临床试验核查项目一览表【模板】
最新药物临床试验检查内容表

适当的医学、药学或相关专业学历并经过必要的培训
监查员人数:医学专业人药学专业人
GCP培训合格,并熟悉药品临床研究审批管理有关法律、法规
参加过GCP或监查员培训的人数:人
熟悉试验药品临床前和临床方面的信息以及临床试验方案
按照GCP(第四十七条)要求制定了标准操作规程
制定日期:年月日第版
每次访视后均向申办者提交临床试验监查的书面报告
根据临床试验方案制定了统计分析计划书
统计分析计划:有□无□ 定稿日期:年月日
与临床试验内容是否一致: 是□ 否□
临床试验的中期分析,应说明理由并有确定的操作程序
中期分析Байду номын сангаас有□ 无□ 时间:年月日
试验方案中有无规定: 有□ 无□有无揭盲:有□ 无□
临床试验中受试者分配必须符合随机化原则
随机分配表:有□无□
接受申办者派遣的监察员或稽查员的监察和稽查
接受SDA的稽查和视察
六、申办者职责
为研究者提供手册,其内容包括按规定应有的试验用药的资料和数据
是
否
不适用
在获得SFDA批准及伦理委员会批准后按方案和GCP组织临床试验
与研究者共同设计临床试验方案并以合同方式确定双方的职责和分工
向研究者提供易于识别及有正确编码的试验药品、对照品和安慰剂
随机分组方式:药物随机编码□随机分组信件□
双盲试验在其试验方案中,应确定保持盲态的方法和保护受试者的措施
药物编盲记录:有□无□应急信件:有□无□
数据和安全监察委员会:有□无□
紧急情况下对个别受试者的破盲,在病例报告表上述明理由
试药期间有无紧急破盲:有□无□破盲病例数:
具有数据管理的系统化程序,且所有实际操作步骤均被记录在案
临床试验结题核查表(专业组用)
第4版
附件 4
临床试验结题核查表(专业组用)
项目名称
项目科室 方案号
批件号
项目负责人 注册分类
申办方/CRO/SMO
分期组长单位Fra bibliotek协议例数 筛选数 筛选失败号
入组数
实际完成数 脱落号
脱落数
退出数
ICF 份数 退出号
CRF 份数
发生 AE 号
项目启动会时间
第 1 例知情同意时间
注: 1. 此表请双面打印于一张纸上
2.项目研究结束,专业组必须进行此结题质控
3.如不适用表中内容,请在备注中标明 NA;如果勾选"否",请写明原因
上海市第一人民医院国家药物临床试验机构
第4版
有生物样本采集、保存、运输记录(含温度记录)
知情、入组、用药、不良事件均完整记录
是否漏记合并用药或住院等
文件记录 和修改
最后 1 例研究结束时间
检查要点
问题
是
否
试验方案
申办者、研究者已签名或盖章 是否最新版且有伦理批准
研 究 者 履 历 研究者均 GCP 培训 及相关文件 研究者均有执业医师资格且被正确授权(关注签名样张一致性)
研究者均参加启动会或补充培训
是否所有入组受试者都签名+日期
是否为受试者本人签署(关注:1.多份签字均类似 2.与研究者签字类似
合并用药记录完整(有明显伴随疾病却没有合并用药) 未合并使用禁用药物
必要时查看 His 是否漏记
实施
方案规定的随访及检查无遗漏或者超窗 检验、检查单均及时打印并且完整
检查单都已签名+日期 有无脱落?说明原因
临床试验信息汇总表

附件415.(2.5.P.)临床试验信息汇总表本套格式申报资料主要适用于以药动学参数为终点评价指标的生物等效性试验资料的申报。
对于需要进行临床试验的特殊制剂等,信息汇总表应参照注册分类3的格式撰写。
