2_15_十六二酮的合成
化妆品25种膏霜类配方大全

化妆品25种膏霜类配方大全.txt45想洗澡吗?不要到外面等待下雨;想成功吗?不要空等机遇的到来。
摘下的一瓣花能美丽多久?一时的放纵又能快乐多久?有志者要为一生的目标孜孜以求。
少年自有少年狂,藐昆仑,笑吕梁;磨剑数年,今将试锋芒。
自命不凡不可取,妄自菲薄更不宜。
化妆品膏霜类配方(25).txt雪花膏化妆品是生活必需品,它对美化容貌,促进身心健康都有很大作用。
膏霜类化妆品,通常品种有雪花膏、营养润肤霜、雀斑霜、粉刺霜、柠檬霜、冰霜、清洁霜等;呈液体状态的有奶液、清洁奶液、营养润肤奶液等。
配方实例原料名称含量% (1) (2) (3) (4) (5) 三压硬脂酸 14.0 15.0 10.0 10.0 12.0 单硬脂酸甘油脂 1.0 1.0 1.5 1.5 1.0 十六醇1.0 1.0 3.2 2.0 3.0 18号白油 2.0 __ __ __ ___ 丙二醇 __ 10.0____ 10 甘油 8.0 __ 10.0 10.0 5.0 苛性钾 0.5 0.5 0.5 0.5 0.6 钛白粉 __ __ __ 2.0 2.0 羊毛醇 __ __ __ __ 2.0 香精适量适量适量适量适量防腐剂适量适量适量适量适量水 73.5 72.35 75 74 64.4 原料名称含量(份) (6) (7) (8) 硬脂酸 180 100 28 甘油 350 40 25 苛性钾 __ 3.8 __ 碳酸钾 18 __ 1.0 硼砂 __ __ 0.6 安息香酸 __ 0.26 __ 蒸馏水 1 500 600 130~150 香精适量适量适量〔工艺流程〕原料加热→混合乳化→搅拌冷却→静止冷却→包装。
〔制备方法〕先将硬脂酸熔化,边搅拌边加入甘油,同时徐徐升温至80℃左右;另将碱性物料溶于蒸馏水内,加热至80℃左右;然后将两种液相物相混,搅拌(单方向不间断),一直搅到接近室温,加入香精和防腐剂,再搅拌均匀。
装袋封存,即为成品。
麝香酮的合成进展(1)

收稿日期: 2007- 07- 02 作 者 简 介 : 王 宁(1982- ), 男 , 山 东 省 烟 台 市 人 , 现 就 读 于 上 海 市 华 东 理工大学化工学院, 硕士研究生。
早 在 1951 年 , Stoll M [16]就 最 早 采 用 乙 酰 乙 酸 乙酯与 1,10- 二溴癸烷反应制得 2,15- 十六烷二酮, 再 经 羟 醛 缩 合 、氢 化 通 过 控 制 单 取 代 反 应 , 经 选 择 性 水解 、纯 化 等 一 系 列 复 杂 操 作 , 最 终 得 到 了 麝 香 酮。其反应方程式为:
4 结论
虽然文献报道的合成麝香酮的方法较多, 但真 正 实现工 业 化 生 产 的 很 少 。综 上 所 述 , 采 用 插 入 甲 基法和扩环法由于原料不易得, 并且合成步骤较烦 琐, 很 难 得 到 较 大 的 发 展 。笔 者 经 过 实 验 认 为 采 用 乙酰乙酸乙酯和 1,10- 二溴癸烷经过相转移催化反 应合成 2,15- 十六烷二酮, 再用 TiCl4- Bu3N 或碘化 乙基锌作为环合剂进一步合成脱氢麝香酮, 然后加 氢制 备 麝 香 酮 的 方 法 是 较 为 经 济 、适 用 的 , 可 以 继 续探索寻求最佳反应条件, 从而实现麝香酮的工业 化生产。 参考文献:
[4] 迟 程. 中 国 药 用 动 物 医 学 文 献 库[M]. 昆 明 : 云 南 民 族
Depres JP[14]等人提出的十三元扩环方法非常独 特新颖, 总产率达到 50%,是一种极有发展潜力的方 法。他们先用格氏试剂制得 1- 甲基环十三碳烯, 然 后与二氯烯酮加成, 氢化脱去卤素就可得到 dl- 麝 香 酮。此 外 以 十 三 元 环 为 原 料 , 通 过 两 碳 扩 环 反 应 合成麝香酮的方法不多, 而且步骤普遍较长, 原料 不易得, 没有实际应用的价值。
利伐沙班合成路线图解

利伐沙班合成路线图解本文综述了已经报道的利伐沙班及其关键中间体4-氨基苯基-3-吗啉酮的合成路线,分别从原料、反应条件、收率等方面对每条路线进行了总结。
根据原料易得程度、路线长短、条件是否温和、收率的高低,得出以N-苯基乙醇胺为起始原料,C路线的合成路线比较适合大量制备,这为大规模工业化生产提供了依据,希望对实际生产有所帮助。
标签:利伐沙班;合成路线;图解利伐沙班(Rivaroxaban,商品名Xarelto)是一种口服抗凝血药物,由拜耳公司研发。
2008年分别在加拿大和欧盟上市,用于预防成年患者髋关节及膝关节置换手术后的静脉血栓栓塞及肺栓塞的形成[1-3]。
2011年7月经过美国FDA 批准上市,同时在静脉血栓患者的二级预防、治疗心房颤动和急性冠状动脉综合症等领域进行临床试验。
利伐沙班通过高选择性直接抑制Xa因子达到抗凝血作用,与传统抗凝血药物相比,具有服用方便、起效迅速、安全性高等特点[4-5]。
根据不同起始原料及工艺,本文对目前已经报道的利伐沙班(1)及其关键中间体4-氨基苯基-3-吗啉酮(19)的合成路线进行了总结,有利于利伐沙班的工业合成。
1 中间体19的合成1.1 以2-(2-氯乙氧基)乙酰氯为起始原料2-(2-氯乙氧基)乙酰氯,在碳酸钾作用下和对硝基苯胺(24)发生酰化反应得23(83.6%),23在碱性条件下发生分子内关环反应得20(97%)[6-7],然后经过催化氢化反应得到19(85%)[11],催化剂为活性镍。
三步反应得到中间体19,总收率较高(69%),但是起始原料2-(2-氯乙氧基)乙酰氯不易得,需要另行制备。
1.2 以对氟硝基苯为起始原料对氟硝基苯(26)与吗啉溶于DMSO中,100℃下反应4 h生成25(98%),然后将25溶于二氯甲烷中,在苄基三乙基氯化铵、高锰酸钾作下,回流10 h后,生成了20(30%)[8-9],雷尼镍将其还原成19[11]。
这种方法第一步收率很高,但是在使用高锰酸钾氧化的时候,副反应多,收率低(30%),有产物不易纯化的问题,不适于大量制备。
二苯乙二酮的制备-2

二苯乙二酮的制备
一、实验目的
1.了解安息香氧化合成二苯基乙二酮的氧化剂的选择
2.熟练掌握回流、重结晶等实验操作
二、实验原理
二苯乙二酮可以由安息香经氧化制得。
氧化剂可以为浓硝酸,但反应生成的二氧化氮对环境污染严重。
也可以使用Fe3+作为氧化剂,铁盐被还原成Fe2+。
本实验改进后采用醋酸铜作为氧化剂。
这样反应中产生的亚铜盐不断被硝酸铵重新氧化成铜盐,硝酸铵本身被还原成亚硝酸铵,后者在反应条件下分解为氮气和水。
改进后的方法在不延长反应时间的情况下可明显节约试剂,且不影响产率及产物纯度。
三、试剂
2.15 g安息香、1 g硝酸铵、冰醋酸、2%醋酸铜溶液、75%乙醇
[注释]
2%的醋酸铜的制备:溶解一水合硫酸铜于100 mL10%醋酸溶液中充分搅拌后过滤去碱性铜盐的沉淀。
实验步骤。
环己酮合成两种方法

