钯催化交叉偶联反应
钯催化交叉偶联反应

钯催化的交叉偶联反应一、偶联反应综述1.交叉偶联反应偶联反应,从广义上讲,就是由两个有机分子进行某种化学反应而生成一个新有机分子的过程。
狭义的偶联反应是涉及有机金属催化剂的碳-碳键生成的反应,根据类型的不同,又可分为自身偶联反应和交叉偶联。
交叉偶联反应是一个有机分子与另一有机分子发生的不对称偶联反应。
2.碳碳键形成的重要性新碳-碳键的形成在有机化学中是极其重要的。
人们了解了天然有机物质的结构和性能,并根据有机物质的结构,通过碳原子组装成链,建立有机分子,最终实现天然有机物质的人工合成。
目前为止,人类已经利用有机合成化学手段创造出几千万种物质,且越来越多的有机物质已经广泛应用到制药、建材、食品、纺织等人类生活领域,我们的生活也几乎离不开有机物了。
合成药物、塑料等有机物质时,需要用小的有机分子将碳原子连接在一起构建新的复杂大分子,因而有机合成中高效的连接碳-碳键的方法是有机合成化学中的重要工具。
从以往该领域诺贝尔化学奖的授予情况也可以看出合成新碳-碳键的重要性:1912年维克多·格林尼亚因发明格林尼亚试剂——有机镁试剂获奖,1950年迪尔斯和阿尔德因发明双烯反应迪尔斯-阿尔德反应获奖,1979年维蒂希与布朗因发明维蒂希反应共同获奖,2005年伊夫·肖万、罗伯特·格拉布、理查德·施罗克因在有机化学的烯烃复分解反应研究方面作了突出贡献获奖。
3.有机合成中的钯催化交叉偶联反应随着时代发展,合成有机化学的研究愈加深入,20世纪后半期,科学家们发现了大量通过过渡金属催化来创造新有机分子的反应,促使有机合成化学快速发展。
特别是赫克、根岸英一和铃木章发现的钯催化交叉偶联反应,为化学家们提供了一个更为精确有效的工具。
三位科学家发现的钯催化交叉偶联反应中都使用了金属钯作为反应的催化剂,当碳原子与钯原子连在一起时,钯原子唤醒了“懒惰”的碳原子但又不至于使它太活泼,于是形成温和的碳-钯键,在反应过程中,钯原子又可以把别的碳原子吸引过来,形成另一个金属-碳键,此时两个碳原子都连接在钯原子上,它们的距离足够接近而发生反应,生成新的碳-碳单键。
第9章 钯催化C-C键交叉偶联反应的机理
C-H活化芳基化反应
1 转移金属化过程是反应的决速步 2 钯不与氢原子直接作用,碱与质子结合后形成卡宾可以 与钯结合形成稳定的环状过渡态 3 Concerted metalation deprotonation (CMD)机 理
实用文档
2.3 还原消除
trans
cis
cis
1 只有顺式产物会进行还原消除 2 还原消除过程不可逆 3 过渡态能垒大小:vinyl < Ph < ethynyl < Me, 马来酰胺 < “empty” < ethylene < PMe3 ≈ MeCN 4 能垒大小与π电子接受能力成反比,因此π电子接受能力差的配体(PMe3)会在还原消除过程前 解离。
实用文档
3. 结论
计算化学 Computational
Chemistry
实验化学 Experimenta l Chemistry
有机反应机理
实用文档
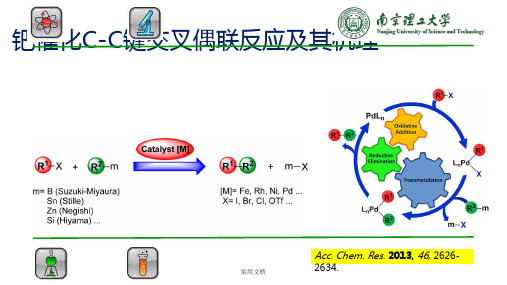
钯催化C-C键交叉偶联反应及其机理
实用文档
Acc. Chem. Res. 2013, 46, 26262634.
1. 钯催化C-C键交叉偶联反应
实用文档
Angew. Chem. Int. Ed. 2012, 51, 50625085.
实用文档
2. 反应机理
1 氧化加成
2 转移金属化
3 还原消除
存在问题:
1 缺乏更为具体深入的理论和验证研究。 2 对于具体的因素和条件对于反应的影响缺乏明确 和系统解释和依据,如配体、金属和底物的类型 对于反应的影响。
实用文档
2.1 氧化加成
协同机理: 1 构型保持 2 气相反应 3 某些极性溶剂
SN2机理: 1 构型反转 2 液相反应
钯催化的交叉偶联反应——2010诺贝尔化学奖简介

产治疗高血压 、 肾脏病等方 面的药 物 。 1本 医药 公司用 3
铃木 反应生产 的 降压 药 .0 9年在 1本 国 内就有 10 20 3 40
亿 1 ( 10 3 约 2 亿人 民币 ) 元 的销售额。电子领域也开始瞩
目“ 钯催 化的交叉偶 联反应 ” 相关成 果不 断被推 出 , , 手
21年 1 00 0月 61瑞典 皇家科学 院诺 贝尔颁奖委员 3 会把今 年 的诺贝尔化学 奖授予 美 国科学家 7 9岁的理 查 德一 赫克 ( i a ek 、 Rc r H c ) 1 hd 3本科学 家 7 5岁 的根岸英 一 ( iih E—ci ei i N g h)和 1本科学家 8 的铃木 章( kr s 3 0岁 A i a
铃木章 。 本公 民。9 0 出生于 1本北海道 。9 9 3 1 13 年 3 15 年在北 海道大学获得博士学位 。16 9 3年一 9 5 , 16 年 铃木 章在美 国普渡大学赫伯特 ・ 布朗教授指导下完成 博士后
研究 。 9 3 17 年起在北海道大学任教授。17 年 , 明“ 99 发 铃 木反应” 。铃木教授 的论文数量不多 。 且大都 以 1文的形 3 式发表在 了 1本 的学术 刊物 上。但 是 . 3 他的研究非 常严 密 , 出来的结果经得起反 复推 敲。为 了证实 自己的研 做 究结果具有可重复性 . 铃木教授不 惜购买全套 的新实验
尔化学奖的赫伯特 ・ 布朗( re rw ) 9 7 , He rB o n 。17 年 发明 bt “ 根岸反应” 。根岸现为美 国普 渡大学化学教授。当根岸 教授在大教 室照常讲授他 的课程 “ 有机化合物 的性质 ”
一
者, 真可谓名师出高徒。
二、 有机合成 中的钯催化交叉偶联反应
钯催化交叉偶联反应

钯催化交叉偶联反应什么是钯催化交叉偶联反应?钯催化交叉偶联反应(Palladium-Catalyzed Cross-Coupling Reaction)是一种重要的有机合成反应。
它是一类碳-碳键构造的反应,是通过将两种不同的碳基官能团或碳碳键连接在一起,以形成新的C-C化合物。
反应机理在钯催化交叉偶联反应中,两个分子的有机基团进行偶联,然后由钯离子起催化作用,生成新的碳碳键。
催化剂形式上是Pd(0)配合物,反应机理如下:1.钯催化剂先通过脱对氢化学计量通常分配Pdcatalyst (I)。
2.钯催化剂进一步和配体形成配合物(PdL2)。
3.配合物和卤代烃发生交换生成过渡态PdL2(RX)。
过渡态中,钯离子与亲电吸引剂的卤素原子形成键;此过程中C-X钩体断裂,形成第一级碳中间体。
4.结合第二个有机基团生成PdL2(RY)介于新的物种。
5.最后的反应产物通常通过还原反应,将钯催化剂还原为Pd(0)。
应用钯催化交叉偶联反应已经成为有机合成中的重要反应之一,广泛应用于制药、化工、材料科学等领域。
其重要应用包括:•制备非对映选择性或对映选择性的C-C连接化合物。
•制备有机材料。
•合成复杂天然产物的合成方法研究。
反应类型钯催化交叉偶联反应可以根据反应物和类型进行分类。
最常用的交叉偶联反应类型是官能团反应 (Functional Group Coupling) 和碳-碳双键偶联反应 (Carbon-Carbon Double Bond Coupling),这些反应分类包括下列:1.骨架化反应 (Fragmentation Reaction)2.偶联反应 (Cross-Coupling Reaction)3.代换反应 (Substitution Reaction)4.重排反应 (Rearrangement Reaction)反应优点由于钯催化交叉偶联反应具有高效性、选择性、重复性和收率高的特点,它已经成为有机化学领域极为重要的反应之一。
钯催化剂催化交叉偶联反应形成P-C键和复杂含磷化合物的研究进展

