GMPbook2
GMP偏差处理中英文

1.PURPOSE 目的Whenever a product, material or system fails to meet the specifications or in the event of a failure to comply with relevant documentation or regulatory requirements, an appropriate investigation must be undertaken, the cause(s) identified and the necessary corrective actions taken当产品、物料或系统不符合质量标准要求或某事件不符合相关文件或法规要求时,必须进行适当的调查,查明原因并采取必要的改正措施。
2.SCOPE 范围This SOP covers all failures and unplanned incidents related to Chemical components, Packaging materials, Drug Products, Processes, Systems, Equipments, Utilities and Facilities used to produce and control them.本SOP适用于处理所有失误及非计划性故障事件,含概用于产品并控制产品的化学成分、包装材料、药品、工艺、系统、设备、公共设施和厂房等。
3.DEFINITIONS/ABBREVIATIONS 定义/缩写Deviation(also known as anomaly):Any unplanned change from a written procedure/document, during manufacturing or testing or a non-conformance to approved specifications or anyfailure on GMP-related systems. Deviations are assessed according to compliance and /orthe risk they present to patient health and/or with regulatory requirements. Deviations areto be classified as “critical or major or minor”偏差(通常也称为异常):在药品生产过程中,任何与既定的程序、文件不符的非计划的变更或与批准的质量标准不符,或与GMP相关的系统失败。
欧盟GMP简介共34页文档

QRM是药品质量风险评 估、控制、沟通和审核的 一个系统程序。可以采用 前摄性或回顾性的方式。
•附录1 无菌药品的生产 •附录2 人用生物制品的生产 •附录3 放射性药品生产 •附录4 兽用非免疫药品的生产 •附录 免疫类兽药制品的生产 •附录6 医用气体生产 •附录7 草药制剂的生产 •附录8 原辅包装材料的取样 •附录9 液剂霜剂和油膏的生产 •附录10 定量吸入式气雾剂的生产
Text
基本要求2 原料药生产质量管理规范
基本要求1 目录
人用药品及兽药生产质量管理规范(基本要求1)2019年10月3日
第一章 质量管理 Quality Management 第二章 人员 Personnel 第三章 厂房与设备 Premises and Equipment 第四章 文件和记录 Documentation 第五章 生产Production 第六章 质量控制Quality Control 第七章 委托生产和委托检验Contract Manufacture and Analysis 第八章 投诉和药品召回Complaints and Product Recall
为了切实实现这一目标, 药品生产企业必须建立 涵盖GMP以及质量控制 (QC)在内的全面的质 量保证系统(QA)。药 品生产企业应以完整的 文件形式明确规定质量 保证系统,并监控其有 效性。
质量保证系统的各个方面 均 应配备足够的称职的人 员、厂房、设备和设施。 药品生产企业及产品放行 责任人(QP)还有其他法 律责任。
美国FDA / 美国注射剂协会 / 国际制药工程协会 / 药品注册标准技术要求国际协调会ICH / 世界卫生组织WHO who.int/
EU-GMP目录
基本要求1 人用药品及兽药制剂生产质量管理规范
药品gmp指南第2版培训资料

一、概述药品GMP(Good Manufacturing Practice)是指药品生产质量管理规范,是确保药品质量的重要标准之一。
为了提高药品生产企业对GMP规范的理解和执行能力,制定了药品GMP指南第2版培训资料。
二、GMP规范的重要性1. GMP规范是保障药品质量的重要保障,直接关系到患者用药的安全和疗效。
2. GMP规范可以有效地规范药品生产企业的生产流程,从源头上控制药品质量。
3. GMP规范对药品生产企业的管理和人员素质提出了严格的要求,有助于企业提升自身的管理水平和技术水平。
三、药品GMP指南第2版培训资料的主要内容1. GMP规范的基本原则和要求:详细介绍GMP规范的主要内容和要求,包括生产设备、生产环境、人员素质等方面的要求。
2. 药品生产流程的管理:介绍药品生产的各个环节的管理要求,包括原材料采购、生产工艺、包装和贮存等环节。
3. 质量管理体系的建立和执行:详细介绍了建立和执行质量管理体系的方法和要求,包括质量控制、质量检测和质量评价等方面的内容。
4. 特殊环境和设备的管理:针对特殊环境和设备的管理要求进行了详细的说明,包括洁净区的管理、设备验证、验证等方面的内容。
5. 不合格品的处理:介绍了不合格品的处理要求和方法,包括不合格品的处置、记录和报告等方面的内容。
四、药品GMP指南第2版培训资料的培训方式1. 培训课程:通过专业的培训课程,向药品生产企业的管理人员和生产人员传授GMP规范的基本知识和要求。
2. 实地考察:组织实地考察,让企业代表亲自感受GMP规范的执行情况,加深对规范要求的理解和认识。
3. 专家讲座:邀请行业资深专家进行讲座,共享GMP规范的实际执行经验,帮助企业更好地理解和执行规范要求。
五、药品GMP指南第2版培训资料的实施效果1. 提升了企业对GMP规范的理解和执行能力,加强了对质量管理的重视和重视。
2. 改善了药品生产环境和设备设施,提高了产品质量和生产效率。
GMP审核项目

