高通量测序样本
一代测序流程

一代测序流程一代测序是指通过一种高通量测序技术,对DNA或RNA样本进行测序,从而获取样本的全基因组或转录组信息。
一代测序技术的发展,为科研工作者和临床医生提供了更多的遗传信息,有助于揭示疾病的发病机制、诊断和治疗方法的研究。
一代测序流程主要包括样品准备、文库构建、测序仪测序、数据分析和结果解读等步骤。
下面将对一代测序流程进行详细介绍。
首先是样品准备。
样品可以是DNA、RNA或其它类型的核酸。
在样品准备阶段,需要对样品进行提取、纯化和定量,确保样品的质量符合测序的要求。
此外,还需要对样品进行质控,检测样品的完整性和纯度,以确保后续步骤的顺利进行。
接下来是文库构建。
文库是指将样品中的DNA或RNA片段连接到测序适配器上,构建成文库,为后续的测序提供样品。
文库构建的关键步骤包括DNA片段的剪切、末端修复、连接适配器和PCR扩增等。
在文库构建过程中,需要严格控制各个步骤的条件和时间,以确保文库的质量和可靠性。
然后是测序仪测序。
文库构建完成后,样品就可以送入测序仪进行测序。
测序仪会根据测序平台的不同,采用不同的测序技术进行测序,如Illumina、Ion Torrent、PacBio等。
在测序过程中,测序仪会逐个读取文库中的DNA或RNA片段,并生成原始的测序数据。
接着是数据分析。
测序仪生成的原始测序数据需要进行数据分析,将原始数据转化为可解读的生物信息学数据。
数据分析的步骤包括序列质量控制、序列比对、变异检测和功能注释等。
通过数据分析,可以获取样品的基因组或转录组信息,识别基因突变、表达差异和功能通路等生物学信息。
最后是结果解读。
在数据分析完成后,需要对分析结果进行解读,理解样品的生物学意义。
结果解读需要结合实验设计和科研目的,对数据分析结果进行综合分析和解释,从而得出科学结论,并为后续的研究或临床诊断提供参考。
总的来说,一代测序流程是一个复杂的过程,涉及样品准备、文库构建、测序仪测序、数据分析和结果解读等多个环节。
高通量测序的生物信息学分析
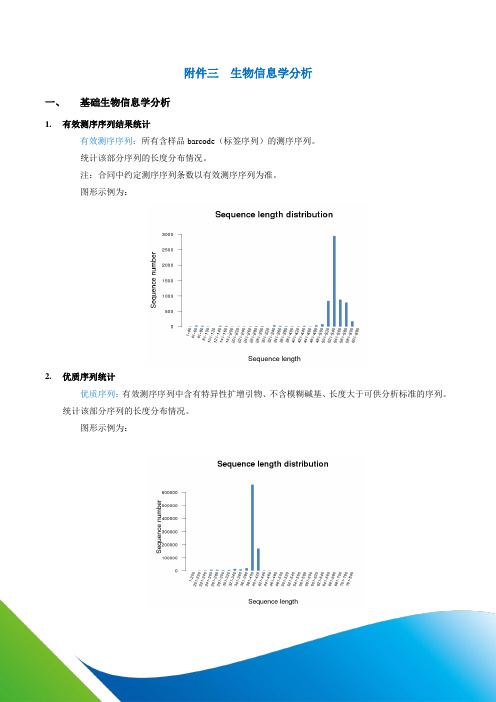
附件三生物信息学分析一、基础生物信息学分析1.有效测序序列结果统计有效测序序列:所有含样品barcode(标签序列)的测序序列。
统计该部分序列的长度分布情况。
注:合同中约定测序序列条数以有效测序序列为准。
图形示例为:2.优质序列统计优质序列:有效测序序列中含有特异性扩增引物、不含模糊碱基、长度大于可供分析标准的序列。
统计该部分序列的长度分布情况。
图形示例为:3.各样本序列数目统计:统计各个样本所含有效测序序列和优质序列数目。
结果示例为:样品有效序列优质序列AB4.OTU生成:根据序列的相似性,将序列归为多个OTU(操作分类单元),以便后续分析。
OTU name A B C D E F G HOTU1 149 410 27 252 45 124 136 101OTU2 0 0 0 0 0 0 0 0OTU3 2 3 14 23 1 5 17 29OTU4 0 47 0 11 0 5 1 7OTU5 19 28 82 9 57 45 303 9OTU6 0 0 0 0 0 0 0 0OTU7 0 182 94 24 14 5 12 60OTU8 0 0 0 0 0 0 0 0...... …………………………………………5.稀释曲线(rarefaction 分析)根据第4条中获得的OTU数据,做出每个样品的Rarefaction曲线。
本合同默认生成OTU相似水平为0.03的rarefaction曲线。
rarefaction曲线结果示例:6.指数分析计算各个样品的相关分析指数,包括:•丰度指数:ace\chao•多样性指数:shannon\simpson•本合同默认生成OTU相似水平为0.03的上述指数值。
多样性指数分析结果示例:注:默认分析以上所列指数,如有特殊需要请说明。
7.Shannon-Wiener曲线利用各样品的测序量在不同测序深度时的微生物多样性指数构建曲线,反映各样本在不同测序数量时的微生物多样性。
高通量测序技术简介

