详细指导--MDD技术文档
MDD技术文档指导文件

发布地点,日期:
签名:
7. 技术文档语言 一个成员国家可能要求技术文档的 A 部分使用官方语言,如果能懂可以不翻译。在需要翻译的情 况,允许文件拥有者额外的时间递交第一部分的内容给检查机构。
而且,关于翻译不能增加更多条件,例如要求有授权的翻译,官方翻译或类似要求。
注:本指令的信息仅作为生产商的指南,生产商仍需仔细阅读并理解 MDD.
2. 技术文档的来历
根据 MDD 93/42/EEC,所有的医疗器械必须符合基本要求附录 I 才能贴 CE 标志。技术文档则提供产 品符合基本要求的文件证据。
然而,技术文档这个词对制造商而言将比较费解,因为在 MDD 中未提到这个词。技术文档是技术 性文件的一个通称,用于证明产品符合基本要求。MDD 的不同附录将会略微不同地描述技术文 档。接下来便介绍每一个 MDD 附录如何描述技术文档。
附录 A) 全部的制造和检验计划 风险分析(见附录 B) 临床数据(见附录 C) 标签,如产品标签,说明书,病人信息,广告材料 合格声明
B 部分包含所有的检测和验证报告,产品有关的质量体系信息,产品的详尽描述如设计图,产品数 据参数,制造过程描述等。如果 B 部分是分散的,应建立一个控制目录,列出每个相关文件。
III 类医疗器械: -附录 II,包括第 4 部分(常指 II.4) -附录 III+附录 IV -附录 III+附录 V
IIb 类医疗器械: -附录 II,不包括第 4 部分(常指 II.3) -附录 III+附录 IV -附录 III+附录 V -附录 III+附录 VI
IIa 类医疗器械: -附录 II,不包括第 4 部分(常指 II.3)
MDD入门讲义

欧盟医疗器械法规要求——IEC 60601-1 解读和介绍主要内容- 欧盟法规介绍 欧盟法规介绍 - 协调标准 协调标准 - IEC 60601家族标准介绍 家族标准介绍 - IEC 60601-1 3.0/3.1版本主要的变化 版本主要的变化 - 全球主要国家标准执行和适用情况 全球主要国家标准执行和适用情况 - Q&A2法规和指令欧盟的主要法律文书(1) 法规(Regulation): - 是一种具有普遍适用性和总约束力的法令。
- 它们适用于所有成员国,包括成员国的自然人。
- 法规一经生效 法规一经生效, 一经生效,各成员国都必须执行, 各成员国都必须执行,没有必要再制定相应的本国法规。
没有必要再制定相应的本国法规。
- 它们可取代或优先于与之冲突的国内法规。
指令(Directive): - 需在成员国制定的转换期之后转变为国家法律的法令。
- 虽然对各成员国均有约束力 虽然对各成员国均有约束力, 对各成员国均有约束力,但对于实施指令的具体方式和方法, 但对于实施指令的具体方式和方法,各成员可以 各不相同, 各不相同,只要能达到指令所要求的目标。
只要能达到指令所要求的目标。
- 指令是针对成员国颁布的,不针对自然人3法规和指令指令(Directive)——解决方案 - 欧洲指令对于所覆盖区域的法规制定提供了法律层面的框架。
- 欧盟成员国必须将指令转化成本国法律,各国法律因此保持协调。
- 在指令覆盖区域的产品必须符合所有适用的欧洲指令的要求, 指令覆盖区域的产品必须符合所有适用的欧洲指令的要求,并附加CE标志 以表明已符合要求 表明已符合要求 - 通过统一的合格评估过程使得产品在全欧洲上市。
CE标志代表了符合性,是一个准入的门框,而不是一个宣称质量的标识或通 标志代表了符合性 过测试的标识。
然而,法规要求仍然存在一些细小的差异,例如:各国对语言的要求。
4医疗器械适用指令医疗器械指令 (MDD) 93/42/EEC (过渡期截至1998年6月) 有源植入医疗器械指令( 源植入医疗器械指令(AIMD) 90/385/EEC (过渡期截至1995年1月) 体外诊断医疗器械指令( 外诊断医疗器械指令(IVDD) 98/79/EC (过渡期截至2003年12月)5MDD指令有三个主要方面需要特别关注: •安全性 (使用者、患者和公众) •有效性 (发挥预期的临床作用) •可重复性(制造过程)6定义和范围设备、 软件、材料或 “医疗器械”是指可单独或组合使用的所有工具、仪器、设备 设备 、软件 其他物品,包括制造商生产的专门用于诊断和/或治疗所必需的软件,制造商意 意 图将其用于人体,以: 图将其用于人体 ——诊断,预防,监测,治疗或减轻疾病, ——诊断,监测,治疗,减轻伤痛或残疾或予以补偿, ——调查,更换或改变解剖或生理过程, ——节育, 以及没有在人体内/上实现其主要效用的药物、免疫或代谢方式,但可通过这种 方式来协助实现其功能。
DOS的MD和RD命令使用说明

