In contrast to the CAR −cell population,the entirety of the CART19CD8+population lacked expression of both CD25and CD127,markers associated with T cell activation and the development of functional memory cells (20).By day 169,although the phenotype of the CAR −cell population remained similar to the day 56cells,the CART19population had evolved to contain a population with features of central memory cells,notably expression of CCR7and higher levels of CD27and CD28,as well as cells that were PD-1−,CD57−,and CD127+.Effector function of CART19cells after 6months in blood In addition to a lack of long-term persistence,a limitation of previous trials with CAR +T cells has been the rapid loss of functional activity of the infused T cells in vivo.The high level of CART19cell persist-ence and surface expression of the CAR19molecule in UPN 03provided the opportunity to directly test anti-CD19–specific effector functions in cells recovered from cryopreserved PB samples.Pe-ripheral blood mononuclear cells (PBMCs)from UPN 03were cultured with target cells that either did or did not express CD19(Fig.4C and fig.S3).Robust CD19-specific effector function of CART19cells was observed by the specific degranulation of CART19cells against CD19+but not CD19−target cells,as assessed by surface CD107a expression.Notably,exposure of the CART19population to CD19+targets induced a rapid internalization of surface CAR19(see fig.S3for constitutive surface expression of CAR19in the same ef-fector cells in standard flow cytometric staining).The presence of costimulatory molecules on target cells was not required for trigger-ing CART19cell degranulation because the NALM-6line,which was used as a target in these studies,does not express CD80or CD86(21).Effector function was evident at day 56after infusion and was re-tained at day 169(Fig.4C).Robust effector function of CAR +and CAR −T cells could also be demonstrated by pharmacologic stimula-tion with phorbol 12-myristate 13-acetate (PMA)and ionomycin.ADay (after infusion)S e r u m c y t o k i n e (f o l d c h a n g e f r o m b a s e l i n e )(f o l d c h a n g e f r o m b a s e l i n e )S e r u m c y t o k i n e (f o l d c h a n g e f r o m b a s e l i n e )BCDay (after infusion)D Day (after infusion)C o n c e n t r a t i o n (p g /m l )αIL-6 IFN-γCXCL10MIP-1βMCP-1CXCL9IL-2R αIL-8 IL-10MIP-1αFig.3.Serum and BM cytokines before and afterCART19cell infusion.(A to C )Longitudinal measure-ments of changes in serum cytokines,chemokines,and receptors in UPN 01(A),UPN 02(B),and UPN 03(C)on the in-dicated day after CART19cell infusion.(D )Serial assessments of the same analytes in the BM from UPN 03.Analytes with agreater than or equal to threefold change are indicated and plotted as relative change from baseline (A to C)or as absolute values (D).In (C)and (D),a subset of the cytokine data (IFN-g ,CXCL10,CXCL9,IL-2R a ,and IL-6)from UPN 03have been pub-lished in (9)and are reprinted here with permission.Absolutevalues for each analyte at each time point were derived from a recombinant protein-based standard curve over a threefold eight-point dilution series,with upper and lower limits of quantification determined by the 80to 120%observed/expected cutoff valuesfor the standard curves.Each sample was evaluated in duplicate with average values calculated and percent CV in most cases less than 10%.To accommodate consolidated data presentation in the context of the wide range for the absolute values,data are presented as fold change over the baseline value for each analyte.In cases where baseline values were not detectable,half of the lowest standard curve value was used as the baseline value.Standard curve ranges for analytes and baseline (day 0)values (listed in parentheses sequentially for UPN 01,02,and 03),all in pg/ml:IL-1R a :35.5to 29,318(689,301,and 287);IL-6:2.7to 4572(7,10.1,and 8.7);IFN-g :11.2to 23,972(2.8,not detected,and 4.2);CXCL10:2.1to 5319(481,115,and 287);MIP-1b :3.3to 7233(99.7,371,and 174);MCP-1:4.8to 3600(403,560,and 828);CXCL9:48.2to 3700(1412,126,and 177);IL-2R a :13.4to 34,210(4319,9477,and 610);IL-8:2.4to 5278(15.3,14.5,and 14.6);IL-10:6.7to 13,874(8.5,5.4,and 0.7);MIP-1a :7.1to 