Document Control Program文件控制程序
1.201
§211.22(a) Does the QA unit have a person or department specifically charged with the responsibility of designing, revising, and obtaining approval for production and testing procedures, forms, and records?
所有培训是否记录有书面资料,表明培训日期,培训类型,参训人员和培训人的签名?
1.305
§211.25 Are training records readily retrievable in a manner that enables one to determine what training an employee has received, which employees have been trained on a particular procedure, or have attended a particular training program?
1.106
If any portion of testing is performed by a contractor, has the Quality Assurance unit inspected the contractor’s site and verified that the laboratory space, equipment, qualified personnel and procedures are adequate?
EIS功能配置说明

EIS 系统功能配置说明(仅供内部使用)文 档 作 者: ______________ 日期:____/____/____ 开发/测试经理: ______________ 日期:____/____/____ 产 品 经 理: ______________ 日期:____/____/____ 文 档 管 理 办: ______________日期:____/____/____深圳市众方信息科技有限公司版权所有 不得复制深圳市众方信息科技有限公司产品名称 产品版本密级EIS内部公开 文档编号:共 38 页修订记录目录1引言 (5)2升级 (5)2.1平台升级说明 (5)2.1.1升级 (5)2.1.2备份数据库 (6)2.2终端升级说明 (6)2.2.1通过平台升级终端 (6)2.2.2在终端上直接升级 (7)2.2.3通过provision加载 (7)3功能配置和查看 (7)3.1平台功能配置 (7)3.1.1更改IP地址 (7)3.1.2Freelink配置 (9)3.1.3修改MGCPSIP注册端口 (9)3.1.4pra配置 (9)3.1.5SIP中继配置 (10)3.1.6呼出呼入路由配置 (11)3.1.7计费服务器配置 (12)3.1.8全局O口配置方法 (12)3.1.9号码替换配置 (12)3.1.10RS路由配置(业务不对外) (12)3.1.11H323配置 (15)3.1.12标准中继对接 (16)3.1.13虚拟业务配置 (16)3.1.14虚拟小交业务配置 (26)3.1.15P2P配置(功能不对外) (26)3.1.16计费管理(半实时计费) (27)3.1.17ADA常用命令 (28)3.1.18信息查看 (31)3.1.19数据库操作命令 (32)3.2终端配置 (33)3.2.1注册配置 (33)3.2.2路由配置 (36)4问题定位跟踪 (36)4.1.1cc跟踪 (36)4.1.2sip跟踪 (37)4.1.3计费跟踪 (37)4.1.4mgcp跟踪 (37)4.1.5H323跟踪 (38)4.1.6pra跟踪 (38)4.1.7新信令跟踪(trace) (38)4.1.8版本单通定位手段(适用于EIGA和EIGB) (38)5其它 (38)EIS系统功能配置说明1引言此文档主要是针对现在EIS系列产品的功能使用的说明书,阐述了如何对平台和终端进行升级、配置和问题定位。
欧洲GMP药品现场检查项目表