DOS的MD和RD命令使用说明导语:今天我们要学的两个命令就是进行目录*作的,它们是md(makedirectory--创建目录)和rd(removedirectory--删除目录)。
下面就由小编为大家介绍一下DOS的MD和RD命令使用说明,大家一起去看看吧!这两个命令很简单,比如我叫小博士,我要把自己的文件都放在一个目录中,我就可以输入mdxbs,建立我的目录。
这时你用dir命令看一看,就会发现根目录下多了一个目录XBS。
你不妨练习一下,建立一个名字叫xyz的目录,再输入dir,是不是可以看到xyz目录啦,如果你看到了,你就成功了(呵呵,这课也就完成一半了)。
现在让我们到这个目录中去,键入cdxyz,注意,提示符是不是变了。
好,输入dir命令。
很奇怪是吧?我们刚刚建立了这个目录,按理说,这个目录中应该什么都没有的。
其实,不管你建立什么目录,这个.和..都会在目录中出现,因为.代表此目录本身,..代表此目录的上一层目录。
显然,一个目录既然已经存在了,就不可能没有本身,也不可能没有上一层目录。
(..你使用过的,记得吗?就是cd..,用来返回上一层目录)。
再练习一次,在xyz目录下建立一个目录:abc,(正确方法是输入mdabc)用dir命令看一下,abc目录显示出来,就说明你已经成功学会建目录了。
既然能建立目录,当然也就可以把它删除,rd命令就是干这活的。
比如想把abc目录删除,输入rdabc就可以了。
不信再用dir命令看看,abc目录是不是没了。
是不是很简单,要删除当前目录下的某个子目录,输入rd空格加上子目录名就可以完成任务。
不过使用RD命令可得注意几点问题(要不然你删一辈子也别想删除某些目录),好好看看下面几点吧:现在你再练习一次(这次你成功了,就胜利出师了,可以安心学下一课了),如果想删除你现在所在的xyz目录,该怎么办呢?正确*:输入cd..命令退回到上一层目录,再输入rdxyz将xyz目录删除。
MDD工作手册

每年9-10月
• •
草拟下一年MDD的AOP; 与各LU沟通渠道发展AOP,包括: – 当年业绩与次年目标 – 问题与机会点 – 策略和战术 – 人员和费用 MDD的AOP在与GM和其它功能部门沟通修正后,最终定稿,; MDD的各渠道拓展经理与MKT的各品牌经理共同进行YTD业务回顾,结合品牌经理提供 的市场促销方案和渠道拓展经理提供的渠道拓展方案制定MU季度行动计划(即QAP),由 GM/UM初审后发给FMDD和FMKT;
分渠道拓展的必要性
如何取得销量和利润的持续增长?
满足客户需求取得双赢
(各种不同渠道客户的集合)
C1公司: •不同渠道对销售系统有不 同的要求; •公司的资源需要整合 P1产品: •不同渠道有各自适合销售 的产品; C2客户: •不同渠道间客户需求不同; C3消费者: •消费者越来越个性化; C4竞争对手: •竞争越来越没有秘密可言;
步骤五: 项目执行/试点
优化销售资源配置
制定渠道拓展策略/战术和方案的 工作方法
Jan 29 5 12 19 26 2 Feb 9 16 23 1 8 Mar 15 22 29 5 Apr 12 19 26 3 May Jun July Aug 16 23 30 6 Sep 13 20 27 4 Oct 11 18 25 1 8 Nov 15 22 29 6 Dec 13 20 27 10 17 24 31 7 14 21 28 5 12 19 26 2 9
So as to cost-effectively reach and sustain maximum channel coverage and VPO. 从而以财务高效的方法达到并保持售点覆盖和单点销量的最大化!
目录
- 市场拓展部工作指导手册 - 市场拓展部的任务 - 市场拓展部组织架构
医疗器械 欧盟技术文档清单 MDD CE Checklist
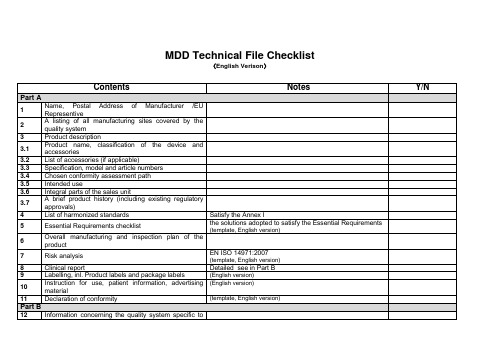
3.7
简要的产品历史(包括现有的管理审批)
4
适用的标准清单
符合医疗器械指令附录I
5
基本要求检查表
符合医疗器械指令附录I的方案
(有固定模板,需提交英文文件)
正在填写
6
产品的总体生产或质量控制方案
7
风险分析
EN ISO 14971:2007
有固定模板,需提交英文文件
正在索要
8
临床报告
详细的临床数据见Part B
9
Labelling, inl.Product labels and package labels
(English version)
10
Instruction for use, patient information,advertising material
(English version)
11
Declaration of conformity
MDDTechnical FileChecklist
(English Verison)
Contents
Notes
Y/N
Part A
1
Name, Postal Address of Manufacturer /EU Representive
2
A listing of all manufacturing sites covered by the quality system
13.2
Packaging and specification
(English version)
13.3
Description of the manufacturing processes
MDD-有关文件的编写

MDD 有关文件的编写
根据MDD中有关符合性评价程序的描述制造商可根据自己的特点选择不同的认证途径获得CE标志¡,主要是选择通过质量体系还是型式试验来得到保证。
对于国内企业来说,由于每批(或每件)产品必须经公告机构或公告机构认可的实验室进行检验,合格后方可以打上CE标志,而且一次性无菌产品不能通过型式实验的方法获得CE标志,因此,在一般情况下,通过质量保证体系方式获得CE标志更适合于绝大部分得国内医疗器械厂家。
附录标准指南
附录II ISO 9001+ ISO 13485E N 724:无源医疗器械
附录V ISO 9002+ ISO 13488p r EN 50103: 有源医疗器械
附录VI I SO 9003+ EN 46003ISO/DIS 14969:
ISO 13485/ ISO 13488 应用指南
第一层次质量手册增加MDD 法规要求简述
第二层次程序文件增加与MDD法规有关的程序文件
(如警戒系统等)
第三层次作业指导书
增加CE 技术文件
检验规范。
mdd-CSS使用技巧大全