数据转换
将采集到的图像数据转换为对应的碱基序列 信息。
质量控制
对转换后的数据进行质量评估和控制,以确 保测序结果的准确性和可靠性。
数据输出
将最终测序结果以FASTQ等格式输出,供后 续生物信息学分析使用。
03
高通量测序技术平台
Illumina平台
伦理规范制定
制定高通量测序技术应用的伦理规范,确保 技术的合理、安全使用。
法规监管和政策支持
加强高通量测序技术的法规监管和政策支持, 推动技术的健康发展。
THANKS
感谢观看
Genia Technologies平台
采用基于光学干涉的测序技术,通过检测DNA分子在光学干涉仪中的干涉信号变化实 现测序,具有高精度、高灵敏度等优势。
04
高通量测序技术在基因组学研究 中的应用
全基因组重测序
定义
全基因组重测序是对已知基因组 序列的物种进行不同个体的基因 组测序,并在个体或群体水平上 进行差异性分析的方法。
该技术能够在短时间内产生大量的序 列数据,为基因组学、转录组学、宏 基因组学等领域的研究提供了有力支 持。
发展历程及现状
第一代测序技术
以Sanger测序为代表,具有读长较长、准确性高的优点, 但通量低、成本高,难以满足大规模测序需求。
第二代测序技术
以Illumina公司的HiSeq系列、Life Technologies公司的 SOLiD系列等为代表,实现了高通量、低成本的目标,广泛应
高通量测序技术简介
• 引言 • 高通量测序技术原理 • 高通量测序技术平台 • 高通量测序技术在基因组学研究中
的应用
• 高通量测序技术在临床医学中的应 用
锐博生物高通量测序样品质量及运输要求

细胞样本 (包括培养微生物样本) 1、 准备:高速离心收集细胞,存于 TRIzol/ RNAlater 中。 2、 贴壁或悬浮细胞:>5X106 细胞/样品。(注:不同类型的细胞 DNA/RNA 产量差别较大,
建库类型 RNA‐Seq ( 包 括普通转录 组及链特异 性转录组)
样品类型
人 、 鼠 Total RNA
检测方法
Agilent 2100/2200 , NanoDrop
检测结果 m≥10μg
5μg≤m<10μg
c≥65 ng/μL RIN≥7.0 OD260/280≥1.8 OD260/230≥1.5
RNA样品要求
1、 完整度:28S/18S>1.5;RIN>8。 2、 纯度:OD 值介于 1.8~2.0;无蛋白质、RNA 或肉眼可见污染。 3、 浓度:>400ng/μl。 4、 备份:接收样品前要求客户分装备份,用于样品检测不合格时分析原因。 5、 运输 RNA 样品时,推荐保存在 70%乙醇中(处理方法如下);如寄送溶解后的
冰上预冷 PBS(无 DNase 及 RNase); 贴壁细胞:去掉培养基后,加入适量预冷 PBS 漂洗一次,按每 10cm2 培养面积加
1mL TRIzol 的比例加入 TRIzol,反复吹打使所有细胞充分消化,转移到离心管或冻 存管中; 悬浮细胞:离心收集细胞,弃培养液,用预冷 PBS 漂洗一次,按每 5X106 细胞加 1mL TRIzol 的比例加入 TRIzol,反复吹打使所有细胞充分消化,转移到离心管或冻存管 中; 液氮速冻后用封口膜封口。
测序方法和流程

测序方法和流程测序方法和流程是现代生物学和基因研究的重要工具之一,它可以帮助科学家确定生物体的基因组序列。
随着科技的进步,测序方法不断发展,其精度和效率得到了显著提升。
本文将介绍常见的测序方法和流程,包括Sanger测序、高通量测序和单分子测序。
Sanger测序是最早被使用的测序方法之一。
它是由Frederick Sanger于1977年发表的一种测序技术。
Sanger测序的核心原理是使用DNA聚合酶在DNA模板上合成新的DNA链。
具体步骤如下:1. DNA样本制备:从生物体中提取DNA样本,并通过PCR扩增或限制性酶切等方法获取所需的DNA片段。
2. 标记引物:在PCR反应中加入标记了荧光染料的引物,引物与待测序片段的一端互补结合。
3. 聚合酶链反应(PCR):加入聚合酶和四种dNTP(脱氧核苷三磷酸)来进行DNA链合成。
当碱基分别为dATP、dCTP、dGTP、dTTP时,会有相应荧光信号释放。
4. 聚丙烯酰胺凝胶电泳:将PCR产物分离到一维聚丙烯酰胺凝胶中,根据碱基分子量的不同进行大小排序。
5. 电泳图分析:使用自动荧光检测仪对凝胶进行扫描,记录荧光信号。
高通量测序(Next-Generation Sequencing, NGS)是目前应用最广泛的测序方法之一。
相较于Sanger测序,高通量测序具有高速、高通量和低成本的特点。
常见的高通量测序方法包括Illumina测序、Ion Torrent测序和Pacific Biosciences测序等。
一般的高通量测序流程如下:1. 文库制备:将DNA样本打断为较小的片段,并在其两端加上适配体序列。
2. 文库扩增:通过PCR扩增文库片段,以生成大量模板DNA。
3. 上机测序:将文库片段固定在测序芯片上,并使用指定的核酸测序试剂盒进行测序。
4. 数据分析:通过计算机软件将测序数据进行处理和分析,包括序列拼接、序列比对、变异位点分析等。
单分子测序是一种新兴的测序技术,其特点是以单个分子为单位进行测序,无需进行PCR扩增。
高通量测序法的流程