钯催化剂催化交叉偶联反应形成P-C键和复杂含磷化合物的研究进展任铁钢;补朝阳;黎桂辉;李勇喆;何志鹏【摘要】综述了利用金属钯催化交叉偶联反应形成P-C键以合成各种复杂的含磷化合物特别是手性膦配体的研究进展;介绍了参加偶联反应的不同价态的P化合物亲核试剂,以及卤代烯烃、卤代芳烃、三氟甲磺酸烯基脂、三氟甲磺酸芳基脂、乙烯基硼酸脂等亲电试剂;探讨了相应的偶联反应的反应条件、反应机理及其在材料、医药、农药等领域的应用.【期刊名称】《化学研究》【年(卷),期】2014(025)006【总页数】7页(P641-647)【关键词】钯催化剂;交叉偶联反应;P-C键形成;磷化物【作者】任铁钢;补朝阳;黎桂辉;李勇喆;何志鹏【作者单位】河南大学化学化工学院,河南开封475004;新乡学院化学化工学院,河南新乡453000;河南大学特种功能材料教育部重点实验室,河南开封475004;河南大学化学化工学院,河南开封475004;河南大学化学化工学院,河南开封475004【正文语种】中文【中图分类】O643.3使用金属及金属配合物催化各种偶联反应如Suzuki反应、Heck反应、Stille 反应和Negishi反应等形成各种C-X键的方法已被广泛地应用于元素有机化合物的合成及研究中. 其中钯类催化剂体系催化交叉偶联反应是形成C-P键的一个非常重要的方法. 而含磷化合物尤其是含手性膦的化合物是开发特殊结构的药物、阻燃剂、催化剂和功能材料等的很普遍的方法.二十世纪八十年代,HIRAO[1]和XU[2-3]分别报道了利用钯配合物催化亚磷酸酯或二烷基亚磷酸酯与卤代芳烃(图1)或芳基三氟甲磺酸脂的交叉偶联来形成P-C键的方法. 此后,钯配合物催化交叉偶联反应形成P-C键的方法受到广泛的关注. 1987年,TUNNEY[4]等人用钯配合物催化芳基卤化物和三甲基甲硅烷二苯基膦的反应,随后IMAMOTO[5] 和LIVINGHOUSE[6]进一步发展了膦-硼烷芳基化反应,且都获得了很高的产率. 对钯类催化剂体系催化交叉偶联反应形成C-P键的研究虽然取得了很好的结果,但这种方法真正得到研究者的重视并应用到有机膦配体特别是手性膦配体的合成中却是在1990年,KURZ [7]报道了在钯催化下1,1′-联萘-二三氟甲磺酸脂和亚磷酸二乙酯或二苯基膦氧化物的P-C偶联反应之后. 此后,钯催化的磷化反应就被广泛地应用于各种膦配体的合成中.钯催化下的亚磷酸二烷基脂与芳基卤化物的偶联反应是合成芳基磷酸酯的一个非常重要以及行之有效的方法,三氟甲磺酸芳基脂也被用作亲电试剂来合成新的P-C 键.在通常的反应中,亚磷酸二烷基脂是过量的,碱一般为三乙胺.在反应过程中,如果底物亚磷酸二烷基脂具有手性,则磷手性中心的构型不发生改变. 而KURZ[7]的研究发现联萘酚双三氟甲磺酸酯与亚磷酸二乙酯反应时,即使亚磷酸二乙酯大大过量,也只生成单取代芳基膦酸酯,当用其他的亚磷酸二烷基脂代替亚磷酸二乙酯时这种情况一样存在. BRAVO-ALTAMIRANO[8]研究了钯催化直接形成H-芳基亚磷酸脂的反应,卤代的芳基、杂芳基、烯基、苄基和烯丙基均可得到较好的收率. 卤代烯基和卤代烯丙基是首次作为此类反应的底物被报道. 作者对底物的范围及可能的反应机理进行了讨论. 反应如图2所示.随后,KALEK等[9]对此类反应的机理进行了进一步的探究(图3),探讨了不同类型的钯催化剂以及不同的配体对反应的影响. 他们发现当使用醋酸根离子与二价的钯形成的配合物作为催化剂时,可以使这类反应获得最大的反应速率. 同时作者采用 31P NMR光谱研究了不同催化剂体系间的差异. 这个发现为亚磷酸二乙酯与卤代的苯基在钯催化下的偶联反应的条件优化提供了理论依据.亚磷酸二烷基脂与乙烯基卤化物或三氟甲磺酸乙烯酯在钯催化下合成乙烯基磷酸酯反应的一个最大的特点是反应前后乙烯基双键的构型保持不变[10]. 反应条件与亚磷酸二烷基脂与芳基卤化物的偶联反应类似,Pd(PPh3)4为催化剂,碱一般为三乙胺,收率较高.反应如图4所示[1].ABBAS等人[11]利用钯催化偶联反应以廉价易得的商品化原料为前体,合成了天然磷酸酯的替代化合物,产物对酸碱敏感,用Pd催化偶联反应时,需要使用环氧丙烷作为缚酸剂来抑制副反应的发生. 该反应是外源合成寡聚核苷酸的重要步骤. 反应如图5所示.当然,手性膦配体的合成一直是研究者关注的重点,早在上世纪九十年代初,UOZUMI[12]就利用KURZ[7]的合成方法得到了手性单齿膦配体,而这些手性单齿膦配体的合成与应用又大大地推动了不对称催化化学的发展[13-14],在Pd催化的烯烃不对称硅烷化,烯丙基甲酸酯的不对称还原,烯丙基的不对称烷基化、氰化、羰基化,金属与烯的不对称反应,二茂锆酰氯对烯酮的不对称1,2-加成以及1,3-烯炔的1,4-不对称硼氢化反应中已经广泛地应用到手性单齿膦配体作为反应物或催化剂配体,并且这些反应产物的ee值均较高. HAYASHI[14]制备了两种手性单齿膦配体(MeO-MOP)与其双蒽基类似物(图6). 这类配体与Pd的配合物在多种不对称催化反应中表现出很强的催化活性和良好的选择性. 作者通过烯烃的不对称硅氢化反应考察了该催化剂的催化活性与选择性. 反应具有很高的收率(>90%),同时选择性也很好(alkyl-substituted terminnal olefins: 92%~97%ee;bicyclo[2,2,1]heptene derivatives: 92%~96%ee; styrene derivatives: 89%~96%ee; cyclic 1,3-dienes: 80%ee). 所得的产物通过对C-Si键的氧化能够转化为手性醇类化合物.CAVAZZINI等[15]在HAYASHI合成方法的基础上,通过在配体尾基位置引入全氟烷基得到新的手性单齿膦配体,而这种全氟烷基的引入有利于配体的循坏使用. 在三氟甲苯溶剂中,这种手性单齿膦配体作为催化剂催化烯丙基的不对称烷基化反应,可以得到高达87%ee值的手性产物,反应后催化剂容易回收再使用.次膦酸酯中磷原子上直接连有两个氢原子,因此在与卤代烃反应的时候可以有选择的与一分子或两分子的卤代烃发生偶联反应进而生成单芳基或双芳基次膦酸酯. 为了抑制双芳基次膦酸脂的生成, 可以适当的提高次膦酸脂的加入量,使得到的单芳基膦酸脂的含量增高[16]. 如果增大反应底物中芳基卤化物的配比,则能够得到较高收率的对称双芳基次膦酸脂.2001年,MONTCHAMP等[17] 研究发现用次磷酸苯胺盐与芳基卤化物在烷氧基硅烷存在下反应,可以很容易地得到高产率的单取代次膦酸脂,避免了双芳基次膦酸脂的生成(图7). 当卤代烃为乙烯基溴代物或三氟甲磺酸乙烯酯时,就得到单乙烯基次膦酸脂. 反应中烯烃的双键在反应前后构型保持不变[18].三芳基或三烷基氧化膦经由二价的硅酸还原后可得到应用广泛的三芳基或三烷基膦. 而三芳基或三烷基氧化膦可以由二芳基或二烷基氧化膦经钯催化与芳基卤化物进行偶联反应得到(图8) [19]. 而由三氟甲磺酸芳基酯与二芳基氧化膦的钯催化的偶联反应则广泛应用于手性膦配体的合成中[19-20].为防止脱保护和五价膦的还原,通常采取直接由伯膦和仲膦与芳基卤代物的偶联反应得到仲膦和叔膦. 而在研究钯催化此类的反应时由于CAI等[21]在研究膦与芳基三氟甲磺酸脂的偶联反应时,认为在反应体系中存在着大量的膦,会导致钯催化剂的毒化而失活从而使得反应无法得到想要的产品,转而使用NiCl2/dppe催化体系来合成仲叔膦. 一段时间里钯催化体系催化此类反应的研究较少. 1996年,HERD等[22-23]发现在钯催化下膦与芳基卤化物的偶联反应能够高产率地合成一系列膦配体(图9),钯催化此类反应的研究重新得到研究者的关注. HERD等合成了一系列的水溶性膦配体. 反应具有很好的官能团耐受性,该研究为制备功能化的三芳基膦提供了一条便利有效的途径. BRAUER等[24]发现钯催化下二苯基膦或苯基膦与溴代碘代苯发生选择性膦化反应,得到溴苯基二苯基膦或二(溴苯基)苯基膦. 产物很容易通过结构修饰后作为功能化的膦配体, 也可以作为起始原料继续和亚磷酸二乙酯发生偶联反应,得到新型的水溶性膦配体,即含有磷酸盐的三芳基膦.随后研究者利用Pd催化的P-C偶联反应制备了多种功能化的新型水溶性膦配体[25],包括分子中含有氨基酸结构的膦衍生物[26]. 此外,KANBARA等[27]以 1,3-二苯基膦丙烷和芳基二卤化物为原料,在 PdCl2催化下合成了主链含三价P原子的有机磷聚合物.当卤代烃为乙烯基卤化物或三氟甲磺酸乙烯酯时,与伯或仲膦通过Pd催化的P-C 偶联反应可以制备乙烯基叔膦化合物. 反应时C=C键的构型保持不变(图10)[28]. KAZANKOVA[29]以PdCl2(PPh3)2为催化剂,在三乙胺存在下,通过二苯基膦与卤代烯烃的偶联反应得到了产率高达97%的叔膦.除了常见的含有P-O键的化合物作为底物形成C-P键外,分子中含有P-Si, P-Sn, P-B键的化合物也被研究者尝试作为底物形成C-P键. 2003年,KABALKA[30]报道了Pd(OAc)2催化亚磷酸三烷基脂与乙烯基硼酸脂的偶联反应(图11),收率为55%~84%,反应前后C=C键构型保持不变. 反应通入O2可提高产物的收率,因为O2的存在能够抑制底物的自身偶联. 当C=C键上连有其他取代基时,也能获得较好的收率. 此外,Ph2PSiMe3[29]、Ph2PSnR3[31]以及膦-硼烷复合物[32]作为底物应用于Pd催化的P-C偶联反应中,在手性膦配体的合成方面受到广泛的关注.经过近几十年的发展,Pd催化的P-C偶联反应在合成各种不同类型的有机膦配体中起到越来越重要的作用. 通过将C-P偶联反应与C-C偶联反应相结合,此类反应的使用范围得到了很大增加. 同时离子液体和超临界CO2[33]等新型反应介质的使用,使得此类反应的效率和选择性都大大提高,环境污染减小. Pd催化的P-C偶联反应在药物、生物农药、新型催化剂、新材料等领域的应用均具有非常广阔的前景.【相关文献】[1]HIRAO T, MASUNAGA T, YAMADA N, et al. Palladium-catalyzed new carbon-phosphorus bond formation [J]. Bull Chem Soc Jpn, 1982, 55(3): 909-913.[2]XU Y Y, LI Z, XIA J Z, et al. Palladium-catalysed synthesis of unsymmetrical alkyl aryl-phenylphosphinates [J]. Synthesis, 1983(5): 377-378.[3]XU Y, WEI H, ZHANG J, et al. An efficient synthesis of chiral, nonracemic isopropyl alkenylmethylphosphinates via palladium route [J]. Tetrahedron Lett, 1989, 30(8): 949-952.[4]TUNNEY S E, STILLE J K J. Palladium-catalyzed coupling of aryl halides with (trimethylstannyl) diphenylphosphine and (trimethylsilyl)diphenylphosphine [J]. J Org Chem, 1987, 52: 748-753.[5]IMAMOTO T, OSHIKI T, ONOZAWA T, et al. Synthesis and reactions of optically active phosphine-boranes [J]. Heteratom Chem,1992, 3(5/6): 563-575.[6]KMASUM M, LIVINGHOUSE T. Pd(0)-Cu(I) cocatalyzed coupling of methylphenylphosphine-borane with aryl halides and aryl nonaflates [J]. Tetrahedron Lett,1999, 40(44): 7731-7734.[7]KURZ L, LEE G, MORGANS J D, et al. Stereospecific functionalization of (R)-(-)-1, 1′-bi-2-naphthol triflate [J]. Tetrahedron Lett, 1990, 31(44): 6321-6324.[8]BRAVO-ALTAMIRANO K, HUANG Z H, MONTCHAMP J L. Palladium-catalyzed phosphorus-carbon bond formation: cross-coupling reactions of alkyl phosphinates with aryl, heteroaryl, alkenyl, benzylic, and allylic halides and triflates [J]. Tetrahedron, 2005,61(26): 6315-6329.[9]KALEK M, STAWINSKI J. Pd(0)-Catalyzed phosphorus-carbon bond formation.mechanistic and synthetic studies on the role of the palladium sources and anionic additives [J]. Organometallics, 2007, 26(24): 5840-5847.[10]MAFFEIA M, BUONOB G. A two step synthesis of 2-oxo-2-vinyl 1,3,2-dioxaphospholanes and-dioxaphosphorinanes [J]. Tetrahedron, 2003, 59(44): 8821-8825.[11]ABBAS S, BERTRAM R D, HAYES C J. Commercially available 5′-DMT phosphoramidites as reagents for the synthesis of vinylphosphonate-linked oligonucleic acids [J]. Org Lett, 2001, 3(21): 3365-3367.[12]UOZUMI Y, SUZUKI N, OGIWARA A, et al. Preparation of optically active binaphthylmonophosphines (MOP′s) containing various functional groups [J]. Tetrahedron, 1994, 50(15): 4293-4302.[13]OGASAWARA M, YOSHIDA K, KAMEI H, et al. Synthesis and application of novel chiral phosphino-oxazoline ligands with 1, 1′-binaphthyl skeleton [J]. Tetrahedron: Asymmetry,1998, 9: 1779-1787.[14]HAYASHI T. Asymmetric hydrosilylation of olefins catalyzed by MOP-palladium complexes [J]. Acta Chemica Scandinavica, 1996, 50: 259-266.[15]CAVAZZINI M, POZZI G, QUICI S, et al. Palladium-catalysed asymmetric allylic alkylation in the presence of a chiral ‘light fluorous’ phosphine ligand [J]. Chem Commun, 2001: 1220-1221.[16]SCHWABACHER A W, STEFANESCU A D, REHMAN A U. Synthesis of a new ligand for metal assembly to a selective receptor [J]. J Org Chem, 1999, 64(6): 1784-1788.[17]MONTCHAMP J L, DUMOND Y R. Synthesis of monosubstituted phosphinic acids: palladium-catalyzed cross-coupling reactions of anilinium hypophosphite [J]. J Am Chem Soc, 2001, 123(3): 510-511.[18]DEPRLE S, MONTCHAMP J L. A novel and convenient preparation of hypophosphite esters [J]. J Organomet Chem, 2002, 643-644: 154-163.[19]HOSHI T, HAYAKAWA T, SUZUKI T, et al. Enantiomerically pure 2-bromo-2′-diphenylphosphinyl-1,1′-binaphthyl as a monophosphorus template for electrophilic functionalization in chiral MOP-type ligand synthesis [J]. J Org Chem, 2005, 70(22): 9085-9087.[20]SAHA B, RAJANBABU T V. Syntheses and applications of 2-phosphino-2′-alkoxy-1,1′-binaphthyl ligands. development of a working model for asymmetric induction in hydrovinylation reactions [J]. J Org Chem, 2005, 72(7): 2357-2363.[21]CAI D W, PAYACK J F, BENDER D R, et al. Synthesis of chiral 2,2′-bis( dipheny1phosphino)-1,1′-binaphthyl (BINAP) via anovel nickel-catalyzed phosphine insertion [J]. J Org Chem, 1994, 59(23): 7180-7181.[22]HERD O, HEΒLER A, HINGST M, et al. Water soluble phosphines VII. Pa lladium-catalyzed P-C cross-coupling reactions between primary or secondary phosphines andfunctional aryliodides-a novel synthetic route to water soluble phosphines [J]. J Organomet Chem, 1996, 522(1): 69-76.[23]KWONG F Y, LAI C W, TIAN Y, et al. A novel synthesis of functionalised tertiary phosphines by palladium catalysed phosphination with triarylphosphines [J]. Tetrahedron Lett, 2000, 41(52): 10285-10289.[24]BRAUER D J, HINGST M, KOTTSIEPER K W, et al. Water soluble phosphines: Part XV. Syntheses of multiply functionalized and chiral phosphine ligands by Pd-catalyzed P-C and C-C coupling reactions [J]. J Organomet Chem, 2002, 645(1/2): 14-26.[25]JIANG J Y, WANG Y H, LIU C, et al. Thermoregulated phase transfer ligands and catalysis XIV: Synthesis of N, N-dipolyoxyethylene-substituted-4-(diphenylphosphino) benzenesulfonamide (PEO-DPPSA) and the catalytic activity of its rhodium complex in hydroformylation of 1-decene [J]. J Mol Catal A: Chem, 2001, 171(1/2): 85-89.[26]GILBERTSON S R, FU Z C. Chiral P,N-ligands based on ketopinic acid in the asymmetric Heck reaction [J]. Org Lett, 2001, 3(2): 161-164.[27]KANBARA T, TAKASE S, IZUMI K, et al. Palladium-catalyzed polycondensation of diiodobenzenes with 1,3-bis(phenylphosphino)propane and preparation of polymer transition-metal complexes [J]. Macromolecules, 2000, 33(3): 657-659.[28]GILBERTSON S R, FU Z C, STARKEY G W. Palladium-catalyzed synthesis of vinyl phosphines from ketones [J].Tetrahedron Lett, 1999, 40(49): 8509-8512.[29]KAZANKOVA M A, CHIRKOV E A, KOCHETKOV A N. Synthesis of vinyldiphenylphosphines by Pd-catalyzed cross-coupling reactions of diphenylphosphine with alkenylhalides [J]. Tetrahedron Lett, 1999, 40(3): 573-576.[30]KABALKA G W, GUCHHAIT S K. Synthesis of (E)-and (Z)-alkenylphosphonates using vinylboronates [J]. Org Lett, 2003, 5(5): 729-731.[31]MARTIN S E, BONATERRA M, ROSSI R A. One-pot palladium-catalyzed phosphination of aryl iodides with Ph2PSnR3 [J]. J Organomet Chem, 2002, 664(1/2): 223-227.[32]LI J, LUTZ M, SPEK A L, et al. Novel phosphite palladium complexes and their application in C-P cross-coupling reactions [J]. J Organomet Chem, 2010, 695(24): 2618-2628.[33]BAILLIE C, CHEN W, XIAO J. Synthesis of biphenyl-based phosphines by Suzuki coupling [J]. Tetrahedron Lett, 2001, 42(51): 9085-9088.。
钯催化剂在有机合成中的应用