Audit Checklist for Drug IndustryDisclaimerThe following checklist is intended to aid in the systematic GMP audit of a facility that manufactures drug components or finished products.The adequacy of any procedures is subject to the interpretation of the auditor. Therefore, the author accepts no liability for any subsequent regulatory observations or actions stemming from the use of this audit checklist.Instructions for Using this Audit Checklist:Before starting an on-site audit, plan the audit. Review past audits, note indications of possible problem areas and items, if any, that were identified for corrective action in a previous audit. If you are not already familiar with this facility, learn the type of product produced here and how it is organized by personnel and function. What does your "customer", ., your superior or senior facility management, expect to learn from this audit?The checklist is to be used with a notebook into which detailed entries can be made during the audit.While the checklist is to guide the auditor, is not intended to be a substitute for knowledge of the GMP regulations.Although a single question may be included about any requirement, the answer will usually be a multi-part one since the auditor should determine the audit trail for several products that may use many different components. Enter details in your notebook and cross reference your comments with the questions.At least three production batches should be selected for thorough analysis to include: (a) traceability of all components or materials used in the subject batches,(b) documentation of raw material or component, in-process, and finished goodstesting for the subject product batches, (c) warehousing and distribution records as they would relate to a possible recall.Responses entered on the checklist should be consistent. "X" is recommended for "NO"; a checkmark for "YES"; "n/a" for not applicable to questions that do not apply. An asterisk and notebook page number should be entered on the checklist to identify where relevant comments or questions are recorded in your notebook.The notebook used should be a laboratory-type notebook with bound pages. the notebook should be clearly labeled as to the audit type, date, and auditor(s). Many auditors prefer to use a notebook for a single audit so it may be filed with the checklist and the final report.The references to sections in the GMP regulation are for your convenience shoulda question arise. In some instances, two or more sections within the GMPregulation may have bearing on a specific subject. The headings in the GMP regulation will usually offer some guidance on the areas covered in each section.A general suggestion for a successful audit is to spend most of your time on major issues and a smaller portion of your time on small issues. there may be observations that you may wish to point out to supervisory personnel that deserve attention, but do not belong in an audit report because they are relatively insignificant. By the same token, too many small items suggests a trend of non-compliance and deserve attention as such. When citing these, be specific.Question Instructions/questions (note any exceptions and comments in notebook).Yes, No, or NA Document N°; archiving areaGeneral Controls。
学习电脑_Book2_V2.00_一卡恢复使用说明
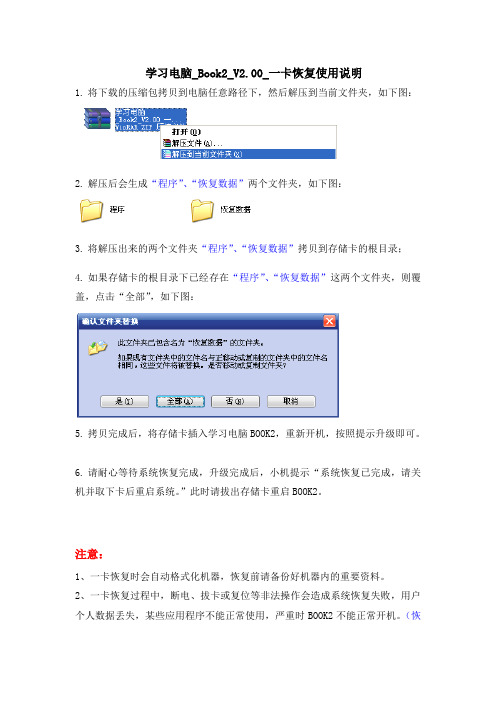
学习电脑_Book2_V2.00_一卡恢复使用说明
1.将下载的压缩包拷贝到电脑任意路径下,然后解压到当前文件夹,如下图:
2.解压后会生成“程序”、“恢复数据”两个文件夹,如下图:
3.将解压出来的两个文件夹“程序”、“恢复数据”拷贝到存储卡的根目录;
4.如果存储卡的根目录下已经存在“程序”、“恢复数据”这两个文件夹,则覆盖,点击“全部”,如下图:
5.拷贝完成后,将存储卡插入学习电脑BOOK2,重新开机,按照提示升级即可。
6.请耐心等待系统恢复完成,升级完成后,小机提示“系统恢复已完成,请关机并取下卡后重启系统。
”此时请拔出存储卡重启BOOK2。
注意:
1、一卡恢复时会自动格式化机器,恢复前请备份好机器内的重要资料。
2、一卡恢复过程中,断电、拔卡或复位等非法操作会造成系统恢复失败,用户个人数据丢失,某些应用程序不能正常使用,严重时BOOK2不能正常开机。
(恢
复失败,可以使用“一卡恢复”重新恢复或使用“超级系统恢复”进行恢复。
)3、如果刚刚开始恢复时因为断电、拔卡或复位等操作造成失败,可能导致无法再次恢复,若出现此现象请使用“超级系统恢复”进行恢复。
4、请使用4G或4G以上存储卡进行恢复。
修改记录:
1.增加同步学习、同步练习、密卷点评;
2.整体优化了所有功能和交互。
确认
2.7.4 确认确认包括设计确认(DQ)、安装确认(IQ)、运行确认(OQ)和性能确认(PQ)。
新版中国GMP对这几种类型确认所应实现的目标有如下要求:《药品生产质量管理规范》2010修订版:第一百四十八条应建立确认和验证的文件和记录,并能以文件和记录证明达到以下预定的目标:1.设计确认(Design Qualification)应证明厂房、辅助设施、设备的设计符合预定用途和本规范要求;2.安装确认(Installation Qualification,IQ)应证明厂房、辅助设施和设备的建造和安装符合设计标准;3.运行确认(Operational Qualification)应证明厂房、辅助设施和设备的运行符合设计标准;4.性能确认(Performance Qualification)应证明厂房、辅助设施和设备在正常操作方法和工艺条件下能持续有效地符合标准要求。
厂房、设施、设备等的生命周期包含设计、采购、施工、测试、操作、维护、变更以及退役,而确认工作应贯穿生命周期的全过程,确保生命周期中的所有步骤始终处于一种受控的状态。
通过下图可以看出确认与生命周期的对应关系。
图2-16 确认与生命周期关系示例确认中的测试项目、范围和程度由风险分析而定。
当发生变更时,应执行变更管理程序并通过风险评估确定是否需要进行再确认。
A.设计确认(DQ)新的厂房、设施、设备确认的第一步为设计确认(DQ).设计确认是有文件记录的对厂房、设施、设备等的设计所进行的审核活动,目的是确保设计符合用户所提出的各方面需求,经过批准的设计确认是后续确认活动(如安装确认,运行确认,性能确认)的基础。
通常,设计确认中包括以下的项目:用户需求说明文件(User Requirement Specification,URS)用户需求说明文件时从用户角度对厂房、设施、设备等所提出的要求。
需求的程度和细节应与风险、复杂程度相匹配,其中可以针对待设计的厂房、设施、设备等考虑以下内容:◆法规方面的要求(GMP要求、环保要求等)◆安装方面的要求和限制(尺寸、材质、动力类型、洁净级别等)◆功能方面的要求◆文件方面的要求(供应商应提供的文件及格式要求,如图纸、维护计划、使用说明、备件清单等)下表为建议的用户需求说明文件模板,具体内容可根据实际需要进行增减。
GMP计算机系统验证(中文39页)

