第四章分子模拟任彦亮剖析
分子模拟的原理与实践

分子模拟的原理与实践分子模拟是指通过模拟分子之间的相互作用以及其运动状态,探究物质的性质和行为。
它是一种全面深入的研究物质结构与性质的手段,已经广泛应用于化学、生物、材料科学等诸多领域。
本文就与大家分享一下分子模拟的原理、方法及其在科学研究中的实践。
一、基本原理分子模拟的基本原理是建立分子在不同环境的各种状态下的量子力学或分子力学模型,依据这些模型来计算物质的结构、动力学和力学性质,从而得到物质性质的定量预测。
分子模型可以从两个方面考虑。
一是通过量子力学来描述分子的电子结构和原子核的运动。
二是通过分子力学来表示分子内部和分子间力的作用以及分子的构象状态和运动。
分子动力学模拟是分子模拟的一种重要方法。
它是基于牛顿力学原理和统计力学原理,模拟分子的运动和实验条件下的动力学行为,来预测它们的结构和性质。
二、模拟方法(一)分子动力学模拟分子动力学模拟是分子模拟中最为常用的方法之一。
它可以通过计算机模拟分子内部的各种物理状态,如位置、速度和位能等,在一定时间内计算出分子内部和分子间的相对位置、角度和速度等信息。
分子动力学模拟需要考虑各种参数,如能量、时间、温度等。
首先需要设置分子初始状态的坐标和速度,然后计算相互作用力和分子运动等参数,最后输出分子的位置和速度等相关信息。
(二)量子力学模拟材料和生物体系具有很强的量子效应,尤其是涉及到电子云的计算,需要使用量子力学方法进行模拟。
量子化学模拟一般使用哈密顿算符来表示能量。
通过求解薛定谔方程来计算体系的波函数,进而计算体系的电子密度和各种分子性质,如键长、键角等。
(三)平衡分子动力学模拟平衡分子动力学模拟是指使用一定温度下的分子动力学方法,模拟出物质在其中的行为和物态,从而使分子和材料结构达到动态平衡状态。
平衡分子动力学模拟可以提供有关热力学性质(如自由能、盐度等)和相对稳定性(比如液体晶体形态等)的信息。
它也可以为材料科学研究提供重要的参考依据。
三、实践案例分子模拟已经被广泛应用于生物、材料科学、纳米科技、药物研究及环境科学等领域。
分子动力学模拟及应用简介

・ 1 0 1 ・
பைடு நூலகம்
分子动 力学模 拟及应用简介
关荣华 周 慧
( 华北电力大学 数理 系, 河北 保 定 0 7 1 0 0 3 1
摘 要: 分子动力 学模拟是一种计算机模拟 实验方 法, 通常利用计算机软件 , 根 据牛顿力 学的基本原理 , 以分子的运动为主要 的模拟 对象 , 研 究体 系中所有粒子的运动状态随时 间的演 变, 分子动力学模拟不仅 可以得到 分子的运动轨迹 , 同时还 可以观 察到原子运动 的微 观 细节。在 统计 力学系综 下, 通过对相 空间取 系综平均 , 进 而获得体 系相应的物理性质 和化 学性质 , 分子动力学模拟是对理论计算和实验 方法的有 力补 充。 关键词 : 牛顿运动方程 ; 能量最小化 ; 能量平衡
的是‘ ' V e r l e t ’ 算 法 + △ r Aw A t  ̄ a ( t ) / 2 ; a ( t + A +At y m ; V ( t + Ao = V 的空间折叠方式决定了蛋 白质的特异性, 蛋白质结构基础决定了蛋白质 A t 【 a +A日 y 2 , 当给出体系在 t = O 时刻的初始速度, 初始坐标刑 用以 的生物学基础。 目 前P D B ( P r o t e i n D a t a B a n k ] 库已经日 月 确了大量蛋白的结 上公式积分就 可以得到任意时亥 J I 体系的坐标, 速度进 而得到体系的运动 晶结构, 这为我们利用分子动力学的方法研究蛋白的空间结构 、构象变 化、 相互作用以及运动趋势提够了结构基础。 为什么要采用分子动力学模 l 2分子动力学模拟的基本过程。分子动力学模拟的基本过程就是 拟的方法研究蛋白质的空间结构、 构象变化以及运动趋势等? 目前, 蛋白 体系能量最/ J 吡 和能量平衡的过程。能量最小化的过程实际上就是求势 质在行使其生物学功能的过程是—1 \ j 奎 续的动态过程, 这是传统的电生 能函数极/ J 值 的过 昆 利用力场函数计算分子内各种可能结构的势能艘 理实验无法实现的, 同时分子动力学模拟的方法可以在分子原子水平来 索体系的势能最低点。 能量最小化是对所有模拟体系探讨的第一步, 即用 察蛋白氨基酸之间的相互作用, 蛋白的构象变化 以及运动趋势等。 系统的方法改变体系的原子坐 通过分子内部运动的势能下降的办法, 我们以钙激活大电导钾通道( B K c h a n n e 1 ) J  ̄ 例珠 介绍分子动力学模 使分子结构逼近稳定结构翊 免不合理 的结构在模拟中出现锗误 。 拟在蛋白模拟方面的具体应用。 首先从 P D B库中获取 B K通道的关闭态 只有当体系完成能量最小化后才能进行真正的分子动力学模拟, 即 的晶体结构( P D B : 3 N A F ) , 因为 3 N A F中的部分氨基酸缺失, 首先利用 同源 能量平衡过程, 能量最小化的过程是求势能 函数极小值的过程, 能量平衡 模建的方法, 构建了完整的蛋白 结构。 接下来, 在 皖整的晶体结构的基础上 的过程实际上就是解这个多粒子体系的牛顿运动方程来描绘体系中所 构建物理模型, 以模拟蛋 白在生物体内的真实生理环境具体步骤为: 对晶 有; } 壶 子 随时间演化的规律的过程。这时就可以根据粒子的位置和动量计 体结构加水盒子模 拟生物体内的液体环境; 在加水体系的基础上添加一 算倒 可 可 则物] 翌 量的j 急 定值。 在, 动 力 莫 拟分析中通 匍 百 过{ 十 算 定物质的量的 K 和C L - 来中和体系内多余的电荷’ 以模拟生物体内中性 就完成了物理模型的构建。 在进行分子动力学模 粒子运动的方均根偏 ̄  ̄( r o o t m e a n s q u a r e d e v i a t i o I 1 , R Ms D l 用来衡量体系 环境。经以上两步骤后, 相对于初始时刻是否能达到平衡,同时也可以反应原子偏离 比对位置的 拟之前, 我们需要构建用来执行动力学模拟的配置文件, 配置文件中包含 程度。R M S D值越大原 子运动的空间范围越大原 子的空间位阻也就越 了一系列的配置参数廊置文件的作用是设定 N A MD分子动力学模拟的 小原子 在时间 N . 内R M S D的求算公式: 过程中的各种参 比如输出文件的频率 、 体系的温度、 计算时长、 P D B文
第四章习题课徐红亮2

2021/6/30
3
➢ 变分原理和线性变分法解H2+
1. 变分原理:
E
*
H d
*d
E0
E0:体系的基态能量 ψ:试探波函数,选取一组满足边界条件且 线性无关的品优函数的线性组合来构造变分 函数。
2021/6/30
5
example
通过变分法计算得到的微观体系的能量总是: (A) 等于真实体系基态能量 (B) 大于真实体系基态能量 (C) 不小于真实体系基态能量 (D) 小于真实体系基态能量
2021/6/30
41
双原子分子的性质
1. 键级:描述原子间的成键程度或化学 键的强弱表示分子键能的大小。
键级 n成键 n反键 2
2021/6/30
42
2. 磁性:
有未成对电子的分子在磁场中呈顺磁性; 无未成对电子分子在磁场中呈反磁性(抗磁 、逆磁)
2021/6/30
43
p137-13
指出O2、O2+和O2-的键长、键能的大小的 顺序,有无磁性,并说明理由。
Saa a2d , Sbb b2d
Sab abd Sba
E
c12 H aa c12 S aa
2c1c2Hab 2c1c2Sab
c22Hbb c22Sbb
Haa2和021/H6/30bb被称为库伦积分,Hab为交换积分,Sab为重叠积分。 9
2) 对ci求导得到久期方程和久期行列式 c1(H aa ESaa ) c2 (H ab ESab ) 0 c1(H ab ESab ) c2 (Hbb ESbb ) 0
多,体系越稳定,这就要求原子轨道重叠时沿 一定的方向,以达到最大重叠。
2021/6/30
04郭江波分子生物学讲义-第四章
错义抑制
错义突变
AUG AGA UAA AUG
错义突变
GGA UAA AUG AGA UAA
UCU
CCU 抑制突变
UCU
Arg
Gly
Gly
图 14-18 反密码子发生突变可抑制错义突变
异亮氨酸(Ile)和缬氨酸 (Val)
异亮氨酸--tRNA合成酶对异亮氨酸的亲和力比缬
氨酸大225倍,体内缬氨酸浓度比异亮氨酸高5
nucleotide present in the third codon
position.