钯催化剂在有机合成中的应用钯催化剂是一种广泛应用于有机合成中的催化剂,具有重要的化学价值。
在有机化学领域,钯催化剂的应用已经被广泛研究,并被成功应用于合成复杂的天然产物和药物分子。
本文将探讨钯催化剂在有机合成中的应用。
一、钯催化剂的优势钯催化剂具有许多优势,例如高催化活性、较低的剂量要求、宽阔的反应适应性和化学选择性等等。
此外,钯催化剂还可以用于对手性分子的合成,这对于药物化学和材料化学领域是非常重要的。
由于这些优势,钯催化剂已被广泛应用于许多有机反应中。
二、钯催化剂在有机合成中的应用1. 交叉偶联反应钯催化的交叉偶联反应是一种常见的有机合成反应。
这种反应可以将两个不同的分子中的芳基、烯基或卤代烷基连接起来,形成一个新的分子。
该反应对于有机化学中的复杂化合物合成非常重要,尤其是在医药领域。
2. 加氢反应加氢反应是将半饱和和饱和的有机化合物进行还原,制备次级和三级醇、脱氧酸、醛气等化合物的重要方法。
钯催化的加氢反应已被广泛应用于合成重要的医药和材料分子,如β-羟基酸、腺苷、卡培他滨、丁二酸等。
此外,钯催化的选择性加氢反应还可以用于制备更有建筑价值的化合物,如乙酸等。
3. 烯烃氧化反应通过钯催化的烯烃氧化反应,可以将烯烃氧化为C—C双键的羰基化合物,在有机合成中具有重要的地位。
该反应被广泛应用于制备各种复杂的有机化合物,包括β-羟基酸、酮、醇和醛等。
此外,烯烃氧化反应还可以制备含氧杂环化合物。
4. 偶氮化反应偶氮化反应是一种将芳香胺转化为富有色彩的偶氮化合物的重要反应。
该反应不仅具有学术研究价值,还可以通过将合成的化合物应用于染料和颜料等领域中。
此外,偶氮化反应还可以用于合成其他富有应用前景的有机化合物。
三、结论综上所述,钯催化剂在有机合成中具有重要的应用价值。
该催化剂已经被广泛研究,并被成功地应用于合成天然产物和药物分子。
随着科技的不断进步和发展,钯催化剂的应用领域也将不断扩大。
钯催化交叉偶联反应

钯催化交叉偶联反应钯催化交叉偶联反应2010-10-26 17:32钯催化交叉偶联反应摘要钯催化交叉偶联反应是一类用于碳碳键形成的重要反应,在有机合成中应用十分广泛。
钯催化交叉偶联反应-简介为制造复杂的有机材料,需要通过化学反应将碳原子集合在一起。
但是碳原子在有机分子中与相邻原子之间的化学键往往非常稳定,不易与其他分子发生化学反应。
以往的方法虽然能令碳原子更加活跃,但是,过于活跃的碳原子却又会产生大量副产物。
而用钯作为催化剂则可以解决这个问题。
钯原子就像"媒人"一样,把不同的碳原子吸引到自己身边,使碳原子之间的距离变得很近,容易结合--也就是"偶联"。
这样的反应不需要把碳原子激活到很活跃的程度,副产物比较少,因此更加精确而高效。
赫克、根岸英一和铃木章通过实验发现,碳原子会和钯原子连接在一起,进行一系列化学反应。
这一技术让化学家们能够精确有效地制出他们需要的复杂化合物。
钯催化交叉偶联反应-应用如今,"钯催化交叉偶联反应"被应用于许多物质的合成研究和工业化生产。
例如合成抗癌药物紫杉醇和抗炎症药物萘普生,以及有机分子中一个体格特别巨大的成员--水螅毒素。
科学家还尝试用这些方法改造一种抗生素--万古霉素的分子,用来灭有超强抗药性的细菌。
此外,利用这些方法合成的一些有机材料能够发光,可用于制造只有几毫米厚、像塑料薄膜一样的显示器。
科学界一些人士表示,依托"钯催化交叉偶联反应",一大批新药和工业新材料应运而生,这三名科学家的科研成果如今已经成为支撑制药、材料化学等现代工业文明的巨大力量。
钯催化交叉偶联反应-诺贝尔奖2010年10月6日在瑞典皇家科学院举行的新闻发布会上,瑞典皇家科学院常任秘书诺尔马克首先宣读了获奖者名单。
他说,赫克、根岸英一和铃木章在"钯催化交叉偶联反应"研究领域作出了杰出贡献,其研究成果使人类能有效合成复杂有机物。
钯催化的交叉偶联反应——2010年诺贝尔化学奖简介

①②博士研究生 , ③博士 , ④教授 , 南京大学化学化工学院 , 南 京 210093
关键词 钯催化 偶联反应 诺贝尔化学奖
2010 年 10 月 6 日 , 瑞典皇家科学院宣布将 2010 年诺贝尔化学奖授予美国科学家 Richard F .Heck , 日本科学家 Ei-ichi Negishi 和 Akira Suzuki。 这三名科学家是因为在有机合成领域中钯催化交叉偶联反应方面的卓越研究而获 奖 。它为化学家提供了一款精致的工具来合成复杂的有机分子 。 这一成果广泛应用于制药 、电子工业和先进材料等 领域 。 笔者对钯催化交叉偶联反应领域作了粗浅的介绍 , 以期起到抛砖引玉之作用 。
· 332 ·
1 早期研究
有机合成化学制 造出的这几 千万种新的 物质绝大 多数都是以碳原子为 主来构建的 。 为了制 备结构更复 杂 、功能更强大的新型材料 , 就要想办法通过各种化学 反应将碳原子连接在 一起 。 然而碳原子本 身是十分稳 定的 , 在化学反应中并不活 泼 , 所以就得想办法来激活 碳原子 , 让它 更容易 参与反 应并 与其 他碳 原子连 接起 来 ,逐步 形成 更 高层 次 的碳 基 骨架 。1912 年 , 法 国人 Grignard 因发明有机镁试剂(格 氏试剂)而 荣获诺贝尔 化学奖 , 可以说是碳基活化史上的第一个里程碑 。随着 时代的发展 , 人们对碳基的研究愈加深入 。在研究的前 期 ,要么无法活化碳基 , 化合物难于参加反应 ;要么使碳 原子过于活 跃 , 虽然 能有效 地制 造出 很多 简单的 有机 物 ,但要是合成复杂分子却有大量的副产物生成 。正如 大家所知 , 在有机合成操 作中提纯是一项繁琐的工 作 。 H eck , Negish i 和 Suzuki 等人 通过实验 发现 , 当 碳原子 和钯原子连接在一 起 , 会形成一种“ 温和” 的碳钯键 , 在 这里钯既活跃了碳基 , 又使 其不至于过于活泼 , 然后又 可以把别的碳原子吸引过来 , 这样使得两个碳原子距离 拉近 , 容易成 键而 偶联 起来 。在 这里 钯原 子就相 当于 “ 媒人”的作用 , 只需使用 催化剂就行 。 所以“ 钯催化交 叉偶联反应”就 是一款精致 的工具 , 让化学 家得以像艺 术家一样来雕刻和拼接类似积木的模块(小的基团), 构 筑令人叹为观 止的艺术品(有机复杂 分子)。 与此同时 还避免了过多不必要副产物的生成 。
钯催化交叉偶联反应

钯催化的交叉偶联反应一、偶联反应综述1.交叉偶联反应偶联反应,从广义上讲,就是由两个有机分子进行某种化学反应而生成一个新有机分子的过程。
狭义的偶联反应是涉及有机金属催化剂的碳-碳键生成的反应,根据类型的不同,又可分为自身偶联反应和交叉偶联。
交叉偶联反应是一个有机分子与另一有机分子发生的不对称偶联反应。
2.碳碳键形成的重要性新碳-碳键的形成在有机化学中是极其重要的。
人们了解了天然有机物质的结构和性能,并根据有机物质的结构,通过碳原子组装成链,建立有机分子,最终实现天然有机物质的人工合成。
目前为止,人类已经利用有机合成化学手段创造出几千万种物质,且越来越多的有机物质已经广泛应用到制药、建材、食品、纺织等人类生活领域,我们的生活也几乎离不开有机物了。
合成药物、塑料等有机物质时,需要用小的有机分子将碳原子连接在一起构建新的复杂大分子,因而有机合成中高效的连接碳-碳键的方法是有机合成化学中的重要工具。
从以往该领域诺贝尔化学奖的授予情况也可以看出合成新碳-碳键的重要性:1912年维克多·格林尼亚因发明格林尼亚试剂——有机镁试剂获奖,1950年迪尔斯和阿尔德因发明双烯反应迪尔斯-阿尔德反应获奖,1979年维蒂希与布朗因发明维蒂希反应共同获奖,2005年伊夫·肖万、罗伯特·格拉布、理查德·施罗克因在有机化学的烯烃复分解反应研究方面作了突出贡献获奖。
3.有机合成中的钯催化交叉偶联反应随着时代发展,合成有机化学的研究愈加深入,20世纪后半期,科学家们发现了大量通过过渡金属催化来创造新有机分子的反应,促使有机合成化学快速发展。
特别是赫克、根岸英一和铃木章发现的钯催化交叉偶联反应,为化学家们提供了一个更为精确有效的工具。
三位科学家发现的钯催化交叉偶联反应中都使用了金属钯作为反应的催化剂,当碳原子与钯原子连在一起时,钯原子唤醒了“懒惰”的碳原子但又不至于使它太活泼,于是形成温和的碳-钯键,在反应过程中,钯原子又可以把别的碳原子吸引过来,形成另一个金属-碳键,此时两个碳原子都连接在钯原子上,它们的距离足够接近而发生反应,生成新的碳-碳单键。
钯催化的偶联反应