高通量测序法的流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!高通量测序法的流程一、样本准备阶段。
在进行高通量测序之前,首先要进行充分的样本准备工作。
高通量测序生物信息学分析(内部极品资料,初学者必看)

基因组测序基础知识㈠De Novo测序也叫从头测序,是首次对一个物种的基因组进行测序,用生物信息学的分析方法对测序所得序列进行组装,从而获得该物种的基因组序列图谱。
目前国际上通用的基因组De Novo测序方法有三种:1. 用Illumina Solexa GA IIx 测序仪直接测序;2. 用Roche GS FLX Titanium直接完成全基因组测序;3. 用ABI 3730 或Roche GS FLX Titanium测序,搭建骨架,再用Illumina Solexa GA IIx进行深度测序,完成基因组拼接。
采用De Novo测序有助于研究者了解未知物种的个体全基因组序列、鉴定新基因组中全部的结构和功能元件,并且将这些信息在基因组水平上进行集成和展示、可以预测新的功能基因及进行比较基因组学研究,为后续的相关研究奠定基础。
实验流程:公司服务内容1.基本服务:DNA样品检测;测序文库构建;高通量测序;数据基本分析(Base calling,去接头,去污染);序列组装达到精细图标准2.定制服务:基因组注释及功能注释;比较基因组及分子进化分析,数据库搭建;基因组信息展示平台搭建1.基因组De Novo测序对DNA样品有什么要求?(1) 对于细菌真菌,样品来源一定要单一菌落无污染,否则会严重影响测序结果的质量。
基因组完整无降解(23 kb以上), OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;每次样品制备需要10 μg样品,如果需要多次制备样品,则需要样品总量=制备样品次数*10 μg。
(2) 对于植物,样品来源要求是黑暗无菌条件下培养的黄化苗或组培样品,最好为纯合或单倍体。
基因组完整无降解(23 kb以上),OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;样品总量不小于500 μg,详细要求参见项目合同附件。
(3) 对于动物,样品来源应选用肌肉,血等脂肪含量少的部位,同一个体取样,最好为纯合。
高通量测序法

附件1肿瘤相关突变基因检测试剂(高通量测序法)性能评价通用技术审查指导原则一、前言本指导原则旨在指导注册申请人对肿瘤相关基因检测试剂分析性能评价注册申报资料的准备及撰写,同时也为技术审评部门对注册申报资料的技术审评提供参考。
本指导原则是针对肿瘤相关基因检测试剂分析性能评价的一般要求,申请人应依据产品的具体特性确定其中内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。
本指导原则是对申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但需要详细阐明理由,并对其科学合理性进行验证,提供详细的研究资料和验证资料,相关人员应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将适时进行调整。
二、适用范围本指导原则所述肿瘤相关基因检测试剂分析性能评价主要是指基于高通量测序(high-throughput sequencing)即下一代测序(next generation sequencing, NGS),又称为大规模平行测序(massively parallel sequencing, MPS),体外检测人体组织中肿瘤细胞中肿瘤相关基因变异。
用于检测体细胞突变的NGS正在广泛用于肿瘤诊疗相关的分子检测,包括对特定基因的DNA/RNA进行测序,以寻找与肿瘤临床诊疗相关的突变基因的改变。
肿瘤基因突变类型包括点突变、插入、缺失、基因重排、拷贝数异常等广义的基因突变。
基于NGS测序原理的IVD检测可能包括以下步骤:样本收集,处理和保存、DNA提取、DNA处理、文库制备、测序和碱基识别、序列比对/映射、变异识别和过滤、变异注释和解读以及检测报告的生成。
同时,某些产品还可能会包括软件部分,但上述相关步骤并不一定被全部包括,应根据产品的具体设计流程来进行判断。
DNA测序技术与高通量测序

DNA测序技术与高通量测序DNA测序技术的发展取得了重大突破,为生物学、医学和农业领域的研究和应用带来了革命性的变化。
其中,高通量测序作为最先进和最广泛应用的测序技术之一,为科学家提供了大规模且高效的DNA 测序解决方案。
本文将介绍DNA测序技术的发展概况,详细阐述高通量测序的原理和应用,以及其在生物信息学、医疗诊断和基因组学研究方面的重要性。
一、DNA测序技术的发展概况DNA测序技术是指通过检测DNA分子的碱基序列,从而确定DNA分子的结构和功能。
早期的DNA测序技术主要依赖于Sanger测序方法,该方法于1977年被发明并获得诺贝尔奖。
然而,Sanger测序方法速度慢、费用高以及需要大量的DNA模板等限制了其在大规模测序中的应用。
随着高通量测序技术的兴起,科学家们实现了对大规模DNA测序项目的高效处理。
高通量测序技术主要分为两类:第一代测序技术和第二代测序技术。
第一代测序技术代表是Sanger测序方法,而第二代测序技术则包括Illumina测序、454测序、Ion Torrent测序和SOLiD测序等。
二、高通量测序的原理和应用高通量测序技术通过将DNA分子分离成大量的片段,并进行并行测序,从而以更快的速度和更低的成本完成DNA测序。
这种技术的核心是构建DNA文库,其中包含了大量的DNA片段,并通过多重并行测序的方法将这些DNA片段进行测序。
测序得到的数据可以通过计算和分析获得DNA分子的碱基序列信息。
高通量测序技术在各领域的应用广泛。
在生物信息学领域,高通量测序技术为基因组学、转录组学和蛋白质组学的研究提供了强有力的工具。
通过大规模测序,科学家们可以获得更多的基因组、转录组和蛋白质组信息,帮助我们更好地理解生物体的基因调控机制、疾病发病机理等重要生物学问题。
在医疗诊断方面,高通量测序技术在个体基因组学和个性化医学方面具有巨大潜力。
通过测序个体基因组,我们可以识别个体携带的疾病相关变异,并为个体提供精确、个性化的医疗方案。
高通量测序技术的原理和应用