基于目前的认识制定本套格式申报资料,可根据化合物的性质和试验特点做适当优化和补充。
须按时间顺序提交所进行的每项生物等效性试验的相关信息。
应以下级目录形式逐个研究项目填写“生物等效性试验信息汇总表”(“如2.5.P.1空腹生物等效性试验;2.5.P.2餐后生物等效性试验”),共用内容集中填写于首个试验项目中,其余项目参见首个试验,并注明标题编号。
2.5.P.1 XX药的空腹生物等效性试验表1(注:依次编号,下同):基本信息表xx:试验药物信息表xx: 处方信息(举例)应提交所有规格的处方信息表xx:体外溶出试验概要精选资料,欢迎下载应提交受试制剂和参比制剂所有规格的溶出试验结果。
精选资料,欢迎下载表xx: 试验设计表xx: 试验报告及生物样品储存信息*: 长期保存稳定性研究应在保存温度范围的上限条件下进行表xx: 完成生物等效性试验的受试者信息(举例)表xx:不良事件发生率统计(举例)表xx: 受试者退出试验情况* 应说明退出时间、所在给药组(受试制剂或参比制剂),退出原因(如果退出原因不是个人原因)等。
表xx: 方案偏离情况表xx: 试验结果表xx:调整评价标准的生物等效性试验数据汇总表(若适用)*经对数转化后的药动学参数表xx:生物样品分析方法学验证结果(举例)上述信息表格请按每个待测物分别提交。
表xx:生物样品分析的标准曲线和质控数据概要*(举例)*如果有多个待测物,应分别提交上述表格表xx :试验样品重复测定情况*重复计算:指样品重复测定后,采用复测值进行计算的情况。
表xx:餐后生物等效性试验中试验餐的组成表xx:浓度数据汇总表(适用于采用体内药代法进行的生物等效性试验)(代码的定义详见后续表格)a)受试制剂组的药物浓度精选资料,欢迎下载精选资料,欢迎下载b)参比制剂组的药物浓度精选资料,欢迎下载精选资料,欢迎下载表xx:药动学参数汇总表(表格中代码的定义详见后续表格)(举例)a)受试制剂药动学参数精选资料,欢迎下载*达峰时间T max的统计值以中位数表示。
临床试验项目结题数据核查表
2.5.3*试验用药品/疫苗接收、保存、发放、使用、回收原始记录的数量一致,核实并记录各环节数量的误差。
2.5.4试验用药品/疫苗运输和储存过程中的温度均符合要求。
2.5.5试验用药品/疫苗批号与药检报告、总结报告等资料一致。
2.4.3 CRF中偏离和/或违背方案相关记录和处理与实际发生例数(门诊/住院病历)及总结报告一致;核实并记录漏填的例数。
2.4.4*CRF中发生的SAE处理和报告记录,与原始病历(住院病历、门诊/研究病历)、总结报告一致;核实并记录瞒填的例数。
2.5
试验用药品的管理过程与记录:
2.5.1*试验用药品/疫苗的来源和药检具有合法性(参比制剂的合法来源证明为药检报告、药品说明书等)。
2.1.5受试者在方案规定的时间内不得重复参加临床试验。
2.2
知情同意书的签署与试验过程的真实完整性:
2.2.1已签署的ICF数量与总结报告中的筛选和入选病例数一致。
2.2.2所有ICF签署的内容完整、规范
2.2.3知情同意签署时间不得早于伦理批准时间,记录违规例数。
2.2.4*知情同意书按规定由受试者本人或其法定代理人签署(必要时,多方核实受试者参加该项试验的实际情况)。
1.2.2委员表决票及审查结论保存完整且与伦理审批件一致。
1.3
临床试验合同经费必须覆盖临床试验所有开支(含检测、受试者营养/交通费补贴、研究者观察费等)。
1.4