环己酮的制备(一)传统实验方法(1)实验原理环己酮的制备可采用浓HNO3、KCrO4 或KMnO4氧化法。
其中最常用的方法是将仲醇用铬酸氧化。
铬酸是重要的铬酸盐和40-50%硫酸的混合物。
酮对氧化剂比较稳定,不易进一步氧化。
铬酸氧化醇是一个放热反应,必须严格控制反应的温度,以免反应过于激烈。
OH Na2Cr2O7/ H2SO4O三、参考步骤1、氧化剂的制备。
在搅拌的条件下,向7.5mL 水和1.3g 重铬酸钠的溶液中慢慢加入1.1mL浓H2SO4,得橙红色铬酸溶液,冷至室温备用。
2、环已酮制备。
向2.5g 环己醇中,分三次加入上述铬酸溶液,每加一次都振摇混匀,并控制反应液温度在55-60℃。
反应约0.5h 后温度开始下降,再放置15min,其间不断振摇,使反应液呈墨绿色为止。
向反应液内加入7.5mL 水,进行简易水蒸气蒸馏,将环己酮与水一起蒸出,收集6mL 馏出液。
用食盐饱和后,分出有机相。
水相用7.5mL 乙醚分两次萃取,萃取液并人有机相。
然后经干燥,空气冷凝管蒸馏,收集151-155℃的馏分。
产0.8-1.0g 左右。
(二)改进方法:以30%H2O2 为氧化剂,用FeCl3 催化氧化环己醇可得到产率(基于环己醇)为75%以上的环己酮,反应中无须加入相转移催化剂,考察了用量、催化剂、反应时间及反应温度对产率的影响.所用催化剂价廉易得且具有极佳的水溶性,分离回收容易,是一条绿色合成环己酮的好途径,克服了目前有机化学实验教材中采用浓HNO3、KCrO4 或KMnO4 氧化法存在污染大、反应时间长等缺点.绿色化学在使用化学药品时遵循4R原则:拒用危险品(Reject),减量使用(Reduce),循环使用(Recycle),重新使用(Reuse)[1].在大学化学教育中渗透和灌输绿色化学思想理念是相当有必要的,而用绿色化学的思想来指导和规范化学实验教学也就显得尤为重要.目前国内有机化学实验教材中环己酮的制备是用浓硫酸催化的重铬酸盐氧化法[2~4],该法存在的主要缺点是:严重污染环境(Cr6+是致癌物),药品较贵,操作繁琐,而且催化剂浓硫酸用量较大,废酸难处理,反应时间长,反应的后处理工作较为复杂困难;而以次氯酸钠作为氧化剂,要用到相转移催化剂四丁基碘化铵,也存在反应副产物和催化剂回收利用难解决的问题[5];也有用有机金属配合物为催化剂、过氧化氢为氧化剂的报道,而且产率高达95%[6],但反应时间达12小时,不适合有机化学实验教学.用30% H2O2作为氧化剂,在55℃~60℃的温度下,采用无毒无害的FeCl3催化剂催化氧化环己醇制备环己酮,反应条件温和,容易控制,氧化剂反应完后只留下水,无毒害废弃物产生,反应时间较短,适宜有机实验教学,而且反应后的产物也极易分离.1实验部分1.1)实验试剂及仪器环己醇(CP)、过氧化氢(30%)、氯化铁(CP)、无水乙醚、氯化钠、无水硫酸镁傅立叶变换红外光谱仪Magua Nicolet 550(II)、阿贝折射仪(ZW AJ)1.2)实验步骤实验按四因子三水平正交法进行,参数如表1.表1正交实验因子水平表在带回流冷凝管、温度计、滴液漏斗的250毫升的三颈烧瓶中加入环己醇、催化剂氯化铁,用滴液漏斗慢慢滴加过氧化氢,水浴控制适宜的反应温度,过氧化氢滴加完后继续反应30分钟,其间不时振摇,使反应完全,反应液呈墨绿色.反应完成后在三颈烧瓶中加入60ml水和几粒沸石,改成蒸馏装置,将环己酮和水一起蒸出来,直至流出液不再浑浊后再多15ml~20ml,约收集50ml流出液.流出液用精盐饱和后,转入分液漏斗,静置分出有机层,水层用15ml无水乙醚萃取一次,合并有机层与萃取液,用无水碳酸钠干燥,然后水浴蒸馏除去乙醚,蒸馏收集152℃~158℃的馏分,称量所得产物的质量.1.3)催化剂单项试验正交实验得到的结果显示,催化剂是影响产率的主要因素,但影响趋势不明显,因此在确定其他条件的情况下,单独考察催化剂用量对环己酮收率的影响.1.4)实验结果的可重复性所有反应条件确定后,进行多次重复性实验,以考察实验结果的稳定性能,以确定能否将这一新的反应体系应用到实验教学中去.1.5)产品分析最后产物用Magua Nicolet 550(II)型FT-IR光谱仪测定其红外吸收.用阿贝折射仪(ZW AJ)测定其折光率.用电子天平称量所得产物的重量.2结果与讨论2.1)反应产物的表征经过处理后,蒸馏收集152℃~158℃所得的馏分为无色透明油状液体,产物的红外光谱显示在1705cm-1~1715cm-1范围有特征吸收峰,说明产物的分子结构中存在羰基;在2800cm-1~3000cm-1范围出现亚甲基特征吸收峰;测得产物折光率为1·4500.所得的红外光谱和折光率均与文献给出的环己酮的数据相符.2.2)系列正交实验产率的直观分析表2是按照四因子三水平正交法安排实验的直观分析.从表中各因子对产物平均收率的贡献来看,A1B1C2D1为最优条件,而从单个实验的产率来看则是A3B3C2D1为最高,由于极值Rj表明过氧化氢对产物平均收率的影响不大,而影响最大的是反应温度,其次是催化剂和反应时间,因此按节约原则选取A1B1C2D1或A1B3C2D1进行下一步实验.表2正交实验结果直观分析表2.3)影响环己酮收率的因素2.3.1过氧化氢的影响图1为过氧化氢与环己醇物质的量比对环己酮平均收率的影响.当二者为1∶1时,平均收率最高,虽然随着过氧化氢的量增加,平均产率有一下降过程随后又逐渐增加,但增加幅度缓慢,而且过氧化氢的多少对平均收率的影响很小,所以从节约的角度出发,尽可能选取用最少的过氧化氢.图1过氧化氢用量对环己酮平均收率的影响2.3.2催化剂FeCl3对环己酮收率的影响图2为催化剂对环己酮平均收率的影响,正交实验所得平均收率显示,取1水平时所得反应结果最好,但就单个实验结果却是3水平的反应产率最高.因此,为了确定催化剂的用量而做了相应的单项实验,结果如表3. 图2催化剂对环己酮平均收率的影响表3FeCl3用量对环己酮产率的影响从表3结果来看FeCl3用量为3g时达到最高产率76.6%.如果从教学意义来说,产率达到70%以上时,现象已经非常明显,此时所得产物有7g以上,足以用各种方法进行的处理和测试,完全能达到教学的目的和要求,因此FeCl3用量为2g~3g都能满足教学实验的要求.2.3.3反应时间及反应温度对环己酮收率的影响图3、图4分别显示反应时间和反应温度对环己酮平均收率的影响.从图中看,反应时间取70min,反应温度取55℃~60℃时反应的平均收率最高.3实验结果的稳定性的考察为了考察实验结果的可重复性,在确定的优化条件下做了多次实验,对结果的稳定性进行了考察,结果如表 4.系列重复试验结果显示,实验的重现性非常好,完全可以用于教学实验.4结论建议用于学生实验的最佳条件为:10·5ml环己醇,3.1ml过氧化氢(30%),2g~3gFeCl3,反应时间70min,反应温度55℃~60℃.该反应时间仅用70min,在规定的实验课时内,学生完全能够完成实验,是一种适用于合成环己酮的教学实验.重要的是该实验方法对学生操作及环境无污染和毒害,催化剂FeCl3分离回收容易,这对改善有机化学实验室的环境、改变学生对有机实验的固有看法及将绿色化学的思想渗透到实验教学中很有意义.100[参考文献][1]Anastas P T,Warner J C. Green Chemistry,Theory and Practice[M].Oxford:Oxford University Press,1998.[2]兰州大学、复旦大学有机化学教研室.有机化学实验(第二版) [M]·北京:高等教育出版社,1994.[3]曾昭琼.有机化学实验(第二版)[M].北京:高等教育出版社,1987 .[4]李霁良.微型半微型有机化学实验[M]·北京:高等教育出版社, 2003.[5]张晓勤,郑柳萍.相转移催化法制备环己酮[J]·福建师范大学学报(自然科学版) ,1999,15(2):56-59.[6]魏俊发,石先莹,何地平,等.无有机溶剂、无相转移催化剂条件下H2O2氧化环己醇为环己酮[J]·科学通报,2002,47(12):1628-1630.[责任编辑黄招扬][责任校对黄少梅]Study on the Preparation of CyclohexanoneDIAO Kai-sheng,LI Yan,QIN Zhi-liu(Chemical and Ecoengineering College, Guangxi University for Nationalities, Nanning530006,China)Abstract:Without phrase transfer catalyst, Cyclohexanone was prepared from cyclohexanol and hy-drogen peroxide. The effect on reaction of factors including the amount of oxidant and catalyst, reaction timeand temperature were accounted and the optimum conditions were found. Compared with that of teachingmaterial in organic chemistry, which is pollutant and poisonous, the new way is more feasible and less poison。
卤代酮的合成-060123