1. 文字的水平居中将一段文字置于容器的水平中点,只要设置text-align属性即可:text-align:center;2. 容器的水平居中先为该容器设置一个明确宽度,然后将margin的水平值设为auto即可。
div#container {width:760px;margin:0 auto;}3. 文字的垂直居中单行文字的垂直居中,只要将行高与容器高设为相等即可。
比如,容器中有一行数字。
1234567890然后CSS这样写:div#container {height: 35px; line-height: 35px;}如果有n行文字,那么将行高设为容器高度的n分之一即可。
4. 容器的垂直居中比如,有一大一小两个容器,请问如何将小容器垂直居中?首先,将大容器的定位为relative。
div#big{position:relative;height:480px;}然后,将小容器定位为absolute,再将它的左上角沿y轴下移50%,最后将它margin-top 上移本身高度的50%即可。
div#small {position: absolute;top: 50%;height: 240px;margin-top: -120px;}使用同样的思路,也可以做出水平居中的效果。
5. 图片宽度的自适应如何使得较大的图片,能够自动适应小容器的宽度?CSS可以这样写:img {max-width: 100%}但是IE6不支持max-width,所以遇到IE6时,使用IE条件注释,将语句改写为:img {width: 100%}6. 3D按钮要使按钮具有3D效果,只要将它的左上部边框设为浅色,右下部边框设为深色即可。
div#button {background: #888;border: 1px solid;border-color: #999 #777 #777 #999;}7. font属性的快捷写法font快捷写法的格式为:body {font: font-style font-variant font-weight font-size line-height font-family;}所以,body {font-family: Arial, Helvetica, sans-serif;font-size: 13px;font-weight: normal;font-variant: small-caps;font-style: italic;line-height: 150%;}可以被写成:body {font: italic small-caps normal 13px/150% Arial, Helvetica, sans-serif;}8. link状态的设置顺序link的四种状态,需要按照下面的前后顺序进行设置:a:linka:visiteda:hovera:active9. IE条件注释你可以利用条件注释,设置只对IE产生作用的语句:< ![endif]-->还可以区分各种不同的IE版本:10. IE6专用语句:方法一由于IE6不把html视为文档的根元素,所以利用这一点,可以写出只有IE6才能读到的语句:/* the following rules apply only to IE6 */* html{}* html body{}* html .foo{}IE7专用语句则要写成/* the following rules apply only to IE7 */*+html .foo{}11. IE专用语句:方法二除了IE6以外,所有浏览器都不能识别属性前的下划线。
MDD文件清单

Listing of required documentation in a Technical Construction File for CE marking acc. to 93/42/EEC按照93/42/EEC进行CE 标志认证所需要提供的技术文档清单1. General description of the device器械的一般描述包括产品的型号、规格,产品在国内的注册和生产状况等。
2. Description of intended use预期用途的描述Class of device,设备的分类applied classification rule and justification选择分类的规则和依据3. Description of accessories (if applicable)附件的描述(如适用)产品的附件清单,附件的更换描述。
4. Description of manufacturing methods and controls:生产方法和控制的描述:a) Description of process过程的描述b) List of procedures and instructions程序和说明的列表c) Provisions to control subcontractors供应商控制的规定5. Answers to essential requirements对基本要求的符合性6. List of applied standards适用标准清单7. Risk analysis风险分析(参考EN 14971:2007)8. Specification of materials材料的详细说明9.Photos, drawings and diagrams照片、图纸和原理图10. Labelling标签(参考EN 980:2007)11. Description and Validation of Packaging包装的描述和确认12. Instructions for use说明书13. Lifetime and/or shelf-life使用期限和/或保存期限14. Sterilization validation (if applicablee)灭菌确认(如适用)15. Software validation软件确认(参考EN 60601-1-4:1996)ability / ergonomics (if applicablee)实用性/人体工学(如适用)(参考EN 60601-1-6:2007)17. Preclinical evaluation (if applicable)临床前评价(如适用)(Test reports electrical / mechanical / biocompatibility / animal testing /…)issued by accredited third party laboratories)由认可的第三方实验室颁发的(电气/ 机械/ 生物相容性/ 动物实验)测试报告aa 18. Clinical evaluation (clinical tests and/or literature with critical evaluation)临床评价(临床测试和/或关键的文献评价)19. Project for EC declaration of conformityEC符合性声明项目20. Only for non-European manufacturers: Contract with EC Representative仅适用于非欧洲制造商:与欧盟授权代表的合同。
mdd工程术语