2、通用性与特殊性
• 蛋白质生物合成的整套密码,从原核生物到 人类都通用。 • 已发现少数例外,如动物细胞的线粒体、植 物细胞的叶绿体。
3、 连续性
• 三个核苷酸编码一个氨基酸。 • 三联子密码是非重叠(non-overlapping)和连
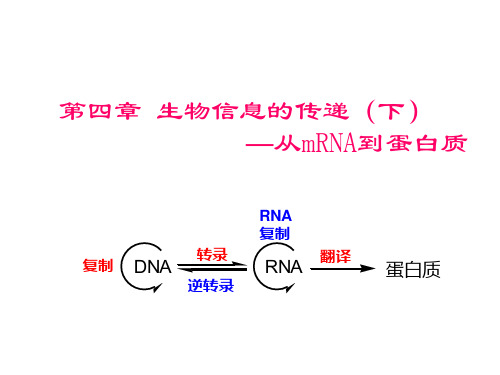
第四章 生物信息的传递(下) —从mRNA到蛋白质
RNA 复制 复制
DNA
转录 逆转录
RNA
翻译
蛋白质
• 蛋白质是生物信息通路上的终产物,一个 活细胞在任何发育阶段都需要数千种不同 的蛋白质。
• 活细胞内时刻进行着各种蛋白质的合成、 修饰、运转和降解反应。 • 蛋白质由20中氨基酸组成。
翻译:指将mRNA链上的核甘酸从一个特定的起始位
• Arg, Ser, Leu 6 codons • Gly, Thr, Ala, Val, Pro 4 codons • Met, Trp 1 codon
Third base degeneracy
describes the lesser
e
点开始,按每三个核苷酸代表一个氨基酸的原则, 依次合成一条多肽链的过程。 蛋白质合成的场所是 蛋白质合成的模板是 核糖体 mRNA tRNA
分子模拟技术的原理及其应用

分子模拟技术的原理及其应用分子模拟是一种重要的计算化学方法,其原理是根据分子的结构和运动特征,利用计算机模拟和计算手段,分析和预测分子间的相互作用和反应过程。
分子模拟技术已经被广泛应用于化学、材料、生物等领域,为科学研究和工业生产提供了有力的支持。
本文将探讨分子模拟的原理及其应用。
一、分子模拟的原理分子模拟技术基于分子动力学模型,将体系中的粒子(原子、分子)看作刚性球体,通过分析其运动状态,预测体系的稳定性、反应性和物理化学性质等。
分子模拟主要包括两种模型:分子动力学模型和蒙特卡罗模型。
其中,分子动力学模型主要用于研究物质的运动和热力学性质,而蒙特卡罗模型则更适用于研究物质的结构和统计学性质。
分子动力学模型中,分子被看作是由原子组成的粒子,原子之间存在弱相互作用力——范德华力和强相互作用力——共价键,这些力使得分子具有各种形态和运动状态。
这些力场可以通过牛顿运动定律的微分方程来描述。
利用分子动力学模型,可以模拟分子在外界刺激下的运动和相互作用,预测分子间的各种物理化学性质,如结构、构象、吸附、扩散等。
蒙特卡罗模型则采用随机模拟原理,将分子的运动看作是自由体运动和碰撞运动,结合分子间的相互作用力,利用随机抽样、概率统计等方法对分子的运动轨迹进行模拟和预测。
这种方法主要用于研究物质的结构和统计学性质,如物质的相变、磁性、化学活性等。
二、分子模拟的应用分子模拟方法在化学、材料、生物等领域中有很多应用,可以模拟和预测物质的结构、性质和活性,为实验研究和工业生产提供了有力的支持。
1. 化学领域分子模拟方法可以用于研究化学反应和化学平衡,以及物质间的相互作用。
通过对反应物分子结构和物理化学性质的预测,可以优化或设计更有效的反应条件和催化剂,提高反应的产率和选择性。
例如,分子动力学模拟可以用于研究液相化学反应中的反应机制和动力学,而蒙特卡罗模拟则可以用于研究气相反应。
2. 材料领域分子模拟技术在材料科学中也得到了广泛应用。
分子动力学

蛙跳(Leap-frog)算法
1. 首先利用当前时刻的加速度,计算半个时间步长后的速度:
1 1 v i (t t) v i (t - t) a i (t) t 2 2 2. 计算下一步长时刻的位置:
1 ri (t t) ri (t) v i (t t) t 2
COMPASS(Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies)
由美国Accelry公司设计的一个专门模拟金属氧化物固体材料的力场。
极化力场(Polarizable force fields)
U U nb U b U U U U el
分子中的各种运动
图1:乙醇分子
力场作用项的一般形式
键伸缩势能项(Bond stretching)
Harmonic函数
2 Ub 1 k ( l l ) 0 2 b i
kb:键伸缩的弹力系数 , l0 :原子间的参考键长, l :原子间的瞬间实际键长 Morse函数
Umorse l De ((1 exp( l l0 ))2 1)
De:键的解离能 β:表示势阱在参考位置平坦程度的参数 l0 :参考键长(其他化学键的力常数均为零时,该化学键在最低势能位置的键长)
Morse势函数与谐振子函数的比较
力场作用项的一般形式
键角弯曲项的一般形式为键角的简谐振荡,即:
U el
i, j
qi q j Drij
一个力场通常包括三个部分: 参数有两种来源: (1)原子类型 (1)实验观测的数据
(2)势函数
《分子模拟方法》课件

加速研发进程
分子模拟可以大大缩短药 物研发、材料合成等领域 的实验周期,降低研发成 本。
揭示微观机制
通过模拟,可以揭示分子 间的相互作用机制和反应 过程,有助于深入理解物 质的性质和行为。
分子模拟的发展历程
经典力学模拟
基于牛顿力学,适用于 较大分子体系,但精度
较低。
量子力学模拟
适用于小分子体系,精 度高,但计算量大,需
详细描述
利用分子模拟方法,模拟小分子药物与生物大分子(如蛋白质、核酸等)的相 互作用过程,探究药物的作用机制和药效,为新药研发提供理论支持。
高分子材料的模拟研究
总结词
研究高分子材料的结构和性能,优化 材料的设计和制备。
详细描述
通过模拟高分子材料的结构和性能, 探究高分子材料的物理和化学性质, 优化材料的设计和制备过程,为新材 料的研发提供理论指导。
分子动力学方法需要较高的计算资源和 精度,但可以获得较为准确的结果,因 此在计算化学、生物学、材料科学等领
域得到广泛应用。
介观模拟的原理
介观模拟是一种介于微观和宏观之间的模拟方 法,通过模拟一定数量的粒子的相互作用和演 化来研究介观尺度的结构和性质。
介观模拟方法通常采用格子波尔兹曼方法、粒 子流体动力学等方法,适用于模拟流体、表面 、界面等介观尺度的问题。
分子模拟基于量子力学、经典力 学、蒙特卡洛等理论,通过建立 数学模型来描述分子间的相互作
用和运动。
分子模拟可以用于药物研发、材 料科学、环境科学等领域,为实 验研究和工业应用提供重要支持
。
分子模拟的重要性
01
02
03
预测分子性质
通过模拟,可以预测分子 的性质,如稳定性、溶解 度、光谱等,为实验设计 和优化提供指导。
分子动力学模拟纳米镍单晶的表面效应[1]
![分子动力学模拟纳米镍单晶的表面效应[1]](https://img.taocdn.com/s3/m/897b7d9170fe910ef12d2af90242a8956becaa76.png)
分⼦动⼒学模拟纳⽶镍单晶的表⾯效应[1]分⼦动⼒学模拟纳⽶镍单晶的表⾯效应Ξ黄 丹陶伟明郭⼄⽊(浙江⼤学⼒学系固体⼒学研究所,杭州,310027)摘 要 对单晶镍纳⽶丝、纳⽶薄膜零温准静态拉伸破坏过程进⾏了分⼦动⼒学模拟.模拟表明表⾯效应对单晶纳⽶材料的原⼦运动及整体⼒学⾏为有显著影响.⾃由表⾯增加纳⽶材料的塑性、降低其强度,影响纳⽶材料的变形机制.受表⾯效应的作⽤,纳⽶镍丝强度与弹性模量均低于纳⽶镍薄膜.纳⽶薄膜的断裂接近脆性断裂,断裂强度符合G riffith 理想晶体脆断理论;纳⽶镍丝在断裂过程中表现出微弱塑性.关键词 分⼦动⼒学,镍,表⾯效应,单晶0 引⾔纳⽶材料由于⽐表⾯⼤,表⾯能、表⾯应⼒和表⾯原⼦运动对材料⼒学⾏为的影响举⾜轻重[1].近年来,⾦属纳⽶⼒学实验主要局限于对晶体硬度、蠕变、延展性等性能的测试[2],且实验结果分散性较⼤,⽽“从头开始”的分⼦动⼒学(M D )⽅法[3]直接根据原⼦间相互作⽤来模拟原⼦的运动过程,可以模拟纳⽶尺度实验很难测试的⼒学⾏为和现象.已有⽂献对纳⽶材料的应变率效应[4]、尺⼨效应[5]、裂纹扩展效应[6]等进⾏了分⼦动⼒学计算.本⽂采⽤适合于描述不规则晶格环境原⼦运动的原⼦镶嵌法(E AM )描述原⼦间作⽤,建⽴了⾦属镍单晶纳⽶丝和纳⽶薄膜模型,并应⽤分⼦动⼒学⽅法对纳⽶丝、纳⽶薄膜准静态单向拉伸破坏过程进⾏了零温模拟.通过⽐较两组模型中原⼦排列、能量、应⼒的变化,揭⽰了不同边界条件下单晶纳⽶材料受拉伸变形破坏的物理本质,讨论了表⾯效应对纳⽶材料弹性模量、拉伸强度等⼒学性能及变形机制的影响.1 分⼦动⼒学模拟1.1 原理与⽅法本⽂的分⼦动⼒学模拟采⽤Voter 等[7]根据镶嵌原⼦法提出的镍的多体势函数,设原⼦总势能E total =∑i12∑jΦ(r ij)+F (ρi )(1)式中Φ(r ij )为相距r ij 的原⼦i ,j 之间的中⼼对势,E (ρi )为到电⼦云ρi 的原⼦镶嵌能,ρi 为原⼦i 处的电⼦云密度,分别表⽰为Φ(r ij )=A 1(r c 1-r ij )2exp (-c 1r ij )(2)F (ρi )=D ρi ln ρiρi =∑jf (r ij )(3)其中f (r ij )=A 2(r c 2-r ij )2exp (-c 2r ij )(4)表⽰离原⼦i 距离为r ij 的原⼦j 对原⼦i 处电⼦云的影响.A 1、A 2、c 1、c 2、D 是由材料物理性能决定的参数.r c 1和r c 2分别表⽰计算原⼦对势和电⼦云密度的截断半径,r c 1=1.65d ,在次近邻原⼦和第三近邻原⼦之间;r c 2=1.95d ,在第三近邻和第四近邻原⼦之间.d 为最近邻原⼦距离,对于⾯⼼⽴⽅晶格⾦属晶体,d =a 0Π2,本⽂中a 0=0.3524nm ,为镍的晶格常数.对动⼒学⽅程的求解通过Verlet 算法[8]的速度形式来进⾏时间积分.为避免原⼦热激活,模拟在零温下进⾏,采⽤Berendsen 等[9]⽅法进⾏等温控制.考虑到表⾯的收缩和截⾯尺⼨的变化,以Parrinello 2Rahman[10]⽅法调节⾃由表⾯应⼒.模拟分两组进⾏,第⼀组为纳⽶镍丝结构,第⼆组为纳⽶镍薄膜结构.1.2 模型的建⽴模型的⼏何构型如图1,其中图1(a )为单胞原⼦排列及晶向与坐标轴对应关系图,x 、y 、z 坐标轴分别对应单胞的[100]、[010]、[001]晶向.图1(b )为整体模型,共6×20×20个单胞,9600个原⼦,原始⼏何尺⼨为2.1144nm ×7.048nm ×7.048nm.分⼦第26卷第2期2005年 6⽉固体⼒学学报ACT A MECHANICA SOL IDA SINICAVol.26No.2J une 2005Ξ国家⾃然科学基⾦(10302025)和教育部回国⼈员启动基⾦(J20030151)资助.2004207207收到第1稿,2004210218收到修改稿.动⼒学迭代的时间步长为3.45fs ,能量单位为1erg.第⼀组模拟将x 、y ⽅向控制为⾃由表⾯,在z ⽅向上施加周期性边界条件,使原⼦模型呈纳⽶丝结构.第⼆组模拟将x ⽅向控制为⾃由表⾯,在y 、z ⽅向上施加周期性边界条件,使原⼦模型呈纳⽶薄膜结构.图1 单晶镍材料原⼦模型1.3 模拟过程⾸先温度初始化,并控制模型原⼦温度始终为绝对零度,避免原⼦热激活.对模型弛豫50000步,使系统有充分时间达到能量最低的稳定状态.沿z 向施加平⾯拉伸应变0.005,进⾏分⼦动⼒学模拟10000步,在34.5ps 内实现对两端原⼦的恒速拉伸加载,然后弛豫50000步,使系统达到平衡态,再增加拉伸应变0.005.重复上述施加应变2分⼦动⼒学模拟2弛豫过程,直⾄模型出现断裂.由纳观加载的应变率效应分析[4]可知本⽂的加载速率处于应变不敏感区,加载属于准静态加载.拉伸过程中调节x 、y ⽅向尺⼨以控制压⼒.