RT-PCR中引物的选择指南
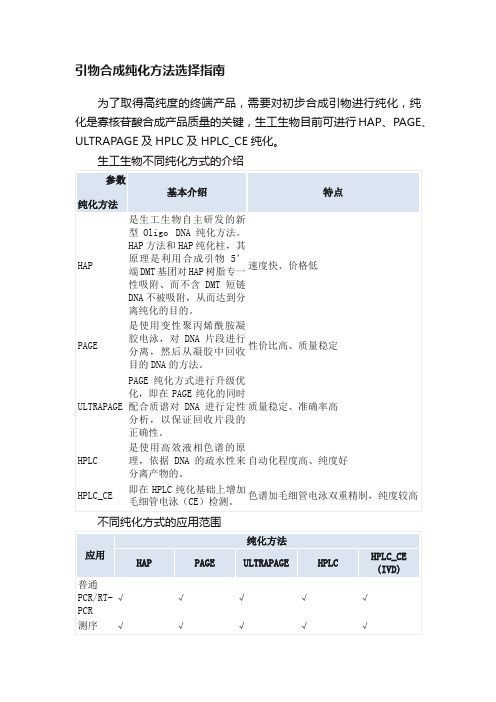
引物合成纯化方法选择指南
√
√
√
基因构建/RNA干扰
√
√
√
√
PCR产物用于克隆表达研究或基因重组等
√
√
√
反义核酸
√
√
修饰或标记引物/化学或物理应用
√
√
体外诊断应用
√
“√” 表示推荐的纯化方式
为了取得高纯度的终端产品需要对初步合成引物进行纯化纯化是寡核苷酸合成产品质量的关键生工生物目前可进行happageultrapage及hplc及hplcce纯化
引物合成纯化方法选择指南
为了取得高纯度的终端产品,需要对初步合成引物进行纯化,纯化是寡核苷酸合成产品质量的关键,生工生物目前可进行HAP、PAGE、ULTRAPAGE及HPLC及HPLC_CE纯化。
性价比高、质量稳定
ULTRAPAGE
PAGE纯化方式进行升级优化,即在PAGE纯化的同时配合质谱对DNA进行定性分析,以保证回收片段的正确性。
质量稳定、准确率高
HPLC
是使用高效液相色谱的原理,依据DNA的疏水性来分离产物的。
自动化程度高、纯度好
HPLC_CE
即在HPLC纯化基础上增加毛细管电泳(CE)检测。
生工生物不同纯化方式的介绍
参数
纯化方法
基本介绍
特点
HAP
是生工生物自主研发的新型Oligo DNA纯化方法。HAP方法和HAP纯化柱,其原理是利用合成引物5’ 端DMT基团对HAP树脂专一性吸附、而不含DMT短链DNA不被吸附,从而达到分离纯化的目的。
速度快、价格低
PAGE
是使用变性聚丙烯酰胺凝胶电泳,对DNA片段进行分离,然后从凝胶中回收目的DNA的方法。
色谱加毛细管电泳双重精制,纯度较高
RT-PCR法则 PCR引物设计

PCR引物设计的11条黄金法则DNA序列的保守区是通过物种间相似序列的比较确定的。
在NCBI上搜索不同物种的同一基因,通过序列分析软件(比如DNAman)比对(Alignment),各基因相同的序列就是该引物长度(primer length)常用的是18-27bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于TaqDNA聚合酶进行反应。
GC含量(composition)过高或过低都不利于引发反应。
上下游引物的............GC..含量不能相差太大..。
另外,上下游引物的Tm值(melting temperature)是寡核苷酸的解链温度,即在一定盐浓度条件下,50%寡核苷酸双链解链的温度。
有效启动温度,一般高于Tm值5~10℃。
若按公式Tm=4(G+C)+2(A+T)估计引物的Tm值,则有效引物的Tm为55~80℃,其Tm值最好接近72℃以使复性条件最佳。
如扩增编码区域,引物3′端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增的特异性与效率。
引物3′端错配时,不同碱基引发效率存在着很大的差异,当末位的碱基为A时,即使在错配的情况下,也能有引发链的合成,而当末位链为T时,错配的引发效率大大降低,G、C错配的引发效率介于A、T之间,所以3′端最好选择T。
引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错误引发(False priming)。
降低引物与模板相似性的一种方法是,引物中四种碱基的分布最好是随机的,不要有聚嘌呤或聚嘧啶的存在。
尤其3′端不应超过3个连续的G或C,因这样会使引物在GC富集序列区错误引发。
引物自身不应存在互补序列,否则引物自身会折叠成发夹结构(Hairpin)使引物本身复性。
这种二级结构会因空间位阻而影响引物与模板的复性结合。
引物自身..不能有连续4个碱基的互补。
两引物之间.....也不应具有互补性,尤其应避免3′端的互补重叠以防止引物二聚体(Dimer 与Cross dimer)的形成。
反转录RT-PCR的内参如何选择?

反转录RT—PCR的内参如何选择?RT—PCR就是将RNA先反转录为cDNA,再将转录得到的cDNA作为模板进行PCR反应扩增目标片段。
用于反转录的引物根据实验的具体情况选择随机引物、Oligo dT及基因特异引物的一种。
对于短的不具有发卡结构的真核细胞mRNA三种都可。
1)随机引物:适用于长的具有发卡结构的RNA。
适用于rRNA、mRNA、tRNA等所有RNA的转录反应。
主要用于单一模板的RT-PCR反应。
2)Oligo dT:适用于具有PolyA尾巴的RNA.(原核生物的RNA、真核生物的Oligo dT rRNA 和tRNA不具有PolyA尾巴。
)由于Oligo dT要结合到PolyA 尾巴上,所以对RNA样品的质量要求较高,即使有少量降解也会使全长cDNA合成量大大减少.oligo(dT) capture多聚胸腺嘧啶-核苷酸捕捉实际上指的是用Oligo(dT)特异结合到mRNA上PolyA尾端的技术,以此技术可以特异地将mRNA从总RNA中分离出来,通常用于mRNA的反转录3) 基因特异引物:与模板序列互补,适用于目的序列已知的情况.实时PCR也就是qPCR,它的基本原理与PCR也是一样的,只是在体系中加入荧光指示,可以对PCR的模板进行定量。
一种是加入荧光染料SYBR Green,SYBR Green会嵌入双链DNA的小沟,且只有在嵌入小沟时才会发出荧光,荧光定量PCR没进行一个循环就会对产物进行一次检测,通过检测每个循环后荧光强度(间接得知DNA的量)从而检测整个PCR的过程,最后通过标准曲线对未知模板进行定量分析。
SYBR Green不具有特异性,对结果稍有影响。
另一种是加入荧光探针T aq man,PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。
探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,T aqman探针在PCR体系中会与目标片段杂交,而在PCR的Extension阶段Taq酶以一条链为模板合成目标片段,此时T aq酶的5'-3’外切酶活性将结合在模板上的探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。
PCR引物设计技巧