经典化学合成反应标准操作α-卤代酮的合成目录1.前言 (2)2. 直接卤化 (2)3.经重氮酮制备 (4)4.从weinreb 酰胺制备 (6)5.傅克酰基化合成卤代酮 (7)6 其他合成α-卤代酮的方法 (9)1.前言α-卤代酮的合成广泛应用于现代有机合成中, 多用于溴的烷基化、合成咪唑及噻唑等杂环类化合物,其合成方法常用直接卤化、经重氮酮制备、经Weinreb 酰胺制备、傅克酰基化等方法合成。
2. 直接卤化酮的α-氢易被取代,可以直接合成α-卤代酮。
一般操作是将酮与卤素于醋酸、氯仿、DMF 或水中反应。
除卤素外, 硫酰氯、五卤化磷、过溴化吡啶氢溴酸盐(C5H5NH.Br 3)、三卤化三甲基苄基铵盐等也可以做卤化试剂。
对称酮或只有一个取代方向的酮卤代时,可以良好产率(80~90%)生成α-卤代酮。
不对称酮卤代,往往生成α-及α’-卤代酮的混合物。
由于酮卤代的决定步骤是酮的烯醇化,因此,易形成烯醇的方向优先卤代。
例 2-甲基环己酮与亚硫酰氯作用, 多取代的α-氢优先氯代1。
OCH 3OCH 3Cl 2485%若利用双(二甲基乙酰胺基)三溴化氢做溴化剂,可使不对称酮在少取代一边溴代2。
OOBr[(Me 2NCOCH 3)2H]Br 3384%若将不对称酮首先转变成为一定构型的烯醇盐,继而卤代,是区域定向卤代的新方法3。
OH 3COH 3CCl1. i -Pr 2NLi, THFPhCOOEtO CH 3PhOCH 3Br 1. NaH, DMSO另外,甲基酮可用甲基格式试剂与相应的Weinreb 酰胺来制备, 如下例即是先合成甲基酮,后溴化来合成α-溴代酮的4。
NBocO HONBocO NO NBocODCC, D MAP, N HMeOMe MeMgI , e t h er合成实例一 5OOOOOOBrBr 2, AcOH2B 2AA suspension of ketone 2A (700 mg, 2.17 mmol) in acetic acid (15 ml) was heated to 70℃, followed by addition of bromine (347 mg, 2.17 mmol). After the mixture was stirred at 70℃ for 3h, the solvent was evaporated and the residue was purified by column chromatography to give the compound 2B (591 mg, 68%).合成实例二6OMe MeOO Br OMeMeOOBr232C2DBromine (7.99 g, 50 mmol) in CHCl 3 (20 ml) was added in a dropwise manner to a stirred solution of 2, 5-dimethoxy-4-bromoacetophenone 2C (12.95 g, 40 mmol) in CHCl 3 (100 ml) at 5℃. After the addition was completed, the reaction mixture was allowed to warm to room temperature and stirred for an additional 2 h. The mixture was poured onto crushed ice, the organic portion was separated and washed with water, saturated NaHCO 3 solution, and again with water. The solution was dried MgSO 4, and evaporated to dryness under reduced pressure to give a crude product. The product was recrystallized from MeOH to yield 14.70 g (87%) of the desired bromoacetophenone 2D as a white solid.ON NH 2NON NH 2NBr AcOH, 48% aq. HBr and Br 2To a solution of 1-(2-aminopyrimidin-4yl) ethanone (412 mg, 3 mmol) in glacial acetic acid (1 mL) and 48% aq. HBr (0.3 mL), bromine (0.153 mL) in acetic acid (0.4 mL) was added and the resulting orange solution was stirred at RT for1.5 hours. After diluting with ethyl acetate (15 mL), the precipitate was filtered and washed with ethl acetate thus affording the target compound as a whitish solid (580 mg, 65%).合成实例四8O OSiOOSi Br 2E 2FBenzyltrimethylammonium tribromide (4.17 g, 10.7 mmol) was added to a solution of Compound 2E (4.00 g, 10.7 mmol) in CH 2Cl 2-MeOH (5:2, 25 mL). The mixture was stirred at RT for 3 h. At this time the reaction mixture was concentrated in vacuo and H 2O (15 mL) was added. The mixture was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with brine (15 mL), dried over MgSO 4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (hexanes:EtOAc, 3:1) to afford to afford Compound 2F (3.97 g, 8.8 mmol, 82%) as a thick yellow oil.3.经重氮酮制备不对称酮卤代时,有时无法得到单卤代产物。
15二羰基化合物合成方法michael加成

15二羰基化合物合成方法michael加成在有机合成领域中具有重要的地位。
本文将针对这一主题进行深入探讨,并就其广度和深度展开全面评估,以便读者能全面、深刻地了解这一合成方法的原理与应用。
1. 了解michael加成的基本原理在有机化学中,michael加成是一种重要的加成反应,其基本原理是通过亲核试剂与α,β-不饱和酮或其他亚醇化合物进行加成反应,生成具有多个功能团的化合物。
这一反应具有较高的立体选择性和效率,因此在有机合成中得到了广泛的应用。
2. 探讨15二羰基化合物的特性与合成方法15二羰基化合物是一类含有两个羰基(C=O)的有机化合物,其合成方法多种多样,其中michael加成反应是一种重要的合成方法之一。
15二羰基化合物在生物医药和材料科学等领域中具有重要的应用价值,因此其合成方法备受关注。
3. 介绍经典的15二羰基化合物合成方法基于michael加成反应的15二羰基化合物合成方法具有较为丰富的多样性,包括使用硫醇、胺类试剂等作为亲核试剂进行加成反应,生成15二羰基化合物。
这些方法在合成化学领域中得到了广泛的应用,并为相关化合物的制备提供了重要的技术支持。
4. 探讨新颖的15二羰基化合物合成方法随着有机合成领域的不断发展,新颖的15二羰基化合物合成方法不断涌现。
一些基于金属催化的michael加成反应及其在15二羰基化合物合成中的应用,以及环状亲核试剂对α,β-不饱和酮的michael加成反应等,为15二羰基化合物合成领域带来了新的发展机遇。
5. 总结回顾15二羰基化合物合成方法michael加成作为有机合成领域中的重要分支,在合成化学的发展历程中扮演着重要的角色。
通过对其原理、传统方法及新颖方法的全面探讨,我们对这一合成方法有了更加深入的了解,并对其在有机合成领域的应用前景有了更清晰的认识。
个人观点和理解:15二羰基化合物合成方法michael加成作为一种重要的有机合成反应,具有广泛的应用前景。
麝香酮的合成