mdd工程术语MDD(Model-Driven Development)是模型驱动开发的缩写,是一种软件开发方法。
在MDD中,软件系统的开发过程主要依赖于从一个或多个模型中自动生成代码和其他开发资源。
以下是一些与MDD相关的术语:1. 模型(Model):在MDD中,模型是软件系统的抽象表示,可以是用于描述系统行为、结构和属性的图形化、文本化或数学化的表示。
2. 模型驱动工程(Model-driven Engineering):模型驱动工程是一种软件工程方法,基于模型,用于系统的设计、分析、实现和维护。
3. 可执行模型(Executable Model):可执行模型是指能够直接运行的模型,通常可以生成代码或其他可执行形式。
4. 模型转换(Model Transformation):模型转换是指将一个模型转换为另一个模型的过程,常用于生成代码、验证模型以及其他模型间的转换。
5. 模型驱动架构(Model-driven Architecture,MDA):MDA是一种MDD的软件开发方法,提供了一种用于描述和转换模型的标准化框架。
6. MDA转换(MDA Transformation):MDA转换是指基于MDA方法的模型转换过程,通常包括从平台无关的模型转换到平台特定的模型。
7. 模型驱动测试(Model-driven Testing):模型驱动测试是一种使用模型驱动方法进行软件测试的技术,通过生成测试用例来验证系统模型的正确性和完整性。
8. 模型驱动工具(Model-driven Tool):模型驱动工具是用于支持MDD开发过程的软件工具,包括模型编辑器、模型转换器、代码生成器等。
9. MDD开发环境(MDD Development Environment):MDD开发环境是指支持MDD方法的集成开发环境(IDE),提供了一系列与模型驱动开发相关的功能。
10. 模型驱动改进(Model-driven Improvement):模型驱动改进是指通过引入MDD方法来提高软件系统开发效率和质量的过程。
MDD附录一基本要求一览表翻译稿
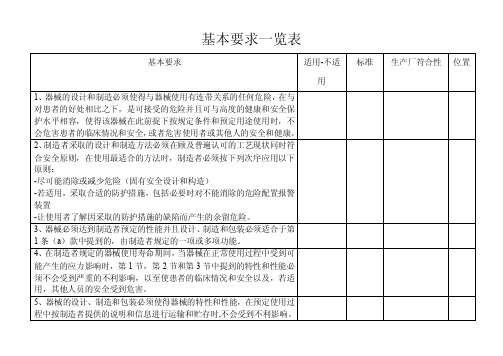
基本要求
适用-不适用
13.5 在只要采取和可行,器械和可拆元部件必须以表达批次的字眼来识 别,以便采取各种适当措施来发现器械和可拆元部件造成的潜在危险。
13.6 若适用,使用说明书必须包含以下内容: A、 第 13.3 节提到的内容,(d)和(e)款除外; B、 第 3 节提到的性能和任何副作用; C、 如果器械必须与其他器械或设备一起安装或连接起来安装才能
标准
生产厂符合性
位置
基本要求
适用-不适用
11.4 说明书 11.4.1 辐射器械的操作说明书必须对辐射的性质、保护患者和使用者的 方法、避免误用和排除设施固有危险的方法给出详细的信息。 11.5 离子辐射 11.5.1 发射离子辐射的器械必须设计和制造得确保能,若可行的话,按 照预定的用途对辐射的数量、几何形状和性质进行改变和控制。 11.5.2 发射离子辐射供放射诊断用的器械应设计和制造得能提供适合 于医疗目的的图象和/或输出质量,同时使患者和使用者受到的辐射照 射降低到最低限度。 11.5.3 发射离子辐射供方射诊断用的器械应设计和制造得能对输出剂 量、射束类型和能量以及,若适用的话,辐射性质进行可靠监视和控 制。 12 对与能源连接或配置能源的医疗器械的要求 12.1 含有电子可编程系统的器械必须设计得确保该系统的重视性、可 靠性和性能符合预定用途。在(该系统中)发生单一故障时,宜采取 合适的方法来排除或尽可能减少跟着发生的危险。 12.2 患者安全取决于内部电源的器械必须配置确定供电状态的装置。
欧盟医疗器械指令MDDEEC培训

- G. 动物移植物或动物组织或细胞,除非器械是利用不能存活的动物组织或从动 物组织中衍生的不能存活的产品制造的
Note 1:Mass-produced devices which need to be adapted to meet the specific requirements of the medical practitioner or any other professional user ►M5shall
· 上市: 指器械首次供应于社会. · 投入使用: 指器械首次按其预定功能被使用.
16
第十六页,共七十六页。
第1章 定义和范围
· 器械子类别
·‘device subcategory’ means a set of devices having common areas of intended use or common technologmeans a set of devices having common areasof intended use or commontechnology;
be custom made devices 大量生产则不应认为是定制器械
12
第十二页,共七十六页。
第1章 定义和范围
制造商
是指在以其名义将器械投放市场前负责器械的设计、制造、包装和标签的自然人或法人 ,无论这些工作是他自己完成的,还是由第三方代表他完成的。本指令规定制造商必须履
行的义务也适用于负责对一件或几件制成品进行装配,包装、加工、全面整修和/或加贴标志和/或
mdd对质量体系的要求_解释说明以及概述

mdd对质量体系的要求解释说明以及概述1. 引言1.1 概述本文旨在探讨面向模型驱动开发(Model-Driven Development,简称MDD)对质量体系的要求,并解释其背后的原因和意义。
质量体系是指为确保产品或服务达到预期质量水平而建立和维护的一系列组织、流程和方法。
MDD作为一种软件开发方法,强调基于模型的系统设计和实现,它对质量的要求在不同层面上有所不同。
1.2 文章结构本文主要分为五个部分。
引言部分是对整篇文章进行概述,介绍MDD对质量体系的要求及其影响和优势。
第二部分将介绍MDD的基本概念,并对质量体系进行解释说明。
接着,在第三部分中详细解释MDD对质量体系的具体要求,包括与质量管理原则的关联、组织结构和责任、过程控制与改进等方面。
第四部分将概述MDD对质量体系带来的影响和优势,包括提高产品质量和符合性能力、增强组织运作效率和灵活性以及实现持续改进与客户满意度提升等方面。
最后,第五部分对全文进行总结,并展望未来发展趋势及提出相应建议。
1.3 目的本文的目的是通过深入研究MDD对质量体系的要求,帮助读者了解MDD方法论与质量管理之间的关联,并认识到采用MDD开发方法所带来的质量改进机会。
同时,本文还将探讨MDD对组织和流程产生的影响,以及经济效益和用户满意度等方面的优势。
通过这些内容的介绍,读者可以更好地理解和应用MDD方法来提高软件开发过程中的质量水平。
2. MDD对质量体系的要求2.1 MDD简介在理解MDD(Model Driven Development,模型驱动开发)对质量体系的要求之前,首先需要了解MDD的基本概念。
MDD是一种软件开发方法论,它强调通过使用抽象模型来指导和支持软件开发过程中的各个阶段,从而提高开发效率和软件质量。
在MDD中,系统设计、实现和测试等活动依赖于构建并维护系统的模型。
2.2 质量体系概念解释质量体系是指以确保产品或服务符合特定标准和要求为目标的组织内部架构和流程。
idea md大纲