模拟过程中每隔500步记录原⼦的应⼒张量,总能量、势能、动能、纳⽶模型的⼏何构型、各⽅向横截⾯原⼦排列和坐标位置.2 模拟结果与讨论2.1 原⼦能量与应⼒演化过程图2、图3分别给出了两组模型中原⼦能量和应⼒演化曲线,图2和图3的纵坐标分别为模型中原⼦平均总能量和平均正应⼒,横坐标均为外加应变.初始弛豫后,纳⽶薄膜的原⼦平均总能量为-6.845463×10-12erg ,⽽纳⽶丝的原⼦平均总能量为-6.845257×10-12erg.施加荷载后,薄膜原⼦能量迅速升⾼,纳⽶丝原⼦能量则增加缓慢.这是因为经过初始弛豫后的系统处于能量最低的稳定状态,内部原⼦处在理想晶格位置,表⾯原⼦由于空间不对称⽽储存了很⾼的表⾯能.纳⽶丝的⾃由表⾯⽐纳⽶薄膜多,因此外部原⼦储存的表⾯能更⾼.在加载初期,模型内部原⼦能量升⾼,⽽表⾯原⼦释放表⾯能,故纳⽶丝原⼦平均能量增加⽐纳⽶薄膜缓慢.当表⾯能的释放⼩于外加能量时,纳⽶丝的能量也开始上升.在加载后期,纳⽶薄膜中由于⼤量的原⼦空位和微缺陷,原⼦结合⼒弱,原⼦滑移主导了薄膜的变形并引起理想晶格破坏,原⼦势能增加;⽽纳⽶丝的⼤量⾃由表⾯发射位错,在静态加载过程中位错消耗晶格的应变能,因此纳⽶丝系统能量低于纳⽶薄膜,上升梯度也低于纳⽶薄膜.图2 原⼦总能量曲线图3 原⼦应⼒曲线经过初始弛豫后,表⾯原⼦受拉,内部原⼦受压,纳⽶薄膜和纳⽶丝中都存在初始应⼒,分别为:纳⽶丝σx =2048Pa ,σy =σz =21470Pa ;纳⽶薄膜σx =19450Pa ,σy =σz =1.657×105Pa ,可见初始应⼒与边界条件即⾃由表⾯数有关.加载过程中,纳⽶薄膜的原⼦应⼒远⾼于纳⽶丝.纳⽶薄膜x 、y 向应⼒较⾼,且持续上升,最⼤值分别为7.46G Pa 和9.47G Pa ;纳⽶丝x 、y 向应⼒很低,在初始加载阶段缓慢上升后,σx 维持在0.3G Pa ,σy 维持在1.7G Pa.这是由于表⾯原⼦⾃由度⾼,原⼦运动剧烈,⼤量的⾃由表⾯降低了纳⽶丝的强度,使其应⼒⽔平下降.2.2 表⾯效应对变形的影响242? 固体⼒学学报 2005年第26卷图4、图5为两组模型在初始弛豫状态和应⼒最⼤时刻(ε=0.148)x 2y 横截⾯原⼦排列图.在初始时刻,表⾯原⼦在空间⽅向失去相邻原⼦,纳⽶丝沿z 向缩短,纳⽶丝横截⾯上四个⾓落原⼦向内收缩,⽽四边中部原⼦向外凸;纳⽶薄膜在x ⽅向的⾃由表⾯原⼦也受到初始拉应⼒的影响,薄膜表⾯在y、z ⽅向均收缩,引起表⾯原⼦在x ⽅向向外凸出.图4 纳⽶镍丝横截⾯原⼦排列形状图5 纳⽶镍薄膜横截⾯原⼦排列形状在初始加载阶段,模型沿拉伸⽅向的缩短逐渐恢复,表⾯原⼦释放表⾯能,运动⽅向向内;荷载升⾼后,模型中⼤量表⾯原⼦由于结合键弱于晶体内部,沿⼀定⽅向出现滑移.由能量演化曲线和图4(b )可见,荷载增加使纳⽶丝在横截⾯四个⾓落沿对⾓线⽅向有明显滑移线,由于x 、y 向均为⾃由边界,表⾯发射位错并向内部晶格深⼊,位错的移动消耗能量,使纳⽶丝表现出⼀定的塑性.从图2和图5(b )可以看出,纳⽶薄膜的变形由原⼦滑移主导,⾃由表⾯发射的少量位错由于y 向的周期性边界条件⽽⽆法滑出边界,限制了位错的后续产⽣.原⼦滑移导致晶格的破坏和空⽳的产⽣,空⽳相互连接、扩展,使薄膜在能量和应⼒持续升⾼后突然断裂,类似于宏观脆性断裂.2.3 弹性模量和强度分析通过对z 向应⼒应变值的最⼩⼆乘法模拟可得,纳⽶镍薄膜的弹性模量为E =186.6G Pa ,略⼩于Shen 等[11]对⽆微孔隙纳⽶镍样品的准静态测试结果(E =217G Pa ),原因在于薄膜表⾯运动⾃由度⾼的原⼦削弱了晶体的强度.受表⾯悬键和表⾯缺陷的影响,纳⽶材料弹性模量可由内部晶格弹性模量、表⾯晶格弹性模量和⾓落晶格弹性模量三部分[12]组成,⾓落晶格弹性模量远远⼩于内部晶格弹性模量.纳⽶丝的⾃由表⾯更多,由相互垂直的⾃由表⾯边缘原⼦构成的⾓落使纳⽶丝整体弹性模量降低.模拟得到纳⽶丝E=98.74G Pa ,约为纳⽶薄膜的⼀半.值得注意的是,考虑到⾯⼼⽴⽅单晶的各向异性,本⽂所得的弹性模量仅为单晶镍在<100>⽅向的弹性模量,对于本⽂中的两组模型,可以进⾏相互⽐较.由图3可知,在ε=0.148时刻,纳⽶丝σz =14.58G Pa ,薄膜σz =21.08GPa.根据G riffith 理想脆断理论,理想晶体断裂的最⼤应⼒应等于晶格的理论强度.Shen 等[11]由实验测得纳⽶镍晶格的理论强度为21.7G Pa (⼀般取0.1E ),本⽂所得纳⽶薄膜的断裂强度(21.08G Pa )已经基本接近镍的晶格强度,符合G riffith 脆断理论,⽽薄膜的应⼒演化曲线也恰恰显⽰,薄膜的破坏类似于宏观脆性断裂.受表⾯效应影响,纳⽶丝的强度远低于纳⽶薄膜.3 结论对纳⽶镍丝、镍薄膜准静态拉伸过程的分⼦动⼒学模拟表明:受表⾯效应影响,纳⽶材料存在本征应⼒和很⾼的表⾯能;纳⽶丝的拉伸强度、弹性模量、原⼦能量和应⼒⽔平低于纳⽶薄膜,⽽延性⾼于纳⽶薄膜.⾃由表⾯改变了晶体的变形机制,纳⽶薄膜的变形以原⼦滑移为主,破坏类似于宏观脆性断裂,断裂强度21.08G Pa ,可以⽤G riffith 理想晶体脆断理论解释;纳⽶丝的变形中位错和原⼦滑移同时起作⽤,使纳⽶丝表现出⼀定的韧性,出现微弱塑性流动,拉伸强度为14.58G Pa.特征尺⼨相同的纳⽶丝弹性模量约为纳⽶薄膜的⼀半.参 考 ⽂ 献1 梁海⼷,倪向贵,王秀喜.表⾯效应对纳⽶铜杆拉伸性能影响的原⼦模拟.⾦属学报,2001,37(8):833~8362 卢柯,卢磊.⾦属纳⽶材料⼒学性能的研究进展.⾦属学报,2000,36(8):785~7893 Ma X inling ,Y ang Wei.M D simulation for nanocrystals.Acta Mech S inica ,2003,19(6):485~5074 徐洲,梁海⼷,王秀喜.纳⽶丝应变率效应的分⼦动⼒学模拟.固体⼒学学报,2003,24(2):229~2345 H orstermeyer M F ,Baskes M I.Atomistic finite deformationsimulation:a discussion on length scale effects in relation to mechaical stresses.J Eng Mater T ech ,1999,121:115~1196 万建松,岳珠峰,耿晓亮,卢智先.⼀种镍基双晶材料疲342?第2期黄 丹等: 分⼦动⼒学模拟纳⽶镍单晶的表⾯效应劳裂纹扩展效应.固体⼒学学报,2003,24(2):148~1547 V oter A F ,Chen S P.Accurate interatomic potentials for Ni ,Al ,and Ni 3Al.Mat Res S oc Symp Proc ,1987,82:175~1828 Allen M P ,T ildesley D J.C omputer S imulation of Liquids.Ox ford :Clarendon Press ,19879 Berendsen H J C ,P ostma J P M ,G unsteren W F V ,et al.P ostma J P M ,G unsteren WFV ,N ola AD ,Haak JR.M olucular dynamics with coupling to an external bath.J Chem Phys,1984,81:3684~369010Parrinello M ,Rahman A.P olym orphic transitions in singlecrystals:a new m olecular dynamics method.J Appl Phys ,1981,52(12):7182~719011Shen T D ,K och C C ,Tsui T Y,et al.On the elastic m oduli ofnanocrystalline Fe ,Cu ,Ni and Cu 2Ni alloys prepared by mechanical milling Πalloying.J Mater Res ,1995,10(11):2892~289612Broughton J Q ,Meli C A ,Vashishta P ,et al.Direct atomisticsimulation of quartz crystal oscillators :bulk properties and mam oscale devices.Phys Rev B ,1997,56(2):611~618MOL ECU LAR DYNAMICS SIMU LATION ON SURFACE EFFECTSOF MONOCR YSTALLINE NICKE LHuang Dan T ao Weiming G uo Y imu(Institute o f Solid Mechanics ,Department o f Mechanics ,Zhejiang Univer sity ,Hangzhou ,310027)Abstract With Embedded Atom Method ,the surface effects of m onocrystalline nickel wire and nickel film under uniaxial tension are studied by M olecular Dynamics.With m ore free surfaces ,the atomic energy and stress ,strength and elastic m odulus of nickel wire are all lower than that of nickel film ,but the plasticity of the former is higher than the latter.The rupture of nano film is m ore like brittle fracture and accords with G riffith theory of rupture ,but that of nano wire is m ore like tough fracture with plastic flow.K ey w ords m olecular dynamics ,nickel ,surface effect ,m onocrystalline442? 固体⼒学学报 2005年第26卷。
分子模拟的原理与应用

分子模拟的原理与应用分子模拟是一种能够通过物理、化学及生物学原理,模拟分子体系中的运动和相互作用的计算方法。
它可以在原子、分子、甚至大分子尺度上,模拟各种物质的运动状态,从而让我们更加深入地理解物质的性质和行为。
本文将介绍分子模拟的基本原理和应用。
一、分子模拟的原理分子模拟的基本原理是牛顿力学和分子动力学理论。
在牛顿力学中,物体的运动状态可以通过它所受到的力来描述。
分子动力学理论基于牛顿力学原理,将物质看作由无数个粒子组成的巨大集合,每个粒子间彼此相互作用,从而产生整体性质。
分子模拟中用来描述分子间相互作用的理论有很多。
最为热门的是分子力学理论和分子动力学理论。
分子力学理论主要关注于分子的结构和力学性质,而分子动力学理论则更加注重粒子间的运动轨迹和相互作用。
二、分子模拟的应用1.新药研究分子模拟技术在新药研究中扮演着重要角色。
通过计算机模拟等手段,科学家们可以在分子水平上,预测化合物与生物分子的相互作用,分析药效、毒性等性质,针对药物副作用进行优化设计。
这不仅可以提高新药研发效率,同时也为疾病治疗提供了更可靠的手段。
2.材料科学分子模拟技术在材料科学领域中也得到了广泛应用。
它可以模拟材料的热力学性能和结构特征,帮助人们了解材料的物理、化学、力学、光学等性质。