麝香酮的合成李茹【摘要】以尼龙-11的下脚料11-溴代十一酸为原料,经4步反应合成了DL-麝香酮,总收率32.1%.产物及部分中间体的结构经元素分析、IR,1H NMR和MS确认.【期刊名称】《合成化学》【年(卷),期】2004(012)003【总页数】3页(P222-224)【关键词】麝香酮;11-溴代十一酸;2,15-十六烷二酮;合成【作者】李茹【作者单位】西安交通大学环境与化学工程学院,陕西,西安,710049;西安工程科技学院环境与化学工程学院,陕西,西安,710048【正文语种】中文【中图分类】O624.4麝香酮(Muscone)即3-甲基环十五酮(3-Methyl-cyclopentadecanone)是麝香的主要成分,麝香不仅是名贵的香料,又是临床上不可缺少的贵重药材。
由于天然资源极其有限,特别是麝作为珍稀动物被保护后,天然麝香来源紧张,价格昂贵,半个世纪以来科学工作者一直在探索人工获得麝香的方法,而麝香酮的合成则是有机合成者追求的目标。
本文以尼龙-11的下脚料11-溴代十一酸作为初始原料,设计出一条步骤少、产率高、可大批量生产的有望工业化的合成麝香酮的新方法。
新合成路线(Scheme 1)由4步构成:(1) 采用洪赛迪克尔—博罗丁银盐脱羧溴化作用的Cristol改良法[1]由11-溴代十一酸(1)合成1,10-二溴癸烷(2);(2) 2与乙酰乙酸乙酯在乙醇钠的作用下反应后,水解生成2,15-十六二酮(6);(3) 6在碘化乙基锌环合剂作用下选择性的发生分子内环合生成去氢麝香酮(7);(4) 7在催化剂(5% Pd-C)作用下催化氢化还原为麝香酮(8)。
1 实验1.1 仪器与试剂FLASH EA1112型元素分析仪;Shimadzu-IR435型红外光谱仪(NaCl涂片);DMX-300型核磁共振谱仪(CDCl3为溶剂,TMS为内标);APEXII型FT-ICR质谱仪。
碘乙烷、锌粉、碘、乙酰乙酸乙酯、乙醇、四氯化碳、甲苯、四氢呋喃、乙酸乙酯、硫代硫酸钠、无水硫酸镁等均为分析纯;红色氧化汞、硫酸铜、盐酸为化学纯。
2,15-十六烷二酮的合成

o )S C 、0m t 1 O 1 5 L无 水 E2 o 2 t 0置 于烧 瓶 中加热 回流 2h 得 金 黄 色 溶 液 。改 为蒸 馏 装 置 , 常 压 蒸 出 , 先 Ez t O和大 部分 S C2再 减压 蒸馏 0 5h除去 残余 O 1, . S C2 得棕 黄色 液体 , O 1, 备用 。I K r , ,m~: R( B ) 一 c
很多 _ , 中 So 的 闭环 法 由于 闭环 和 甲基 引 入 2其 J tl l
同步进 行 而备受关 注 。闭环 法研 究重 点之 一是 中 间体 2 1. 六 烷 二 酮 的合 成 。So 等 E 最 早 是 ,5十 tl 3 l J 用 1 1. 溴癸 烷 和 乙酰 乙酸 乙酯 反 应后 水 解脱 ,0二 羧制 2 l. ,5十六烷 二 酮 ; 嫒等 _ 以十 四碳 二 酸 和 郭 4 J
密封 。将 5m ( .8m 1C 3 和 3 L无 水 E2 L 0 0 o) H I 0m t 0
混合 液 加 入 漏斗 , 滴 加 几 毫升 , 拌 引 发反 应 , 先 搅
进行 取代 反应 后 水 解脱 羧 的方 法 得 到 了 2 1. ,5十
六烷二 酮 , 以十 四碳 二 酰氯 和 金 属 有机 化 合 物 但
碘化锌 试剂 反应 , 水解 后获 得 2 1. ,5 十六烷 二酮 :
叫 : :
1 实验部 分 11 主要 仪器 与试剂 .
a 一
加入 C n 溶 液 中 , 冰 盐 浴 冷 却 到 0℃ 以 下 , H ZI 用
B OR D FB15型 傅 里 叶 红 外 光 谱 仪 ; o L .A 1 6 N. v.0 H型核 磁共 振பைடு நூலகம் (0 z 。 a30 4 0MH )
麝香酮 有机结合

麝香酮的合成研究进展摘要:麝香酮(3- 甲基- 环十五烷酮)是麝香具有生理活性的重要组分,是麝香香味的主要来源,不仅可用作高级定香剂,而且还可用于医药。
关键词:麝香酮;合成麝香是中国的特产,《神农本草经》将其列为上品。
梁陶弘景《本草集注》、明李时珍《本草纲目》等历代著作均有记载。
麝香以通诸窍、开经络、透肌骨,内治中风、中气、中恶及小儿惊痫,外治跌打损伤及疮毒等症而著称[1]。
麝香酮是天然麝香的主要功能成分,化学名为3- 甲基环十五烷酮,分子式为C16H30O,分子量为238。
其外观为无色或淡黄色结晶体,具有麝香的特殊香味,熔点为33 ℃,熔化后呈黏稠状液体,沸点为130 ℃/1.6 kPa,相对密度为0.920 0~0.926 8,折光系数为1.479~1.489,不溶于水,溶于乙醇和油类溶剂。
雄性麝鹿生殖腺的分泌物(即天然麝香)经水蒸气蒸馏可分离出麝香酮,干燥后为红棕色的粒状物[1]。
麝香酮可作为香料的重要组成成分,具有沉柔、令人愉快的强烈麝香香气,而且能使香精具有高雅、润和的香气,是不可多得的高级调香料。
同时,麝香酮还具有天然麝香的某些重要药理作用。
麝香酮在临床上的应用始于20 世纪70 年代,北京医药界首先把麝香酮单独制成气雾剂和含片在临床上用来治疗冠心病,缓解心绞痛。
1.麝香酮的合成麝香酮的用途广泛,但天然品来源有限且价格昂贵,远不能满足人们的需要,在这种情况下,势必需要人工合成来加以补充才能满足日益扩大的需求。
于是,麝香酮的人工合成吸引了众多化学家的兴趣,对麝香酮的人工合成进行了各种各样的尝试,取得了许多理论和应用成果。
同时,开发可应用于生产的光学活性麝香酮合成工艺,仍然具有极大的挑战性。
1.1消旋麝香酮的合成研究1906 年Walbaum 从天然麝香中分离出一个具有麝香香气的大环酮类化合物,引起了众多化学家的兴趣。
1926 年Ruzicka 确定了这一个具有麝香香气的大环酮类化合物的结构为3- 甲基环十五酮,俗名麝香酮。
合成1_2_环十二二酮的新方法