13,778(57.6,57.3,and 48.1).o n F e b r u a r y 20, 2012s t m .s c i e n c e m a g .o r g D o w n l o a d e d f r o mProfound antitumor clinical activity of CART19cellsThere were no significant toxicities observed during the4days after the infusion in any patient other than transient febrile reactions. However,all patients subsequently developed significant clinical and laboratory toxicities between days7and21after the first infusion. With the exception of B cell aplasia,these toxicities were short-term and reversible.Of the three patients treated to date,there are two complete responses and one partial response lasting greater than8months after CART19infusion according to standard criteria(22).Details of past medical history and response to therapy are described in Table1. The clinical course of UPN03has been described in detail(9).In brief,patient UPN02was treated with two cycles of bendamus-tine with rituximab,resulting in stable disease;he received a third dose of bendamustine as lymphodepleting chemotherapy before CART19 cell infusion.After CART19infusion,and coincident with the onset of high fevers,he had rapid clearance of the p53-deficient CLL cells from his PB(Fig.5A)and a partial reduction of adenopathy.He de-veloped fevers to40°C,rigors,and dyspnea requiring a24-hour hos-pitalization on day11after the first infusion and on the day of his second CART19cell boost.Fevers and constitutional symptoms per-sisted,and on day15,he had transient cardiac dysfunction;all symp-toms resolved after corticosteroid therapy was initiated on day18.His BM showed persistent extensive infiltration of CLL1month after therapy despite marked PB cytoreduction.He remained asymptomatic at the time of publication.Patient UPN01developed a febrile syndrome,with rigors and transient hypotension beginning10days after infusion.The fevers persisted for about2weeks and resolved;he has had no further consti-tutional symptoms.He achieved a rapid and complete response(Fig.5, B and C).Between1and6months after infusion,no circulating CLL cells were detected in the blood by deep sequencing(Table2).His BM at1,3,and6months after CART19cell infusions showed sustainedA1.40.67.890.223.58.02939.5CCR7CD28CD127CCR7CD28CD127C0.999.187.97.10.34.65.165.726.72.60.746.551.71.265.814.34.1715.71.81236.949.49.317.644.129CD4Day 169CCR7CD28CD127CCR7CD28CD127CD45RACD27CD25CD45RACD27CD25CD45RACD27CD25CD45RACD2744.017.34.234.4CD57CD5731.932.111.424.648.218.55.128.2CD57CD57PD-1PD-1PD-1PD-150.111.01127.927.841.716.613.874.719.81.73.8CD57CD57CD57CD5735.933.610.220.352.738.94.63.936.09.814.239.974.9230.61.5CD57CD57CD57CD57CD27CD28CD27CD28CD27CD28CD27CD28CD2565.00.60.433.719.959.411.39.231.551.66.610.030.147.910.811.12.459.635.42.639.59.28.842.5CCR7CD28CD127CD57CD57CD5779.516.70.90.36.833.234.925.110.928.041.319.82542.926.16.014.244.524.516.93.7 2.025.468.875.623.70.20.56.040.339.314.316.328.831.922.937.242.318.12.414.338.222.225.36.0 4.530.359.296.6 3.414.67.41464.15.115.760.418.89.411.464.614.60.60.12.596.818.473.03.05.697.10.70.062.111.2 3.111.873.95.58.052.833.77.8 6.454.231.635.559.21.33.927.264.316.779.20.200.898.97.500.891.73.60.10.395.97.60.40.191.90.10.213.885.90.20.312.387.2C00.284.715.100.969.329.80.10.21188.60.20.56.692.74.00.21.194.73.396.6Fig.4.Prolonged surface CAR19expressionand establishment of functional memoryCART19cells in vivo.(A and B)T cell immuno-phenotyping of CD4+(A)and CD8+(B)T cellsubsets.Frozen peripheral blood(PB)samplesfrom UPN03obtained at days56and169afterT cell infusion were subjected to multipara-metric immunophenotyping for expression ofmarkers of T cell memory,activation,and ex-haustion;data are displayed after biexponentialtransformation for objective visualization ofevents.(C)Functional competence of persistingCAR cells.Frozen PB samples from UPN03ob-tained at days56and169after T cell infusion were evaluated directly ex vivo for the ability to recognize CD19-expressing target cells using CD107 degranulation assays.Presented data are for the CD8+gated population.The gating strategies for these figures are presented in fig.S2.onFebruary2,212stm.sciencemag.orgDownloadedfrom。
Gliclazide 说明书

to which dose needs to be adjusted.