PCR引物设计技巧PCR(聚合酶链反应)是一种重要的分子生物学技术,用于扩增目标DNA序列。
PCR的成功与否,很大程度上依赖于引物的设计质量。
一个好的PCR引物设计可以确保反应的特异性、有效性和稳定性。
以下是一些PCR引物设计的技巧。
1.引物长度和GC含量:一般而言,引物长度应在18-24个碱基之间。
引物的GC含量应在40-60%左右。
过低的GC含量可能导致反应特异性不足,而过高的GC含量可能导致引物相互结合和非特异性扩增。
2.引物序列选择:引物序列应选择在目标序列上具有一定变异性的区域。
引物的序列不应包含重复序列、自身互补序列或多聚性序列,以免导致引物间的相互结合或非特异性扩增。
3.引物特异性检测:在设计引物时,应进行引物特异性的检测。
可以使用生物信息学工具,如BLAST,检查引物序列是否与其他非目标序列有任何匹配。
特别是在设计引物用于复杂的基因组中时,应特别关注引物的特异性。
4. 引物互补性检测:在进行引物设计时,应检查引物之间的互补性。
引物之间的互补性可能导致引物相互结合,影响扩增效率和特异性。
可以使用生物信息学工具,如OligoAnalyzer,检查引物之间的互补性。
5.引物长度和跨越剪切位点:在设计用于检测RNA的引物时,应特别注意引物的长度和是否跨越了剪切位点。
过长的引物可能跨越剪切位点导致扩增产物的长度不一致,降低扩增特异性。
6.引物末端修饰:PCR引物的末端可以根据需要进行一些修饰,如磷酸化、生物素化、荧光标记等。
这些修饰可以用于后续的检测和分离。
7.引物浓度和混合物:在PCR反应中,引物的浓度对反应的成功与否至关重要。
一般而言,两个引物的浓度应保持一致。
此外,在进行多重PCR反应时,不同引物的混合物也需要进行优化,以确保特异性和扩增效率。
8.引物的核酸相互作用力:引物的核酸相互作用力是指引物与模板DNA之间的结合力。
引物的核酸相互作用力可以通过计算引物的熔解温度(Tm)来评估。
设计RTPCR引物方法

设计RTPCR引物方法设计RT-PCR引物方法要鉴定某一个基因在mRNA水平上的表达,需要使用Q-PCR的方法。
引物设计是很重要的一环,要进行Q-PCR时,获得引物的途径有很多种,其中最为方便的是使用NCBI和primer bank,首先介绍这一方法:比如你要检测某药物对小鼠细胞中IL-2的表达影响,通过Q-PCR检测,要设计引物。
①登陆NCBI,选择gene,在选框中输入 IL-2 mus(搜索小鼠IL-2基因)点击搜索获得结果如下:选择第一个基因,打开,可以获得gene的ID利用primer bank和ncbi的gene ID就可以获得前人已经设计好的引物②登陆primer bank (),按下图选择NCBI gene ID 选择物种mouse ,在for text中输入前面获得的你要检测基因的ID (NCBI的编号)点击submit 查询,获得如下结果可以看到其它人设计的好几对引物,基本上是通过实验验证的,做Q-PCR的话直接拿来用就行了,点击绿色高亮的链接可以看到其他人使用这对引物做Q-PCR获得的结果。
二、自己设计Q-PCR引物方法比如要检测CXCR3的表达,做Q-PCR的扩增目的条带在100bp 左右,如果半定量的话(通过跑胶定量)扩增产物一般400bp左右①登陆NCBI,选择gene,在选框中输入 CXCR3 mus(搜索小鼠CXCR3基因)点击搜索点击打开将网页往下拖到NCBI reference sequence 选择箭头所示mRNA 序列打开链接往网页下方拖动,可以看到CDS,点击CDS(编码蛋白区)出现如下界面,高亮部分为CDS去ATG起始密码子TAA终止密码子复制CDS区及其附近部分序列,如果要提取基因的话需要完全包括CDS区,如果只是检测的话,比如半定量的话就没必要全部包括。
打开primer 5 软件,新建一个DNA序列将复制的序列粘贴到选项框中,点击oK序列就导入了primer 5中,然后单击箭头所指primer,进入引物设计界面界面如下,点击search,使用primer自动寻找引物功能进入界面后,分别按下图选取所需要的参数,PCR product size 使用默认值即可,primer length一般在20bp左右比较合适点击OK后进入如下界面,点击OK出现如下界面,点击rating,按打分排列,打分是100分制,分值越高引物越好,根据自己试验目的,选择打分高的,产物片段400bp左右的引物对,并且Tm温度最好在60以上,有义链和反义链Tm值靠的越近越好,最好不要超过2摄氏度,下图中Tm行中蓝色字体表示有义链的Tm值,红色字体表示反义链Tm值,PCR 退火温度一般设置比Tm值低5摄氏度。
RTPCR引物设计原则和方法