第26卷第11期应用化学V o.l26N o.11 2009年11月 C H I NESE J OURNAL OF A PPL I ED C HE M IS TRY N ov.2009合成1,2 环十二二酮的新方法陈韶蕊a,b 马吉海b 周雪琴a 刘东志a*(a天津大学化工学院 天津300072;b河北科技大学理学院 石家庄)摘 要 以十二烷二酸(1)为原料,与甲醇酯化生成十二烷二酸二甲酯(2),收率为86 5%。
化合物2经偶联缩合得1,2 二三甲基硅氧基环十二烯(3),收率为63 1%。
利用溴氧化化合物3得到1,2 环十二二酮(4)淡黄色晶体,收率79%,全过程收率达43 1%,是合成1,2 二酮的新方法。
目标化合物结构经红外光谱、核磁共振氢谱和碳谱表征确认。
关键词 十二烷二酸,偶联缩合,环十二二酮中图分类号:O622.4 文献标识码:A 文章编号:1000 0518(2009)11 1374 03二酮是苯酰二苯基乙醇酸重排[1]和二磷烯合成[2]等特殊反应的重要中间体。
二酮还可应用于 氧代内酰胺如千里光碱[3]、鸭嘴花碱[4]等的合成以及 内酰胺[5]如青霉素和头孢霉素衍生物[6]的制备。
1,2 环十二二酮是一个典型的环状 二酮,广泛用于稠杂环类化合物、碳桥类化合物和某些具有特殊结构化合物的合成,而且是合成大环1,6 二酮及麝香酮等扩环反应的重要中间体[7]。
文献曾报道由 羟基酮、环十二酮以及十二烯为原料经氧化合成1,2 环十二二酮[10]。
但这些方法中由 羟基酮的氧化反应需低温及产物分离操作复杂(需柱层析分离),由环十二酮氧化时氧化试剂(高氯酸银或Se O2等)昂贵;而由环十二烯氧化时产率低,只有20%左右。
因此,研究1,2 环十二二酮的新合成方法有重要意义。
本文通过溴水氧化烯醇式硅醚合成了1,2 环十二二酮,合成路线如Sche m e1所示。
Sche m e1 Synt hesis route o f1,2 cyclododecand i one十二烷二酸(工业级),三甲基氯硅烷(工业级),甲醇、甲苯、苯、浓H2SO4、无水碳酸钠、无水硫酸镁、四氢呋喃、溴素均为分析纯试剂,金属钠为化学纯试剂。
青霉烷酸二苯甲酯1-氧化物的合成
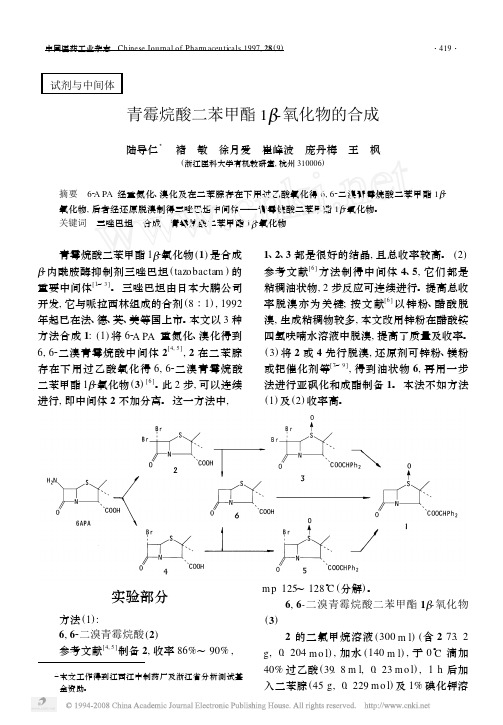
试剂与中间体青霉烷酸二苯甲酯1Β-氧化物的合成陆导仁3 褚 敏 徐月爱 崔峰波 庞丹梅 王 枫(浙江医科大学有机教研室,杭州310006)摘要 62A PA 经重氮化、溴化及在二苯腙存在下用过乙酸氧化得6,62二溴青霉烷酸二苯甲酯1Β2氧化物,后者经还原脱溴制得三唑巴坦中间体——青霉烷酸二苯甲酯1Β2氧化物。
关键词 三唑巴坦 合成 青霉烷酸二苯甲酯1Β2氧化物△本文工作得到江西江中制药厂及浙江省分析测试基金资助。
青霉烷酸二苯甲酯1Β2氧化物(1)是合成Β2内酰胺酶抑制剂三唑巴坦(tazobactam )的重要中间体[1~3]。
三唑巴坦由日本大鹏公司开发,它与哌拉西林组成的合剂(8∶1),1992年起已在法、德、英、美等国上市。
本文以3种方法合成1:(1)将62A PA 重氮化、溴化得到6,62二溴青霉烷酸中间体2[4,5],2在二苯腙存在下用过乙酸氧化得6,62二溴青霉烷酸二苯甲酯1Β2氧化物(3)[6]。
此2步,可以连续进行,即中间体2不加分离。
这一方法中,1、2、3都是很好的结晶,且总收率较高。
(2)参考文献[6]方法制得中间体4、5,它们都是粘稠油状物,2步反应可连续进行。
提高总收率脱溴亦为关键;按文献[6]以锌粉、醋酸脱溴,生成粘稠物较多,本文改用锌粉在醋酸铵四氢呋喃水溶液中脱溴,提高了质量及收率。
(3)将2或4先行脱溴,还原剂可锌粉、镁粉或钯催化剂等[7~9],得到油状物6,再用一步法进行亚砜化和成酯制备1。
本法不如方法(1)及(2)收率高。
实验部分方法(1):6,62二溴青霉烷酸(2)参考文献[4,5]制备2,收率86%~90%,m p 125~128°C (分解)。
6,6-二溴青霉烷酸二苯甲酯1Β-氧化物(3)2的二氯甲烷溶液(300m l )(含273.2g ,0.204m o l ),加水(140m l ),于0°C 滴加40%过乙酸(39.8m l ,0.23m o l ),1h 后加入二苯腙(45g ,0.229m o l )及1%碘化钾溶液(14m l);继续反应30m in,再加40%过乙酸(39.8m l,0.23m o l)及10%硫酸(16m l),室温反应3h。
稳定的环丙酮等价物的合成及应用