ideamd大纲如何在IntelliJ IDEA中使用Markdown编辑器生成大纲摘要Markdown是一种轻量级的标记语言,可以用简单的语法来编写文档,并将其转换为HTML格式。
IntelliJ IDEA是一款功能强大的集成开发环境,支持多种编程语言和技术,包括Markdown。
IntelliJ IDEA提供了一个专用的Markdown编辑器,可以让用户在编写文档的同时,实时预览HTML效果,并且可以生成文档的大纲,方便用户查看和导航文档的结构。
本文介绍了如何在IntelliJIDEA中使用Markdown编辑器生成大纲,包括如何创建和编辑Markdown文件,如何查看和调整大纲,以及如何导出和打印大纲等,旨在为有意使用IntelliJ IDEA编写Markdown文档的用户提供指导。
关键词IntelliJ IDEA;Markdown;大纲正文一、创建和编辑Markdown文件要在IntelliJ IDEA中创建和编辑Markdown文件,需要先安装并启用Markdown支持插件。
该插件是IntelliJ IDEA的默认插件,一般不需要手动安装。
如果没有启用该插件,可以在设置/首选项(Ctrl+Alt+S)中的插件页面,找到Markdown支持插件,并勾选启用。
创建Markdown文件的方法有以下几种:- 在项目中的任意目录上右键,选择新建-文件,输入文件名,以.md或.为后缀,例如readme.md。
- 在项目中的任意目录上右键,选择新建-Markdown文件,输入文件名,不需要输入后缀,例如readme。
- 在编辑器中的任意位置,按Alt+Insert,选择Markdown文件,输入文件名,不需要输入后缀,例如readme。
编辑Markdown文件的方法有以下几种:- 在项目工具窗口(Alt+1)中,双击要编辑的Markdown文件,打开编辑器。
- 在编辑器中的任意位置,按Ctrl+Shift+N,输入要编辑的Markdown文件的名称或路径,选择要编辑的Markdown文件,打开编辑器。
mdd认证计划书

mdd认证计划书篇一:iSo13485设计开发控制程序iSo13485设计开发控制程序1目的对产品设计和开发全过程进行控制,确保设计能满足合同及顾客的要求,以及政府有关的法令规定、国家标准、mdd93/42/EEc和directive20XX/47/Ec欧盟指令等要求。
2范围本程序适用于新产品的设计和定型产品的改进活动。
3职责3.1技术部:负责编制和执行产品设计开发计划,对设计和开发全过程进行组织、协调和管理工作,组织设计评审、设计验证、设计确认工作。
3.1.1负责处理生产过程中发生的产品设计问题,生产工艺的编制,工装夹具的设计与制作。
3.1.2负责制定风险管理计划,提交风险管理报告。
3.2生产部:负责组织试产,参与相关过程评审。
3.3采购部:负责试产过程中的物料采购。
3.4经营部:负责市场调研并参与相关的设计评审。
3.5品质部:负责试产中产品的检验与测试。
4内容4.1设计开发策划4.1.1设计项目来源4.1.1.1经营部、技术部根据国内外的市场动向,有针对性的做市场调研,收集市场情报。
例如电子报刊杂志、展览会等,在需要时购回参考样机,以供技术部参考之用。
4.1.1.2顾客委托设计与定型产品改良的产品,由经营部与顾客充分沟通,并收集相关资料。
在情况允许的条件下,由顾客提供参考样机,以供技术部参考之用。
4.1.1.3经营部通过对市场调查和分析结果,提出“设计开发建议书”,报总经理批准后,连同有关资料转交技术部。
4.2设计开发输入4.2.1技术部根据新产品“设计开发建议书”或参考样机,编制“设计任务书”,“设计任务书”应规定对设计的要求,内容包括:a.根据预期用途和使用说明,规定产品的功能、性能、结构和软件的要求。
B.iSo13485:20XX标准、mdd93/42/EEc和directive20XX/47/Ec欧盟指令、iSo14971:2000等相关的法律和法规的要求,以及使用者和患者的要求。
c.过去类似设计的有关信息。
详细指导--MDD技术文档

技术文档指导(医疗器械指令93/42/EEC)1.介绍大部分新的指令需要制造商提供能证明产品符合指令基本要求的技术性文件。
在指令中,技术性文件通常指技术文档。
尤其值得注意的是,在MDD中,每个医疗器械都应有技术文档。
虽然在MDD中对技术文档的内容有所描述,这里还将提供更为详尽的描述以帮助生产商理解和编制技术文档。
而且,符合本指令的技术文档将有助于公告机构和国家检测机构的评审。
注:本指令的信息仅作为生产商的指南,生产商仍需仔细阅读并理解MDD.2.技术文档的来历根据MDD 93/42/EEC,所有的医疗器械必须符合基本要求附录I才能贴CE标志。
技术文档则提供产品符合基本要求的文件证据。
然而,技术文档这个词对制造商而言将比较费解,因为在MDD中未提到这个词。
技术文档是技术性文件的一个通称,用于证明产品符合基本要求。
MDD的不同附录将会略微不同地描述技术文档。
接下来便介绍每一个MDD附录如何描述技术文档。
附录II附录II的4.1部分提到“制造商必须向公告机构提交申请产品相关的设计文档的检查”4.2部分提到“申请必须包括3.2(c)部分所说的文件,以评价产品是否符合指令要求,”3.2(c)部分要求“产品的一般描述……包括适用的标准……以及为符合基本要求采取的方案的描述。
”设计文档是III类医疗器械的技术性文件,公告机构会全面审查。
附录III附录III的第2部分提到“第3部分提到的文件需要评价抽样样品的符合性”第3部分提到“需要采用风险分析结果和标准清单(参考第5章)以满足基本要求”附录VII附录VII的第2部分提到“制造商必须提供第3部分所说的技术文档”,第3部分进一步要求“需要采用风险分析结果和标准清单(参考第5章)以满足基本要求”因此,为简化起见,MDD每个附录要求的用来证明满足基本要求技术性文件简称为技术文档。
3.11章及合格评审途径的回顾完全理解技术文档需要快速浏览MDD的11章(合格评审途径)。
MDD协调标准
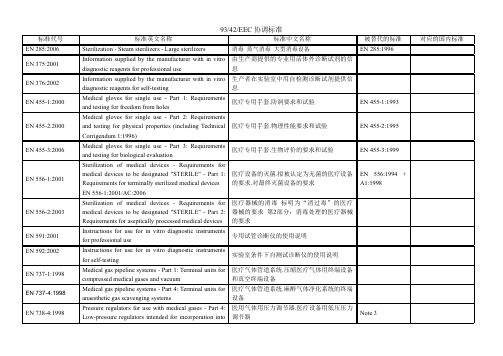
注射器、针头和其它特定医疗器械用有6%(路 厄)锥度的圆锥形配件.锁定配件 气管和连接件 麻醉贮袋
救护车里的担架和其他病人运送设备规范
diagnostic reagents for professional use
息
Information supplied by the manufacturer with in vitro 生产者在实验室中用自检测诊断试剂提供信
diagnostic reagents for self-testing
Note 3 Note 3 Note 3 Note 3 EN 980:1996 Note 3
Note 3
EN 1060-4:2004
EN 1089-3:2004
EN 1280-1:1997
ENห้องสมุดไป่ตู้1281-2:1995
EN 1282-2:2005 EN 1422:1997 EN 1618:1997 EN 1639:2004 EN 1640:2004 EN 1641:2004 EN 1642:2004
medical devices to be designated "STERILE" - Part 2: 器械的要求 第2部分:消毒处理的医疗器械
Requirements for aseptically processed medical devices 的要求
Instructions for use for in vitro diagnostic instruments 专用试管诊断仪的使用说明
02-详细设计说明书(MSD-OA-DES-DD)V1.0