第一章 概 述...................................................................................................................................2 第二章 范 畴...................................................................................................................................3 第三章 名词解释.............................................................................................................................4 第四章 英文缩写解释.....................................................................................................................7 第五章 计算机系统分类.................................................................................................................7 第六章 计算机系统发展及验证生命周期.....................................................................................8 第七章 验证实施过程...................................................................................................................10
CAPA(cGMP培训系列2)_2

and corrective action plan required.
• Warning Letter– more serious, available
to the public
• Fines, stop product
4
What are the symptoms of an eCAfPfAecstyisvteem? 好的预防纠偏系统有 那些特征?
• A reduction in quality i•sAsureesduction in the severity of i•sMsourees preventive actions over t•iMmoere consistent p•rIomdpurcotvse/dprcoucsetsosmeesr s•aBteitstfearctbiuosniness results
auodbistervations, ianutdeirtnsaland external audits
• Quality Records
▫ Periodic r▫evPireowcess c▫onTtersotling results, trends,
• Peotsct Market
dat▫a Frequency and
used
• Document
▫ Dates of i▫nvDeasttaigraetviioenwed (data
sources, records, dates)
Cause and Effect, 6 M’s 5 Why’s, etc.
▫ Corrections or
GMP计算机化系统管理规程

计算机化系统管理规程目的:规范我公司在药品生产质量管理过程中应用的计算机化系统管理,确保其运行稳定、准确、真实、可靠,计算机化系统代替人工操作时,不会对产品的质量、过程控制和其质量保证水平造成负面影响,不增加总体风险。
范围:适用于我公司在药品生产质量管理过程中应用的计算机化系统的管理。
责任:设备部、质量保证部、质量控制部、生产研发技术中心、综合制剂生产部、原料药生产部、物料部等相关部门计算机化系统验证、使用、维护、管理人员均应对本规程的实施负责。
内容:1. 制订依据1。
1 中国法规:《药品生产质量管理规范(2010版修订)》及GMP附录1 计算机化系统1。
2 ISPE国际制药工程协会:《ISPE GAMP5 良好自动化生产实践指南》1。
3 美国法规:21 CFR Part 211,药品制剂cGMP;21 CFR Part 11,电子记录和电子签名1.4 欧盟法规:欧盟GMP附录11,计算机化系统2. 相关部门职责设备部:对公司计算机化系统全面负责,负责协同相关部门对公司主要计算机化系统的规划、选型、购置、安装、调试、验收、验证、登记、配备、维修、检查、改造、退役和更新的全过程进行综合管理.协同质量保证部、信息部从技术角度完成供应商审计工作,包括供应商技术能力评估、软件开发标准及软件测试能力审核、程序编制人员的资格审定、硬件开发及制造能力评估等。