申办者/合同研究组织(CRO)按照药物临床试验管理规范(GCP)原则、方案及合同承担相应职责的文件和记录(如合同或方案中规定的项目质量管理责任及监查、稽查相关记录等)。
临床试验核查项目一览表【模板】

完成试验受试者编码目录
受试者各信息与筛选入组表中的信息一致
3
其他类文件
以下文档质控时若无可不查
3.1
分中心小结表
本中心的信息如入组数量、SAE数量等总结无误
3.2
统计分析报告
查看各方面信息是否准确无误
3.3
总结报告
查看各方面信息是否准确无误
3.4
至伦理委员会的结题报告
查看各方面信息是否准确无误
CFDA批件号:______
CFDA批准日期:______
试验应在批件有效期内前开始实施(目前为3年内)
1.2
伦理委员会相关文件
本院伦理委员会共______次会审,______次快审。
首次伦理批准时间:______
有本院伦理委员会(及组长单位)历次批件、委员会成员表等复印件
各批件信息准确
1.3
研究者手册
4
4.1
原始医疗记录(包括住院、门诊病历、化验单、检查单、患者日志、医生科研病历记录、AE/合并用药表格等等)
1、病历中应有参与该临床试验的记录
2、受试者原始文件中相关信息应符合入选/排除标准,查看各种检查/检验结果(项目是否有遗漏,是否在窗口内,结果是否符合方案要求)
3、受试者入组、随机、访视、合并用药、不良事件均需准确、完整记录(查看是否由授权的医师进行;各种原始记录是否有不一致)
实际完成病例数
第一例受试签署ICF时间
最后一例受试者完成时间
试验过程中发生SAE发生例数
2.核查记录(因各项目不同,包括但不限于以下文件)
序号
文件名称
检查要点
检查结果
备注
有
无
NA
1
研究者文件夹
药物临床试验数据核查表

药物临床试验数据核查表(浙大一院)药物名称:注册分类:申办者及联系方式:CRO 及联系方式:含各方在临床试验项目中职责落实)1.1* 临床试验单位承担药物临床试验的条件与合规性:1.1.1 临床试验须在具有药物临床试验机构资格的医院内进行 (含具有一次性临床试验机构资格认定的批件),落实临床试验条件是否支持试验项目实际的实施过程。
1.1.2 具有合法的《药物临床试验批件》。
1.1.3 核对项目开始实施时间与国家食品药品监督管理总局《药物临床试验批件》时间相符性。
1.2 伦理审查批件及记录的原始性及完整性:1.2.1 试验过程的所有伦理审查活动,包括初始审查、加快审查、持续审查等。
1.2.2 核对伦理审查时相应提交资料的完整性。
1.3 临床试验合同经费必须覆盖临床试验所有开支(含检测、受试者营养/交通费补贴、研究者观察费等)。
1.4 申办者/合同研究组织(CRO)按照药物临床试验管理规范(GCP) 原则、方案及合同承担相应职责的文件和记录(如合同或者方案中规定的项目质量管理责任及监查、稽查相关记录等)。
以研究数据的真实完整性为关注点)2.1 受试者的筛选/入组相关数据链的完整性:2.1.1*申报资料的总结报告中筛选、入选和完成临床试验的例数与分中心小结表及实际临床试验例数一致,若不一致须追查例数修改的环节。
2.1.2*方案执行的入选、排除标准符合技术规范(如实记录体检、血尿常规、血生化、心电图等详细内容),其筛选成功率为多少?(含有证据的初筛受试者例数)。
2.1.3*受试者代码鉴认表或者筛选、体检等原始记录涵盖受试者身份鉴别信息(如姓名、住院号/门诊就诊号、身份证号、联系地址和联系方式等),由此核查参加临床试验受试者的真实性。