RT—PCR引物设计原则与方法近刚开始做PCR,参考了很多与引物设计相关得帖子,结合自己使用primer5与oligo得体会,想谈谈自己设计引物得方法与步骤,恳请各位前辈指正、ﻫﻫRT—PCR引物设计原则与方法ﻫﻫ在NCB I上搜索到该基因,找到该基因得mRNA,在CDS选项中,找到编码区所在位置,在下面得origin 中,Copy该编码序列作为软件查询序列得候选对象。
ﻫ打开Primer Premier5,点击sequence, 出现输入序列窗口,Copy目得序列在输入框内(选择As),此窗口内,序列也可以直接翻译成蛋白。
点击Primer,进入引物窗口。
ﻫﻫ此窗口可以链接到“引物搜索”、“引物编辑”以及“搜索结果"选项,点击Search按钮,进入引物搜索框,选择“PCR pri mers”,“Pairs”,设定搜索区域与引物长度与产物长度。
在Search Parameters里面,可以设定相应参数。
一般若无特殊需要,参数选择默认即可,但产物长度可以适当变化,因为100~200bp得产物电泳跑得较散,所以可以选择300~500bp。
ﻫ点击OK,软件即开始自动搜索引物,搜索完成后,会自动跳出结果窗口,搜索结果默认按照评分(Rating)排序,点击其中任一个搜索结果,可以在“引物窗口"中,显示出该引物得综合情况,包括上游引物与下游引物得序列与位置,引物得各种信息等。
ﻫ对于引物得序列,可以简单查瞧一下,避免出现下列情况:3’端不要以A结尾,最好就是G或者C,T也可以;3'不要出现连续得3个碱基相连得情况,比如GGG或 CCC,否则容易引起错配。
此窗口中需要着重查瞧得包括:Tm应该在55~70度之间,GC%应该在45%~55%间,上游引物与下游引物得Tm 值最好不要相差太多,大概在2度以下较好。
该窗口得最下面列出了两条引物得二级结构信息,包括,发卡,二聚体,引物间交叉二聚体与错误引发位置。
引物纯化方式选择指南

引物纯化方式选择指南2012-2-16 10:24:14容导读一、DNA合成的方法和原理二、引物纯化的方法原理及其效果三、纯化方法与应用指南四、常见问题的原因分析及相应的对策一、DNA合成的方法和原理目前引物合成主要采用固相亚磷酰胺三酯法进行。
基于该方法的DNA合成仪有多种,由ABI/PE 公司生产的高通量DNA自动合成仪得到了广泛的应用。
各合成仪进行引物合成的原理基本相同,主要区别在于合成产率的高低、试剂消耗量和单个循环用时等。
生工公司采用的合成仪主要机型为全新的ABI3900高通量合成仪。
固相亚磷酰胺三酯法合成DNA片段,具有高效、快速偶联以及起始反应物比较稳定的特点。
该方法是在固相载体上完成DNA链的合成的,DNA化学合成不同于酶促的DNA合成过程从5’ →3’方向延伸,而是由3’端开始,相邻的核苷酸通过3’→ 5’磷酸二酯键连接。
具体的反应步骤如图一。
1、脱保护基 (Deblocking)用三氯乙酸 (Trichloroacetic Acid,TCA) 脱去连结在CPG (Controlled Pore Glass) 上的核苷酸的保护基团DMT (二甲氧基三苯甲基),获得游离的5'-羟基端,以供下一步缩合反应。
2、活化 (Activation)将亚磷酰胺保护的核苷酸单体与四氮唑活化剂混合并进入合成柱,形成亚磷酰胺四唑活性中间体(其3'-端已被活化,但5'-端仍受DMT保护),此中间体将与GPG上的已脱保护基的核苷酸发生缩合反应。
3、连接 (Coupling)亚磷酰胺四唑活性中间体遇到CPG上已脱保护基的核苷酸时,将与其5'-羟基发生亲合反应,缩合并脱去四唑,此时合成的寡核苷酸链向前延长一个碱基。
4、封闭 (Capping)缩合反应后,为了防止连在CPG上的未参与反应的5'-羟基在随后的循环反应中被延伸,常通过乙酰化来封闭此端羟基,一般乙酰化试剂是用乙酸酐和N-甲基咪唑等混合形成的。
RTPCR的实验原理与操作步骤

提取组织或细胞中的总RNA,以其中的mRNA作为模板,采用Oligo(dT)或随机引物利用逆转录酶反转录成cDNA。
再以cDNA为模板进行PCR 扩增,而获得目的基因或检测基因表达。
RT-PCR使测的灵敏性提高了几个数量级,使一些极为微量RNA样品分析成为可能。
该技术主要用于:分析基因的转录产物、获取目的基因、合成cDNA探针、构建RNA高效转录系统。
(一) 反转录酶的选择1. Moloney鼠白血病病毒(MMLV)反转录酶:有强的聚合酶活性,RNA酶H活性相对较弱。
最适作用温度为37℃。
2. 禽成髓细胞瘤病毒(AMV)反转录酶:有强的聚合酶活性和RNA酶H活性。
最适作用温度为42℃。
3. Thermus thermophilus、Thermus flavus等嗜热微生物的热稳定性反转录酶:在Mn2 存在下,允许高温反转录RNA,以消除RNA模板的二级结构。
4. MMLV反转录酶的RNase H-突变体:商品名为SuperScript 和SuperScriptⅡ。
此种酶较其它酶能多将更大部分的RNA转换成cDNA,这一特性允许从含二级结构的、低温反转录很困难的m模板合成较长cDNA。
(二) 合成cDNA引物的选择1. 随机六聚体引物:当特定mRNA由于含有使反转录酶终止的序列而难于拷贝其全长序列时,可采用随机六聚体引物这一不特异的引物来拷贝全长mRNA。
用此种方法时,体系中所有RNA分子全部充当了cDNA第一链模板,PCR引物在扩增过程中赋予所需要的特异性。
通常用此引物合成的cDNA中96%来源于 rRNA。
2. Oligo(dT):是一种对mRNA特异的方法。
因绝大多数真核细胞mRNA具有3’端Poly(A )尾,此引物与其配对,仅mRNA被转录。
由于Poly(A )RNA仅占总RNA 的1-4%,故此种引物合成的cDNA比随机六聚体作为引物和得到的cDNA在数量和复杂性方面均要小。
3. 特异性引物:最特异的引发方法是用含目标RNA的互补序列的寡核苷酸作为引物,若PCR反应用二种特异性引物,第一条链的合成可由与mRNA 3’端最靠近的配对引物起始。
引物设计