高通量测序技术的原理和应用随着基因组学研究的不断深入,对基因组的了解也越来越深入。
而为了更好地研究基因组,人们已经开发出了很多种测序技术。
其中,高通量测序技术便是一种效率和精准度都很高的测序技术。
这篇文章将针对高通量测序技术的原理和应用进行讲述。
一、高通量测序技术的原理1.端点测序和鸟枪法测序端点测序是第一种测序技术,它是通过将DNA的一端连接到一种特殊的引物上,然后引物与DNA的另一端连接,最后利用酶开放区域,加入dNTPs和DNA聚合酶进行扩增,然后进行测序。
而鸟枪法测序则是利用两串寡聚核苷酸将DNA分成一小段一小段,然后进行扩增,在完成扩增后,通过比较不同反应组严格高精的测序结果,我们可以得出完整序列。
2.震荡式测序(Sanger测序)震荡式测序(Sanger测序)是目前使用较多的一种测序方法,它通过将所需的DNA样本进行扩增,得到多个特异性片段。
然后将这些片段进行分离电泳,得到A、T、C和G四个碱基片段的信号。
最后,根据各个碱基标记的强度,推算出大概的有机物组成,根据机组运转偏测结果进行判断,从而得到DNA的序列。
3.Pyrosequencing技术Pyrosequencing技术是一种比较新颖的测序技术,它基于酶反应来测序。
在这种技术中,DNA序列是通过酶反应来完成的,从而得到相应的序列信息。
二、高通量测序技术的应用1.基因组重测序基因组测序是目前较为常见的一种DNA测序方法,它可以对整个基因组的信息进行测定和分析。
基因组重测序技术是一种利用高通量测序技术的方法,通过对基因组中的所有区域进行大规模的测序,比对得到一份更加准确的基因组数据。
这种技术具有处理样本齐全、成本低廉、得到准确数据等优势。
而应用于此类测序的高通量测序技术,则可以大量试用高效的测序数据,使数据分析更加准确。
2.转录组测序转录组测序是一种较为常用的RNA测序方法。
它可以对一个生物体中所有的mRNA进行大规模的测序,并得到DNA序列信息。
tngs的样本类型

tngs的样本类型TNGS(全称为“下一代测序技术”)是一种高通量测序技术,它的出现极大地推动了基因组学和生物学研究的发展。
在TNGS技术中,样本的选择和准备是非常重要的一步,不同的样本类型可以提供不同的信息,因此对于研究者来说,选择合适的样本类型至关重要。
首先,常见的样本类型之一是DNA样本。
DNA是生物体内的遗传物质,它携带着生物体的遗传信息。
通过对DNA样本进行测序,我们可以了解到生物体的基因组结构、基因组变异以及基因功能等信息。
DNA样本可以来自于不同的生物体,包括人类、动物、植物等。
在人类基因组计划中,研究者们使用了大量的人类DNA样本,通过对这些样本的测序,揭示了人类基因组的组成和变异情况,对于人类疾病的研究和诊断起到了重要的作用。
除了DNA样本,RNA样本也是TNGS中常见的样本类型之一。
RNA是DNA的转录产物,它在细胞内起着重要的调控作用。
通过对RNA样本进行测序,我们可以了解到细胞内基因的表达情况,即哪些基因在特定条件下被转录和表达。
RNA样本可以来自于不同的组织和细胞类型,通过对不同组织和细胞的RNA样本进行测序,我们可以了解到不同组织和细胞的功能和特性。
例如,通过对肿瘤组织中的RNA 样本进行测序,可以发现哪些基因在肿瘤发生和发展过程中起到了关键的作用,从而为肿瘤的治疗提供新的靶点。
此外,蛋白质样本也是TNGS中常见的样本类型之一。
蛋白质是细胞内的重要功能分子,它们参与了几乎所有的生物过程。
通过对蛋白质样本进行测序,我们可以了解到细胞内蛋白质的组成和功能。
蛋白质样本可以来自于不同的组织和细胞类型,通过对不同组织和细胞的蛋白质样本进行测序,我们可以了解到不同组织和细胞的蛋白质组成和功能差异。
例如,通过对癌细胞和正常细胞的蛋白质样本进行比较,可以发现哪些蛋白质在癌细胞中过度表达或缺失,从而为癌症的治疗提供新的靶点。
综上所述,TNGS的样本类型包括DNA样本、RNA样本和蛋白质样本。
高通量基因测序实验报告