2.1.4 对受试者的相关医学判断和处理必须由本机构具有执业资格的医护人员执行并记录,核查医护人员执业许可证及其参与临床试验的实际情况。
2.2 2.3 2.1.5 受试者在方案规定的时间内不得重复参加临床试验。
医疗器械临床试验核查项目汇总表

医疗器械临床试验核查项目汇总表XX年医疗器械临床试验核查项目汇总表试验用医疗备案号器械名称器械类别试验用医疗临床试验名称临床试验机构同时注明代理人)申办者吉林省中医药科学院第一临床医院吉林省赛凯生物技术有限公司β-葡聚糖液体敷料境内Ⅱ类β-葡聚糖液体敷料治疗急慢性鼻炎临床试验吉林邦安宝医用设备有限公司β-葡聚糖阴道灌洗液β-葡聚糖阴道灌洗液长春中医药大学附属医院治疗细菌性阴道炎、外境内Ⅱ类阴阴道假丝酵母菌病吉林省中医药科学院临床试验第一临床医院β-葡聚糖液体敷料治疗生殖器疱疹的临床试验长春中医药大学附属医院吉林省中医药科学院第一临床医院吉林邦安宝医用设备有限公司β-葡聚糖液体敷料境内Ⅱ类吉林邦安宝医用设备有限公司1脂蛋白相关磷脂脂蛋白相关磷脂酶A2北华大学第一附属医酶A2活性测定院境内Ⅱ类活性测定试剂盒试剂盒(速率法)临床吉林市人民医院(速率法) 试验长春恒晓生物科技有限责任公司中性粒细胞载脂蛋白检测试剂盒中性粒细胞载脂蛋白检测试剂盒视黄醇结合蛋白测定试剂盒中性粒细胞载脂蛋白境内Ⅱ类检测试剂盒临床试验中国人民解放军第二零八医院长春博德生物技术有限公司中性粒细胞载脂蛋白境内Ⅱ类检测试剂盒临床试验吉林大学第一医院长春博德生物技术有限公司中国人民解放军第视黄醇结合蛋白测定吉林普阳医用科技医院境内Ⅱ类试剂盒临床试验葛洲坝集团中心医院中国人民解放军第血清淀粉样蛋白A测吉林普阳医用科技医院境内Ⅱ类定试剂盒临床试验葛洲坝集团中心医院血清淀粉样蛋白A测定试剂盒2中性粒细胞明胶酶相关脂质运载蛋白测定试剂盒境内Ⅱ类抗环瓜氨酸肽抗体测定试剂盒胱抑素C测定试剂盒中性粒细胞明胶酶相关脂质运载蛋白测定试剂盒临床试验中国人民解放军第医院葛洲坝集团中心医院吉林普阳医用科技有限公司中国人民解放军第抗环瓜氨酸肽抗体测吉林普阳医用科技医院境内Ⅱ类定试剂盒临床试验葛洲坝集团中心医院中国人民解放军第胱抑素C测定试剂盒吉林普阳医用科技医院境内Ⅱ类临有限公司床试验葛洲坝集团中心医院β2微球蛋白测β2微球蛋白测定试剂定试剂盒α1-微球蛋白测定试剂盒境内Ⅱ类盒葛洲坝集团中心医院临床试验α1-微球蛋白测定试剂盒临床试验中国人民解放军第医院葛洲坝集团中心医院。
药物临床试验检查内容表

药物临床试验检查内容表药物临床试验是指在受控环境中对新药物进行安全性和有效性评价并确定最佳用药方案的过程。
临床试验需要一系列检查来评估患者的身体状况,确保试验的可靠性和患者的安全。
以下是药物临床试验中常见的检查内容表。
基本信息试验的基本信息需要在受试者入组之前记录:•患者的基本情况,如姓名、年龄、性别、身高、体重等;•疾病的诊断情况和病程;•患者的用药史和过敏史;•患者是否接受其他试验项目;•患者是否有心理和认知障碍,是否需要翻译或口译。
生命体征检查生命体征检查是用来评估患者的身体状况, includes:•血压、脉搏、呼吸等生命体征;•体温;•血液生化检查,包括血糖、血脂、肝肾功能等指标。
疾病诊断与评估为确保受试者符合试验入组标准,还需要进行诊断和评估:•完整的疾病史记录;•确认疾病的诊断和严重程度;•检测疾病相关的生物标志物,如血清学检查、肿瘤标志物等。