例如,科学家们可以通过分子模拟技术,模拟纳米材料的形态、尺寸、表面性质等,从而在材料设计中起到重要的作用。
3.环境污染分子模拟技术在环境污染领域中有着重要的应用价值。
例如,科学家们可以利用分子模拟技术,模拟污染物在不同环境中的行为,包括其迁移途径、分布、反应和生物转化等。
此外,分子模拟还可以用来预测空气、水和土壤中有害化学物质的扩散、生物降解和生物累积。
三、分子模拟的发展趋势随着计算机速度和计算技术的不断提高,分子模拟技术将具有更高的精度和更大的模拟规模。
在细胞分子层面上的模拟也将成为未来的热点,模拟更加复杂的化学、生物体系将是分子模拟技术的一个重要方向。
分子模拟课件

ME346–Introduction to Molecular Simulations–Stanford University–Winter2007 Handout1.An Overview of Molecular SimulationWei Caic All rights reservedSeptember26,2005Contents1Examples of molecular simulations1 2What consists of a molecular dynamics?2 3Newton’s equation of motion2 4A simple numerical integrator:Verlet algorithm5 1Examples of molecular simulationsPictures and movies of modern large scale molecular simulations can be downloaded from the course web site /∼me346(click“Course Materials”).Exam-ples include plastic deformation in metals containing a crack,phase transformation induced by shock waves,nano-indentation of metalfilm,laser ablation on metal surface,etc.In each of these examples,molecular simulations not only produce beautiful pictures,but they also offer new understanding on the behavior of materials at the nano-scale.Many times such understanding provides new insights on how the materials would behave at the micro-scale and nano-scale.With unique capabilities to offer,molecular simulation has become a widely used tool for scientific investigation that complements our traditional way of doing science: experiment and theory.The purpose of this course is to provide thefirst introduction to molecular level simulations—how do they work,what questions can they answer,and how much can we trust their results—so that the simulation tools are no longer black boxes to us.Students will develop a set of simulation codes in Matlab by themselves.(A simple Matlab tutorial written by Sigmon[5]may help you refresh your memory.)We will also give tutorial and exercise problems based on MD++,a molecular simulation package used in my research group.MD++will be used for illustration in class as well as tools to solve homework problems.2What consists of a molecular dynamics?In a molecular simulation,we view materials as a collection of discrete atoms.The atoms interact by exerting forces on each other and they follow the Newton’s equation of motion. Based on the interaction model,a simulation compute the atoms’trajectories numerically. Thus a molecular simulation necessarily contains the following ingredients.•The model that describes the interaction between atoms.This is usually called the interatomic potential:V({r i}),where{r i}represent the position of all atoms.•Numerical integrator that follows the atoms equation of motion.This is the heart of the ually,we also need auxiliary algorithms to set up the initial and bound-ary conditions and to monitor and control the systems state(such as temperature) during the simulation.•Extract useful data from the raw atomic trajectory pute materials properties of interest.Visualization.We will start our discussion with a simple but complete example that illustrates a lot of issues in simulation,although it is not on the molecular scale.We will simulate the orbit of the Earth around the Sun.After we have understood how this simulation works(in a couple of lectures),we will then discuss how molecular simulations differ from this type of “macro”-scale simulations.3Newton’s equation of motionThroughout this course,we limit our discussions to classical dynamics(no quantum mechan-ics).The dynamics of classical objects follow the three laws of Newton.Let’s review the Newton’s laws.Figure1:Sir Isaac Newton(1643-1727England).First law:Every object in a state of uniform motion tends to remain in that state of motion unless an external force is applied to it.This is also called the Law of inertia.Second law:An object’s mass m,its acceleration a,and the applied force F are related byF=m a.The bold font here emphasizes that the acceleration and force are vectors.Here they point to the same direction.According to Newton,a force causes only a change in velocity(v)but is not needed to maintain the velocity as Aristotle claimed(erroneously).Third law:For every action there is an equal and opposite reaction.The second law gives us the equation of motion for classical particles.Consider a particle (be it an atom or the Earth!depending on which scale we look at the world),its position is described by a vector r=(x,y,z).The velocity is how fast r changes with time and is also a vector:v=d r/dt=(v x,v y,v z).In component form,v x=dx/dt,v y=dy/dt,v z=dz/dt. The acceleration vector is then time derivative of velocity,i.e.,a=d v/dt.Therefore the particle’s equation of motion can be written asd2r dt2=Fm(1)To complete the equation of motion,we need to know how does the force F in turn depends on position r.As an example,consider the Earth orbiting around the Sun.Assume the Sun isfixed at origin(0,0,0).The gravitational force on the Earth is,F=−GmM|r|2·ˆe r(2)whereˆe r=r/|r|is the unit vector along r direction.