Remember the time-action profile of Gliclazide:
If BG readings are consistently high from breakfast to the evening meal consider increasing the morning dose.
The starting dose is usually 80mgs with a meal but consider starting with 40mgs if there are any particular concerns about hypoglycaemia. A blood glucose profile will help you identify whether to initiate a dose with breakfast or the evening meal.
If BG readings are high after evening meal and on waking consider increasing the evening dose.
Dose Increments:
Increments of 40-80mgs can be made depending on how high the BG levels are:
levels or hypoglycaemia- so to titrate Gliclazide it is more appropriate to use a BG profile.
The BG profile and information regarding the patient’s activity and diet allows an informed decision to be made as
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Product Name:
Gliclazide CAS No.:
21187-98-4Cat. No.:
HY-B0753Product Data Sheet
MWt:
323.41Formula:
C15H21N3O3S Purity :>98%
Solubility:
DMSO 65 mg/mL; Water <1 mg/mL
y Mechanisms:Biological Activity:
Gliclazide is a whole cell beta cell ATP sensitive potassium currents blocker with an IC50of 184nM Pathways:Membrane Tranporter/Ion Channel; Target:Potassium
Channel g ;g Gliclazide is a whole-cell beta-cell ATP-sensitive potassium currents blocker with an IC50 of 184 nM.
Target: Potassium Channel gliclazide further characterize its mechanism of hypoglycemic effect: the observed improvements in insulin sensitivity and in GLUT4 translocation indicate that gliclazide counters the hydrogen peroxide-induced insulin resistance in 3T3L1 adipocytes and also would further augment the
hypoglycemic effect of this drug as insulinotropic sulfonylurea [1]. Gliclazide blocked whole-cell beta-cell KATP currents with an IC50 of 184 +/- 30 nmol/l (n = 6-10) but was much less effective in
cardiac and smooth muscle (IC50s of 19.5 +/- 5.4 micromol/l (n = 6-12) and 37.9 +/- 1.0 micromol/l (n =5-10),respectively).In all three tissues,the action of the drug on whole-cell KATP currents was References:
[1]. Shimoyama, T., et al., Gliclazide protects 3T3L1 adipocytes against insulin resistance induced
by hydrogen peroxide with restoration of GLUT4 translocation. Metabolism, 2006. 55(6): p. 722-30.[2]. Lawrence, C.L., et al., Gliclazide produces high-affinity block of KATP channels in mouse
(n 510), respectively). In all three tissues, the action of the drug on whole cell KATP currents was rapidly reversible. In inside-out patches on beta-cells, gliclazide...
isolated pancreatic beta cells but not rat heart or arterial smooth muscle cells. Diabetologia, 2001.
44(8): p. 1019-25.Caution: Not fully tested. For research purposes only
Medchemexpress LLC
18W i l k i n s o n W a y , P r i n c e t o n , N J 08540,U S A
E m a i l : i n f o @m e d c h e m e x p r e s s .c o m W e b : w w w .m e d c h e m e x p r e s s .c o m。