最近刚开始做PCR,参考了很多与引物设计相关的帖子,结合自己使用primer5和oligo的体会,想谈谈自己设计引物的方法和步骤,恳请各位前辈指正。
RT-PCR引物设计原则和方法在NCBI上搜索到该基因,找到该基因的mRNA,在CDS选项中,找到编码区所在位置,在下面的origin中,Copy该编码序列作为软件查询序列的候选对象。
打开Primer Premier5,点击File-New-DNA sequence, 出现输入序列窗口,Copy 目的序列在输入框内(选择As),此窗口内,序列也可以直接翻译成蛋白。
点击Primer,进入引物窗口。
此窗口可以链接到“引物搜索”、“引物编辑”以及“搜索结果”选项,点击Search按钮,进入引物搜索框,选择“PCR primers”,“Pairs”,设定搜索区域和引物长度和产物长度。
在Search Parameters里面,可以设定相应参数。
一般若无特殊需要,参数选择默认即可,但产物长度可以适当变化,因为100~200bp的产物电泳跑得较散,所以可以选择300~500bp。
点击OK,软件即开始自动搜索引物,搜索完成后,会自动跳出结果窗口,搜索结果默认按照评分(Rating)排序,点击其中任一个搜索结果,可以在“引物窗口”中,显示出该引物的综合情况,包括上游引物和下游引物的序列和位置,引物的各种信息等。
对于引物的序列,可以简单查看一下,避免出现下列情况:3’端不要以A结尾,最好是G或者C,T也可以;3’不要出现连续的3个碱基相连的情况,比如GGG 或CCC,否则容易引起错配。
此窗口中需要着重查看的包括:Tm应该在55~70度之间,GC%应该在45%~55%间,上游引物和下游引物的Tm值最好不要相差太多,大概在2度以下较好。
该窗口的最下面列出了两条引物的二级结构信息,包括,发卡,二聚体,引物间交叉二聚体和错误引发位置。
若按钮显示为红色,表示存在该二级结构,点击该红色按钮,即可看到相应二级结构位置图示。
PCR引物设计的一般原则

PCR引物设计的一般原则PCR(聚合酶链式反应)是一种常用的分子生物学技术,它用于扩增DNA片段。
在PCR过程中,引物扮演着重要的角色。
引物的设计直接影响PCR反应的准确性和可靠性。
本文将介绍PCR引物设计的一般原则。
引物长度引物的长度通常应在18到30个碱基对之间。
过短的引物可能产生非特异性扩增,而过长的引物可能导致扩增效率下降。
对于大多数PCR实验,推荐使用20到25个碱基对长度的引物。
碱基组成引物的碱基组成应该尽可能均衡,避免过多的GC或AT含量。
过高的GC含量可能导致引物的二聚体形成,而过高的AT含量可能导致不稳定的引物结构。
一般来说,推荐使用40-60%的GC含量。
温度引物的熔解温度(Tm值)应该在50-65°C之间。
具体的Tm值可以通过以下公式估算:Tm = 2(A + T) + 4(G + C)其中A、T、G和C分别代表引物中的腺嘌呤、胸腺嘧啶、鸟嘌呤和胞嘧啶的数量。
该公式仅供初步估算,实际引物设计时还需要考虑其他因素。
特异性引物在目标DNA序列上应具有高特异性,避免与非目标DNA序列发生扩增。
为了确保引物的特异性,可以使用引物设计工具进行检测,例如NCBI的Primer-BLAST工具。
末端特性引物的末端应尽量避免包含相同的碱基序列,特别是相同的碱基重复序列。
这样可以减少由于引物间的二聚体形成而导致的非特异性扩增。
引物浓度在PCR反应中,引物的浓度应适中。
过高的引物浓度可能导致非特异性扩增,而过低的引物浓度可能导致扩增效率下降。
一般来说,引物的浓度应在0.1-0.5 μM之间。
引物设计工具为了帮助引物设计,有许多在线工具可供选择。
例如,Primer3、Primer-BLAST、OligoAnalyzer等工具都是常用的引物设计工具,它们能够根据输入的序列和参数提供最优的引物设计方案。
PCR引物设计的一般原则可帮助实验者提高PCR扩增的成功率和准确性。
根据目标序列的特点合理设计引物,选择适当的引物长度、碱基组成和温度,确保引物的特异性和稳定性,是进行PCR实验的基本要求。
RT-PCR中引物的选择指南

91-110
57.5 60.4 517
human GAPDHBC004109
Rat GAPDHBC059110
Mouse GAPDHM32599 Rat 18S rRNAM11188 human 18S rRNAM10098 mouse 18S rRNAK013364
表一:三种引物应用特点
名称
特异性引物
Random 引物
oligo(dT)
序列
该类引物与模板序列特异性 结合。
多个任意的碱基连接而成的一套寡聚 多个 T 碱基连接而成的寡聚引
引物组,常见的是 random(6),random 物, oligo(dT)10-18,
(9),random(10),
特点
比较短小,随机组合丰富,与模板结合 针对 Poly(A)尾巴设计,与使用随机引物相
反转录酶,又称逆转录酶,是依赖 RNA 为模板的 DNA 聚合酶的统称。美国科学家 H.M.Temin 和 D.Baltimore 在 1970 年发现了反转录酶,并因此获得了 1975 年的诺贝尔生理学医学奖。目 前常用的反转酶主要有 AMV(avian myeloblastosis virus)和 M-MULV(molomey murine leukemia virus)两种反转录酶。AMV 来源于禽成髓细胞瘤病毒,最适反应温度在 42℃,具有 较强的反转录活性和 RNase-H 活性, RNase H 是一种核糖核酸内切酶,它能够特异性地水解 杂交到 DNA 链上的 RNA 磷酸二酯键,故能分解 RNA/DNA 杂交体系中的 RNA 链,从而限制了 cDNA 的合成长度。 与之对应的 M-MULV 该酶基因来源于莫洛尼氏小鼠白血病病毒,在具有较 强的反转录活性的同时,却具有较低的 RNase-H 活性,所以它可以获得较长的 cDNA 片段。目 前通过基因工程的方法降低 RNase-H 活性,使得 M-MULV 的反转录活性大大提高,提高酶的 热稳定性,因此目前 M-MULV 应用最广。
TR-PCR