AgNO3/KF作用下的Pd催化2-溴噻吩S原子邻位上的C-H键选择性偶联反应摘要:溴噻吩的衍生物与芳基碘在加入了钯的硝酸银/氟化钾催化剂的催化下发生C—H键的偶联反应,而C—Br键未发生变化。
这些含有C —Br键的偶联产物在钯的进一步催化下使溴噻吩和芳基碘的C—C键相连接从而得到理想的产量。
引言:狭义上的偶联反应是涉及由基金属催化剂的C-C键生成的反应,根据类型不同,可分为交叉偶联反应和自身偶联反应。
交叉偶联反应是一个有机分子与另一有机分子发生的不对称偶联反应。
例如:烯丙基锂与2-氯辛烷可以发生交叉偶联反应生成4-甲基-1-癸烯。
格利雅试剂、有机铝、有机锌、有机锡、有机铜、有机铅、有机汞等多种有机金属化合物也都可以与卤化烷等烃基化试剂发生交叉偶联反应,生成相应的不对称烃,是合成不对称烃,特别是单烷基芳烃和含有三级碳原子的链烃的有效方法。
交叉偶联反应的范围很广,像芳烃重氮盐与苯酚或N,N-二甲基苯胺的偶联反应,也属于交叉偶联反应。
正文:芳香族化合物与有机卤化物的C-H键取代反应和那些含金属试剂与相同的有机卤化物的偶联反应相比,在有机合成中更有前景。
【1】相比之下,C-H键上的直接反应将有利于含有不同种类的官能团的衍生物的合成,并且,反应也会加强合成中原子的效应。
我们注意到噻吩衍生物的偶联反应是发生在C-H键上,从而形成了联噻吩。
在添加了AgF后,反应效率得到了提高。
【2】当噻吩与2-溴噻吩反应生成正联溴噻吩时,仍然是C-H键发生偶联,而C-Br键未发生变化。
我们的注意力集中到溴噻吩衍生物C-H键的交叉耦合上,来介绍噻吩环上的取代基。
【3】溴噻吩上的C-H键偶联,如果可以通过C-Br键的反应而进一步改变偶联产物,那么C-H键和C-Br键的偶联反应的相互结合将得到一种新的合成取代噻吩的方法。
这将把人们的注意力都吸引到设计更先进的有机金属材料来揭示液晶、光发射和有机半导体的特点。
【4】在此,我们报告一个新的催化剂系统—AgNO3/KF,它有助于提高钯催化下溴噻吩衍生物C-H键的取代反应发的效率。
钯催化交叉偶联反应及其在有机合成中的应用

第16卷第12期江苏技术师范学院学报JOURNAL OF JIANGSU TEACHERS UNIVERSITY OF TECHNOLOGY Vo l.16,No.12Dec .,20102010年12月0引言2010年的诺贝尔化学奖颁发给了三个有机化学家,理查德·海克(Richard Heck )、根岸英一(Ei-ichi Negishi)和铃木章(Akira Suzuki ),他们发现的钯催化交叉偶联反应具有高度的选择性,在相对温和的条件下,形成碳-碳单键。
在过去的40年里,这些反应成为有机化学家主要的非常有效的工具。
该反应作为五个被授予诺贝尔化学奖反应之一,其重要性已不言而喻。
前四个反应分别是Grignard 反应(1912),Diels-Alder 反应(1950),Wittig 反应(1979)和烯烃复分解(olefin metathesis )反应(2005)。
由Heck 、Negishi 和Suzuki 研究的钯催化碳-碳单键的成键反应给有机合成带来了巨大的影响,在靶向合成方面有很多的应用。
这三个反应之所以被广泛应用于合成大量的天然化合物和具有生物活性的复杂分子结构的物质,主要是因为反应条件温和,大量的官能团在如此温和的条件下,能够不被破坏而保留下来。
这三个反应同样也广泛应用于精细化学品工业和制药工业中。
1钯催化交叉偶联反应———Heck 反应的发现20世纪50年代过渡金属开始在有机化学中发挥了相当大的作用,出现了大量的由过渡金属催化的构建有机化合物分子的反应。
一家德国公司Wacker Chemie AG 以钯作为催化剂,以空气氧化乙烯生成乙醛,这一重要的方法在工业上称为Wacker 法[1]。
Richard Heck 当时就职于美国Delaware 的一家化学公司,由于受到对化学生产中Wacker 法成功应用的日渐强烈的好奇心的驱使,他开始使用钯作为催化剂进行实验。
1968年,Heck 就其成功的研究工作,以唯一作者的身份发表了一系列研究论文[2-6]。
钯催化交叉偶联反应的发展历程简介_但世辉

图6 似蛇霉毒结构简式(R=CH3或C2H5)(椭圆内化学键即为偶联处)
成路线。 但纵观所有的钯催化交叉偶联反应的实例, 它 们的应用还是十分有限的, 例如, 现在参与钯催化交叉 偶联反应的底物主要是卤代烃, 而很多卤代物难以制 备、 价格高昂, 且制备过程中产生大量酸, 对环境不友 好; 此外, 如何扩大偶联反应的应用范围、 提高催化效 率也是需要长期探索的课题。 因此, 钯催化交叉偶联反 应还要在将来的发展中不断得以改进和创新, 才能让 实际的生产和生活受益更大。 参考文献:
·76·
弄醒碳原子, 使它变得勤快起来, 愿意与其他碳原子反 应。 1901 年, 法国科学家格林纳发明了一种试剂, 他将 镁屑放在无水乙醚中, 滴加卤代烷, 卤代烷与镁作用生 成有机镁化合物, 这种有机镁化合物称为格林纳试剂, 简称格氏试剂, 一般用 RMgX 表示。 实际上, 格林纳是 将一个镁原子接在了一个他希望使之活性增强的碳原 子上。 由于镁最外层有 2 个电子且极想失去, 因此在和 碳原子相遇时它便强行将电子塞给了碳原子, 使碳原子 核的正电荷与核外电子云的负电荷数不均, 活性大大增 强, 可以与很多的碳原子相互反应以合成新物质。 格氏试剂的发明将有机合成技术向前推进了一大 步, 使得合成烷烃、 醇、 醛、 羧酸等有机物变得十分简单。 为此, 格林纳也获得了 1912 年的诺贝尔化学奖。 格氏 试剂的另一重大用途是将碳原子进行偶联以增长碳链, 为大分子的合成拉开了序幕。 但是, 格林纳的方法让碳 原子变得太活泼了 , 反应无法控制, 在制造大分子的过 们曾这样赞誉福井所走过的化学求索之路: 他用心灵去拥抱大自然, 他以锲而不舍的精神和勤劳向大自然求索; 他凭借学习与思考, 双翼起飞, 终于走上了一条化学创新之路, 获取了丰收的硕果! 参考文献:
有机合成中钯催化下的交叉偶联反应

有机合成中钯催化下的交叉偶联反应-2010年诺贝尔化学奖简介陈明华( 兴义师范学院化学生物系,贵州兴义 562400)摘要:介绍了2010年诺贝尔化学奖的科学背景,即“有机合成中钯催化下的交叉偶联反应”的产生、发展和应用,体现了有机化学已经发展成为一门艺术形式,在这个形式下,科学家们在试管里创造性的产生出不可思议的化学物质的过程。
关键词:钯催化剂;交叉偶联反应;赫克反应;铃木反应;根岸反应Palladium-Catalyzed Cross Couplings in Organic SynthesisCHEN Ming-Hua(Department of Chemistry and Biological, Xingyi Normal College, Xingyi, Guizhou 562400)Abstract: This paper introduces scientific background of the Nobel Prize in Chemistry for 2010, it’s palladium-catalyzed cross couplings in organic synthesis.And this fack had been presents that “Organic chemistry has developed into an art form where scientists produce marvelous chemical creations in their test tubes”.Key words: palladium catalyst; cross-coupling reaction; heck reaction; suzuki reaction; negishi reaction2010年10月6日,瑞典皇家科学院决定授予美国特拉华大学(University of Delaware) 理查德-赫克(Richard F. Heck), 普渡大学(Purdue University)根岸荣一(Ei-ichi Negishi)和日本北海道大学(Hokkaido University)的铃木彰(Akira Suzuki)三位教授2010年的诺贝尔化学奖,以表彰他们在“有机合成中钯催化下的交叉偶联反应”作出的贡献[1]。
钯催化的交叉偶联反应——2010年诺贝尔化学奖获奖工作介绍