无锡NIIT软件开发有限公司文档编号: MSD-OA-DES-DD详细设计说明书V1.0编写:牛金瑞审核:徐晓明批准:日期:2013.5.10 日期:3013.5.10 日期:2013.5.10变更履历版本文件内容描述编写日期编写审核批准1.0 经过评审确认为正式版本2013.5.10牛金瑞徐晓明钱庭荣目录1引言 (1)1编写目的 (1)2背景 (1)3定义 (2)4参考资料 (2)2程序描述 (3)1首页 (3)1.1模块描述 (3)1.2功能 (3)1.3性能 (3)1.4实体类描述 (3)1.5算法与程序逻辑 (8)2消息面板 (8)2.1模块描述 (8)2.2功能 (8)2.3性能 (9)2.4实体类描述 (9)2.5算法与程序逻辑 (12)3后台管理 (12)3.1模块描述 (12)3.2功能 (12)3.3性能 (12)3.4算法与程序逻辑 (13)3复用与外购 (13)1复用分析 (13)2外购分析 (14)4接口设计 (15)1内部接口 (15)2外部接口 (15)5界面设计 (16)1登录页面 (16)2系统首页页面 (17)3消息面板页面 (17)4事务管理页面 (17)5计划管理页面 (17)6客户跟踪页面 (17)7会议管理页面 (18)8人力资源页面 (18)9个人设置页面............................................................................................ 错误!未定义书签。
10意见与建议页面 ................................................................................... 错误!未定义书签。
11后台管理页面 ....................................................................................... 错误!未定义书签。
DMC文档重要内容

独立定位控制器可以对所有各轴同时或分别发送BG 指令,需要用A,B,C,D 来选择所要运动的轴。
当未指定轴号时,就意味着使所有轴开始运动。
在运动期间,可以随时改变运动速度(SP)和加速度(AC),不过,直到运动完成后,才可以改变减速度(DC)和位置值(PR或PA)。
请记住,控制器不是在实际电机处于指定目标位置时认为运动完成,而是规划中的“指令位置”到达目标位置时,也就是电机应该到达时。
在到达终点位置之前,可以随时发送停止指令(ST)使电机减速停止。
只要附加运动与正在执行的运动方向相同,在运动期间就可以指定增量位置(IP),此时用户只指定新的位置增量n,新的目标位置就等于原有目标位置+增量n。
一旦接收到IP指令,就会重新规划运动轨迹,使运动达到新的终点位置。
IP指令不需要BG。
注意:如果电机不在运动中,IP指令就等效于PR和BG 指令的组合。
位置跟踪在独立轴定位控制方式下,在运动过程中可以改变运动速度、加速度但不能任意改变目标位置。
而在一些应用中,电机在运动中跟踪一个随机位置,无论一个轴是否在运动,都要即刻改变最终位置。
位置跟踪模式同样需要指定速度(SP)、加速度(AC)和减速度(DC)指令。
启动位置跟踪模式后,随时可以指定目标位置(PA)。
不需要BG 指令来开始运动,指定位置后,控制器就会根据当前的位置、速度和目标位置来产生或修改位置规划。
启动位置跟踪所用的指令为PT a,b,c,d。
当a,b,c,d为1 时,启动对应轴的位置跟踪模式;当a,b,c,d为0 时,关闭对应轴的位置跟踪模式。
位置跟踪模式下,AM、MC 指令无效。
直线插补指定直线段用指令LI a,b,c,d,e,f,g,h来为各轴指定增量运动距离,BGS指令之前,可以给出511个增量线段。
一旦开始运动,还可以给控制器发送附加LI线段。
在运动启动之前,能用清除线段指令CS 来删除存储在缓冲区中的LI 线段。
要想停止运动,就使用指令STS 或AB。
项目MD设计指引书V10