逆转录-聚合酶链反应 (Reverse Transcription-Polymerase Chain Reaction,RT-PCR)的原理是:提取组织或细胞中的总RNA,以其中的mRNA作为模板,采用Oligo(dT)或随机引物利用逆转录酶反转录成cDNA。
再以cDNA为模板进行PCR 扩增,而获得目的基因或检测基因表达。
RT-PCR使RNA检测的灵敏性提高了几个数量级,使一些极为微量RNA样品分析成为可能。
该技术主要用于:分析基因的转录产物、获取目的基因、合成cDNA探针、构建RNA高效转录系统。
一、反转录酶的选择1. Money 鼠白血病病毒(MMLV)反转录酶:有强的聚合酶活性,RNA酶H活性相对较弱。
最适作用温度为37℃。
2.禽成髓细胞瘤病毒(AMV)反转录酶:有强的聚合酶活性和RNA酶H活性。
最适作用温度为42℃。
3.Thermus thermophilus、Thermus flavus等嗜热微生物的热稳定性反转录酶:在Mn2 存在下,允许高温反转录RNA,以消除RNA模板的二级结构。
4.MMLV反转录酶的RNase H-突变体:商品名为SuperScript 和SuperScriptⅡ。
此种酶较其它酶能多将更大部分的RNA转换成cDNA,这一特性允许从含二级结构的、低温反转录很困难的mRNA模板合成较长cDNA。
二、合成cDNA引物的选择1.随机六聚体引物:当特定mRNA由于含有使反转录酶终止的序列而难于拷贝其全长序列时,可采用随机六聚体引物这一不特异的引物来拷贝全长mRNA。
用此种方法时,体系中所有RNA分子全部充当了cDNA第一链模板,PCR引物在扩增过程中赋予所需要的特异性。
通常用此引物合成的cDNA中96%来源于rRNA。
2. Oligo(dT):是一种对mRNA特异的方法。
因绝大多数真核细胞mRNA具有3’端Poly(A )尾,此引物与其配对,仅mRNA可被转录。
由于Poly(A )RNA 仅占总RNA的1-4%,故此种引物合成的cDNA比随机六聚体作为引物和得到的cDNA在数量和复杂性方面均要小。
rt pcr的常用方法

rt pcr的常用方法rt-PCR( real-time PCR )是一种用于检测和定量新型冠状病毒(SARS-CoV-2)核酸的技术。
下面是 rt-PCR 的常用方法及其拓展:1. 设计引物:rt-PCR 的第一步是设计引物。
引物是探针分子,用于结合目标核酸,并用于在 rt-PCR 中扩增目标核酸。
引物的质量和准确性对 rt-PCR 的结果至关重要。
通常,引物由两个互补的短链组成,分别位于引物的两端。
引物的长度和结构可以影响 rt-PCR 的反应时间和结果。
2. 提取核酸:从患者或样本中提取足够的核酸是 rt-PCR 的第一步。
通常,需要使用核酸提取试剂盒,将患者或样本中的核酸提取出来。
核酸提取的准确性和效率对 rt-PCR 的结果也有很大的影响。
3. 混合核酸:将提取的核酸与引物一起混合,并使用 rt-PCR 试剂盒进行扩增。
扩增过程中,引物将与目标核酸结合,并生成一个反应物。
这个反应物可以在rt-PCR 中测量,并计算出病毒的数量。
4. 控制温度和湿度:rt-PCR 的反应需要一定的温度和湿度条件。
通常,需要使用专门的 rt-PCR 设备,并将设备放置在适当的温度和湿度环境中。
这有助于提高 rt-PCR 的准确性和效率。
5. 数据分析:一旦扩增了目标核酸,就需要对数据进行分析。
通常,使用rt-PCR 数据分析软件对数据进行处理和可视化。
这可以帮助确定扩增是否成功,并确定病毒的数量。
rt-PCR 是一种快速、准确和灵敏的方法来检测和定量新型冠状病毒。
通过设计合适的引物,提取足够的核酸,并将核酸与引物一起混合,并控制温度和湿度,可以提高 rt-PCR 的准确性和效率。
逆转录-聚合酶链(RT-PCR)实验的具体步骤及方法

逆转录-聚合酶链(RT-PCR)实验的具体步骤及方法提取组织或细胞中的总RNA,以其中的mRNA作为模板,采用Oligo(dT)或随机引物利用逆转录酶反转录成cDNA,再以cDNA为模板进行PCR扩增,从而获得目的基因或检测基因表达。
一、总RNA的提取见总RNA的提取相关内容。
二、cDNA第一链的合成目前试剂公司有多种cDNA第一链试剂盒出售,其原理基本相同,但操作步骤不一。
现以GIBICOL公司提供的SuperScriptTM Preamplification System for First Strand cDNA Synthesis 试剂盒为例。
1.在0.5 ml微量离心管中,加入总RNA 1-5 ug,补充适量的DEPC H2O使总体积达11 ul。
在管中加10 uM Oligo(dT)12-18 1 ul,轻轻混匀、离心;2.70℃加热10min,立即将微量离心管插入冰浴中至少1min;3.取0.5 ml PCR管,依次加入下列试剂:第一链cDNA 2 ul;上游引物(10 pM)2 ul;下游引物(10 pM)2 ul;dNTP(2mM) 4 ul;10x PCR buffer 5 ul;Taq 酶(2 u/ul)1 ul。
轻轻混匀,离心。
42℃孵育2-5 min;4.加入SuperscriptⅡ1 ul ,在42℃水浴中孵育50 min;5.于70℃加热15 min以终止反应;6.将管插入冰中,加入RNase H 1 μl ,37℃孵育20 min,降解残留的RNA。
-20℃保存备用。
三、PCR1.取0.5 ml PCR管,依次加入下列试剂:第一链cDNA 2 ul;上游引物(10 pM)2 ul;下游引物(10 pM)2 ul;dNTP(2 mM) 4 ul;10x PCR buffer 5 ul;Taq 酶(2 u/ul)1 ul;2.加入适量的ddH2O,使总体积达50 ul。
RT-PCR引物的选择