一、实验目的本实验旨在通过高通量基因测序技术,对某特定样本进行基因检测,分析其基因表达水平、基因突变等信息,为疾病诊断、基因治疗等提供科学依据。
二、实验材料1. 样本:某疾病患者的组织样本2. 试剂:高通量测序试剂盒、DNA提取试剂盒、PCR试剂、荧光定量PCR试剂等3. 仪器:高通量测序仪、PCR仪、凝胶成像系统、离心机、核酸分析仪等三、实验方法1. 样本DNA提取(1)将组织样本加入DNA提取试剂盒,按照说明书进行操作,提取样本DNA。
(2)使用核酸分析仪检测提取的DNA浓度和质量。
2. PCR扩增(1)根据目的基因序列设计特异性引物,进行PCR扩增。
(2)使用荧光定量PCR试剂对PCR产物进行定量分析,确保扩增效率。
3. 高通量测序(1)将PCR产物进行文库构建,包括适配子连接、指数富集等步骤。
(2)将文库进行高通量测序,获得大量测序数据。
4. 数据分析(1)使用生物信息学软件对测序数据进行质控、比对、基因表达水平分析等。
(2)根据分析结果,筛选出与疾病相关的基因突变和差异表达基因。
四、实验结果1. 样本DNA提取提取的DNA浓度为500 ng/μL,A260/A280比值约为1.8,符合实验要求。
2. PCR扩增PCR扩增产物经荧光定量PCR检测,扩增效率达到90%以上。
3. 高通量测序高通量测序获得大量测序数据,测序深度达到100倍。
4. 数据分析(1)基因表达水平分析:与对照组相比,实验组某基因表达水平显著升高。
(2)基因突变分析:在实验组样本中,发现某基因存在突变,与疾病发生密切相关。
五、实验讨论1. 本实验通过高通量基因测序技术,成功检测出与疾病相关的基因突变和差异表达基因,为疾病诊断和基因治疗提供了科学依据。
2. 高通量测序技术具有高通量、高灵敏度、高准确度等优点,在基因检测领域具有广泛应用前景。
3. 本实验中,样本DNA提取、PCR扩增、高通量测序等步骤均严格按照实验操作规范进行,保证了实验结果的可靠性。
高通量测序

什么是高通量测序?高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。
什么是Sanger法测序(一代测序)Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。
直到掺入一种链终止核苷酸为止。
每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。
每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequencing)全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。
通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
什么是de novo测序de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。
ngs高通量测序流程

ngs高通量测序流程英文回答:Next-generation sequencing (NGS) is a high-throughput sequencing technology that has revolutionized genomics research. It allows for the rapid and cost-effective sequencing of millions of DNA fragments simultaneously, providing researchers with a wealth of information about the genetic makeup of an organism.The NGS workflow consists of several key steps, including sample preparation, library construction, sequencing, and data analysis. Let me walk you through each of these steps.1. Sample preparation: This step involves obtaining the DNA or RNA samples that will be sequenced. The quality and integrity of the samples are crucial for accurate sequencing results. Various methods can be used to extract DNA or RNA from different sample types, such as blood,tissue, or cells.2. Library construction: Once the DNA or RNA samples are obtained, they need to be converted into a library of fragments that can be sequenced. This involves several enzymatic reactions, including fragmentation, adapter ligation, and amplification. The resulting library contains millions of DNA fragments, each with a unique adapter sequence.3. Sequencing: The prepared library is loaded onto a sequencing platform, such as an Illumina sequencer. The fragments are then amplified and immobilized on a solid surface, forming a DNA cluster. Fluorescently labeled nucleotides are added one at a time, and the emitted light is captured by a camera. This process is repeated multiple times to generate millions of short DNA sequences, called reads.4. Data analysis: The raw sequencing data needs to be processed and analyzed to extract meaningful information. This involves several bioinformatics steps, includingquality control, read alignment, variant calling, and functional annotation. The resulting data can provide insights into various aspects of genomics, such as gene expression, genetic variation, and epigenetic modifications.NGS has numerous applications in research and clinical settings. For example, it can be used to study the genetic basis of diseases, identify disease-causing mutations, and monitor the response to treatment. It can also be used in agriculture to improve crop yields and develop disease-resistant varieties.中文回答:NGS(下一代测序)是一种高通量测序技术,彻底改变了基因组学研究的方式。
对于不同测序长度、不同测序样本量的最佳测序方案比较