2011年第31卷 有 机 化 学V ol. 31, 2011 * E-ma i l: nxwang@ma i l.iReceived December 9, 2010; revised and accepted March 10, 2011.·学术动态·钯催化的交叉偶联反应——2010年诺贝尔化学奖获奖工作介绍王乃兴(中国科学院理化技术研究所 北京 100190)摘要 钯催化的交叉偶联反应是非常实用的合成新方法. 文章给出了Heck 反应、Negishi 反应和Suzuki 反应的概念, 对其反应机理作了详细的说明, 并对其在复杂化合物和天然产物全合成中的应用作了评价. 关键词 钯催化; Heck 反应; Negishi 反应; Suzuki 反应Palladium-Catalyzed Cross-Coupling Reactions —Introduction of Nobel Prize in Chemistry in 2010Wang, Naixing(Technical Institute of Physics and Chemistry , Chinese Academy of Sciences , Beijing 100190)Abstract Palladium-catalyzed cross-coupling reactions provide chemists with a more precise and efficient new methodologies. The concepts of the Heck reaction and Negishi reaction as well as Suzuki reaction are given, the reaction mechanisms are proposed, and applications of these reactions in the total synthesis of natural products are commented.Keywords palladium-catalyzed; Heck reaction; Negishi reaction; Suzuki reaction2009年10月6日, 瑞典皇家科学院宣布, 美国科学家Richard F. Heck(理查德 赫克)、日本科学家Ei-ichi Negishi(根岸英一)和Akira Suzuki(铃木章)共同获得今年的诺贝尔化学奖. 美国教授Richard F. Heck, 1931年出生于美国的斯普林菲尔德, 1954年在美国加利福尼亚大学洛杉矶分校获得博士学位. 随后他进入瑞士苏黎世联邦工学院从事博士后研究, 后在美国特拉华大学任教, 于1989年退休. Richard F. Heck 现为特拉华大学名誉教授. Ei-ichi Negishi 教授是日本人, 1935年出生于中国长春, 1958年从东京大学毕业后进入帝人公司, 1963年在美国宾夕法尼亚大学获得博士学位, 现任美国普渡大学教授. Akira Suzuki 也是日本人, 1930年出生于日本北海道鹉川町, 1959年在北海道大学获得博士学位, 随后留校工作了一段时间. 1963年到1965年, Akira Suzuki 在美国普渡大学从事了两年的博士后研究工作. Akira Suzuki 于1973年任北海道大学工学系教授, 现在是北海道大学名誉教授.钯催化的交叉偶联反应是一种可靠而又实用的工具, 对有机合成具有长久和深远的影响力, 该反应得到了合成化学工作者的普遍应用.笔者于2004年在《有机反应——多氮化物的反应及有关理论问题(第二版)》的第4.13节中列举了5个较新的人名反应[1], 其中有Heck 反应、Negishi 反应和Suzuki 反应. 对其定义分别为: Heck 反应是钯催化下, 不饱和有机卤化物或三氟磺酸酯与烯烃进行的偶联反应. Negishi 反应是钯催化下的不饱和有机锌试剂和芳基或乙烯基卤化物等进行偶联的反应. Suzuki 反应是钯催化下不饱和有机硼试剂和芳基或乙烯基卤化物等进行偶联的反应. 这是钯催化的交叉偶联反应的基本概念. 最初的Suzuki 反应还需要在无氧无水的条件下来进行, 后来发展的一些反应条件已经无需无氧无水操作了.这几种钯催化的交叉偶联反应机理不尽相同, 对机1320有 机 化 学 V ol. 31, 2011理的说明也不止一种, 一些可能的机理对研究生也较难接受. 如Heck 反应, 即使一些已经出版的专门论述人名反应的专著也较为简略[2,3] Heck 本人最先提出的Heck 反应机理是应该接受的.1 反应机理1.1 Heck 反应机理目前关于Heck 反应机理描述较多, 但一些机理过于简单, 一些机理的描述很难让有机化学家接受. 笔者认为Jutand 等[4]最近在Heck 反应的专门著作中总结的Heck 反应机理最为贴切和容易接受(Scheme 1). 这个详细的反应过程实际上是Heck 首先建议的.Scheme 1理解各步过程并不困难. 关键是整个机理中左下角画箭头处, 表示出一个负氢迁移过程, 双键上的电子是由钯直接提供的.Heck 反应的机理主要分为四个步骤:(1)氧化加成. 上式催化循环的第一步是芳基卤和Pd(0)的氧化加成, Titton 报道的芳基卤和Pd 0(PPh 3)4的作用支持了氧化加成步骤的机理, Titton 还报道了芳基卤活性次序: ArI >ArBr >>ArCl.(2)烯烃插入. 氧化加成给出反式的σ芳基Pd(II)卤化物ArPdXL 2, 脱去一个PPh 3配体后与烯烃配位, 再经过烯的顺式插入, 得到σ烷基Pa(II)卤化物[5,6], 读者可以参照上述催化循环机理图.(3) β负氢消除. 上述催化循环机理图中的σ烷基Pa(II)卤化物有一个C —C 键内旋转, 结果使得β氢原子(与sp 3碳原子相连)和Pd 原子处于顺式位置, 接着产生了顺式的β负氢消除. 这个顺式的β负氢消除反应会是一个可逆的过程.(4)还原消除. 钯催化的偶联反应产物(与芳基直接相连的烯烃衍生物)游离产生以后, H —Pd(II)的卤化物再经过一个可逆还原消除过程, 再生出具有催化活性的Pd(0)的络合物. 碱性的辅助催化剂通过粗灭产生的卤化氢, 促使还原消除过程向Pd(0)络合物催化剂方向移动.Heck 不仅发现了这个钯催化的偶联反应, 而且对其机理做出了透彻的阐述. Heck 提出的氧化加成、烯烃插入、β负氢消除、还原消除这四个主要步骤在实验中都得到了证实. β负氢消除是一个重要过程, 钯提供了一对电子形成了双键. 最近认为β负氢消除通过一个顺式消除过程. 实际上Heck 反应不能仅看作交叉偶联反应, 它只是偶联反应一种.机理中涉及一些不同的Pd(0)和Pd(II)的中间体, 这些中间体的结构和活性依靠实验条件, 钯催化剂可以是Pd(0)的络合物, 如Pd(PPh 3)4, 可以是Pd(OAc)2等. 当Pd(OAc)2作为催化剂时, 需要加入1,3-二(二苯基膦基)丙基(dppp), 首先形成Pd(OAc)2(dppp), 再得到离子型络合物Pd 0(dppp)(OAc)- [7], Pd 0(dppp)(OAc)-分解得到Pd(0)络合物Pd(dppp). 1.2 Suzuki 反应机理笔者[8]曾研究过Suzuki 反应, 利用苯硼酸和2,2'-二溴-5,5'-二噻吩通过催化量的金属钯络合物Pd(PPh 3)4进行交叉偶联反应(Eq. 1).当时采用的反应条件还是无氧无水操作[8], 产物熔点是145 ℃, 产率为51%.笔者在文献的基础上[9~11], 提出了一个离子型的反应机理, 该论文发表在一个国外化学期刊上[8]. 该反应可能的机理由三个主要步骤完成的: (a)氧化加成; (b)硼试剂参与; (c)还原消除.(1)氧化加成. 反应过程中, Pd(0)被加到有机卤化物中间, 有机卤化物中的碳原子通过极性转换由原来荷正电变为荷负电, 钯原子被氧化为Pd(II) (Scheme 2). 氧化加成的过程是速率决定步骤, 反应中, 有机卤化物的活性按卤原子如下次序递减: I >Br >>C.N o. 8王乃兴:钯催化的交叉偶联反应——2010年诺贝尔化学奖获奖工作介绍1321Scheme 2(2)硼试剂参与. 接着, 硼试剂中的C —B 键异裂, 碳原子荷负电, 形成的芳基负离子与钯正离子结合为ArPdAr', 而游离出来的卤离子(X -)与硼正离子配位得到XB(OH)2 (Eq. 2).(3)消除反应. 最后是还原消除过程, 钯有机物分解, 形成新的C —C 键, 金属钯游离出来, 再与PPh 3络合, 再生出活性钯催化剂Pd(PPh 3)4, 完成了催化过程.Scheme 3笔者在当时研究苯硼酸和2,2'-二溴-5,5'-二噻吩通过Pd(PPh 3)4催化进行的交叉偶联反应, 发现该反应采用弱碱Ba(OH)2作为辅助催化剂比其它强碱反应快, 收率高, 甚至用碳酸钾代替Ba(OH)2也往往引起副产物增加. 笔者采用了甲醇和甲苯(V ∶V =1∶1)的混合溶剂. 就溶剂效应而言, 甲醇溶剂对反应有利. 在反应过程中的氧化加成阶段, 甲醇产生的烷氧基负离子MeO -能够置换配位在钯上的卤负离子, 容易生成ArPdOR 中间体(Scheme 4).Scheme 4RPdOMe 的形成被认为是一个重要的中间体, 曾被分离得到过[12,13]. 1.3 Negishi 反应机理笔者曾制备了有机锌试剂[8], 采用一锅反应方法, 利用溴锌苯和2,5-二溴噻吩通过催化量的金属钯络合物Pd(PPh 3)4进行交叉偶联反应(Scheme 5).Scheme 5与Suzuki 反应相比, 利用Negishi 反应合成目标化合物, 产率没有Suzuki 反应高[8,14].Negishi 反应的机理与Suzuki 反应非常类似, 也是通过氧化加成、有机锌试剂(亲核试剂)参与和还原消除的三个主要步骤进行的, 下面用离子反应历程作以描述(Eq. 3, Schemes 6, 7):(1)氧化加成Scheme 6(2)有机锌试剂参与(3)消除反应Scheme 7Pd(0)游离出来, 再与PPh 3络合再生出催化剂Pd(PPh 3)4, 完成了催化循环.另外需要说明的是, 交叉偶联反应有许多种, 一些虽然没有得到诺贝尔化学奖, 但应用价值还是比较高, 例如Songashira 反应. Sonogashira 反应是钯配合物催化的卤代芳烃或者卤代烯烃与末端炔烃的交叉偶联反应, 它是一种合成芳炔、烯炔和炔酮等化合物的有效方法. 其反应如Eq. 4.Sonogashira 反应的本质是PdCl 2与CuI 复合催化剂催化末端炔烃与碘、溴代芳或者烯烃的交叉偶联反应. 2007年发表在Chem. Rev.上的Sonogashira 反应机理, 说明了铜盐作为助催化剂的过程, 是一个容易接受的机理(Scheme 8)[15].铜盐作为助催化剂的作用一些文献也作了报道[16]. 近年来Sonogashira 反应的应用报道较多, 读者可以参考相关文献[17~19].2 结束语Heck 反应、Negishi 反应和Suzuki 反应, 代表了钯催化的交叉偶联反应的最高成就, 反应非常新颖独特,1322有机化学V ol. 31, 2011Scheme 8确实在有机合成方法学的最前沿取得了重大突破, 这些原创性的成就卓有建树, 这些新方法首先在有机合成领域得到了普遍应用, 对发展有机合成的策略和技巧产生了长久和深远的影响.人类健康对特效新药的发展不断提出更高的要求, 天然产物作为先导药, 在这方面寄托了人们的无限期望[20]. 近年来, 海洋天然产物的生物医学活性引起了人们的高度重视. 海绵、珊瑚以及海洋微生物的次生代谢的天然产物, 结构新颖而活性显著, 已经成为人们挖掘具有自主知识产权的创新先导抗肿瘤等新药的战略新领域. 人工全合成这类复杂的化合物和天然产物对人类来说是一种艰难的挑战, Heck反应、Negishi反应和Suzuki反应的新方法无疑在这方面会发挥出巨大的作用.近年来, 围绕Heck反应、Negishi反应和Suzuki反应, 化学家发展了一些新的反应方法和条件, 如Ni代替Pd进行催化的交叉偶联反应. Iyer等[21]报道了Cu催化(CuI催化剂)的Heck反应, 相对Pd和Ni催化剂更为经济. 最近, Darcel等[22]报道了Fe催化的Suzuki反应, 产率较高. Nakamura等[23]不久前报道了Fe催化的Negishi 反应, 产物收率高且有立体专一性. 反应条件已经从开始需要无氧无水操作到现在可以在水相反应体系中进行. Chao等[24]报道了在水合溶剂中进行Suzuki反应的研究结果, 产物能够获得中等以上的收率. Bach等[25]在Tetrahedron的一篇文章中(其参考文41), 对笔者关于Suzuki反应和Negishi反应的报道作了一些介绍. 相信以后还会有一些新的关于催化的交叉偶联反应的研究论文不断发表出来.Heck不仅开创了著名的Heck反应, 而且他提出的有机化学反应机理也非常之透彻和精到, 可见他的有机化学之功底和对该方法的深刻的理解. 可是, Heck在完成Heck反应研究之后, 一度连科研经费都没有, 甚至不得不离开科学界. 笔者刚在德国应用化学刊物(Angew. Chem. Int. Ed. 2010,49, 2092)看到一篇关于德国合成化学教授H. Kunz的作者介绍, H. Kunz教授列出了他的五篇文章, 其中第二篇文章发表在Tetrahedron 上, 第三篇发表在Synthesis上. 笔者在中国科学院研究生院为硕博连读生讲授“有机反应”专业课, 学生对Heck反应、Negishi反应和Suzuki反应的兴趣非常浓厚. 希望年青一代的学者, 通过研究和借鉴Heck反应、Negishi反应和Suzuki反应, 提升我国在有机合成方法学方面的整体水平.在这篇简介文章结束时, 笔者再介绍几篇关于钯催化的交叉偶联反应的代表性综述文章. 一篇是Suzuki本人1995年在Chem. Rev.上的综述[26], 希望有兴趣的读者参阅. 另外3篇对相关钯催化的交叉偶联反应最新进展作了详细综述[27~29], 希望读者特别是青年学者能够继续深入学习和掌握这一研究领域. 最近, Suzuki和Negishi[30~31]还分别发表了他们的诺贝尔化学奖获奖演说. 就在这篇文章付印之际,作者又读到了一篇最新的关于非对映选择性的Negishi反应的论文, 该方法为此类反应的立体控制开拓了又一个新生面[32]. References1 Wang,N.-X. Organic Reac tions—The Reac tion of Polyni-trogen Compounds and Some Theoretic Questions, 2nd ed., Chemical Industry Press, Beijing, 2004, pp. 165~171 (in Chinese).(王乃兴, 有机反应—多氮化物的反应及有关理论问题(第二版), 化学工业出版社, 北京, 2004, pp. 165~171.)2 Li, J. J. Name Reactions, Springer, New York, 2006, p. 285.3 Kürti, L.; Czakó, B. Strategic Applications of Named Reac-tions in Organic Synthesis, Elsevier Academic Press, 2005, p. 196.4 Jutand, A. In The Mizoroki-Hec k Reac tion, Ed.: Oestreich,M., Wiley, United Kingdom, 1999, pp. 1~5.5 Dieck, H. A.; Heck, R. F. J. Am. Chem. Soc.1974, 96, 1133.6 Ziegler, C. B.; Heck, R. F. J. Org. Chem. 1978, 43, 2941.7 Kozuch, S.; Shaik, S.; Jutand, A.; Amatore, C. Chem. Eur. J.2004, 10, 3072.8 Wang,N.-X. Synth. Commun. 2003, 33, 2119.9 Anderson, C. B.; Burreson, B. J.; Michalowski, J. T. J. Org.Chem. 1976, 41, 1990.10 Zask, A.; Helquist, P. J. Org. Chem. 1978, 43, 1619.11 Aliprantis, A. O.; Canary, J. W. J. Am. Chem. Soc. 1994,116, 6985.12 Yoshida, T.; Okano, T.; Otsuka, S. J. Chem. Soc., DaltonTrans. 1976, 993.13 Grushin, V. V.; Alper, H. Orgnometallics1993, 12, 1890.14 Wang,N.-X. Chin. . Chem. 2004, 24, 350 (in Chi-N o. 8 王乃兴:钯催化的交叉偶联反应——2010年诺贝尔化学奖获奖工作介绍1323nese).(王乃兴, 有机化学, 2004, 24, 350.)15 Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874.16 Doucet, H.; Hierso, J. C. Angew. Chem., Int. Ed. 2007, 46,834.17 Gelman, D.; Buchwald, S. L. Angew. Chem., Int. Ed. 2003,42, 5993.18 Saha, D.; Dey, R.; Ranu, B. C. Eur. J. Org. Chem. 2010,6067.19 Karpov, A. S.; Merkul, E.; Rominger, F.; Müller, T. J. J.Angew. Chem., Int. Ed. 2005, 44, 6951.20 Harmata, M. Strategies and Tac tic s in Organic Synthesis,Elsevier, Oxford, 2010.21 Iyer, S.; Ramesh, C.; Sarkar, A.; Wadgaonkar, P. P. Tetrahe-dron Lett. 1997, 38, 8113.22 Bźziera, D.; Darcela, C. Adv. Synth. Catal. 2009, 351, 1732.23 Hatakeyama, T.; N akagawa, N.; N akamura, M. Org. Lett.2009, 11, 4496.24 Cho, S. Y.; Kang, S. K.; Ahn, J. H.; Ha, J. D.; Choi, J. K.Tetrahedron Lett. 2006, 47, 5237.25 Schröter, S.; Stock, C.; Bach, T. Tetrahedron2005, 61,2245.26 Norio Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.27 Roglans, A.; Pla-Quintana, A.; Moreno-Mañas, M. Chem.Rev. 2006, 106, 4622.28 Martin, R.; Buchwald, S. L. Acc. Chem. Res. 2008, 41,1461.29 Denmark, S. E.; Regens, C. S. Acc. Chem. Res. 2008, 41,1486.30 Suzuki, A. Angew. Chem., Int. Ed.2011, 50, 6723.31Negishi, E. Angew. Chem., Int. Ed.2011, 50, 6738.32 Seel, S.; Thaler, T.; Takatsu, K.; Zhang, C.; Zipse, H.;Bernd, F.; Straub, B. F.; Mayer, P.; Knochel, P. J. Am. Chem.Soc. 2011, 133, 4774.(Y1012093 Li, L.)。
有机合成钯催化交叉偶联反应(精选文档)