信息部:配合设备部负责计算机化系统软件方面的管理,参与计算机化系统软件的规划、选型、购置、安装、调试、验收、验证、维修、检查、改造及供应商管理等管理工作,并在计算机化系统使用及维护过程中提供技术支持.质量保证部:监督计算机化系统的管理工作,按照《计算机化系统验证总计划管理规程》规定的职责管理计算机化系统验证工作.各使用部门:在设备部、信息部指导下正确验证、使用、维护计算机化系统。
3. 具体内容:3.1 计算机化系统定义计算机化系统由硬件、系统软件、应用软件以及相关外围设备组成的,可执行某一功能或一组功能的体系。
欧盟GMP简介

Conte nts 欧盟概况欧盟药品审评及检查欧盟GMP基本要求1 附录1无菌药品的生产经济领域 公共卫生 社会方针和政策科研消费者和环保 合作程序(共同 决定程序)条约〜法令、法规〜指导方针、指南d 951-7-25欧洲煤钢共同体d 958-1-1欧洲经济共同体 欧洲原子能共同体1967-7-1 欧洲共同体d 993-11-1欧共体更名欧盟截止2004年1月25成员国,4.56亿丿欧盟概况・EU欧洲联盟•EuropeanCouncil欧洲理事会•Council of•Commission ofEU欧盟委员会•European Parliament 欧洲议会•Court of justice欧洲法院•支柱产业之一•占全球40%Ministers欧盟理事会•欧盟委员会企业理事会下属药品部•EMEA•EDQM药品安全性评价药品有效性评价EMEA使命(Eu「opean Agency for the Evaluation of Medicinal Prod欧盟药品审评及检查用药品委员兽药委员会用药品委员草药委员会4个专家委员会•由EMEA 颁布实施 的一些技术性指南(Guidelines)和 对一些法规条款所 做出H ? E ; I 0欧盟药品审评及检查欧盟药事法规三层次品注册监督管理程GMP 指南技术性指南 我术注释•由欧盟委员会依? 有关法冬和法规H 颁布实施的药品哲 册监督管理程序牙 GMP 指南•由欧盟议会和 理事会颁布实才 少部分由欧盟M 会颁布实施•Directives •Regulations的技术注释(Notes)是国家药品监督管理部" 门认可,负责确保每批11 药品己经按国家相关法律法规生产、检验和放行的人员。
-------- X 法规保证2001/83/EC 第48、49> 5152条。
既强化了企业对药品质量的个人责任,也有助于保证产品相关信息的可追溯性。
产品放行责任人- —---- QPQualified Person合成、提取或发酵获得的有机 聾无机原料药(活性成分)或组分一 在药品生产或制备过程中,使用有传染 _性克雅氏症病原体风险的任何产品-任何草药相关的制剂tothe monoqraphs of the Euj^Dean Pharmacopoeia)评及检查H ? E :I欧盟药品审 EDMF 欧洲药品主档案文件对专论控制原料药化学纯度和微生物质量适用性的评估 T 根据新的通则,对降低TSE 风险进行评估 一或对以上二方面同时评估欧盟常用网址一览表洲药晶管理局 http://www.emea.eu.int药品检查合作组织PIC/S •・.•・・1 欧洲药品质量理事会Tjlfp:〃www.pheur.og))品注册标准技术要求国际协调会ICH /丿Q 美国FDA /•••“•6美国注射剂协会 /O国际制药工程协会/•世界卫生组织WHO http://www.who.int/基木要求1人用药品及兽药制剂生产质量管理规范•附录1无菌药品的生产•附录2人用生物制品的生产•附录3放射性药品生产•附录4兽用非免疫药品的生产•附录5免疫类兽药制品的生产•附录6医用气体生产•附录7草药制剂的生产•附录8原辅包装材料的取样,附录9液剂霜剂和油膏的生产Q 付录io定量吸入式气雾剂的生•附录11计算机系统•附录12药品生产中电离辐射的应用•附录13临床试验用药的生产附录14人血液或血浆制品的生产•附录15确认和验证•附录16药品放行责任人签发证书和放行批产品•附录17参数放行[附录19对照样品和留样驸录20质量风险管理基木要求2原料药生产质量管理规范EU-GMP目录欧盟GMP附录Mil厂欧盟GMP附录•:•人用药品及兽药生产质量管理规范(基本要求1) 2005年10月3日彳勺第一章质量管理Quality Managemento 第二章人员Personnel ;第三章厂房与设备Premises and Equipment第四章文件和记录Documentation11 第五章生产Production __________________________________彳号第六章质量控制Quality Control __________________________(爭第七章委托生产和委托检验Contract Manufacture and Analysis彳爭第丿l章投诉和药品召回Complaintsand Product Recall暫勒療则:药品生产企业必须确保所生产的产品适用于预定的用途, 符合药品注册批准的要求,并不使患者承担安全、质量和疗效的风险。
数据完整性和计算机管理GMP要求