对于不同测序长度、不同测序样本量的最佳测序⽅案⽐较对于不同测序长度、不同测序样本量的最佳测序⽅案⽐较(2013-09-05 00:10:56)转载▼标签:⾼通量测序服务⼆代测序服务hiseq扩增⼦测序sanger测序对于不同测序长度、不同测序样本量的最佳测序⽅案⽐较1.根据对于单个样本所需检测的基因范围分三档a.测序范围<= 100Kb,推荐⽤Sanger法测序2.根据上⾯的分法,经济⽐较a.Sanger法测序,适⽤范围:测序范围长度 <= 100Kbi.⽤传统sanger法测序,1.每500bp测⼀对来回,每个测序20元(PCR + sanger测序)。
2.每个样本的测序成本:100000/500 * 20 = 4000元/样本。
3.数据分析,500元/样本4.再加⼀点⼈⼯,总成本4500元/样本以内就完成了。
是优势选择。
ii.⽤⾼通量测序,建库2000元/样本,再加测序的上机费1000~3000元/样本,还有⼆代测序必须的⽣物信息分析费,成本上不占优势。
⾼通量测序在此的唯⼀的优势,是可以在⼀个试管中完成PCR,减少起始样本(DNA)⽤量b.扩增⼦+ ⼆代测序,适⽤范围:500Kb <= 测序范围长度< 100Kbi.⽤扩增⼦(Amplicon)法测序1.以200Kb,100样本为例2.引物及PCR,设计多重PCR引物,每个扩增⼦约150Bp的长度,总120000元,分摊到每样本,1200元3.PCR反应,成本20元/样本4.建库,2000元/样本5.测序,a.Miseq平台,5G数据/run,26500元/runb.如果⼀次跑⼀个run,测24个样本,每个样本的测序部分的成本:26500/24=1104元/样本c.如果⼀次跑超过24个样本,测序成本可以进⼀步分摊6.数据分析,500元/样本7.总成本:1200 + 20 + 2000 + 1104 + 500 = 4824元ii.数据的覆盖深度:1.Miseq平台,a.PE 2*250 测序模式b.总共5G数据/Flowcell,c.分到24个样本,最多可到96个样本d.被测范围,200Kbe.覆盖深度,5,000,000,000/24/200,000 = 1041倍覆盖;如果是96个样本,5,000,000,000/96/200,000 = 260倍2.PGM平台a.314芯⽚(也可以选⽤316/318等芯⽚,只有通量差别,没有质量差别)b.分到4个样本c.每个314芯⽚的数据产量0.1Gd.被测范围200Kbe.覆盖深度,100,000,000/4/200,000=125倍iii.扩增⼦测序是100Kb~500Kb测序长度范围内的优势选择iv.⽤sanger法测序,200000 / 500 * 2 *30=24000,成本过⾼,不可⾏v.⽤捕获测序,约5000~6000元,较贵,且操作复杂,分析也复杂,vi.扩增⼦测序的限制因素:扩增⼦测序前⼀步要做多重PCR,到⽬前为⽌,多重PCR扩增效率的⼀致性⼀直是⼤难题。
高通量测序及分析

高通量测序及分析高通量测序与功能分析微生物群落测序是指对微生物群体进行高通量测序,通过分析测序序列的构成分析特定环境中微生物群体的构成情况或基因的组成以及功能。
借助不同环境下微生物群落的构成差异分析我们可以分析微生物与环境因素或宿主之间的关系,寻找标志性菌群或特定功能的基因。
对微生物群落进行测序包括两类,一类是通过16s rDNA,18s rDNA,ITS区域进行扩增测序分析微生物的群体构成和多样性;还有一类是宏基因组测序,是不经过分离培养微生物,而对所有微生物DNA进行测序,从而分析微生物群落构成,基因构成,挖掘有应用价值的基因资源。
以16s rDNA扩增进行测序分析主要用于微生物群落多样性和构成的分析,目前的生物信息学分析也可以基于16s rDNA的测序对微生物群落的基因构成和代谢途径进行预测分析,大大拓展了我们对于环境微生物的微生态认知。
目前我们根据16s的测序数据可以将微生物群落分类到种(species)(一般只能对部分菌进行种的鉴定),甚至对亚种级别进行分析,几个概念:16S rDNA(或16S rRNA):16S rRNA基因是编码原核生物核糖体小亚基的基因,长度约为1542bp,其分子大小适中,突变率小,是细菌系统分类学研究中最常用和最有用的标志。
16S rRNA基因序列包括9个可变区和10个保守区,保守区序列反映了物种间的亲缘关系,而可变区序列则能体现物种间的差异。
16S rRNA基因测序以细菌16S rRNA基因测序为主,核心是研究样品中的物种分类、物种丰度以及系统进化。
OTU:operational taxonomic units (OTUs)在微生物的免培养分析中经常用到,通过提取样品的总基因组DNA,利用16S rRNA或ITS的通用引物进行PCR 扩增,通过测序以后就可以分析样品中的微生物多样性,那怎么区分这些不同的序列呢,这个时候就需要引入operational taxonomic units,一般情况下,如果序列之间,比如不同的 16S rRNA序列的相似性高于97%就可以把它定义为一个OTU,每个OTU对应于一个不同的16S rRNA序列,也就是每个OTU对应于一个不同的细菌(微生物)种。
高通量测序结果分析流程

ESP6500 cosmic HGMD
OMIM Risk
病
信
息
数
值
据 库
疾
突 变 类 型
息 数 据 库 信
息 数 据 库 信
本信
地息
数
据
库
数
信
据
息
库
ExAC Clinvar
基
项
标
因
目
准
名
简 写
命 名
据 库 信 息数
出本 现批 次次 数试
验
据
库
信 息
数
数千 据人 库基 信因 息组
二、筛选基因与位点------临床表现
据库收录;
地数据库MAF > 10%
0
其它突变类型或HGMD/clinvar数 1000g/ESP6500 中 MAF>1% 或 本 地 良性
据库未收录;
数据库MAF > 2%
注:a,预计致病突变指无义突变、移码突变、剪接突变、影响翻译起始位点的突变等公认可能严重影响蛋白质结 构的突变;b,MAF=Minor Allele Frequency,即最小等位基因频率;c,VUS=variant of unknown clinical significance,临床意义未明。
Well-established
功能学 研究
functional studies show no deleterious effect BS3
Nonsegregation with 共分离 disease BS4 家系研究
新发突变
等位基因上 的其他变异
其他数据库
In-frame indels in repeat w/out
atac-seq 每个样本文库具体的质控标准