稳定的环丙酮等价物的合成及应用摘要环丙酮是一类重要的小环化合物,但它自身的不稳定性限制了其应用。
本文首次合成了一个新的稳定的环丙酮等价物-环丙酮苯亚磺酸,这个等价物具有容易制备、稳定性好以及反应活性高等三方面的优点。
同时,本文研究了它在有机合成中的应用,主要包括以下三方面的工作:1、发展了水相中 AuCl 催化的环丙酮苯亚磺酸、末端炔烃和二级胺的三组分偶3联反应,得到了一系列环丙基炔胺类化合物。
2、以环丙酮苯亚磺酸为原料,利用可控的连续亲核加成反应合成了一系列环丙基炔基 1,3-二醇类化合物。
3、将可控的连续亲核加成反应这一方法学扩展到普通醛酮,实现了由简单原料合成具有多个官能团的 1,3-二醇类化合物。
关键词:环丙酮;环丙酮苯亚磺酸;AuCl 催化;可控的连续亲核加成反应31AbstractCyclopropanone is an important small-ring compound, but its synthetic utility wasdwarfed by its instability. In this paper, we developed a new stable cyclopropanoneequivalent and explored its synthetic application1. A series of 1-alkynyl cyclopropylamine derivatives were synthesized from the coupling reaction of the cyclopropanone equivalent with terminal alkyne anddisubstituted amine in water with AuCl as catalyst32. A series of 1-cyclopropyl-3-alkynyl-1,3-diols was prepared from the reaction ofthe cyclopropanone equivalent with acetaldehyde enolate and lithium alkynilidethrough consecutive, competitive nucleophilic addition3. This new methodology was successfully extended to the reaction of simple aldehydes and ketones to afford a series of alkynyl substituted 1,3-diolsKeywords: Cyclopropanone; 1-arylsulphonylcyclopropanol; Auric catalysis;Consecutive Competitive Nucleophilic Reaction2目录第1 章绪论11.1 环丙酮化学 11.1.1 环丙酮的制备方法. 21.1.2 环丙酮的反应性质. 41.1.3 小结. 81.2环丙酮半缩酮化学. 81.2.1 环丙酮半缩酮的制备 81.2.2 环丙酮半缩酮的化学反应性质 101.2.3 环丙酮半缩酮在合成中的应用 141.2.4 小结171.3 具有环丙基结构的天然产物及药物 17第2 章新的稳定的环丙酮等价物的合成..19 2.1 新的稳定的环丙酮等价物的发现. 192.2 环丙酮苯亚磺酸的化学反应性质. 192.2.1 与有机锂试剂的反应. 202.2.2 与格氏试剂的反应202.2.3 与酰氯的反应. 202.3 实验部分202.3.1 环丙酮苯亚磺酸的合成 212.3.2 环丙酮苯亚磺酸与炔锂试剂的反应. 22 2.3.3 环丙酮苯亚磺酸与格氏试剂的反应. 222.3.4 环丙酮苯亚磺酸与酰氯的反应 222.4 本章小结23第3 章利用环丙酮苯亚磺酸合成环丙基炔基胺243.1 研究背景243.2 反应条件的优化253.3底物普适性研究 263.4 关于反应机理 283.5 实验部分283.5.1 对氯苯乙炔的合成283.5.2 环丙基炔基胺类化合物的合成 293.6 本章小结291第4 章利用环丙酮苯亚磺酸合成环丙基炔基 1,3-二醇类化合物..30 4.1 研究背景304.2 反应机理304.3 反应条件的优化314.4 底物普适性研究324.5 应用研究344.5.1 合成α,β-不饱和酮类化合物. 344.5.2 合成氧杂八元环酮类化合物. 344.6 实验部分354.6.1 合成环丙基炔基 1,3-二醇类化合物354.6.2合成α,β-不饱和酮类化合物354.6.3合成氧杂八元环酮类化合物354.7 本章小结36第5 章可控的连续亲核加成反应合成炔基1,3-二醇..37 5.1 研究背景375.2 反应条件的优化375.2.1 醛作底物的反应条件. 375.2.2 酮作底物的反应条件. 395.2.3反应的非对映选择性395.3 底物普适性研究405.4应用研究 415.5 实验部分425.5.1由醛合成炔基 1,3-二醇类化合物. 425.5.2 由酮合成炔基 1,3-二醇类化合物 425.5.3 三氯化金催化的重排反应425.6 本章小结43第6 章化合物谱图数据..446.1 第 2章的化合物数据 446.2 第 3章的化合物数据 466.3 第 4章的化合物数据 526.4 第 5章的化合物数据 57参考文献68发表文章72致谢. 732图表索引图 1.1昀先报道的几个环丙酮化合物. 1图 1.2环丙酮衍生物1图 1.3 烯酮与重氮甲烷反应合成环丙酮2图 1.4 光化学法合成环丙酮. 3图 1.5 脱卤素法合成环丙酮的衍生物 3图 1.6 Favorskii 反应法合成环丙酮4图 1.7 环丙酮与亲核试剂的反应4图 1.8 环丙酮在酸性和碱性条件下发生不同的开环反应. 5 图 1.9 环丙酮参与的扩环反应 5图 1.10 环丙酮参与的电环化反应 6图 1.11 环丙酮参与[4+3]电环化反应的机理. 6图 1.12 [3+2]环加成反应. 7图 1.13 [2+2]环加成反应. 8图 1.14 环丙酮聚合物8图 1.15几种环丙酮等价物. 8图 1.16 首次合成环丙酮半缩酮的方法. 9图 1.17 合成的一系列环丙酮半缩酮9图 1.18 合成环丙酮半缩酮的简便方法. 9图 1. 19 利用 Simmon-Smith反应合成环丙酮半缩酮. 9图 1.20 其他的合成环丙酮半缩酮的方法. 10图 1.21 环丙酮在金属离子作用下发生氧化开环. 10图 1.22 利用氧化开环反应来延长碳链10图 1.23 取代环丙酮与氧气发生反应.11图 1.24 与过氧叔丁醇发生开环反应.11图 1.25 在酸性条件下发生开环反应.11图 1.26苯基环丙酮半缩酮在酸性条件下开环.11图 1.27苯基环丙酮半缩酮在酸性条件下开环的机理及氘代实验. 12 图 1.28 碱性条件下环丙酮半缩酮的开环反应12图 1.29 两种环丙酮半缩酮之间互相转变. 12图 1.30 环丙酮半缩酮与苯胺反应. 13图 1.31 环丙酮半缩酮与烯基格氏试剂和炔基格氏试剂反应13图 1.32 环丙酮半缩酮被格氏试剂活化13图 1.33 环丙酮半缩酮活化后可发生一些列反应. 14图 1.34 具有内酰胺结构的药物14图 1.35 氨基环丙醇发生扩环反应. 15图 1.36 环丙酮半缩酮与叠氮化钠反应153图 1.37 乙烯基环丙醇扩环生成环丁酮15图 1.38 炔基环丙醇扩环生成烯基环丁酮. 15图 1.39 π-1,1-二亚甲基烯丙基钯络合物的反应. 16图 1.40 由亚甲基环丙烷可以合成多种天然产物. 16图 1.41 一些含有环丙基的药物17图 1.42 环丙基结构对药物活性起到关键作用18图 2.1 环丙酮苯亚磺酸的合成. 19图 2.2 环丙酮苯亚磺酸的单晶结构19图 2.3 环丙酮苯亚磺酸与炔锂反应20图 2.4环丙酮苯亚磺酸与格氏试剂反应. 20图 2. 5 环丙酮苯亚磺酸与各种酰氯. 20图 2.6环丙酮苯亚磺酸的合成路线 21图 2.7环丙酮苯亚磺酸与苯乙炔的反应. 22图 2.8环丙酮苯亚磺酸与苄基格氏试剂的反应. 22图 2.9 环丙酮苯亚磺酸与苯乙酰氯的反应23图 3.1 醛、末端炔和二级胺的三组分偶联反应 24图 3.2 环丙酮苯亚磺酸参与的三组分偶联反应 24图 3.3 三组分偶联反应的机理. 28图 3. 4对氯苯乙炔的合成路线. 28图 3.5 环丙酮苯亚磺酸、苯乙炔和哌啶发生的偶联反应29 图 4.1 意外得到的二醇化合物. 30图 4.2 四氢呋喃与丁基锂发生裂解反应 30图 4.3 反应可能经过的机理31图 4.4 反应的底物普适性 33图 4.5 钯催化环丙烷开环 34图 4.6 三氟甲磺酸铝催化的 Petasis-Ferrier 重排34图 5.1 对硝基苯甲醛参与的可控连续亲核加成反应. 37图 5.2 C1H和 C3H的 NOE. 39图 5.3 反应可能的过渡态 40图 5.4 可控连续亲核加成反应的底物普适性. 41图 5.5 三氯化金催化的 1,3-二醇重排反应 42表 1.1 一系列环丙酮化合物的合成方法2表 1.2 含取代基的环丙酮与各种呋喃发生电环化反应6表 1.3 含不同取代基的环丙酮与呋喃发生电环化反应的速率常数 7表 3.1 三组分偶联反应条件的优化25表 3.2 三组分偶联反应的底物普适性. 26表 4.1 反应条件的优化..32表 5.1 芳香醛作底物时的反应条件优化 384表 5.2 正丁醛作底物时的反应条件筛选 38表 5.3酮作底物时的反应条件优化 395稳定的环丙酮等价物的合成及应用第1章绪论1.1 环丙酮化学环丙酮作为一个小环化合物,具有很高的反应活性。
麝香酮的药理与合成研究进展