数据的生命周期:数据包括原始数据自初始生产和记录,到处理和 (包括转移或移植)、使用、数据保留、存档/恢复和重建的整个 生命阶段。
源(元)数据(Metadata): 指描述其他数据属性的数据,提供 语境和含义。一般来说,这些数据描述数据的结构、要素、内在 关系和其他数据特征。 它还允许数据追踪至个体。如产品A批号 267, 有2kg等;没有源数据,数据就没有意义。
20
数据管理基本要求
数据必须符合 ALCOA 原则: 1. A -- 可追踪至生产数据的人 2. L -- 清晰、在数据的整个生命周期内均可以获得,必要时能
永久保存 (如长达30年) 3. C -- 同步 4. O -- 原始(或真实复制): 可根据原始数据对数据生产的整
个活动进行重现 5. A --准确性
18
法规要求
欧盟GMP简介

EU-GMP目录
基本要求1 人用药品及兽药制剂生产质量管理规范 欧盟GMP附录
附录1 无菌药品的生产 •附录2 人用生物制品的生产 •附录3 放射性药品生产 •附录4 兽用非免疫药品的生产 •附录5 免疫类兽药制品的生产 •附录6 医用气体生产 •附录7 草药制剂的生产 •附录8 原辅包装材料的取样 •附录9 液剂霜剂和油膏的生产 •附录10 定量吸入式气雾剂的生产
thinkbook plus 2使用手册

thinkbook plus 2使用手册
1.触控式主页按钮
在Eink应用程序或壁纸界面,按此按钮可返回E-ink主页。
在Eink主页上,按此按钮可显示壁纸。
按住该按钮5秒钟可重置E-ink屏幕。
注:如果LCD屏幕已打开,您可以按此按钮启用E-ink屏幕。
2.电源指示灯、指纹读取器指示灯
电源指示灯
快速白色闪烁2次:计算机已开启。
缓慢白色闪烁:计算机处于睡眠模式。
白色长亮:计算机正在使用中。
熄灭:计算机处于关闭或休眠模式。
指纹读取器指示灯
绿色闪烁然后绿色长亮:指纹读取器已准备好扫描您的指纹。
白色长亮:指纹扫描成功。
琥珀色闪烁然后绿色长亮:指纹扫描已失败(前三次以内)。
琥珀色闪烁然后白色长亮:指纹扫描已失败(超过三次)。
3.E-ink触控式屏幕
做笔记、素描、阅读内容或操作某些Windows应用程序。
屏幕支持触控功能。
gmp指南