一、引言随着生物信息学的迅速发展,高通量测序技术在基因组学、转录组学和表观基因组学等领域得到了广泛应用。
其中,atac-seq(assay for transposase-accessible chromatin using sequencing)作为一种测定染色质开放区域的技术,可以揭示细胞内基因组的开放性和可及性,广泛应用于转录调控和疾病机制的研究中。
在进行atac-seq实验时,样本文库作为实验数据产生的基础,其质控标准的制定对于数据分析和结果解释至关重要。
二、样本质控标准1. DNA质量评估在进行atac-seq实验之前,首先需要对DNA样本的质量进行评估。
DNA质量的评估可以通过比色法、琼脂糖凝胶电泳或者生物分析仪等多种方法进行。
确定DNA的纯度和完整性对于后续的操作至关重要。
2. 转座子插入效率atac-seq技术的核心是通过转座子将开放的染色质区域进行标记,因此转座子的插入效率直接影响到实验结果的可信度。
在进行样本文库构建时,需要对转座子的插入效率进行评估,通常可以通过PCR扩增和定量PCR等方法进行。
3. 文库大小选择文库大小选择的合理性对于atac-seq实验的结果有着重要的影响。
文库大小的选择应基于研究目的和测序评台等因素进行权衡,通常情况下,合适的文库大小应在100-1000bp之间。
4. 实验重复性为了确保实验结果的可靠性,atac-seq实验中通常会进行实验重复,评估在不同条件下的一致性。
实验重复性的评估可以通过Pearson相关系数、Spearman相关系数等统计学方法进行,确保实验结果的稳定性和可重复性。
5. 负对照实验为了排除技术误差和外部干扰因素的影响,atac-seq实验中通常会设置负对照实验。
在样本文库构建过程中,可以引入对照组进行比较,评估实验的特异性和可信度。
三、结论atac-seq作为一种广泛应用于基因组学研究的高通量测序技术,样本文库的质控标准对于实验结果的可信度和可重复性至关重要。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2、Paired-end library
Pai点,在第 一轮测序完成后,去除 第一轮测序的模板链, 用对读测序模块(PairedEnd Module)引导互补 链在原位臵再生和扩增, 以达到第二轮测序所用 的模板量,进行第二轮 互补链的合成测序(图2)。
测序样本制备 的基本流程
样本核酸准备
预处理(片段化, 测序模板制备(单分子多拷贝) 上机测序 进一步处理测序模板
样本核酸的获取r-end Mate-pair DNA/mRNA/miRNA ChIP-Seq/MeDIP-Seq/转录组/表达谱等 等 ……
PCR Amplification
1、Fragment Library/single-read Library
3、HydroShear
The Hydroshear is used for fragmenting DNA to sizes larger than 1KB. The HydroShear offers the simplest, most reproducible, and most controllable method available for generating random DNA fragments. Virtually any source of DNA at any concentration in volumes as small as 40µ l can be randomly fractured to within a two-fold size distribution. For instance, if your target size is 2kb, you can calibrate the HydroShear to produce a distribution centered on 2kb with 90% of your DNA falling between 1.3 kb and 2.6kb
一、样本DNA等的片段化技术
1、超声破碎技术
不足之处: 体积大 易污染 精度不够
能量水平,时间,体积等参数
Covaris S-series
2010年1月5日报道——安捷伦科技公司(NYSE: A)与 Covaris 公司于今日宣布了签订一项合作开发市场协 议,该协议表明,未来Covaris S2 DNA剪切技术将 与安捷伦 SureSelect 靶向序列捕获系统一起销售,以 提高新一代测序实验的效率。
3、Mate-pair Libr段 包含基因组中较大跨度(2-10 kb)片段两端的序列,更具体 地说:首先将基因组DNA随 机打断到特定大小(2-10 kb范 围可选);然后经末端修复, 生物素标记和环化等实验步骤 后,再把环化后的DNA分子 打断成400-600 bp的片段并通 过带有链亲和霉素的磁珠把那 些带有生物素标记的片段捕获。 这些捕获的片段再经末端修化
通常采用的DNA溶液体系为:1μg-5μg经纯化的基因组DNA溶解 于50 μL 的TE,并加入700 μL nebulization buffer。对DNA进行 雾化6分钟,气体压力为32--35psi(Pounds per square inch。P 是磅pound,S是平方square,I是英寸inch),雾化环境为冰浴。 所得的典型结果为DNA打碎至0–1200 bp,峰值为5–600 bp 雾化法与更早期的酶切法相比,可重复性相对较好,并且对于所处 理的DNA序列没有选择性,非常快速并且便宜。但是所得的DNA分 子片段长度的区间非常大,使得处于可用区间200 ± 20bp 的片段 仅占10%左右。为了满足新一代测序的要求,必须要选择长度较为集 中的片段,增加的纯化筛选造成了样本DNA的大量流失。这一缺点 不仅使成本增加,而且使得痕量DNA的研究难以实现,因而商业推 广价值较小。通量较低也是其显著缺点。目前已经基本被CovarisTM 超声仪进行超声打碎替代。