|r|=√x2+y2+z2is the magnitudeof r.m is the mass of the Earth(5.9736×1024kg),M is mass of the Sun(1.9891×1030kg), and G=6.67259×10−11N·m2/kg2is gravitational constant.If we introduce a potential field(the gravitationalfield of the Sun),V(r)=−GmM|r|(3)Then the gravitational force can be written as minus the spatial derivative of the potential,F=−dV(r)d r(4)In component form,this equation should be interpreted as,F x=−dV(x,y,z)dxF y=−dV(x,y,z)dyF z=−dV(x,y,z)dzNow we have the complete equation of motion for the Earth:d2r dt2=−1mdV(r)d r(5)in component form,this reads,d2x dt2=−GMx|r|3d2y dt2=−GMy|r|3d2z dt2=−GMz|r|3In fact,most of the system we will study has a interaction potential V(r),from which the equation of motion can be specified through Eq.(5).V(r)is the potential energy of the system.On the other hand,the kinetic energy is given by the velocity,E kin=12m|v|2(6)The total energy is the sum of potential and kinetic energy contributions,E tot=E kin+V(7) When we express the total energy as a function of particle position r and momentum p=m v, it is called the Hamiltonian of the system,H(r,p)=|p|22m+V(r)(8)The Hamiltonian(i.e.the total energy)is a conserved quantity as the particle moves.To see this,let us compute its time derivative,dE tot dt =m v·d vdt+dV(r)d r·d rdt=md rdt·−1mdV(r)d r+dV(r)d r·d rdt=0(9)Therefore the total energy is conserved if the particle follows the Newton’s equation of motion in a conservative forcefield(when force can be written in terms of spatial derivative of a potentialfield),while the kinetic energy and potential energy can interchange with each other.The Newton’s equation of motion can also be written in the Hamiltonian form,d r dt =∂H(r,p)∂p(10)d p dt =−∂H(r,p)∂r(11)Hence,dH dt =∂H(r,p)∂r·d rdt+∂H(r,p)∂p·d pdt=0(12)Figure2:Sir William Rowan Hamilton(1805-1865Ireland).4A simple numerical integrator:Verlet algorithmA dynamical simulation computes the particle position as a function of time(i.e.its trajec-tory)given its initial position and velocity.This is called the initial value problem(IVP). Because the Newton’s equation of motion is second order in r,the initial condition needs to specify both particle position and velocity.To solve the equation of motion on a computer,thefirst thing we need is to discretize the time.In other words,we will solve for r(t)on a series of time instances t ually the time axis is discretized uniformly:t i=i·∆t,where∆t is called the time step.Again,the task of the simulation algorithm is tofind r(t i)for i=1,2,3,···given r(0)and v(0).The Verlet algorithm begins by approximating d2r(t)/dt2,d2r(t) dt2≈r(t+∆t)−2r(t)+r(t−∆t)∆t2(13)Thusr(t+∆t)−2r(t)+r(t−∆t)∆t2=−1mdV(r(t))dr(14)r(t+∆t)=2r(t)−r(t−∆t)−∆t·1m·dV(r(t))dr(15)Therefore,we can solve r(t+∆t)as along as we know r(t)and r(t−∆t).In other words,if we know r(0)and r(−∆t),we can solve for all r(t i)for i=1,3,4,···.Question:Notice that to start the Verlet algorithm we need r(0)and r(−∆t).However, the initial condition of a simulation is usually given in r(0)and v(0).How do we start the simulation when this initial condition is specified?References1.Alan and Tildesley,Computer Simulation of Liquids,(Oxford University Press,1987)pp.71-80.2.Frenkel and Smit,Understanding Molecular Simulation:From Algorithms to Applica-tions,2nd ed.(Academic Press,2002).3.Bulatov and Cai,Computer Simulation of Dislocations,(Oxford University Press,2006)pp.49-51.ndau and Lifshitz,Mechanics,(Butterworth-Heinemann,1976).5.Kermit Sigmon,Matlab Primer,Third Edition,/Matlab/matlab-primer.pdfME346–Introduction to Molecular Simulations–Stanford University–Winter2007 Handout2.Numerical Integrators IWei Caic All rights reservedJanuary12,2007Contents1Which integrator to use?1 2Several versions of Verlet algorithm2 3Order of accuracy3 4Other integrators4 1Which integrator to use?In the previous lecture we looked at a very simple simulation.The Newton’s equation of motion is integrated numerically by the original Verlet algorithm.There are many algorithms that can be used to integrate an ordinary differential equation(ODE).Each has a its own advantages and disadvantages.In this lecture,we will look into more details at the numerical integrators.Our goal is to be able to make sensible choices when we need to select a numerical integrator for a specific simulation.What questions should we ask when choosing an integrator?Obviously,we would like something that is both fast and accurate.But usually there are more issues we should care about.Here are a list of the most important ones(Allen and Tildesley1987):•Order of accuracy•Range of stability•Reversibility•Speed(computation time per step)•Memory requirement(as low as possible)•Satisfying conservation law•Simple form and easy to implementItem1and2(especially2)are important to allow the use of long time steps,which makes the simulation more efficient.2Several versions of Verlet algorithmThe Verlet algorithm used in the previous lecture can be written