RT-PCR引物的选择
点击次数:977 作者:佚名发表于:2009-05-29 00:00转载请注明来自丁香园
来源:互联网
随机引物
适用于长的或具有发卡结构的RNA.适用于rRNA、mRNA、tRNA 等所有RNA的反转录反应。
主要用于单一模板的RT -PCR反应。
Oligo dT
适用于具有PolyA尾巴的RNA.(原核生物的RNA、真核生物的Oligo dT rRNA和tRNA不具有PolyA尾巴。
)由于Oligo dT要结合到PolyA 尾巴上,所以对RNA样品的质量要求较高,即使有少量降解也会使全长cDNA合成量大大减少。
基因特异性引物
与模板序列互补的引物,适用于目的序列已知的情况。
天为时性引物代公司的SuperScript One-Step System特别适合于与基因特异性引物连用。
PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。
4. 操作多份样品时,制备反应混合液,先将dNTP、缓冲液、引物和酶混合好,然后分装,这样即可以减少操作,避免污染,又可以增加反应的精确度;
5. 最后加入反应模板,加入后盖紧反应管。
BIORAD荧光定量PCR原理和方法介绍

分子信标(Molecular Beacons)
3’ 5’ 5’ 3’
Primers
d.NTPs Taq酶
Molecular Beacon
反应组分
R
Q
Taq
R Q
变
性
l
Q R
5’
3’
退 火
Q
R
Taq
5’ Molecular Beacon 3’
1. 链替换
Taq
R Q 5’ 5’ 3’
2. 聚合完成
Taq
ID
相对杂交探针方法,插入染料法相对较便宜 不需要特别设计探针 SYBR Green I, 最常用的插入染料 SYBR Green 染料插入双链DNA,检测灵敏度 提高1000倍。 和杂交探针方法相比,插入染料法特异性低 。 不能做多重PCR。
插入染料能够和引物二聚体和非特异性扩增产物结合
提
纲
• 荧光定量PCR的原理和应用 • 荧光定量PCR实验的设计和优化 • 引物和探针的设计 • 仪器软件的操作和仪器的维护 • 常见问题及解决方案
荧光定量 PCR的原理
PCR 反应过程
3’ 5’ 5’ 3’
Primers
d.NTPs
Taq 酶
反应组分
变
性
3’
5’
5’
3’
退
火
3’
Taq
Taq
R
5’
3’
解
Taq
5’
5’ 3’
3. 聚
合
l Taq
5’
R
5’ 3’
4. 检测荧光
产生很强的荧光信号 容易设计和合成 可以做多重检测 能够进行SNP (Single Nucleotide Polymorphism) 检测
引物选择——精选推荐

一、引物的合成和纯化1. 引物是如何合成的?目前引物合成基本采用固相亚磷酰胺y三酯法。
该方法具有高效、快速的偶联以及起始反应物比较稳定的特点。
主要是将DNA固定在固相载体上完成DNA链的合成的,合成的方向是由待合成引物的3'端向5'端合成的,相邻的核苷酸通过3'→5'磷酸二酯键连接。
固相亚磷酰胺三酯法合成引物的具体步骤如下:1) 将预先连接在固相载体CPG上的活性基团被保护的核苷酸与三氯乙酸反应,脱去其5'-羟基的保护基团DMT,获得游离的5'-羟基。
2) 合成DNA的原料,亚磷酰胺保护核苷酸单体,与活化剂四氮唑混合,得到核苷亚磷酸活化中间体,它的3'端被活化,5'-羟基仍然被DMT保护,与溶液中游离的5'-羟基发生缩合反应。
3) 带帽(capping)反应,缩合反应中可能有极少数5'-羟基没有参加反应(少于2%),用乙酸酐和1-甲基咪唑终止其后继续发生反应,这种短片段可以在纯化时分离掉。
4) 在氧化剂碘的作用下,亚磷酰形式转变为更稳定的磷酸三酯。
经过以上四个步骤,一个脱氧核苷酸被连接到固相载体的核苷酸上。
再以三氯乙酸脱去它的5'-羟基上的保护基团DMT,重复以上步骤,直到所有要求合成的碱基被接上去。
合成过程中可以观察TCA处理阶段的颜色判定合成效率。
通过氨水高温处理,连接在CPG上的引物被切下来,通过OPC、PAGE等手段纯化引物,成品引物用C18浓缩,脱盐,沉淀。
沉淀后的引物用水悬浮,测定OD260定量,根据定单要求分装。
2. DNA合成粗产物中含有什么杂质?主要是合成反应过程中产生的失败片段以及脱保护基团时产生的铵盐。
3. 引物纯化方式有哪些?引物常用的纯化方式C18脱盐、OPC纯化、PAGE纯化、HPLC纯化。
纯化方式详细说明C18柱脱盐又称为简易反相柱,对DNA有特异性吸附,可被有机溶液洗脱,但不会被水洗脱,因此能有效地去除盐分,但不能有效去除比目的片段短的小片段。
RT-PCR引物设计及注意事项