◦ AMPure Beads ◦ AMPure XP Beads
NEB next generation series
Agencourt AMPure XP Beads
SPRI works Fragment Library System
Beckman Coulter has created a simplified automated workflow that utilizes Solid Phase Reversible Immobilization (SPRI) paramagnetic bead based technology to create an automated system for fragment library preparation for the Illumina Genome Analyzer.
接头自连接问题?
Forked adaptor(illumina)
Single reads adaptor
Pair-end adaptor
连接酶
尼克酰胺单磷酸
DNA连接酶 RNA连接酶
DNA连接酶的作用机制
腺苷基
细菌的DNA连接酶以 NAD为能源,动物细胞 和噬菌体的连接酶以ATP 为能源。T4的DNA连接 酶可以连接平末端。
片段化方法比较
数字表达谱
二、Fragment Recovery(Size selection)
1、传统切胶回收
◦ ◦ ◦ ◦ 操作复杂 专门试剂盒 效率低 高效试剂盒开发?
2、改进型的凝胶回收(E-gel)
7分钟内完成DNA分离(50bp10kb) 选择琼脂糖浓度为0.8%、1.2%或 2%且结合有SYBR Safe或溴化乙 啶DNA染料的E-Gel 无需紫外线即可实时显示DNA迁移, 灵敏度超高 快捷简单的DNA条带提取 适合低、中、高不同通量
测序模板制备
◦ 乳液PCR ◦ 桥式PCR ◦ 滚环扩增
的质控标准
DNA: 完整性 Total RNA: 完整性(Agilent) miRNA Pretreatment 某测序公司原始样本需求
1. 所需处理过的水中、75%的乙醇、异丙醇中,具体以什么方式保存请注明。 2. 如提供实验材料为动物组织材料,样品质量需大于2g; 3. 如提供实验材料为植物样品,样品质量需大于4g; 4. 如提供实验材料为培养细胞,请提供1×107培养好的细胞; 5. 如提供实验材料为血液样品,请提供≥2ml的样品。
6、Target Capture Sequencing / Selection library
Molecular Inversion Probe (MIP)
靶标富集
为了在大样本量中充分发挥新一代测序技 术的潜能,科学家们开发出几种方法,来 选择性富集目标区域。与全基因组测序相 比,富集后再测序降低了成本,也减轻了 后续数据分析的压力,让研究人员能够轻 松利用新一代测序的通量。目前有几种靶 向富集方法,包括分子倒臵探针连接测序 (MIPS)、基于寡核苷酸杂交 三种富集技术:罗氏NimbleGen的 SeqCap寡核苷酸杂交型芯片捕获,安捷 伦的SureSelect寡核苷酸杂交溶液型捕获 以及Raindance的多重PCR方法的方法。
HydroShear
HydroShear
我们的结果
可以实 现自动 清洗的 新设备
4、雾化法(Nebulization)
DNA雾化仪的工作原理如下: 高压氮气从顶端压入,通过两 侧的进气道对底部的DNA缓冲 液混合物产生压力,液体顺着 中部细管上升至顶部。顶部出 液口处有筛子之类的东西, DNA通过“筛子”,被打成小 片段。通过调节压力大小,可 以把DNA打成所需要的bp长度。 在使用之前DNA必须经过纯化。
3、新的仪器设备
Caliper的Labchip XT全自动DNA片段 回收仪等 前期投入大 耗材昂贵 回收载量低
4、新的试剂 —bead-based size selection
磁珠法纯化试剂:可以进行某特定片段区域的选择
E-Z 96® Mag-Bind® Oligonucleotide Purification Kit Agencourt Bioscience
Fragmentase(NEB)
Generates dsDNA breaks in a time-dependent manner to yield 100–800 bp DNA fragments depending on reaction time.
损失相对较小 集中程度不高,需要后续的片段选择
Covaris Adaptive Focused Acoustics™ (AFA)
浓度不同
长度不同
物种不同
一些比较理想的结果
体会: 有很大的损失 不同的样本需要不同的优化,没有现成可直接用的条件
2、片段化酶
最初是利用限制性内切酶(Endodeoxyribonuclease)或DNA 酶I(Deoxyribonuclease I)配合Mn2+将DNA大片段切割成小 片段 针对新一代的高通量测序平台,已经有多家公司开发了配套的酶 切试剂,多数是利用几种酶组合而成,其中包含一种可以在双链 DNA上有随机识别位点的酶将双链DNA制造切口,然后由另外一 种酶将切口处的单链DNA切断形成双链DNA片段 Fragmentase:该片段化酶产品包含着两种不同功能的酶,其一 可以实现在DNA双链分子上随机制造切口(nick),另一种酶可 以识别这种切口并且在互补链对应处制造缺口,从而完全从该处 断开双链DNA,从而实现只需根据反应时间的长短获得不同长度 的DNA片段,范围为100–800 bp DNA。不同的处理反应时间 获得的长度有所差异,但是这种处理方法对原始样品的需求较大, 而且对于目前主流的二代测序技术而言,还需要进一步切胶选择 特定的片段,而且该商品对于不同样品的反应效果均一性有待进 一步验证