1.格氏试剂——拉开钯催化交叉偶联反应的序幕
有机合成化学所构造出来的物质大部分都是以碳胳为骨架所构建起来的,然而碳原子本身十分稳定,在化学反应中并不活泼。因此化学家们希望通过各种化学反应,来激活碳原子,使其更容易参与到反应中并与其它碳原子相连,构造更
复杂的有机物。通过多年的尝试与努力, Grignard (格林尼亚发明了有机镁试剂(即格氏试剂,并利用其活化了碳原子,成功将碳原子连接在一起。以下为利用格氏试剂所进行的烷基化反应:
赫克反应与格氏反应相比,具有更好的化学选择性,减少反应的副产物,而且赫克反应在常温下进行,反应条件温和,对于工业生产具有重要的应用价值。不过赫克反应的局限之处在于,它往往只能用于有机合成中碳碳单键的合成,在合成一些更大的分子时会显示出其缺陷及产生较多的副产物。化学家们并不满足于停留在当前的成果中,而是孜孜不倦地进一步改进钯催化交叉偶联反应。
为铃木反应。
铃木反应与根岸反应相
似,均经历了三个过程:氧
构建单键最重要的反应之
一,并以他自己的名字赫克对
反应命名。
赫克反应的反应机理如
下(见右图:反应开始,活泼
的钯Pd(0催化剂与卤代烃发
生被称为氧化-加成的反应,
在这步反应中,生成了R-Pd-X ,钯的氧化态形式上从(0转化为(Ⅱ ,也就意味着生成了Pd-C键;第二步,烯烃与钯配位,此时烯烃和R基团同时与钯连接,这样就使它们能够相互发生反应;第三步, R基团迁移到烯烃的碳原子上,而钯同时与烯烃的另一个碳原子相连,这一步称为迁移-插入,结果生成了C-C键;第四步, R替换了底物烯烃上的一个氢原子,即通过消除烯烃的β-H得到了一个新的取代烯烃,同时还生成了HPdX ,它随即失去HX得到Pd(0,进入另一次催化循环。
贵金属钯Pd催化的偶联反应

通过调整钯催化剂的反应条件(温度、溶剂、配体、碱和其他添加剂),可使钯催化成为有机化学合成中用途广泛的工具。
其中,钯催化的交叉偶联反应彻底改变了分子的构造方式。
从有机合成和药物化学领域,到材料科学和聚合物化学,交叉耦合已经影响到多个科学领域。
在偶联反应中,钯催化剂不但可以形成C-C、C-O、C-N和C-F等碳键,而且对各种官能团具有很高的耐受性,通常能够提供良好的空间和区域特异性,可以不用引入保护基团。
常用的偶联反应包括Heck偶联、Suzuki偶联、Stille偶联、Hiyama偶联、Sonogashira偶联、Negishi偶联、Buchwald-Hartwig胺化等等。
具体反应如下所示:1、Negeshi偶联反应(C-C) [1](其中,R/R’可以是烷基、烯基、芳基、烯丙基、炔基或炔丙基,X/X’可以是氯、溴、碘或其他基团,催化剂是钯)2、Suzuki偶联反应(C-C) [2](其中,R可以是烯基,芳基或烷基,R’可以是烯基,芳基、炔基或烷基,Y可以是烷基,羟基或者氧烷基,X可以是氯、溴、碘或三氟甲磺酸)3、Stille偶联反应(C-C) [3](其中,R可以是烯基、芳基、酰基,R’可以是烯基、芳基或者烷基,R’’可以是烷基,X可以是氯、溴、碘或者三氟甲磺酸)4、Buchwald–Hartwig偶联反应(C-N/C-O)[4](其中,R是芳基,R’可以是邻、间芳基或烷基,R”可以是烷基或芳基,X可以是氯、溴、碘或者三氟甲磺酸)5、Heck偶联反应(C-C) [5](其中,R可以是烯基、芳基和不含有β氢的烷基,R’可以是烯基,芳基和烷基,X可以是氯、溴、碘、三氟甲磺酸、对甲基苯磺酰氯或者N2+)6、Sonogashira偶联反应(C-C) [6](其中,R可以是烯基或者芳基,R’可以是H、炔基、芳基、烷基或者硅烷基,X可以是氯、溴、碘或者三氟甲磺酸)。
钯催化Stille交叉偶联反应研究新进展