身体状况评估对于不同的药物临床试验,需要进行不同的身体状况评估:•心血管系统评估:包括心电图、超声心动图等检查;•呼吸系统评估:包括肺功能测试、肺部影像学检查等;•皮肤评估:包括皮肤过敏性反应等;•肝功能评估:包括肝功能检查等;•肾功能评估:包括肾功能检查等。
药物代谢与排泄评估药物代谢与排泄评估是通过了解药物在人体中的代谢和排泄情况,评估药物的安全性和有效性:•药物代谢酶的基因多态性检测;•药物代谢酶和药物转运蛋白的酶活性检测;•尿液和粪便中药物代谢产物的检测。
不良反应记录药物临床试验需要记录患者在试验过程中的不良反应和不良事件,以评估药物的安全性和可耐受性。
常见的不良反应包括:•恶心、呕吐、腹泻等消化系统反应;•皮肤过敏、荨麻疹、药疹等皮肤反应;•头痛、头晕、失眠等神经系统反应;•血清学反应等。
药物临床试验的检查内容多种多样,根据不同药物的特性和试验的特殊要求而有所不同。
不过,无论是何种试验,都需要对受试者进行全方位的身体评估和检查,以确保试验的安全和可靠性。
常用临床试验检验检查目录
52.5
丙肝抗体
丙型肝炎抗体测定
LIS2853
50
丙型肝炎标志物检测
丙型肝炎抗体测定、丙型肝炎核心抗原测定
LIS0017
105
梅毒螺旋体特异性抗体检测
LIS0081
49
人免疫缺陷病毒抗体测定
LIS0113A
54
乙型肝炎病毒DNA定量
LIS0183
72
丙型肝炎病毒(HCV)RNA检测外检自费
常用临床试验检验检查目录
医嘱名称
明细
收费代码2
单价
血液常规分析
全血细胞计数+五分类
LIS0168
14.5
血液常规+网织红细胞计数
全血细胞计数+五分类、网织红细胞计数(Ret)
LIS0141
37.5
凝血检查四项
血浆凝血酶原时间测定(PD)、活化部分凝血活酶时间测定(APTT)、血浆纤维蛋白原测定、凝血酶时间测定(TT)
数字化X光
按部位
130.5
磁共振平扫(1T以上,不含T1)
按部位
520
磁共振增强扫描(1T以上,不含T1)
按部位,耗材和药物另行计算
560
心脏彩色多普勒超声(含报告)
XG00023
120
脑电图
36
填写注意:
因为检验检查项目需在HIS系统后台录入,试验过程中才能够免费开单,请务必填写完整。如果涉及增强CT、增强MRI、介入检查等项目,请将检查所必须的药品和材料等条目查询完整。
LIS0101
42
常规肝功
血清总蛋白测定、血清Y谷氨酰基转移酶测定、血清碱性磷酸酶测定、血清白蛋白测定、血清总胆红素测定、血清直接胆红素测定、血清内氨酸氨基转移酶测定、血清天门冬氨酸氨基转移酶测定
临床试验项目基本信息表
临床试验分期
□药物
○ Ⅰ期○Ⅱ期○Ⅲ期○Ⅳ期○其它
□医疗器械
○临床验证○临床试用○上市后再评价○其它
□不适用
是否
方案设计类型
□干预性研究□观察性研究(○回顾性研究○前瞻性研究)
资金来源于
□企业□政府□学术团体□本单位□其它
研究总例数
本中心例数
临床试验项目基本信息表
项目名称
申请类型
□药物注册临床试验
□申办方发起的非注册性临床研究
□医疗器械注册临床试验
□研究者发起的临床研究
产品种类
□药物分类
○中药、天然药物(类)
○生物制品(类)
○进口药物类
○化学药品(类)
○放射性药物
○其它
□医疗器械
(复选)
○一类
○植入
○二类○三类
○非植入
□体外诊断试剂
○一类○二类○三类
申办方和CRO信息
申办方
申办方指定联系人
电话/邮箱
CRO公司
监查员姓名
电话/邮箱
研究者信息
姓名
技术职称
最近一次GCP培训时间
职责分工
签名
主要研究者签名
日期