D
1
一、步骤 1.查找目的基因序列
D
2
D
3
D
4
D
5
D
6
D
7
D
8
目的序列的选择
D
9
TNFalpha用全称
D
10
小鼠的TNFalpha序列
D
11
大鼠的TNFalpha序列
D
12
一、步骤 2.在线设计引物
D
13
D
14
D
15
D
16
D
17
rt-pcr引物设计及注意事项 (定量100-300,定性300-500bp) 飞跃科技 分享于 2020-10-14 12:40:38.0 rt-pcr引物设计及注意事项 文档格式: .ppt 文档页数: 39页 文档大小: 5.53m 文档热度: 文档分类: 高等教育 -- 教育学 文档标签: rt-pcr引物设计及注意事项 系统标签: pcr tnfalpha 事项 注意 gdna 序列D来自18D19
D
20
D
21
D
22
D
23
一、步骤 3.在线验证引物
D
24
D
25
D
26
D
27
D
28
D
29
D
30
D
31
二、注意事项
1. 找对靶序列(要cDNA,不要gDNA) 2. 引物的位置(5’调控区、阅读框、3‘调控区) 3. 目的片段的长度(定量100-300,定性300-
500bp)
4. 注意剪接异构体(P63为例)
D
32
RT-PCR引物设计原则和方法