as,r(t+∆t)=2r(t)−r(t−∆t)+a(t)·∆t2(1)wherea(t)=−1mdV(r(t))d r(t)(2)While this is enough to compute the entire trajectory of the particle,it does not specify the velocity of the particle.Many times,we would like to know the particle velocity as a function of time as well.For example,the velocity is needed to compute kinetic energy and hence the total energy of the system.One way to compute velocity is to use the followingapproximation,v(t)≡d r(t)dt≈r(t+∆t)−r(t−∆t)2∆t(3)Question:if the initial condition is given in terms of r(0)and v(0),how do we use the Verlet algorithm?Answer:The Verlet algorithm can be used to compute r(n∆t)for n=1,2,···provided r(−∆t)and r(0)are known.The task is to solve for r(−∆t)given r(0)and v(0).This is achieved from the following two equations,r(∆t)−r(−∆t)2∆t=v(0)(4)r(∆t)−2r(0)+r(−∆t)∆t2=−1mdV(r(t))d r(t)t=0(5)Exercise:express r(−∆t)and r(∆t)in terms of r(0)and v(0).Therefore,r(−∆t)and r(∆t)are solved together.During the simulation,we only know v(t)after we have computed(t+∆t).These are the(slight)inconveniences of the(original) Verlet algorithm.As we will see later,in this algorithm the accuracy of velocity calculation is not very high(not as high as particle positions).Several modifications of the Verlet algorithms were introduced to give a better treatment of velocities.The leap-frog algorithm works as follows,v(t+∆t/2)=v(t−∆t/2)+a(t)·∆t(6)r(t+∆t)=r(t)+v(t+∆t/2)·∆t(7) Notice that the velocity and position are not stored at the same time slices.Their values are updated in alternation,hence the name“leap-frog”.For a nice illustration see(Allen and Tildesley1987).This algorithm also has the draw back that v and r are not known simultaneously.To get velocity at time t,we can use the following approximation,v(t)≈v(t−∆t/2)+v(t+∆t/2)2(8)The velocity Verlet algorithm solves this problem in a better way.The algorithm reads,r(t+∆t)=r(t)+v(t)∆t+12a(t)∆t2(9)v(t+∆t/2)=v(t)+12a(t)∆t(10)a(t+∆t)=−1mdV(r(t+∆t))d r(t+∆t)(11)v(t+∆t)=v(t+∆t/2)+12a(t+∆t)∆t(12)Exercise:show that both leap-frog and velocity Verlet algorithms gives identical results as the original Verlet algorithm(except forfloating point truncation error).3Order of accuracyHow accurate are the Verlet-family algorithms?Let’s take a look at the original Verletfirst. Consider the trajectory of the particle as a continuous function of time,i.e.r(t).Let us Taylor expand this function around a given time t.r(t+∆t)=r(t)+d r(t)dt∆t+12d2r(t)dt2∆t2+13!d3r(t)dt3∆t3+O(∆t4),(13)where O(∆t4)means that error of this expression is on the order of∆t4.In other words, (when∆t is sufficiently small)if we reduce∆t by10times,the error term would reduce by 104times.Replace∆t by−∆t in the above equation,we have,r(t−∆t)=r(t)−d r(t)dt∆t+12d2r(t)dt2∆t2−13!d3r(t)dt3∆t3+O(∆t4),(14)Summing the two equations together,we have,r(t+∆t)+r(t−∆t)=2r(t)+d2r(t)dt2∆t2+O(∆t4)≡2r(t)+a(t)∆t2+O(∆t4)(15)r(t+∆t)=2r(t)−r(t−∆t)+a(t)∆t2+O(∆t4)(16) Therefore,the local accuracy for the Verlet-family algorithms is to the order of O(∆t4). This is quite accurate for such a simple algorithm!The trick here is that,by adding t+∆t and t−∆t terms,the third order term cancels out.Notice that the accuracy analysis here deals with the local error.That is,assuming r(t)is exact,how much error we make when computing r(t+∆t).In practice,the total error of computed r at time t is the accumulated (and possibly amplified)effect of all time steps before time t.Because for a smaller timestep ∆t more steps is required to reach a pre-specified time t,the global order of accuracy is always local order of accuracy minus one.For example,the global order of accuracy for Verlet algorithms is3.Since all Verlet-family algorithms generate the same trajectory,they are all O(∆t4)in terms of particle positions.But they differ in velocity calculations.How accurate is the velocity calculation in the original Verlet algorithm?Let us subtract Eq.(13)and(14),r(t+∆t)−r(t−∆t)=2v(t)∆t+13d3r(t)dt3∆t3+O(∆t4),(17)v(t)=12∆tr(t+∆t)−r(t−∆t)−16d3r(t)dt2∆t3+O(∆t3)(18)v(t)=r(t+∆t)−r(t−∆t)2∆t+O(∆t2)(19)Thus the velocity is only accurate to the order of O(∆t2)!This is much worse than O(∆t4) of the positions.A reason for the lose of accuracy is that the velocities are computed by taking the difference between two large numbers,i.e.r(t+∆t)and r(t−∆t),and then divide by a small number.1Intuitively,higher order algorithms allows the use of bigger time steps to achieve the same level of accuracy.However,the more important determining factor for the choice of time steps is usually not the order of accuracy,but instead is the algorithm’s numerical stability (see next section).4Other integratorsBefore closing this lecture,let us briefly look at two other well known integrators.Gear’s predictor-corrector method is a high-order integrator.It keeps track of several time deriva-tives of the particle position.Definer0(t)≡r(t)(20) 1Strictly speaking,Eq.