麝香是中国的特产,《神农本草经》将其列为上品。
梁陶弘景《本草集注》、明李时珍《本草纲目》等历代著作均有记载。
麝香以通诸窍、开经络、透肌骨,内治中风、中气、中恶及小儿惊痫,外治跌打损伤及疮毒等症而著称[1]。
在国家药典中,有10%的中成药需用麝香,北京和上海应用麝香配伍的中成药达1/4以上。
目前京沪两地医药界用麝香酮代替天然麝香与其他中药配伍制成的中成药主要有苏合香丸、紫雪散、周氏回生丹、牛黄清心丸、第一丹、西黄丸、六神丸等。
这些用麝香酮代替天然麝香制成的中成药用于治疗冠心病、小儿高烧、急性肠胃炎、跌打损伤、五宫科炎症、乳腺炎、淋巴结核、扁桃腺炎、腮腺炎等疾病均收到良好疗效。
麝香酮是天然麝香的主要功能成分,化学名为3-甲基环十五烷酮,分子式为C16H30O,分子量为238。
其外观为无色或淡黄色结晶体,具有麝香的特殊香味,熔点为33℃,熔化后呈黏稠状液体,沸点为130℃/1.6kPa,相对密度为0.9200~0.9268,折光系数为1.479~1.489,不溶于水,溶于乙醇和油类溶剂。
雄性麝鹿生殖腺的分泌物(即天然麝香)经水蒸气蒸馏可分离出麝香酮,干燥后为红棕色的粒状物[2]。
麝香酮可作为香料的重要组成成分,具有沉柔、令人愉快的强烈麝香香气,而且能使香精具有高雅、润和的香气,是不可多得的高级调香香料。
同时,麝香酮还具有天然麝香的某些重要药理作用。
麝香酮在临床上的应用始于20世纪70年代,北京医药界首先把麝香酮单独制成气雾剂和含片在临床上用来治疗冠心病,缓解心绞痛。
1麝香酮药理活性1.1对中枢神经系统的作用实验表明,大鼠多次灌服麝香酮5mg/kg剂量能明显地缩短戊巴比妥钠引起的睡眠时间。
这并非是直接兴奋中枢,而是由于麝香酮激活肝微粒体药物转化酶的作用,加速肝内戊巴比妥钠代谢失活的结果。
与上述结果相反,如果麝香酮剂量为100~500mg/kg,可使戊巴比妥钠引起的小鼠睡眠时间延长。
故麝香酮小剂量兴奋中枢,大剂量则抑制中枢[3]。
碘催化合成2,2-二甲基-1,3-二恶烷-4,6-二酮

6二 酮 的合成 最经 典 的方 法是 由浓硫 酸 催 化 丙二 一 酸 与丙 酮在 乙酸 酐 中缩 合 制 得 l 。由 于 浓硫 酸 5 棚 具 有 强氧化 性 致 使 产 品往 往带 有 浅 黄 色 杂 质 , 纯 度 不 高 , 处 理 工序 复 杂 , 境 污 染严 重 , 后 环 不利 于 工 业化 生产 。为 了简化 其合 成工 艺并 使 工艺 绿色 化 , 年来有 人 开展 了对 甲基 苯 磺酸 l 、 近 _ 固体 超强 7 ] 酸 催化 合 成 2 2 甲基一 ,- 口 烷一 ,一 酮 的研 ,一 1 3二 恶 4 6二 究 , 都 取得 了较 好 的实 验效 果 。碘 作 为 一 种温 并 和 的 L wi 催化 剂 在 合成 中应 用 广 泛 , 催 化 e s酸 碘 的反应具 有 温 和 、 高效 、 定 、 稳 环境 友好 、 于操 作 易 等 优点 。笔 者报 道 以碘 为 催 化 剂 , 由丙 二 酸 和 丙 酮 直接缩 合 合成 2 2 甲基一 , - 恶 一 ,一 酮 , ,一 1 3二口 烷 4 6二 并对 较优 工 艺条 件进 行 了探 索 。
酸 ): ( 酮 ) 1 0: . , 化 剂 用 量 为 0 2g 0 1mo 丙 二 酸 , 应 温 度 3 丙 一 . 11催 . / . l 反 O℃ , 应 时 间 为 3 0 h 产 物 收 率 反 . ,
达 7 . 。 71 关 键 词 : 2二 甲基 一 ,- 口 一 6 二 酮 2, 1 3 二 恶烷 4,一 碘 丙二 酸 丙 酮
合 成 _ 。它 还是 合成 农用 化 学 品材料 、 料 、 4 ] 颜 特殊
功 能 材 料 的 关 键 原 料 。 2 2 甲 基 一 ,一 口 一 , ,一 1 3二 恶烷 4
2,15-十六烷二酮的合成方法[发明专利]
![2,15-十六烷二酮的合成方法[发明专利]](https://img.taocdn.com/s3/m/0be0c3016294dd88d1d26b18.png)
专利名称:2,15-十六烷二酮的合成方法专利类型:发明专利
发明人:焦克芳,陈望忠,李新胜
申请号:CN91109897.6
申请日:19911029
公开号:CN1059709A
公开日:
19920325
专利内容由知识产权出版社提供
摘要:本发明属于一种合成麝香酮的关键中间体-2, 15-十六烷二酮(2,15-Hexadecanedione)的方法,合 成麝香酮的主要难题之一是关键中间体难得,合成步 骤过长、产率较低,工业生产价值不大。
本发明中,所 用原料为1,10-二溴癸烷和乙酰乙酸乙酯,采用相转 移催化法,不经分离中间体,直接水解和重结晶即可 得到较高产率(接近80%)的2,15-十六烷二酮,为大 量的工业化生产提供了一个比较好的合成方法。
申请人:中国人民解放军军事医学科学院毒物药物研究所
地址:100850 北京市太平路27号
国籍:CN
代理机构:中国人民解放军总后勤部专利服务中心
更多信息请下载全文后查看。
2,2-二甲基-1,5-二氧杂菲-6-酮的微波合成研究

第 3 卷 第 4期 1 21 0 0年 8月
化 学 工业 与工 程 技 术 Jo r a f h mia n u ty & En n e ig u n l C e c lI d sr o giern
Vo131 N O 4 . .
Au ,2 0 g. 01
2 2一 甲基一1 5一 氧 杂菲一6一 的微 波合 成研 究 , 二 , 二 酮
体 , ] 目前 其 合成 方 法 主要 是通 过香 豆 素衍 生 物经 醚化 和 Casn重排 得到 , l e i 大致有 以下几 种 : 方法 一 : 乙酰氧 基香豆 素与 2一 7一 甲基一3 丁炔 一
一
利 用季 铵盐 Al u u 作 为催 化 剂 成 功地 合 成 了双 i at g
丁 恒 良 , 宏顺 , 孙 徐 宏
( 京化工职业技术学院 , 苏 南京 南 江 204) 1 0 8
15二羰基化合物合成方法michael加成