RT—PCR引物设计原则和方法近刚开始做PCR,参考了很多与引物设计相关的帖子,结合自己使用primer5和oligo的体会,想谈谈自己设计引物的方法和步骤,恳请各位前辈指正。
RT-PCR引物设计原则和方法在NCBI上搜索到该基因,找到该基因的mRNA,在CDS选项中,找到编码区所在位置,在下面的origin中,Copy该编码序列作为软件查询序列的候选对象。
打开Primer Premier5,点击File-New—DNA sequence,出现输入序列窗口,Copy目的序列在输入框内(选择As),此窗口内,序列也可以直接翻译成蛋白。
点击Primer,进入引物窗口。
此窗口可以链接到“引物搜索”、“引物编辑”以及“搜索结果”选项,点击Search按钮,进入引物搜索框,选择“PCR primers”,“Pairs”,设定搜索区域和引物长度和产物长度.在Search Parameters里面,可以设定相应参数。
一般若无特殊需要,参数选择默认即可,但产物长度可以适当变化,因为100~200bp的产物电泳跑得较散,所以可以选择300~500bp。
点击OK,软件即开始自动搜索引物,搜索完成后,会自动跳出结果窗口,搜索结果默认按照评分(Rating)排序,点击其中任一个搜索结果,可以在“引物窗口”中,显示出该引物的综合情况,包括上游引物和下游引物的序列和位置,引物的各种信息等。
对于引物的序列,可以简单查看一下,避免出现下列情况:3’端不要以A结尾,最好是G或者C,T 也可以;3’不要出现连续的3个碱基相连的情况,比如GGG或 CCC,否则容易引起错配。
此窗口中需要着重查看的包括:Tm应该在55~70度之间,GC%应该在45%~55%间,上游引物和下游引物的Tm值最好不要相差太多,大概在2度以下较好。
该窗口的最下面列出了两条引物的二级结构信息,包括,发卡,二聚体,引物间交叉二聚体和错误引发位置.若按钮显示为红色,表示存在该二级结构,点击该红色按钮,即可看到相应二级结构位置图示。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1.2 上游或者下游引物或者两者,引物本身在两个不同的外显子上 注意引物在两个外显子上分布,在“内侧”要少些,少于 8bp 左右。这样的引物就不能从基因
RT-PCR 中引物的选择指南
一.用于 RT-PCR 的引物 1.特异性引物(跨内含子设计引物) 2.随机引物(Random primer) 3. oligo(dT)引物
二.反转录酶 M-MULV 和 AMV 三.常用内参引物的参考序列
一.用于 RT-PCR 的引物 一般做 RT-PCR 进行第一链 cDNA 合成的起始可以使用特异性引物、随机引物(Random
91-110
57.5 60.4 517
human GAPDHBC004109
Rat GAPDHBC059110
Mouse GAPDHM32599 Rat 18S rRNAM11188 human 18S rRNAM10098 mouse 18S rRNAK013364
构的 CDNA 的合成。4.可用于
如果目标序列位于基因的 5’端,那么推荐
步法检测目标基因,一般建议
Northern/Southern 杂交、原位杂交和 采用随机六聚体引物或随机六聚体与寡
添加特异性引物。一般第一链
克隆、噬菌斑杂交的标记探针。 5.mRNA (dT)n 引物的混合物用于逆转录步骤。一般
反应的敏感度和特异性。
解也会是全长 cDNA 合成量大大减少。
适用实验
1.RT-PCR 实验中,合成带有或不带有
poly(A)的 cDNA。2.长片断的 mRNAS 5’
的 RT-PCR。3.模板 RNA 具有较多二级结 适合于模板是 Poly(A)+RNA 的 RT-PCR 实验,
但是如果以 RNA 为模板进行一
因为体系中所有 RNA 分子全部充当了 cDNA 第一链模板,所以随机引物法是三种方法中特 异性最低的,通常用此引物合成的 cDNA 中 96%来源于 rRNA。所以随机引物介导制备的 cDNA 产物复杂程度高,从而降低了后续 PCR 反应的敏感度和特异性。为了获得最长的 cDNA,需要 按经验确定每个 RNA 样品中引物与 RNA 的比例。随机引物的起始浓度范围为 50 到 250ng 每 20μl 反应体系。 2.1 随机引物优先应用范围: ü RT-PCR 实验中,合成带有或不带有 poly(A)的 cDNA。 ü 长片断的 mRNA5’—区域的 RT –PCR。 ü 模板 RNA 具有较多二级结构的 CDNA 的合成。 ü 可用于 Northern/Southern 杂交、原位杂交和克隆/噬菌斑杂交的标记探针。mRNA 差异显示。
的 CDNA,42-45℃范围具有最佳活性。55℃仍有活性,因
修饰后
仍有活性,因此可以适合具有大量二级结构的模板,提高反
此可以适合具有大量二级结构的模板,提高反应的褪火温
应的褪火温度。
度。
适用实
基因比较复杂、有二级结构或 GC 含量较高的 RT 反应,如果
RT-PCR 反应中如果使用来做检测,价格比 AMV 稍便宜。
primer)、oligo(dT)三种不同的方法,各种方法的相对特异性影响了所合成 cDNA 的量、种类及 效果(如表一)。 1.特异性引物(跨内含子设计引物)
对于 RT-PCR,引物设计质量影响 RT 的结果,而且不同引物退火温度本来就不相同,所以 第一链 cDNA 合成不推荐使用。但是如果以 RNA 为模板进行一步法检测基因表达情况,一般建 议添加特异性引物,这样设计的主要目的是消除 RT-PCR 时基因组 DNA 污染对结果的影响。跨 内含子设计引物有两种做法(如下图 A 和 B): 1.1 在两个不同的外显子上设计引物,上下引物分别位于不同的外显子上
52.6
BC002409
F1379 agcgagcatcccccaaagtt 1379-1398 57.3
54
285
R1663 gggcacgaaggctcatcatt 1663-1644 56.3
Rat actin betaNM_031144
F18 R224
cacccgcgagtacaaccttc cccatacccaccatcacacc
表二:RT-PCR 中常见的反转录酶 M-MULV、AMV 的应用
名称
M-MULV
AMV
莫洛尼氏小鼠白血病病毒
来源
禽成髓细胞瘤病毒 (avian myeloblastosis virus)
(molomey murine leukemia virus)
特点
DNA-RNA 依 赖 的 聚 合 酶 活
DNA-RNA 依赖的聚合酶活性。DNA-RNA 解旋活性,RNase-H 活
组 DNA 扩增,从而消除基因组 DNA 污染的影响(这要求目的基因具有较多的外显子,从而有较 多的选择,引物本身质量可能不好。
2.随机引物(Random primer) 在两步法 RT-PCR 中,mRNA 必须先转录为 cDNA。在这种情况下,采用不依赖序列的引
物,例如随机引物和 知的,可以消除 位点多,在 RT 种是含有二级结构模板 比,cDNA 的数量和复杂度要少得多,但同时,
RT-PCR 时基因组 DNA 污染对结 的最佳组合,但同时导致制备的 cDNA 对于比较长的 mRNA,他经常不能延伸到 5’
果的影响。
产物复杂程度高,从而降低了后续 PCR 端。但对 RNA 样品要求较高,即使有少量降
验
是用来扩增目的片段,可选用 AMV。
三.常用内参引物的参考序列:
内参基因名称 基因库序列号
引物名称 序列
引物位置
引物 Tm
最佳退火 扩增长 温度 ℃ 度
Human actin beta
F305 Ctgggacgacatggagaaaa 305-324 52.3
59.4 564
R868 aaggaaggctggaagagtgc 868-849
随机引物,一般指的是多个任意的碱基连接而成的一套寡聚引物组,常见的是 random(6),random (9),random(10),如果 6 个碱基的随机序列,则是 5’-d(NNNNNN)-3’,共有 4 6 条引物。
原核生物由于没有 poly(A),因此在进行总 mRNA 的 RT 时要用到随机引物,由于其比较短 小,且随机组合丰富,所以在 mRNA 上的结合也很多,产生短的,部分长度的 cDNA,因此也 可以进行有效的 RT。这种方法经常用于获取 5’末端序列及从带有二级结构区域或带有逆转录酶 不能复制的终止位点的 RNA 模板获得 cDNA。因此一般适用于长的或具有发卡结构的 RNA;适 用于 rRNA、mRNA、tRNA 等所有单一模板 RNA 的反转录反应。在 RT-PCR 用随机引物制备 CDNA 时,要去除总 RNA 中的 tRNA 、rRNA,只保留 mRNA。
性。
RNase-H 活性较低,
性较强,有较强的反转录活性,最适 45-50℃.在 45-60℃很
有较强的反转录活性,42 范围具有最佳活性。制备 CDNA
宽的范围具有活性。制备 CDNA 最长可 13Kb。
最长可 9Kb。
点突变后 RNase-H 活性完全消失,因此可以扩增长达 13Kb
可以扩增长达 13Kb 的 CDNA,42-45℃范围具有最佳活性。55℃
CDNA 合成不推荐使用。
差异显示。6.病毒鉴定,因为病毒序列 采用 60μM 随机六聚体和 2.5μM 寡(dT)n。
大多数是未知的,在 RNA 病毒的研究中
一般用随机引物进行扩增并进行鉴定。
纯化方式 HAP, ULTRAPAGE, HPLC
HAP
HAP
二. 反转录酶 M-MULV 和 AMV
通常 M-MULV 用于普通的 RT 反应,AMV 用于基因比较复杂、有二级结构或 GC 含量较高 的 RT 反应,如果 cDNA 要用于克隆表达,建议使用 M-MULV 或 AMV。
反转录酶,又称逆转录酶,是依赖 RNA 为模板的 DNA 聚合酶的统称。美国科学家 H.M.Temin 和 D.Baltimore 在 1970 年发现了反转录酶,并因此获得了 1975 年的诺贝尔生理学医学奖。目 前常用的反转酶主要有 AMV(avian myeloblastosis virus)和 M-MULV(molomey murine leukemia virus)两种反转录酶。AMV 来源于禽成髓细胞瘤病毒,最适反应温度在 42℃,具有 较强的反转录活性和 RNase-H 活性, RNase H 是一种核糖核酸内切酶,它能够特异性地水解 杂交到 DNA 链上的 RNA 磷酸二酯键,故能分解 RNA/DNA 杂交体系中的 RNA 链,从而限制了 cDNA 的合成长度。 与之对应的 M-MULV 该酶基因来源于莫洛尼氏小鼠白血病病毒,在具有较 强的反转录活性的同时,却具有较低的 RNase-H 活性,所以它可以获得较长的 cDNA 片段。目 前通过基因工程的方法降低 RNase-H 活性,使得 M-MULV 的反转录活性大大提高,提高酶的 热稳定性,因此目前 M-MULV 应用最广。
表一:三种引物应用特点
名称
特异性引物
Random 引物
oligo(dT)
序列
该类引物与模板序列特异性 结合。
多个任意的碱基连接而成的一套寡聚 多个 T 碱基连接而成的寡聚引
引物组,常见的是 random(6),random 物, oligo(dT)10-18,
(9),random(10),
特点
比较短小,随机组合丰富,与模板结合 针对 Poly(A)尾巴设计,与使用随机引物相