可能做厚一点;机壳破大孔,孔边缘距离rubber至少留0.2mm防撞 4、使用rubber方式,rubber整体高度(也就是PCB到TP油墨开孔区底部位置)建议不超过3.5mm,有客户
? 堆叠中的dome,摄像头组件,其他支架等需要组装到主板上的物料, 需要MD工程师最终确认结构设计。
? 喇叭,听筒,马达,MIC等电声器件,具体的工作高度、导线长度等 需要MD根据供应商规格书最终确定给出。
整机类型
基本尺寸 LCD TP
摄像头
喇叭 听筒 MIC 马达 传感器 天线 其它
T89基本配置信息
电池内部添加温度检测电路示意图
为避免主板充电电路的温升影响电芯温度的测量,NTC电阻在电池包装中应远离电池金手指pin脚, 靠近电池电芯,见下图。
NTC在电池包装中的推荐位置
如果电池内部没有NTC电阻,即无法实现温度检测功能。此时要求电池内部用普通10k电阻代替。
如果普通10k电阻都没加,请告知我司项目经理,我们会在软件上关闭温度检测相关功能。
注意不能做反 。
T卡座: 1. 注意出卡方向和距离; 2.卡座与周围壳料间隙 0.5mm ,
方便取卡 ;
T卡
大SIM 卡MIC
SIM 卡二合一
USB、耳机插头设计要点
USB、耳机插头与壳料单边间隙 0.2mm 以上。耳机开孔 φ 4.1mm 以上
USB插头请根据贵司具体长度核 对干涉。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
技术文档指导(医疗器械指令93/42/EEC)1.介绍大部分新的指令需要制造商提供能证明产品符合指令基本要求的技术性文件。
在指令中,技术性文件通常指技术文档。
尤其值得注意的是,在MDD中,每个医疗器械都应有技术文档。
虽然在MDD中对技术文档的内容有所描述,这里还将提供更为详尽的描述以帮助生产商理解和编制技术文档。
而且,符合本指令的技术文档将有助于公告机构和国家检测机构的评审。
注:本指令的信息仅作为生产商的指南,生产商仍需仔细阅读并理解MDD.2.技术文档的来历根据MDD 93/42/EEC,所有的医疗器械必须符合基本要求附录I才能贴CE标志。
技术文档则提供产品符合基本要求的文件证据。
然而,技术文档这个词对制造商而言将比较费解,因为在MDD中未提到这个词。
技术文档是技术性文件的一个通称,用于证明产品符合基本要求。
MDD的不同附录将会略微不同地描述技术文档。
接下来便介绍每一个MDD附录如何描述技术文档。
附录II附录II的4.1部分提到“制造商必须向公告机构提交申请产品相关的设计文档的检查”4.2部分提到“申请必须包括3.2(c)部分所说的文件,以评价产品是否符合指令要求,”3.2(c)部分要求“产品的一般描述……包括适用的标准……以及为符合基本要求采取的方案的描述。
”设计文档是III类医疗器械的技术性文件,公告机构会全面审查。
附录III附录III的第2部分提到“第3部分提到的文件需要评价抽样样品的符合性”第3部分提到“需要采用风险分析结果和标准清单(参考第5章)以满足基本要求”附录VII附录VII的第2部分提到“制造商必须提供第3部分所说的技术文档”,第3部分进一步要求“需要采用风险分析结果和标准清单(参考第5章)以满足基本要求”因此,为简化起见,MDD每个附录要求的用来证明满足基本要求技术性文件简称为技术文档。
3.11章及合格评审途径的回顾完全理解技术文档需要快速浏览MDD的11章(合格评审途径)。
11章要求制造商满足MDD的某个附录要求以获得CE标志。
使用哪一个附录取决于仪器的分类。
合格评审途径总结如下:III类医疗器械:-附录II,包括第4部分(常指II.4)-附录III+附录IV-附录III+附录VIIb类医疗器械:-附录II,不包括第4部分(常指II.3)-附录III+附录IV-附录III+附录V-附录III+附录VIIIa类医疗器械:-附录II,不包括第4部分(常指II.3)-附录VII+附录IV-附录VII+附录V-附录VII+附录VII类医疗器械,含无菌和/或测量功能:-附录VII+附录IV-附录VII+附录V(无菌仪器必须采用这一途径)-附录VII+附录VII类医疗器械,无无菌和/或测量功能:-附录VII合格评审途径强调了为何附录II,III,VII涉及了技术文档(请见本文件2.技术文档的来历)。
名义上,只有附录II,III,VII涉及了技术文档是因为所有的医疗器械必须使用附录II,III或VII以满足11章要求并使用CE标志。
4.评审技术文档的公告机构的角色令一个制造商迷惑不解的是公告机构在技术文档评审中的角色。
决定公告机构在技术文档评审中的角色的最简单的方法是进行医疗器械分类。
III类医疗器械:技术文档必须递交公告机构评审和批准。
IIb类医疗器械(采用附录III及IV,V,VI途径):技术文档必须递交公告机构评审和批准。
IIb类医疗器械(采用II.3),IIa类医疗器械,I类医疗器械(含无菌和/或测量功能):技术文档必须在合格评定过程中经公告机构审核。
5.技术文档和权威机构技术文档必须受权威机构支配以便于其检查。
这个要求来自欧盟1985年5月8日采纳的“技术协调性和标准的新方案”,并促进了新指令的产生。
而且,MDD 的附录中包含了如下要求:-附录II的4.1提到“在产品生产后至少5年期限内,制造商必须确保4.2中所提到的文件受国家权威机构的支配”-附录III的7.3提到“制造商和授权代表必须在产品生产后至少5年期限保留技术文档”-附录VII的2提到“制造商和授权代表必须编制文件,包括合格声明,在产品生产后至少5年期限保留技术文档,便于国家权威机构检查”产品在欧洲市场销售后,不管产品的最初产地,制造商或欧盟代表有义务保证自产品销售后可以获得技术文档。
如果一个权威机构需要技术文档,技术文档的A部分必须立刻能获得。
考虑到B部分的容量和格式,可以稍微缓一点时间。
技术文档A部分和B部分的详细内容将在本文第6部分进行说明。
6.技术文档的内容和形式概要简而言之,技术文档所要求的信息包括:(i)证明产品符合基本要求的必要的技术信息(ii)质量管理体系和产品质量流程的简要描述(iii)符合声明技术文档可以根据产品系列的组分相似性,制造过程,预期用途来进行编制。