伦理审查委员会形式审查
受理号
受理人
受理日期
受理人签字
注:①职责分工中,请注明本项目的院内联系人。
药物临床试验质量核查记录表项目专用
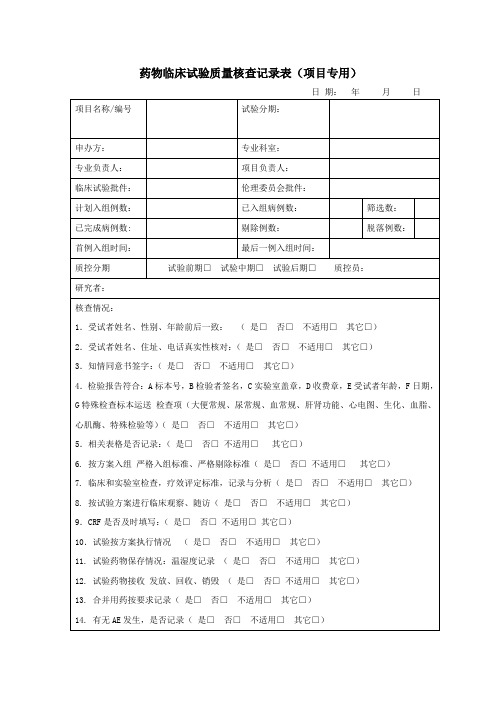
日期:年月日
项目名称/编号
试验分期:
申办方:
专业科室:
专业负责人:Leabharlann 项目负责人:临床试验批件:
伦理委员会批件:
计划入组例数:
已入组病例数:
筛选数:
已完成病例数:
剔除例数:
脱落例数:
首例入组时间:
最后一例入组时间:
质控分期
试验前期□试验中期□试验后期□质控员:
研究者:
17.文件保存是否符合GCP规定(是□否□不适用□其它□)
18.其它:
检查情况说明(可附页):
处理情况(是否申报或撤回及其理由):
检查者签字:
日期:
研究者签字:
日期:
项目负责人签字:
日期:
机构办公室签字:
日期:
11.试验药物保存情况:温湿度记录(是□否□不适用□其它□)
12.试验药物接收发放、回收、销毁(是□否□不适用□其它□)
13.合并用药按要求记录(是□否□不适用□其它□)
14.有无AE发生,是否记录(是□否□不适用□其它□)
15.有无发生SAE?是否按规定记录、报告与处理(是□否□不适用□其它□)
16.文件目录是否清晰(是□否□不适用□其它□)
核查情况:
1.受试者姓名、性别、年龄前后一致:(是□否□不适用□其它□)
2.受试者姓名、住址、电话真实性核对:(是□否□不适用□其它□)
3.知情同意书签字:(是□否□不适用□其它□)
4.检验报告符合:A标本号,B检验者签名,C实验室盖章,D收费章,E受试者年龄,F日期,G特殊检查标本运送检查项(大便常规、尿常规、血常规、肝肾功能、心电图、生化、血脂、心肌酶、特殊检验等)(是□否□不适用□其它□)
临床试验项目信息表
____例
与药物相关例数
有关____例 无关____例
严重不良事件描述
数据处理单位名称
数据处理人员
£委托专业医学统计人员£经过统计培训的研究者
数据处理软件
纳入总例数
完成例数
脱失例数
机构质量检查
£是£否
科室质控
£是£否
协议签署日期
年 月 日
本研究支付费用
元(人民币)
总结报告盖章日期
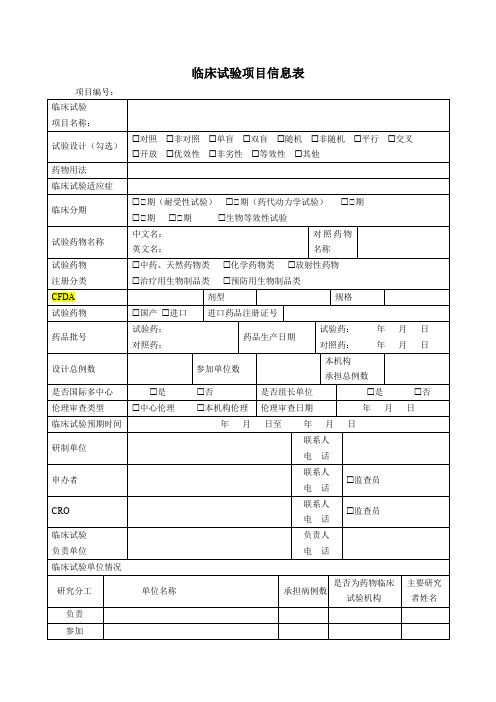
临床试验项目信息表
项目编号:
临床试验
项目名称:
试验设计(勾选)
¨对照¨非对照¨单盲¨双盲¨随机¨非随机¨平行¨交叉
¨开放¨优效性¨非劣性¨等效性¨其他
药物用法
临床试验适应症
临床分期
£Ⅰ期(耐受性试验)£Ⅰ期(药代动力学试验)£Ⅱ期
£Ⅲ期£Ⅳ期£生物等效性试验
试验药物名称
中文名:
英文名:
对照药物名称
试验药物
£中心伦理£本机构伦理
伦理审查日期
年 月 日
临床试验预期时间
年 月 日至 年 月 日
研制单位
联系人
电 话
申办者
联系人
电 话
£监查员
CRO
联系人
电 话
£监查员
临床试验
负责单位
负责人
电 话
临床试验单位情况
研究分工
单位名称
承担病例数
是否为药物临床试验机构
主要研究者姓名
负责
参加
参加
参加
参加
本机构专业科室
1
项目负责人
注册分类
£中药、天然药物类£化学药物类£放射性药物
临床试验项目质控检查表
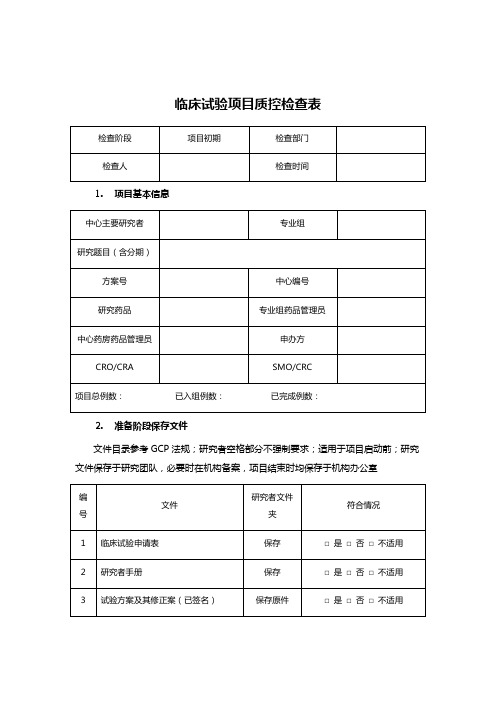
临床试验项目质控检查表
1. 项目基本信息
2. 准备阶段保存文件