15二羰基化合物合成方法michael加成摘要:一、引言1.简介二羰基化合物2.介绍Michael加成反应二、二羰基化合物的合成方法1.通过醇解法2.通过酰氯法3.通过Michael加成反应三、Michael加成反应的实验步骤1.准备试剂2.反应条件3.产物分离与纯化四、反应的影响因素1.试剂比例2.反应温度3.催化剂五、产物的应用1.有机合成中间体2.药物合成正文:在有机化学领域,二羰基化合物是一种重要的化合物类型。
它们广泛存在于天然产物、药物以及有机材料中。
二羰基化合物的合成方法有很多,其中一种较为常见且具有较高实用价值的方法是Michael加成反应。
Michael加成反应是一种高效的合成二羰基化合物的方法。
该反应通常发生在具有活泼α-氢的酮或醛与具有活泼α-卤素的烯烃之间。
在适当的催化剂作用下,这两个分子通过加成反应形成一个稳定的碳负离子,随后失去一个质子,生成二羰基化合物。
具体实验步骤如下:1.准备试剂:首先需要准备合适的酮或醛、烯烃、催化剂以及溶剂。
常见的催化剂有碘、金属钠、锂等,溶剂通常为惰性溶剂,如乙醚、氯仿等。
2.反应条件:Michael加成反应一般在室温下进行,反应过程中需要严格控制温度,避免过高或过低。
此外,反应体系需要保持惰性,避免与空气中的氧气发生副反应。
3.产物分离与纯化:反应完成后,可以通过蒸馏、萃取等方法将产物与反应物分离。
然后,通过柱层析等纯化方法对产物进行纯化,得到高纯度的二羰基化合物。
反应的影响因素主要有:1.试剂比例:合适的试剂比例可以提高反应的产率。
通常,酮或醛与烯烃的摩尔比例为1:1,催化剂的用量约为反应物的5%-10%。
2.反应温度:温度对反应速率和平衡位置有很大影响。
一般而言,较低的温度有利于反应向生成二羰基化合物的方向进行。
3.催化剂:催化剂的选择和用量对反应的产率和选择性具有重要影响。
不同催化剂在反应中的活性顺序为:碘>钠>锂。
合成的二羰基化合物具有广泛的用途,既可以作为有机合成中间体,也可以用于药物合成。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2,152十六二酮的合成
李 全 刘复初 林 军 朱洪友
(云南大学合成化学研究所 昆明 650091)
李全 男,29岁,从事有机合成的研究、应用与开发。
1999209214收稿,2000204205修回
摘要 以廉价易得的102十一碳烯酸为原料经两步反应合成了人工合成麝香酮的关键中间体2,152十六二酮
关键词 麝香酮 2,152十六二酮 双三苯基膦二氯化钯
Abstract T he key inter m ediate fo r synthesis of m uscone w as synthesized by tw o step s from 102un 2deceno ic acid .
Key words M uscone ,2,152hexadecanedi one ,B is (tri phenylpho sph ine )palladium (II )ch lo ride
麝香酮(R 232甲基环十五酮)是名贵中药材麝香的主要有效成份,天然来源稀少。
近半个世纪以来,许多学者为寻求一廉价的人工合成路线做了大量的研究工作,并有许多评论[1~3],其中以2,152十六二酮为中间体的合成路线最引人瞩目,因为2,152十六二酮在分子内闭环时Β位同时引入甲基,具有一定的优势
:
对其闭环及随后的加氢反应已有许多研究[4~7],较为成熟。
因此,2,152十六二酮的合成为人们所关注,已有许多文献报道[6~10]。
作者通过钯(II )[11]及锰(III )[12]的催化反应用廉价易得的102十一碳烯酸和丙酮为原料,经两步反应较好地合成了2,152十六二酮
:
COOH
A c 2O Pd (PPh 3)2C l
2+O
M n (III )O 0
+ O 羧酸降解烯化反应中双三苯基膦二氯化钯的用量仅为十一碳烯酸的1 2000(质量比)左右,锰(III )催化反应中锰(III )以四水合醋酸锰(II )的形式回收,1,92癸二烯与丙酮单边加成产物122烯222十三酮还可与丙酮在锰(III )的催化下再次加成得到2,152十六二酮。
该合成路线未见文献报道,成本较低,易于操作。
1 实验部分
1.1 1,92癸二烯的合成
按摩尔比1∶1配制102十一碳烯酸与醋酐混合物并放置一周得混酐。
于250mL 的三口烧瓶中・
571・h ttp : ch ina .chem istrym ag .o rg 化学通报 2001年第3期
加入双三苯基膦氯化钯011g 、三苯基膦1g 后,再加入50mL 上述混酐。
加热蒸馏,同时滴入混酐,滴加速度与蒸出速度相等,当馏份温度升至200℃时停止滴加,继续蒸馏至反应烧瓶内残留液约为2~3mL 后停止加热(此时共消耗102十一碳烯酸200.4g (1.089m o l ))。
馏份于大约120℃下减压重蒸后,倒入5%盐酸中,摇动,放置过夜,分出上层,无水硫酸钠干燥得1,92癸二烯113.2g ,收率
75.3%。
b .p .158.5℃(81434Pa ),n 20D 1.4349。
文献值[15]:b .p .169℃,n 20D 1.4320。
M S :M z 138。
I R
(液膜),Μm ax c m -1:3090,2980,2930,2860,1826,1643,1460,1441,992,966,909。
1.2 2,152十六二酮的合成
于500mL 烧瓶中加入80g M n (OA c )3・2H 2O 、200mL 丙酮,搅拌下回流,同时滴入1,92癸二烯20g (0.145m o l )与丙酮混合物100mL ,反应48h 后,过滤,沉淀回收,滤液蒸去丙酮和其它低沸点物质得黄色固体,用50%乙醇重结晶得白色固体2,152十六二酮7.4g ,收率20%。
m .p .82~83℃(文献值[10]82~84℃)。
重结晶时的不溶有机层经柱层析得无色液体122烯222十三酮9.6g 。
2,152十六二酮波谱数据如下(与文献值[10]一致):
M S :M z 254;I R (KB r 压片),Μm ax c m -1:1704,1716;1H NM R ∆
:2.13(s ,6H ,COCH 3),2.34~2.42(t ,4H ,CH 2CO ),1.45~1.68(m ,4H ,CH 2),1.26(m ,16H ,CH 2)。
122烯222十三酮波谱数数据如下:
M S :M z 196。
I R (液膜),Μm ax c m -1:1710,1639,989,960,904。
1H NM R ∆
:1.30(m ,10H ,CH 2),1.35(m ,2H ,CH 2),1.55(m ,2H ,CH 2),1.90(m ,2H ,CH 2),2.11(s ,3H ,CH 3CO ),2.4(t ,2H ,COCH 2),4.9(m ,2H ,=CH 2),5.8(m ,1H ,—CH =)。
参
考文献
[1] 贾春华,施达常.化学通报,1983,(8):6.
[2] 陈望忠,陈邦华,焦克芳.中国医药工业杂志,1992,23(1):37.
[3] 李全,刘复初.合成化学,1998,6(1):19.
[4] T suji J ,Yam ada T ,Kaito M et al .Bull .Chem .Soc .Jpn .,1980,53(5):1417.
[5] N i ppon M ining Co L td .JP 59157047,1984.
[6] 王永坚,王宫,刘亚森.医药工业,1988,19(7):291.
[7] H uell m nn M ,Kuekenhoehner T ,B renner K et al .U S :5120880,1992.
[8] Sto llM ,Rouv éA .H elv .Ch i m .A cta .,1947,30:2019.
[9] 梁利华.药学学报,1984,9:30.
[10] 焦克芳,陈望忠,李新胜.CN :1059709,1992.
[11] M iller J A ,N elson J A ,Byrne M P .J .O rg .Chem .,1993,58:18.
[12] M elikyan G G .Synthesis ,1993,(9):833.
[13] 黄枢,谢如刚,田宝芝等.有机合成试剂制备手册.成都:四川大学出版社,1988:50.
[14] H eiba W E I ,D essau R M et al .J .Am .Chem .Soc .,1969,91:138.
[15] A ldrich H andbook of fine chem icals and labo rato ry equi pm ent ,2000~2001:489.[CJ I 论文摘要](V o l .03N o .2Page 008:h ttp : www .chem istrym ag .o rg cji
2001 032008pe .h tm )有机结构波谱中二级结构效应机理的新探索(III )
——羟基伸缩振动频率中的p →p 配位效应
李润卿 范国梁 刘翠华 李玉荷
(天津大学材料科学与工程学院,300072,天津)
摘要 H 2O 和ROH 中的O 原子轨道为不等性sp 2杂化的,并且其2p 孤对电子和H 原子的2p 空轨道之间存在着一定程度的p →p Π配价键。
这个配价键在O —H 键伸缩振动频率随取代基电子效应的变化中起着主导作用。
关键词 羟基,红外光谱,p →p Π配位・671・化学通报 2001年第3期 h ttp : www .chem istrym ag .o rg。