形式可以集中也可以分散(除了III类和部分IIb类技术文档应递交公告机构)必须提醒的是,技术文档必须受控,在公告机构审核过程中应随时可以得到。
格式为了有效评审并降低另外的文字工作,建议技术文档分为两个部分-A部分和B部分A部分生产地必须在制造商处,在合格机构评审时在制造商处可以获得。
如果制造商不在欧盟范围内,A部分必须在授权的欧盟代表处可以获得。
B部分可以独立于A部分,它的生产地接近A部分,可以是制造商的另一个生产点,或位于海外的OEM供应商。
A部分包含了大多数重要的与合格评定程序有关的技术数据的概括,包括:∙制造商和欧盟代表名称和地址∙质量管理体系覆盖的生产地点∙产品描述,包括产品标识(型号等)预期用途的描述产品附件(适用情况下)销售单元的一体性部件(适用情况下)产品及其附件的分类选择的合格评审途径产品的简要历史(包括已有的批准文件)∙制造商采用的协调化标准清单和/或满足基本要求的解决方案(例如,基本要求清单-见附录A)∙全部的制造和检验计划∙风险分析(见附录B)∙临床数据(见附录C)∙标签,如产品标签,说明书,病人信息,广告材料∙合格声明B部分包含所有的检测和验证报告,产品有关的质量体系信息,产品的详尽描述如设计图,产品数据参数,制造过程描述等。
如果B部分是分散的,应建立一个控制目录,列出每个相关文件。
基本要求:毫无疑问,技术文档的灵魂是能证明符合基本要求的技术信息。
根据MDD的第5章,符合基本要求的前提是符合协调化标准。
然而,实际上,并不是所有的医疗器械存在协调化标准。
因此,在协调化标准不存在时,使用其它存在的国际性标准,国家标准,一些贸易组织标准,例如ISO, IEC, AFNOR, DIN, ANSI, AAMI, ASTM等。
制造商可以选择任何可接受的技术方法以符合基本要求。
然而必须注意,利用技术基本原理而非存在标准,将导致公告机构和合格机构的详细审查和时间上的拖延。
一个证明符合基本要求的简易方法是通过检查表,它系统列出了每个基本要求和证明符合性的方法。
这个检查表减轻了制造商,公告机构,合格机构的压力。
因此,附录A包含了基本要求检查表和另外的解释要求的指南。
附录A的检查表可以扩展到更多栏目,包括要求的应用,证明符合性所使用的标准,支持文件的位置,部分符合标准的技术原理,评论等。
标准欧洲标准委员会,CEN,CENELC采用的标准用前缀EN进行识别。
一旦一个标准在欧盟官方杂志发表,就称为协调化标准。
协调化标准清单将定期在欧盟官方杂志发表。
因为非欧洲的制造商对跟踪这些欧盟特殊标准存在困难,我们建议他们利用欧洲贸易组织,例如IAPM,EUCLMED,或标准服务以获得这些信息。
附录D包含标准组织,欧盟贸易组织,标准服务,欧盟官方杂志来源的名称和地址,他们将帮助提供标准的现有状态和标准拷贝。
合格声明根据附录II的第2部分,附录VI的第2部分, 附录VII的第1部分,制造商必须发表一份符合MDD要求的声明。
这份文件必须经公司管理者代表批准。
可以是一个产品型号,或一个系列产品;无需为每一批产品准备。
合格声明的格一个成员国家可能要求技术文档的A部分使用官方语言,如果能懂可以不翻译。
在需要翻译的情况,允许文件拥有者额外的时间递交第一部分的内容给检查机构。
而且,关于翻译不能增加更多条件,例如要求有授权的翻译,官方翻译或类似要求。
8.技术文档控制一旦编制结束,技术文档应受控。
虽然不必在文件受控中,但仍需某些程度的控制。
文件必须随时包含产品和支持数据的正确描述。
因此,它必须联系于制造商工程变化系统,就如510(K)和PMA的文件。
如果产品发生变化,技术文档需要更新或评估。
技术文档控制失败将导致递交给合格机构的文件过期或数据矛盾。
这将导致制造商因技术文档不充分而错误使用CE 标志而面临惩罚,。
如上所说,技术文档的A部分将置于制造商处。
如果制造商不在欧盟,A部分的复印件必须在授权代表处可以获得。
因为B部分分散,授权代表处不必一定有复印件。
9.保密成员国必须确保所有涉及申请MDD的相关方在执行任务过程中对所获信息进行保密(见MDD的20章)。
特别是成员国授权的公告机构,对技术文档的信息评审等过程保密尤为重要。
附录A 基本要求检查表附录B 风险分析附录C 临床数据风险分析1.风险分析要求风险分析是许多EU和FDA法规的要求,例如指令93/42/EEC,FDA QSR.风险分析的标准已制定,如:EN1441 医疗器械-风险分析ISO/DIS 14791-1“医疗器械-风险分析”第1部分-医疗器械风险分析的应用IEC 601-1-4:1996(EN60601-1-4,DIN EN60601-1-4) 可编程的电子医疗器械的安全要求风险分析不仅是法规要求,也是研发过程中确定安全要求的有效工具,因而是研发过程的一部分。
2.已存在产品的风险分析在给一个医疗器械贴CE标志之前,MDD需要进行风险分析,不管产品是新的还是已上市的。
对于已上市产品,风险分析必须进行回顾分析。
如不幸发现上市产品的回顾分析显示了风险是不可接受的,那么必须对产品重新设计再进行CE贴标。
3.风险分析和FMEA风险分析从大的方面看,可用两个不同方法进行。
一种是从上而下,符合EC指令的风险分析要求。
另一种是从下而上,或失败模式效果分析(FMEA),它们相互补充。
他们在设计的不同阶段,不同的目的都能发挥作用。
“从上而下”的风险分析从上而下的方法从病人(或操作人)有关的医学危险(如失血,电休克)开始。
每一个风险它都列出了可能的原因,发生的可能性。
必要时制定好对策。
这种风险应在产品开发前进行,因为这是产品的安全要求。
“从下而上”的风险分析(FEMA)从下而上的方法开始于已形成的产品设计,对任何一个子系统,任何一个组分,它列出了潜在的失败模式,分析了后果。
这也常称作FMEA,是证明设计要求的有效工具(如失败-安全设计要求,通过自测检测失败的要求)。
FMEA的输入是产品的设计输出(至少是文件上的),因而不能在设计输入阶段完成。