文件目录参考GCP法规;研究者空格部分不强制要求;适用于项目启动前;研究文件保存于研究团队,必要时在机构备案,项目结束时均保存于机构办公室
3. 研究者
4. 受试者
5. 知情同意
6. 原始资料填写
7. 病例报告表
8. 试验用药物
9. CRA和CRC
10. 不良事件(AE)与严重不良事件(SAE)管理
备注:
1、检查不只限于表中内容,项目运行中出现任何问题都可以记载于该表中,且须详细记载。
2、医疗器械临床试验质量检查参照药物临床试验进行。
临床试验项目质控检查表
11. 项目基本信息。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
受试者招募广告及其它提供给受试者的书面文件
本试验共______版招募广告获得本院伦理委员会批准
1.8
保险和赔偿措施或相关文件
注意保险的时限是否涵盖整个试验周期
保险起止时间:______
1.9
协议(可单独保存)
有本试验相关的所有协议,共______份。
主协议签署日期:______
1.10
研究者履历及相关文件(GCP证书、从业资格证书)
2、所有人员均有培训记录、培训时间应在其参与临床试验之前(注意新增人员的培训、注意方案和知情更新后的培训)
3、查看在试验过程中是否各司其职
4、各培训的证书、账号核实(如EDC,IVRS)
1.12
医学或实验室操作的质控证明
1、试验涉及的操作均有相应质控证明(如离心机,心电图机,温度计、CT机、PET-CT机、体重器、试验所进行的各项血液学检查项目)等等(根据试验需要)
2.9
试验药物的药检证明(含对照药)
不同批号需要有相应的药检证明
本试验所有药品批号:___________
2.10
试验用药品登记表
1.研究者分工表中应有专人负责药物管理
2.接收记录应与试验药物运送单信息一致
3.应有药物发放/回收记录,发药日期不早于ICF签署日期和入组/随机日期
4.发放/回收数量应与受试者日记卡、CRF等记录一致,并符合方案要求
1、应有研究者签字及日期
授权的研究者为:__________________
2、所有研究者简历、GCP培训证书按授权分工表的姓名顺序排列(GCP证书需是2003年之后获得的)
3、注意查看新增研究者授权时间在其他签字文件之前
1.11
研究者授权表/签名样张/培训记录
1、分工应与研究者实际工作内容一致.并有主要研究者的签字授权、各研究人员签字
5.最终使用数量应与各受试者发放/回收/遗失数量吻合
本试验总计药品接收______支、
CFDA批件号:______
CFDA批准日期:______
试验应在批件有效期内前开始实施(目前为3年内)
1.2
伦理委员会相关文件
本院伦理委员会共______次会审,______次快审。
首次伦理批准时间:______
有本院伦理委员会(及组长单位)历次批件、委员会成员表等复印件
各批件信息准确
1.3
研究者手册
2.6
不良事件
经HIS系统溯源及所有原始文件确认,不良事件记录准确、及时、完整
共发生______例AE
2.7
合并用药
经HIS系统溯源确认,合并用药记录真实、完整
注意方案违禁药品的使用情况
注意外院合并用药的记录
合并用药总计______种
2.8
试验用药品与试验相关物资的运货单
接收记录应与运货单信息一致
本试验总计运输______次
2、临床研究批件上的申办方信息与备案的申办方资质文件一致
1.16
监查单位资质文件
监查负责单位资质文件应在有效期内;
2
试验过程记录
2.1