GMP指南什么是GMP?GMP(GNU Multiple Precision Arithmetic Library)是一个用于高精度计算的C 语言库。
它提供了一组函数,用于执行精确的整数和浮点数运算。
GMP允许程序员处理比标准数据类型更大的数值,甚至可以进行高精度的计算。
安装GMP要在您的计算机上使用GMP库,您需要先安装它。
下面是在不同操作系统上安装GMP的步骤。
在Linux上安装GMP在大多数Linux发行版中,您可以使用包管理器来安装GMP。
例如,在Ubuntu上,可以使用以下命令安装:sudo apt-get install libgmp-dev如果您使用的是其他Linux发行版,请查阅相关文档以了解如何安装GMP。
在Windows上安装GMP在Windows上安装GMP可以通过编译源代码或使用预编译的二进制包。
编译源代码要编译GMP源代码,请按照以下步骤操作:1.下载GMP源代码压缩包(.tar.gz格式)并解压缩。
2.打开命令提示符或PowerShell,并进入解压缩后的目录。
3.运行以下命令来配置安装选项:./configure4.运行以下命令来编译GMP:make5.最后,运行以下命令来安装GMP:make install使用预编译的二进制包如果您不希望编译源代码,可以使用预编译的二进制包。
您可以从GMP官方网站下载适用于您的Windows版本的二进制包,并运行安装程序进行安装。
使用GMP进行高精度计算一旦您成功安装了GMP,您就可以在您的程序中使用它进行高精度计算了。
下面是一个简单的例子,展示了如何使用GMP进行整数加法:```c #include <stdio.h> #include <gmp.h>int main() { mpz_t num1, num2, sum;mpz_init(num1);mpz_init(num2);mpz_init(sum);mpz_set_ui(num1, 1234567890);mpz_set_ui(num2, 987654321);mpz_add(sum, num1, num2);gmp_printf(\。
IBM THINKPAD X22笔记本电脑