(17)is valid when we have exact values for r(t+∆t),r(t)and r(t−∆t).But in doing the order-of-accuracy analysis,we can only assume that we have exact values for r(t)and r(t−∆t) and we know that we are making an error in r(t+∆t).Fortunately,the error we make in r(t+∆t)is also O(∆t4).Thus Eq.(17)still holds.r1(t)≡∆t d r(t)dt(21)r2(t)≡∆t22!d2r(t)dt2(22)r3(t)≡∆t33!d3r(t)dt3(23)···Sincer(t+∆t)=r(t)+∆t d r(t)dt+∆t22!d2r(t)dt2+∆t33!d3r(t)dt3+O(∆t4)=r(t)+r1(t)+r2(t)+r3(t)+O(∆t4)r1(t+∆t)=r1(t)+2r2(t)+3r3(t)+O(∆t4)r2(t+∆t)=r2(t)+3r3(t)+O(∆t4)r3(t+∆t)=r3(t)+O(∆t4)we can predict the values of r and r i at time t+∆t simply based on their values at time t,r P(t+∆t)=r(t)+r1(t)+r2(t)+r3(t)r P1(t+∆t)=r1(t)+2r2(t)+3r3(t)r P2(t+∆t)=r2(t)+3r3(t)r P3(t+∆t)=r3(t)Notice that we have not even calculated the force at all!If we compute the force at time t+∆t based on the predicted position r P(t+∆t),we will obtain the second derivative of r at time t+∆t—let it be r C(t+∆t).Mostly likely,r C(t+∆t)will be different from the predicted value r P(t+∆t).Thefinal results are obtained by adding a correction term to each predictor,r n(t+∆t)=r Pn (t+∆t)+C n[r C2(t+∆t)−r P2(t+∆t)](24)The Gear’s6th order scheme(uses r0through r5)takes the following C n values,C0=3 16C1=251 360C2=1C3=11 18C4=1 6C5=1 60The global accuracy is of the order O(∆t)6,hence the method is called the Gear’s6th order algorithm.Exercise:show that the local truncation error of the above scheme is of the order of O (∆t 7).In a way,the Runge-Kutta method is similar to predictor-corrector methods in that the final answer is produced by improving upon previous predictions.However,the Runge-Kutta method requires multiple evaluation of forces.The Runge-Kutta method is most conveniently represented when the equation of motion is written as a first order differential equation,˙η=g (η)≡ω∂H (η)∂η(25)whereη≡(r ,p )(26)andω= 0I−I 0 (27)and I is the 3×3identity matrix and H is the Hamiltonian.The fourth-order Runge-Kutta method goes as (from ),k 1=∆t ·g (η(t ))k 2=∆t ·g η(t )+12k 1 k 3=∆t ·g η(t )+12k 2 k 4=∆t ·g (η(t )+k 3)η(t +∆t )=η(t )+16k 1+13k 2+13k 3+16k 4(28)Exercise:show that the local truncation error of the above scheme is of the order of O (∆t 5)and the global accuracy is of the order of O (∆t 4).Suggested reading1.(Alan and Tildesley 1987)pp.71-84.2.J.C.Butcher,The numerical analysis of ordinary differential equations:Runge-Kutta and general linear methods ,(Wiley,1987),pp.105-151.ME346–Introduction to Molecular Simulations–Stanford University–Winter2007 Handout3.Symplectic ConditionWei Caic All rights reservedJanuary17,2007Contents1Conservation laws of Hamiltonian systems1 2Symplectic methods4 3Reversibility and chaos5 4Energy conservation7 1Conservation laws of Hamiltonian systemsThe numerical integrators for simulating the Newton’s equation of motion are solvers of ordinary differential equations(ODE).However,the Newton’s equation of motion that can be derived from a Hamiltonian is a special type of ODE(not every ODE can be derived from a Hamiltonian).For example,a Hamiltonian system conserves it total energy,while an arbitrary ODE may not have any conserved quantity.Therefore,special attention is needed to select an ODE solver for Molecular Dynamics simulations.We need to pick those ODE solvers that obeys the conservation laws of a Hamiltonian system.For example,if an ODE solver predicts that the Earth will drop into the Sun after100cycles around the Sun,it is certainly the ODE solver to blame and we should not immediately start worrying about the fate of our planet based on this prediction.A conservation law is a(time invariant)symmetry.A Hamiltonian system possess more symmetries than just energy conservation.Ideally the numerical integration scheme we pick should satisfy all the symmetries of the true dynamics of the system.We will look at these symmetries in this and following sections.When discussing a Hamiltonian system,it is customary to use letter q to represent the particle position(instead of r).Recall that the equation of motion for a system with Hamiltonian H(q,p)is,˙q=∂H(q,p)∂p(1)˙p=−∂H(q,p)∂q,(2)where˙q≡d q/dt,˙p≡d p/dt.If the system has N particles,then q is a3N-dimensional vector specifying its position and p is a3N-dimensional vector specifying its momentum. (In the above Earth orbiting example,N=1.)Notice that in terms of q and p,the equation of motion is a set of(coupled)first-order differential equations(whereas F=m a is second order).The equation can be reduced to an even simpler form if we define a6N-dimensional vectorη≡(q,p)(3) Then the equation of motion becomes,˙η=ω∂H(η)∂η(4)whereω=0I−I0(5)and I is the3×3identity matrix.The6N-dimensional space whichη≡(q,p)belongs to is called the phase space.The evolution of system in time can be regarded as the motion of a pointηin the6N-dimensional phase space following thefirst-order differential equation(4).(a)(b)Figure1:(a)Motion of a point(q,p)in phase space is confined to a shell with constant energy.