2006年第26卷有 机 化 学V ol. 26, 2006 第1期, 19~26Chinese Journal of Organic ChemistryNo. 1, 19~26 * E-mail: jhli@ Received January 17, 2005; revised April 15, 2005; accepted May 24, 2005.国家自然科学基金(No. 20202002)资助项目.此后Stille 等[3,4]对该类反应做了大量研究, 对其反应的机理也做了初步研究. 此后, 随着人们对Stille 反迄今为止, 有关Stille 反应的报道主要集中在有机锡与卤代芳香烃、卤代烷烃以及酰氯的反应等方面. 为了让读者更好地了解Stille 反应的最新研究动态, 本文20有机化学V ol. 26, 2006反应都用三苯基膦作配体[11,12]. 最近, Littke等[13]用三叔丁基膦作配体的钯催化体系催化Stille反应, 成功地解决了上述难题(Eq. 3).作者首先利用上述反应对碱进行筛选, 发现用底物量的2.2倍的CsF效果最理想. 于是, Littke及其合作者们利用这一条件进行了一系列反应, 效果很好(Eq. 4).对于带钝化基团(供电子基团)的氯代芳香烃以及位阻极大的2.6-二甲基氯苯都取得了较高的产率, 达到90%以上. 更难能可贵的是作者利用这一条件, 首次将氯代芳烃的Stille偶联在室温条件下得以实现, 产率达到86% (Eq. 5).总之, 利用P(t-Bu)3为配体的钯催化体系催化Stille 反应有相当宽的底物适用范围, 反应条件更加优越, 产率很高, 从而进一步拓展了Stille反应的应用范围.Simon等[14]用CuI协同钯催化溴代及氯代芳香烃, 效果也不错, 对底物的兼容性也较好, 但对氯代芳香烃的催化效果不理想, 而且必须用DMF作溶剂, 产物后处理较麻烦.但是这些配体一个最大的缺陷是对空气和水敏感, 从而造成操作困难. 虽然后来Fu等[15]利用对空气稳定的[(t-Bu)3PH]BF4来代替上述配体, 取得类似的结果, 但该配体同样存在合成困难和价格昂贵等不足. 最近, Yoshifuji等[16]尝试使用另一种对对空气和水均稳定的DPCB代替上述配体(Eq. 6). 但是在该配体作用下只有溴代芳烃可以与有机锡试剂反应, 而氯代芳烃则不能发生该反应.最近Verkade等[17]报道了以P(i-BuNCH2CH2)3N为配体, 不但溴代芳烃可以顺利地与有机锡试剂反应, 氯代芳烃包括带吸电子基团而且带供电子基团的同样可以顺利发生反应. 不过对于不活泼的带供电子基团氯代芳烃需要更强的含氟的碱即Me4NF (Eq. 7).然而所有上述配体均合成困难, 从而价格昂贵. 因此发展高效和价廉的配体仍具有重要意义. 我们发现叔胺可以作为钯催化Stille交叉偶联反应配体[18]. 研究结果表明: 当Dabco(三乙二胺)作为钯催化Stille交叉偶联反应的配体时催化效果最好. 在3 mol%到0.0001 mol%醋酸钯和6 mol%到0.0002 mol% Dabco存在下, 卤代芳烃包括芳基碘、溴和氯均可以顺利地与不同类型的有机锡化合物反应得到非常好的产率, 并且最高转换率达到九十八万(Eq. 8).虽然Chiappe等[19a]实现了在绿色介质中无配体条件下的Stille反应, 但底物仅局限于碘代芳烃, 意义有限. 最近, Park等[19b]利用可回收重复使用的SiO2/TEG/ Pd和TiO2/TEG/Pd等钯催化剂来催化Stille反应, 但是底物同样局限于碘代芳烃.2 饱和碳原子上的Stille反应2.1 卤代烷烃的Stille偶联反应最近, 用一种新型的配体钯催化体系成功地催化了卤代烷烃与有机锡的Stille偶联(Eq. 9)[20], 使Stille偶联反应的应用范围更加广泛.这一反应对溴代烷烃在室温条件下就能进行, 对底物的兼容性也较好, 但对于氯代烷烃则不反应.No. 1王德平等:钯催化Stille 交叉偶联反应研究新进展212.2 苄基卤的Stille 反应近年来, 苄基卤与有机锡的反应逐渐为人们所关注. Crawforth 等[21]用一种含P 和N 配体的钯试剂作催化剂, 对于苄基溴的催化效果比较好(Eq. 10).Crawforth 等[22]还详细研究了这一反应, 利用他们的反应条件, 不仅对苄基卤效果较好, 对于烯丙基溴的Stille 偶联的催化效果也很不错. 作者还对苄基溴及溴苯与有机锡作了一个对照实验(Eq. 11), 结果, 用Ligand A 得到的苄基溴偶联反应产物高达86%, 而溴代芳烃则没有反应.3 其它类型的Stille 偶联反应3.1 合成胺的反应最近, 一种合成饱和胺的反应通过Stille 偶联得以实现[23]. 这个反应条件十分优越, 在室温下就能进行, 选择性也非常好(Eq. 12).作者对这个反应的机理也进行了研究后认为, 首先是胺与酰氯及钯加成, 再与有机锡反应, 得到目标产物(图1).作者首次利用Stille 反应, 采用一锅法, 一步合成了α-取代饱和胺. 这个反应的实现对于许多具有高生物活性的α-取代胺及其衍生物的合成无疑具有重大的现实借鉴意义.3.2 有机磺酰氯的Stille 反应有机磺酰氯的Stille 反应[24]最近也有报道(Eq. 13).图1 反应的机理Figure 1 Mechanism of the reaction并且通过一系列实验, 得出一个Stille 反应的活性顺序: ArI >ArSO 2Cl >ArBr >>ArCl, 作者对Stille 系列的羰基化也作了研究, 效果较好(Eq. 14).3.3 羰基化反应Mazzola 等[25]对于Stille 类型的羰基化反应作了比较详细的研究(Eq. 15).Mazzola 与其合作者们发现这一反应必须用铜盐作助催化剂, 否则, 产率很低. Alphonse 等[26]在用杂环芳香烃硫醚为原料进行Stille 反应时发现, 他们的反应也必须加铜盐作助催化剂, 否则反应不发生.4 Stille 交叉偶联反应机理4.1 反应机理的研究随着人们对钯催化的Stille 偶联反应研究的不断深化, Stille 反应机理也日趋完善. 有关Stille 反应机理的文献报道[27]较多, 1986年, Stille 等[4]在综合大量实验数据的基础上提出了它的反应机理, 他认为, Stille 反应分四步进行: (1)氧化加成; (2)金属转移; (3)分子内异构化;22有 机 化 学 V ol. 26, 2006(4)还原消除(图2).图2 Stille 反应的机理Figure 2 Mechanism of the Stille reaction作者认为, 从1到2是决定反应速率的关键步骤, 其中这一过程是通过形成一个过渡态4得以实现的, 上述机理对于大多数实验结果可以进行较为完善的解释.后来, Falk 等[28,29]在通过Stille 偶联合成手性化合物时, 发现手性保持不变(Eq. 16-2), 与Stille 等[30]的研究(Eq. 16-1)相冲突. 显然, 这一现象用上述机理是无法解释的. 按照上述机理, 应当发生构型翻转.为了解释上述现象, Espinet 等[31,32]提出了双过渡态理论, 即环状过渡态与开放式过渡态理论(图3). 他认为, 当形成环状过渡态A 时, 构型保持不变, 当形成开放式过渡态B 时, 构型翻转. 在图(3)所示状过渡态机理中, 作者认为在氧化加成后的金属转移步骤时, 不是卤族元素X 被有机锡上的α碳原子取代, 而是形成了一个Pd —X —Sn 桥键环状状过渡态A . 在开放式过渡态机理理论中, 作者认为有两种可能: (1)氧化加成后卤族元素X 直接被有机锡化合物的α碳原子取代, 形成过渡态B; (2)氧化加成后卤族元素X 被(S)取代, 得到一种复合盐, 再与有机锡化合物反应完成金属转移. 这一理论较好地解释了上述两种不同的实验现象, 一直以来, 为人们所认同.Christian 等[33]以及Espinet 等[34]在上述反应机理的图3 Stille 反应的机理Figure 3 Mechanism of the Stille reaction基础上对于钯催化的Stille 反应机理也作了十分详细的阐述和补充.4.2 铜盐对Stille 反应的影响人们在研究[PdL 4]催化的Stille 偶联反应时, 发现加入CuI 或其它铜盐可以明显加快反应速率, 缩短反应时间[26,35~38]. Farina 等[35]在1991年发现这一现象后, 又进行了一番细致的研究, 1994年, Farina 等又在研究基础上建设性地提出了CuI 促进Stille 反应机理[39]. 他们认为, 对于反应的决速步骤是从中间体1到中间体2(图2中)时, CuI 具有促进1与配体L(如PPh 3等)分裂, 从而快速与有机锡反应.后来的研究证明, CuI 不能直接参与[PdRXL 2]与L 的分裂, 但CuI 却具有捕获中性配体L 能力, 在进行氧化加成以及决速步骤从中间体1到中间体2(图2中)过程中释放出的配体L 都可以被铜盐捕获, 由于上述氧化加成以及决速步骤从中间体1到中间体2是可逆的, 而这些配体被铜盐捕获后, 促使反应朝预定方向发展, 且CuI 对[PPh 3]的捕获能力比[AsPh 3]要强很多, 而在解吸附时[PPh 3]又比[AsPh 3]快, 从而很好地解释了加入铜盐对两种不同配体[PPh 3]及[AsPh 3]的影响力不同的原 因[40].Farina 等[36]还发现另一现象, 在极性很强的溶剂中, 能够发生Sn/Cu 之间的交换, 这一发现预示着可以用铜盐催化Stille 反应, 很快, Piers 等[41]利用当量的CuCl 成功催化合成了分子内的Stille 偶联, 后来, 有关铜盐催No. 1王德平等:钯催化Stille 交叉偶联反应研究新进展23化的报道也越来越多[42,43]. 例如, Savall 等[44]就报道了一例铜盐催化的反应(Eq. 17).最近, Kim 等[45]又报道了一例奇怪的现象(Eq. 18), 他们在用3,5-二溴吡喃酮进行Stille 偶联时, 发现加入CuI 后反应的选择性发生了改变(表1).表1 3,5-二溴吡喃酮进行Stille 偶联反应时在不同极性溶剂中有无CuI 的选择性Table 1 Regioselectivity of the coupling of 3,5-dibromo-2- pyrone in the presence/absence of CuI EntryConditionsRatio of C ∶D 1 Toluene, 100 ℃ 100∶0 2 Toluene, 100 ℃, CuI (1 equiv.) 100∶0 3 DMF, 50 ℃ 100∶04 DMF, 50 ℃, CuI (1 equiv.)30∶704.3 氟离子对Stille 反应的影响Fu 组[13,46a]和Nolan [46b]组分别发现氟离子对Stille 偶联反应具有促进作用. 例如Fu 等[13,46a]以对氯甲苯和乙烯基锡的反应为模板, 研究了一系列含氟离子的碱和不含氟离子的碱对Stille 偶联反应的影响(Eq. 19), 实验结果见表2. 实验结果表明, 虽然不含氟离子的碱对Stille 偶联反应同样有促进作用, 但效果没有含氟离子的碱的好, 其中CsF 作用下的Stille 偶联反应产率最高(50%, 表2中Entry 5), 增加CsF 的量到2.2 equiv., Stille 偶联反应的产率升高到59%(表2中Entry 6).Nolan 和Grasa 等[46b]得到类似的结果, 并且他们从机理上进一步证实了氟离子对Stille 偶联反应具有促进作用(Eq. 20). 他们当场通过19F 谱确定了六价态的锡中间体1的生成, 从而揭示了氟离子的作用Stille 偶联反表2 碱对Stille 偶联反应的影响Table 2 The effect of bases on Stille cross-coupling reactionEntryAdditives (1.1 equiv.)Yield/%1 None 122 TAS-F 43 TBAF 244 KF 285 CsF 506CsF (2.2 equiv.)597 NEt 3 16 8 Cs 2CO 3 409 NaOH 42应的机理.5 Stille 交叉偶联在有机合成中的应用近年来, Stille 交叉偶联反应得到了广泛的应用. 许多天然产物及医药原料都通过Stille 反应来合成. 5.1 11C 标记的PET 示踪剂的合成利用正电子扫描进行体层摄影的方法称为PET, 该方法广泛用于生命性的脑、心脏等重要器官的扫描. PET 扫描的实现需要一种PET 示踪剂来协助进行. 最近, 一种快速且简单的合成PET 示踪剂的方法通过Stille 反 应[47]得以实现(Eq. 21).反应采用{Pd 0[P(t -Bu 3)]4}作催化剂, CsF 或KF 作碱, DMF 作溶剂在60 ℃条件下只需5 min 便可达到95%的产率. 这个反应条件十分温和, 速度快, 产率高, 因此, 此类反应具有非常高的实际应用作值. 5.2 α-取代吡喃酮的合成α-取代吡喃酮的合成是一类具有高生物活性的有机化合物, 它的合成也成为有机化学及医学界的注目焦点[48]. 长期以来, 它的合成是利用过渡金属(Ag, Hg, Rh 和Pd)等来催化, 用炔酸与有机锡发生加成反应而得 到[49,50], 这些反应选择性很低, 且生成较多副产物, 产 率低, 难以分离. Thibonnet 等通过Stille 偶联制α-取代24有 机 化 学 V ol. 26, 2006吡喃酮的[51]方法引起了人们的关注(Eq. 22).该反应条件温和, 产率可以达到85%. 作者认为反应是通过如下过程进行的(Eq. 23):即通过两次Stille 偶联一步合成α-取代吡喃酮, 这一过程的实现对于天然产物α-取代吡喃酮及其衍生物的合成具有十分重要的意义.5.3 分子内的Stille 偶联合成天然产物天然产物通常具有高生物活性、药用价值较高、市场需求量较大. 从自然界中直接提取天然产物, 产量有限, 且费用较高. 因此, 通过化学方法合成天然产物近年来逐渐流行[52]. 然而, 天然产物通常结构复杂, 相连着大量高活性基, 通过一般的有机反应通常难以得到目标产物, 而通过Stille 偶联则可以实现[53]. 例如, 一种从深海植物中提取的大环内酯化合物 Macrolactin A 的合成通过Stille 偶联反应得以实现(Scheme 1)[54].Scheme 1Entwistle 等[55,56]报道了通过Stille 偶联反应合成具有抗焦虑、抗癌功效的威里霉素(Virginiamycins) (Eq.24).Lett 和Quéron 等[57]首次报道通过分子内的Stille 偶联反应合成具有生理活性的bafilomycin A 1. 该反应条件温和, 总产率达到33% (Eq. 25).同样Vogel 等[58]巧妙地利用Stille 偶联反应和Stille 偶联羰基化反应高效选择性合成了具有阻止酶水解等生理功能的C -Glycosides (Eq. 26).当然上面的所提到的只是Stille 偶联反应在具有生理活性的天然产物和药物合成中应用的众多例子中的一小部分. 我们有理由相信, 随着包括新配体、底物及催化剂的发现和应用等在内的Stille 偶联反应的新技术和方法的发展, 将大大拓宽Stille 偶联反应在有机合成的应用途径.No. 1 王德平等:钯催化Stille交叉偶联反应研究新进展256 结论与展望综上所述, 钯催化Stille交叉偶联反应适用范围广, 产物易分离, 对底物的兼容性较好, 产率一般较高. 因此, Stille交叉偶联反应已经越来越为人们所关注, 在医药以及天然产物合成中得到了广泛的应用.目前, 钯催化Stille交叉偶联反应还存在以下几个关键问题: (1)有机锡化物是一种剧毒化合物, 且造价高昂, 因此, 使催化量的锡通过置换反应, 从而减少有机锡化合物的用量无疑将是一个值得进一步探讨的课题;(2)进一步寻找适合钯催化的高效、稳定的配体, 发现新的反应渠道; (3)优化条件进一步拓宽Stille交叉偶联反应的应用范围. 我们相信, 越来越多的研究者将对这一方向产生兴趣, 解决这些问题, 从而使Stille交叉偶联反应得到更加广泛的应用.References1 For selected reviews, see: (a) Diederich, F.; Stang, P. J.Metal-Catalyzed Cross-coupling Reactions, Wiley-VCH, Weinheim, 1998.