IBM THINKPAD X22笔记本电脑
范岳
【期刊名称】《电子测试》
【年(卷),期】2002(000)004
【摘要】@@ T HINKPAD X22笔记本电脑的外形方正,颜色沿用IBM笔记本电脑的传统黑色,右下角有醒目的IBM商标.整机的体积并不大,整个机器分为主机和扩展坞两部分,如果拿掉扩展坞,主机变得更加轻巧,大小和重量都减小了许多.显示屏的打开方式为两侧侧开式,需要双手同时动作才能打开机器.
【总页数】1页(P70)
【作者】范岳
【作者单位】无
【正文语种】中文
【相关文献】
1.4个配角1台戏:打造无线简报套装——NEC LT265+无线投影机 IBM ThinkPad X40笔记本电脑 IOGEAR简报鼠标 SHURE SM58+ULX无线麦克风[J],
2.自有一T IBM ThinkPad T43笔记本电脑 [J],
3.小黑新生——IBM ThinkPad X41笔记本电脑 [J],
4.IBM ThinkPad X22笔记本电脑 [J], 范岳;
5.无线安全便携——全新IBM ThinkPad X22笔记本电脑 [J], ;
因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
变更控制
• 定义:
– 改变现行工艺参数,生产条件,设备,测试方法 或标准,人员操作,流程等而又不在SOP允许 范围内的称为变更.其目的是为了技术更新 或运行更合理.
• 举例:
– 降低顶盖和勺子的损耗率
如何变更?
• • • • 提议人填写[变更申请表] 部门内审批 3个工作日内QA,相关部门及总监审批 变更实施部门一个月内提交相应数据,评 价结果 • QA确认结果,保存申请表
• 始终按要求穿工衣,保持扣紧,经常检 查是否有破损,坚持每天换洗 • 始终按要求工鞋,定期清洗 • 按要求戴发帽,将头发和耳朵全部包住 • 按要求戴口罩,要遮盖口和鼻子 • 按要求洗手,戴手套或消毒手部 • 当你有感染,感冒,咳嗽,腹泻或开放 性伤口,必须立即向你的上级报告 • 避免抓搔,抠鼻子和嘴等不良习惯
• 使用设备,吸尘管前确保其清洁并已消毒
按规定配置和使用洗涤剂,消毒剂 工作时小心,避免造成粉尘或喷洒 坚持执行生产前清场并记录 坚持执行[洁净区的卫生程序] 始终保持良好的个人卫生
1.4 防止人的污染----讲卫生
是每个人的职责 是去除赃物而不是隐藏之 是一种纪律 养成习惯
2. 如何做好记录
2.1 即时
• 进行每一步操作,读数和检查后必须立即记录并 签名. • 不能是: ----几项工作完成后 • ----一天工作结束后 • ----更不能一周以后
2.2 真实
必须有第二个人签名证实: 某项工作已经做了 亲自检查该工作是否正确 不能够:
相信你的同伴 不及时签字
千万别麻痹大意!
是非题: • 千万不能用手直接接触产品或原辅料 • 操作中不要在地上捡东西 • 尽量减少谈话,不能打闹,嘻耍,唱歌 或吹口哨 • 生产操作中不要用手碰其他的操作工 • 时刻保持警惕,发现任何可能造成污染 的迹象立即报告
问题
• 如何在预进间更衣?
• 戴发帽的要求?
• 为什么要戴口罩?有要求吗? • 如何洗手?
GMP 培训
• II. 卫生
• • • • 防止污染 讲卫生 保持清洁的规则 洁净区的卫生
1. 污染的来源
物料:如被污染的原料,包装材料等 环境:如开门,开窗 生产:如一个房间同时生产两种产品 人本身
最大污染源——人体和人手!
1.1 防止物料污染
生产中必须密切注意——
原料包装是否完好 原料包装外表面是否清洁 观察原料是否有异常 包装材料外观是否良好 与产品接触的包材内表面是否清洁
偏差控制
• 定义:
– 实际工艺参数,生产条件,设备运行状态,测试 方法或标准,原材料,人员操作,记录等与现行 SOP或标准要求不一致而未经变更批准的称 为偏差.
• 举例:
– 生产半批MMS
发生偏差时怎么办?
• • • • • • 岗位人员向主管报告 填写[偏差报告] 24小时内评估和批准(包括QA和总监) 偏差操作记录, 按要求检查记录数据 QA一个月内对偏差的影响确认核实 QA保存[偏差报告]及记录
有程序吗?
• 生产区附近配备合适清洗设施,如自来 水,肥皂,干手器及卫生间 • 水的质量控制及质量跟踪 • 地漏,排水,防止溢流 • 及时处理工艺废物 • 生产区不允许进食,吸烟及不卫生行为 • 穿戴工衣,帽 • 定期培训员工
讨论
• 我们有哪些清洁设施? 如何使用?
• 生产过程中的废物有哪些?
• 如何处理废物?
永远保持整洁(包括你自己)
• 保持个人卫生包括哪些方面?
• 包括身体,头发,手,指甲和衣服,勤 剪指甲,坚持便后洗手,进入生产车间 前洗手, 穿工衣, 戴发帽, 口罩, 穿工鞋, 勤 换洗, 避免抓搔,抠鼻子和嘴等不良习惯, 有感染立即报告
3. 保持清洁防止污染的规则
是非题: • 生产操作时不使用化妆品,不戴戒指, 饰物, 但可带手表,掌握时间. • 生产操作区不允许进食,饮水,咀嚼; 也不能将食物和药品带入生产区. • 所有公司的资料都可带入操作岗位.
GMP 培训
• III.记录
• • • • • 记录 做好记录的规则 偏差控制 变更控制 验证
1. 记录
1.1 清楚,准确地做好工作记录 并确保复核----GMP的基本原 则
• 1.2 为什么要做记录? • 记录所有关键操作,便于确认和 追溯
1.3 我们有哪些记录? 1.4 生产批记录包括什么?
讨论
• 如何防止生产过程中的污染?
• 请举例说明.
2. GMP的卫生要求
• 保持厂房清洁,防止昆虫,鼠类进入
如何防止昆虫, 鼠类进入? 那么6号门和北门呢?
• 书面程序规定清洁卫生负责人及清洁周 期,所用设备,工具,方法和材料
有程序吗?
• 规定清洁剂或消毒剂的使用,避免污染 设备和产品
1.2 保持环境清洁
保证生产区有稳定的温湿度环境
• 生产前确认温湿度符合要求
保持地面干净,门窗,设备表面不积聚 粉尘,天花,墙壁无脱落物 生产物料堆放整齐,生产工具,计量仪 器,真空管等放在规定的地清洁及执行清场
坚持执行设备清洁和消毒程序
验证
• 定义:
– 指能证实任何程序,生产过程,设备,物料,活动 或系统确实能导致预期结果的有文件证明的 行为.
• 范围:
– 生产工艺, 厂房与设施, 生产设备, 清洁系统, – 消毒方法及效果, 控制区环境, 检验方法等
2.3 准确
必须有统一的计量单位 精确的数值和统一的有效数值
2.4 整洁, 清楚
– – – – 字体要端正, 清晰 格式要对齐 记录表格要保持干净, 不能乱图乱画 想一想:
• 记录后谁看? • 记录保存多少年?
3. 如何修改错误的记录?
– 3.1 在错误处划一横线(还能看清原来的 数据) – 3.2 在旁边写上正确的文字或数字 – 3.3 填写日期和签名 • 例如: 重量:360.25公斤 • 360.55 lily 13/12/2000