受试者筛选表与入选表
1、筛选例数应不少于入组例数
2、筛选时间应不晚于入组时间/随机号发放时间
3、应注明未入组受试者的筛选失败原因
2.2
受试者鉴认代码表
受试者鉴认代码表或原始记录中涵盖以下受试者身份鉴别信息:受试者姓名、身份证号、地址、联系电话、门诊就诊号或住院号等。
与筛选表/入选表比对核查一致性
2.3
已签署知情同意书
本试验共签署______份ICF。
若受试者签署多份ICF,请列出其签署的ICF的版本号、版本日期、签署日期。
每份ICF均有受试者本人或其法定代理人、见证人签字及签字日期。(注意签署是否正确、笔迹)
每份ICF均有被授权研究者的签字及签字日期
签署完整
2.4
1.5
病例报告表(样表)
本试验使用eCRF。
本试验共______版CRF获得本院伦理委员会批准。
有所有CRF样本,请按照批准日期顺序整理或者刻盘
1.6
知情同意书
(样表)
本试验共______版ICF获得本院伦理委员会批准。有所有ICF样本,请按照批准日期顺序整理。
1.6
知情同意书
(样表)
各时间段使用的ICF版本与该时段伦理委员会批准版本一致
临床试验核查项目一览表
核查日期
核查 员
1.项目基本信息:
项目名称:
负责科室
主要研究者
责任医师SI
试验类型
药物□器械□
试验启动时间
药物类型
化药☐生物制品☐免疫治疗☐其他☐NA☐
药物类别
新药☐仿制药☐CART☐其他☐
监查负责单位
监查方联系人及电话/邮箱
申办方
申办方联系人及电话/邮箱
筛选病例数
实际入组病例数
1、应及时报告(得知SAE的24小时内)、信息准确、完整(核对病程和SAE报告表),并由授权研究者填写、签字和日期
2、按要求上报相关部门(5个部门:申办者、SFDA、市卫计委、伦理委员会、药理机构),并跟踪随访
3、报告过程应有证明文件(传真回执或者快递底单)
本院共发生______例SAE
请填写SAE自查表
2、质控证明书时限需要涵盖整个试验期限(即注意更新)
若涉及中心实验室,需核查中心实验室资质证书信息
1.13
临床试验有关的实验室检测正常值范围
1、需要PI或者实验室主任签字确认
2、注意更新、查看是否与现行的化验单正常值一致
1.14
药品标签
核对标签上的信息是否准确
1.15
申办方资质文件
1、备案的申办方资质文件应在有效期内;
实际完成病例数
第一例受试签署ICF时间
最后一例受试者完成时间
试(因各项目不同,包括但不限于以下文件)
序号
文件名称
检查要点
检查结果
备注
有
无
NA
1
研究者文件夹
1.1
国家食品药品监督管理局批件
临床试验信息应与批件信息(如药品名称、适应症、试验药品规格)一致,审批结论应为同意开展临床试验
方案执行记录
所有入组的受试者均符合入排条件
所有受试者随机符合方案要求(记录随机过程)
本试验方案违背总计___________例次,其中重大方案违背___________例次
所有访视均按计划进行。
请附方案违背报告
研究者对所有异常检查/检验结果的临床意义进行判断,并记录处理措施。
是否存在破盲情况
2.5
严重不良事件
本试验共______版IB获得本院伦理委员会批准
各版本研究者手册均已存放在TCF(或者刻盘)
1.4
试验方案及其修正案
本试验共______版试验方案、______版方案修正案、______版方案增补获得本院伦理委员会批准。
序号
版本号
版本日期
伦理批准日期
各版本方案均有研究者签字(注意签字日期)
注意方案执行时是否使用正确版本