(b) Farina, V.; Scott, W. J. Organic Reactions, Vol. 50, Ed.:Paquette, L. A., Wiley, New York, 1997.(c) Shirakawa, E.; Hiyama, T. J. Organomet. Chem.1999,576, 169.(d) Miyaura, N. Cross-Coupling Reaction, Springer, Berlin,2002.(e) Hegedus, L. S. In Organometallics in Synthesis, Ed.:Schlosser, M. J., Wiley & Sons, Chichester, 2002, p. 1123.(f) Negishi, E. Handbook of Organopalladium Chemistryfor Organic Synthesis, Wiley-Interscience, New York, 2002.(g) Littke, A. F.; Fu, G. C. Angew. Chem., Int. Ed.2002, 41,4176.(h) Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Le-maire, M. Chem. Rev.2002, 102, 1359.2 Kosugi, M.; Shimizu, Y.; Mifita, T. Chem. Lett.1977, 1423.3 Milstein, D.; Stille, J. K. J. Am. Chem. Soc. 1978, 100,3636.4 Stille J. K. Angew. Chem., Int. Ed. Engl.1986, 25, 508.5 Nicolaou, K. C.; Sorensen, E. J. Classics in Total Synthesis,VCH, Weinheim, 1996.6 Nicolaou, K. C.; Snyder, S. A. Classics in Total Synthesis,VCH, Weinheim, 2003.7 Shair, M. D.; Yooin, T. Y.; Mosny, K. K.; Chou, T. C.;Danishefsky, S. J. J. Am. Chem. Soc.1996, 118, 9509.8 Pattenden, G.; Sinclair, D. J. J. Organomet. Chem. 2002,653, 261.9 Kosugi, M.; Sasazawa, K.; Shimizu, Y.; Mifita, T. Chem.Lett.1977, 301. 10 Li, J. J.; Gribble, G. W. Palladium in Heterocyclic Chemis-try, Pergamom, New York, 2000.11 Kosugi M.; Ogata T.; Sano H.; Migita T. Chem. Lett.1984,1255.12 Kosugi, M.; Ogata, T.; Ohhashi, K.; Sano, H.; Migita, T.Chem. Lett.1985, 997.13 Littke, A. F.; Schwarz, L.; Fu, G. C. J. Am. Chem. Soc.2002, 124, 6343.14 Simon, P. H. M.; Lee, V.; Baldwin, J. E. Angew. Chem., Int.Ed.2004, 43, 1132.15 Netherton, M. R.; Fu, G. C. Org. Lett.2001, 3, 4295.16 Gajare, A. S.; Jensen, R. S.; Toyota, K.; Yoshifuji, M.;Ozawa, F. Synlett2005, 144.17 Su, W.; Urgaonkar, S.; Verkade, J. G. Org. Lett.2004, 6,1421.18 Li, J.-H.; Liang, Y.; Wang, D.-P.; Liu, W.-J.; Xie, Y.-X.;Yin, D.-L. J. Org. Chem.2005, 70, 2832.19 (a) Chiappe, C.; Imperato, G.; Napolitano, E.; Pieraccini, D.Green Chem.2004, 6, 33.(b) Kim, N.; Kwon, M. S.; Park, C. M.; Park, J. Tetrahe-dron Lett. 2004, 45, 7057.20 Tang, H. F.; Menzel, K.; Fu, G. C. Angew. Chem., Int. Ed.2003, 42, 5079.21 Crawforth, C. M.; Burling, S.; Fairlamb, I. J. S.; Taylor, R.J. K.; Whitwood, A. C. Chem. Commun.2003, 2194.22 Crawforth, C. M.; Fairlamb, I. J. S.; Taylor, R. J. K. Tetra-hedron Lett. 2004, 45, 461.23 Davis, J. L.; Dhawan, R.; Arndtsen, B. A. Angew. Chem.,Int. Ed.2004, 43, 590.24 Dubbaka, S. R.; Vogel, P. J. Am. Chem. Soc.2003, 125,15292.25 Mazzola, Jr. R. D.; Giese, S.; Benson, C. L.; West, F. G. J.Org. Chem.2004, 69, 220.26 Alphonse, F. A.; Suzenet, F.; Keromnes, A.; Lebret, B.;Guillaumet, G. Org. Lett.2003, 5, 6.27 Ricci, A.; Sterzo, C. L. J. Organomet. Chem.2002, 653,177.28 Ye, J.; Bhatt, R. K.; Falck, J. R. J. Am. Chem. Soc.1994,116, 1.29 Ye, J.; Bhatt, R. K.; Falck, J. R. Tetrahedron Lett.1993, 34,8007.30 Labadie, J. W.; Stille, J. K. J. Am. Chem. Soc.1983, 105,6129.31 Casado, A. L.; Espinet, P.; Gallego, A. M. J. Am. Chem.Soc.2000, 122, 11771.32 Casado, A. L.; Espinet, P. J. Am. Chem. Soc. 1998, 120,8978.33 Amatore, C.; Bahsoun, A. A.; Jutand, A.; Meyer, G.; Ntepe,A. N.; Ricard, L. J. Am. Chem. Soc.2003, 125, 4213.34 Espinet, P.; Echavarren, A. Angew. Chem., Int. Ed.2004,43, 4704.35 Farina, V.; Krishnan, B. J. Am. Chem. Soc.1991, 113, 9585.36 Farina, V. Pure Appl. Chem.1996, 68, 73.37 Gelman, D.; Buchwald, S. L. Angew. Chem., Int. Ed.2003,26有机化学V ol. 26, 200642, 5993.38 Soheili, A.; Albaneze-Walker J.; Murry, J. A.; Dormer, P.G.; Hughes, D. L. Org. Lett.2003, 43, 1132.39 Farina, V.; Kapadia, S.; Krishnan, B.; Wang, C.; Liebe-skind, L. S. J. Org. Chem.1994, 59, 5905.40 Casado, A. L.; Espinet, P. Organometallics2003, 22, 1305.41 Piers, E.; Wong, T. J. Org. Chem. 1993, 58, 3609.42 Falck, J. R.; Bahtt, R. K.; Ye, J. J. Am. Chem. Soc.1995,117, 5973.43 Mohapatra, S.; Bandyopadhyay, A.; Barma, D. K.; Cap-devila, J. H.; Falck, J. R. Org. Lett.2003, 5, 4759.44 Savall, B. M.; Blanchard, N.; Roush, W. R. Org. Lett.2003,5, 377.45 Kim, W.-S.; Kim, H.-J.; Cho, C.-G. J. Am. Chem. Soc.2003, 125, 14288.46 (a) Littke, A. F.; Fu, G. C. Angew. Chem., Int. Ed.1999, 38,2411.47 (b) Grasa, G. A.; Nolan, S. P. Org. Lett.2001, 3, 119.48 Hosoya, T.; Wakao, M.; Kondo, Y.; Doi, H.; Suzuki, M.Org. Biomol. Chem.2004, 2, 34. 49 Posner, G. H.; Nelson, T.; Kinter, C.; Johnson, N. J. Org.Chem.1992, 57, 4803.50 Xu, C.; Negishi, E. Tetrahedron Lett.1999, 40, 431.51 Ma, S.; Shi, Z. J. Org. Chem.1998, 63, 6387.52 Thibonnet, J.; Abarbri, M.; Parrain, J. L.; Duchene, A. J.Org. Chem.2002, 67, 3941.53 For a selected review on the applications of cross-couplingreaction in total synthesis, see: Nicolaou, K. C.; Sorensen,E. J. Classics in Total Synthesis, Wiley-VCH, 1996, p. 565.54 Duncton, M. A. J.; Pattenden, G. J. Chem. Soc., PerkinTrans.11999, 1235.55 Boyce, R. J.; Pattenden, G. Tetrahedron Lett.1996, 37,3501.56 Entwistle, D. A.; Jordan, S. I.; Montgomery, J.; Pattenden,G. J. Chem. Soc., Perkin Trans. 11996, 1315.57 Entwistle, D. A.; Jordan, S. I.; Montgomery, J.; Pattenden,G. Synthesis 1998, 603.58 Lett, R.; Quéron, E. Tetrahedron Lett.2004, 45, 4539.59 Dubbaka, S. R.; Steunenberg, P.; Vogel, P. Synlett2004,1235.(Y0501174 LI, L. T.)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
钯催化交叉偶联反应
钯催化交叉偶联反应是一类用于碳碳键形成的重要化学反应,在有机合成中应用十分广泛。
简介:
为制造复杂的有机材料,需要通过化学反应将碳原子集合在一起。
但是碳原子在有机分子中与相邻原子之间的化学键往往非常稳定,不易与其他分子发生化学反应。
以往的方法虽然能令碳原子更加活跃,但是,过于活跃的碳原子却又会产生大量副产物,而用钯作为催化剂则可以解决这个问题。
钯原子就像“媒人”一样,把不同的碳原子吸引到自己身边,使碳原子之间的距离变得很近,容易结合——也就是“偶联”。
这样的反应不需要把碳原子激活到很活跃的程度,副产物比较少,因此更加精确而高效。
赫克、根岸英一和铃木章通过实验发现,碳原子会和钯原子连接在一起,进行一系列化学反应。
这一技术让化学家们能够精确有效地制出他们需要的复杂化合物。
发展阶段:
一、大约100年前,法国化学家维克多·格林尼亚发现,将一个镁原子同一个碳原子偶联在一起,会将额外的电子推向这个碳原子,使得它能够更容易同另外一个碳原子连接在一起。
不过,科学家们发现,这样的方法在创造简单的分子时起到了效果,但是在对更为复杂的分子进行合成时,却在试管里发现了很多并不需要的副产品。
二、早在上世纪60年代,赫克就为钯催化交叉偶联反应奠定了基础,1968年,他报告了新的化学反应——赫克反应,该反应使用钯作为主要的催化剂来让碳原子连接在一起。
三、1977年,根岸英一对其成果进行了精练,他使用一种有机氯化物作为催化剂;两年后,铃木章发现使用有机硼化合物的效果会更好。
应用:
如今,“钯催化交叉偶联反应”被应用于许多物质的合成研究和工业化生产。
例如合成抗癌药物紫杉醇和抗炎症药物萘普生,以及有机分子中一个体格特别巨大的成员——水螅毒素。
科学家还尝试用这些方法改造一种抗生素——万古霉素的分子,用来灭有超强抗药性的细菌。
此外,利用这些方法合成的一些有机材料能够发光,可用于制造只有几毫米厚、像塑料薄膜一样的显示器。
科学界一些人士表示,依托“钯催化交
叉偶联反应”,一大批新药和工业新材料应运而生,这三名科学家的科研成果如今已经成为支撑制药、材料化学等现代工业文明的巨大力量。
钯催化反应的应用:对制药工业举足轻重
诺贝尔化学奖委员会主席拉尔斯·哲兰德说:“有人告诉我说,目前25%的合成药品都由这三种反应中的一种制成,因此,他们的发明对制药工业具有举足轻重的影响。
钯具有非常神奇的属性,它可以让两个不同的碳原子连接在一起,使得它们更靠近并且在非常温和的环境下就能发生反应。
”
瑞典皇家科学院表示,科学家已经使用钯催化交叉偶联反应人工合成了最开始在海绵中发现的抗癌药物,目前,科学家已经开始进行临床测试。
而且,科学家也使用该方法制造出了新的抗体和许多其他有用的药物,包括抗炎镇痛药物萘普生(NAPROXEN)等等。
诺贝尔奖:
2010年10月6日在瑞典皇家科学院举行的新闻发布会上,瑞典皇家科学院常任秘书诺尔马克首先宣读了获奖者名单。
他说,赫克、根岸英一和铃木章在“钯催化交叉偶联反应”研究领域作出了杰出贡献,其研究成果使人类能有效合成复杂有机物。
释疑:
以钯为“媒”撮合碳原子
碳原子化学性质不活泼,不愿相互结合。
怎么让这些懒洋洋的碳原子活跃起来,好将它们凑作一堆?一百多年前人们已经想到办法,法国科学家格林尼亚发明了一种试剂,利用镁原子强行塞给碳原子两个电子,使碳原子变得活跃。
这是一项非常重要的成果,使格林尼亚获得了1912年的诺贝尔化学奖。
但是这样的方法在合成复杂大分子的时候有很大局限。
人们不能控制活跃的碳原子的行为,反应会产生一些无用的副产物。
在制造大分子的过程中,副产物生成得非常多,反应效率低下。
用钯作为催化剂可以解决这个问题。
钯原子就像“媒人”一样,把不同的碳原子吸引到自己身边,使碳原子之间的距离变得很近,容易结合——也就是“偶联”,而钯原子本身不参与结合。
这样的反应不需要把碳原子激活到很活跃的程度,副产物比较少,更加精确而高效。
碳氢键活化
这是有机化学里的一个概念,是最近几年兴起地一个有机化学中重要的领域。
按反应机理上来讲,一般为氧化还原型的和复分解型的,而从催化剂来看,可以分为金属催化的和非金属催化的。
由于有机化学可认
为是定向地断裂和生成C-X键的过程,因此在反应中通常需要定位基团,可以高效地构筑C-X键,但在在些过程中也有这样一个问题,那就是定位基团的引入使得反应原子经济性不高,而如果不引入定位基团,而是直接选择性地断裂C-H键,进一步地发生其它反应,这样就能高效地构筑化合物,提高反应效率和原子经济性。