(b)Area of a phase space element(shaded)is conserved.Because the Newton’s equation of motion conserves total energy,the motion of a point in phase space must be confined to a subspace(or hyperplane)with constant energy,i.e.a constant energy shell,as illustrated in Fig.1(a).Think about all the points in the constant energy shell.As time evolves,the entire volume that these points occupy remains the same —the points simply rearrange within the constant energy shell.Let us now consider the evolution of a small element in phase space over time,as illus-trated in Fig.1(b).Think of this as the ensemble of trajectories of many Molecular Dynamics simulations with very similar initial conditions.We will show that the area enclosed by this element remains constant,even though the element inevitably experiences translation anddistortion.1Let the element at time t be a hypercube around pointη,whose area is,|dη|=|d q|·|d p|=dq1···dq3N dp1···dp3N(6) For a pointηat time t,letξbe its new position at time t+δt.In the limit ofδt→0,wehave,ξ=η+˙ηδt=η+ω∂H(η)∂ηδt(7)Because the above equation is true only in the limit ofδt→0,we will ignore any higher order terms(e.g.δt2)in the following discussions.Let M be the Jacobian matrix of the transformation fromηtoξ,i.e.,M≡∂ξ∂η=1+ω∂2H(η)∂η∂ηδt(8)For clarity,we write M explicitly in terms of q and p,M=1+0I−I0·∂2H∂q∂q∂2H∂q∂p∂2H∂p∂q∂2H∂p∂p·δt(9)=1+∂2Hδt∂2Hδt−∂2H∂q∂qδt1−∂2H∂q∂pδt(10)The area of the new element is related to the the determinant of the Jacobian matrix,|dξ|=|dη|·|det M|=|dη|·(1+O(δt2))(11) Because thefirst order term ofδt in det M vanishes,we can show that the area of the element remains constant for an arbitrary period of time(i.e.forever).2This is an important property of a Hamiltonian system.Because the area of any element in phase space always remains constant,the evolution of phase space points is analogous to that in an incompressiblefluid.Exercise:Write down the explicit expression for the Jacobian matrix M for problem of the Earth orbiting around the Sun.The Hamiltonian dynamics has even more symmetries.Notice that the transpose of M is,M T=1−∂2H∂η∂ηωδt(12)1It then follows that the area enclosed by an arbitrarily shaped element in the phase space also remains constant.2To show that area remains constant after afinite period∆t,we only need to divide the time interval into N subintervals,each withδt=∆t/N.The area change per subinterval is of the order of1/N2.The accumulated area change over time period∆t is then of the order of1/N,which vanishes as N→∞.we haveM ωM T = 1+ω∂2H ∂η∂ηδt ω 1−∂2H ∂η∂ηωδt =ω+ω∂2H ∂η∂ηωδt −ω∂2H ∂η∂ηωδt +O (δt 2)(13)=ω+O (δt 2)(14)(15)Therefore the Jacobian matrix M thus satisfies the so called symplectic condition (up to O (δt 2)),M ωM T =ω(16)Again,we can show that the symplectic condition is satisfied for an arbitrary period of time.To be specific,let ξbe the point in phase space at time t ;at time 0this point was at η.ξcan then be regarded as a function of t and η(initial condition),i.e.ξ=ξ(η,t ).Define M as the Jacobian matrix between ξand η,we can show that M satisfies the symplectic condition Eq.(16).3Obviously,the Hamiltonian dynamics is also time reversible.This means that if ηevolves to ξafter time t .Another phase space point η that has the same q as ηbut with reversed p (momentum)will exactly come to point ξafter time t .To summarize,the dynamics of a system that has a Hamiltonian has the following symmetry properties:•Conserves total energy•Conserves phase space area (incompressible flow in phase space)•Satisfies symplectic condition Eq.(16)•Is time reversibleIdeally,the numerical integrator we choose to simulate Hamiltonian system should satisfy all of these symmetries.In the following,let us take a look at how specific integrators are doing in this regard.In fact,if an algorithm satisfies the symplectic condition,it automatically conserves phase space area 4,is reversible,and should have good long-term energy conserva-tion properties.This is why symplectic methods are good choices for simulating Hamiltonian systems.2Symplectic methodsWhat do we mean if we ask whether a numerical integrator satisfies the symplectic condition or not?For a numerical integrator,the system moves forward in time by discrete jumps.Let3Again,we devide the time period t into N subintervals,each with δt =∆t/N .Let M i be the Jacobian matrix from time (i −1)δt to iδt .We have M = N i =1M i .We can show that M T ωM =ωin the limit of N →∞.4Since M T ωM =ω,det ω=det(M T ωM )=(det M )2det ω.Thus det M =±1,which means area conservation.η(i)=(q(i),p(i))be the place of the system in the phase space at time step i,i.e.t i=i∆t. At the next time step,the algorithm brings the system to pointη(i+1)=(q(i+1),p(i+1)). Let M be the Jacobian matrix that connectsη(i)withη(i+1).The method is symplectic if MωM T=ω.The good news is that the Verlet-family algorithms are symplectic.To show this is the case,let us consider the leap-frog algorithm.Let q(i)correspond to q(i·∆t)and p(i) correspond to p((i−12)·∆t).Then the leap-frog algorithm is,p(i+1)=p(i)−∂V∂q(i)∆t(17)q(i+1)=q(i)+p(i+1)m∆t=q(i)+1mp(i)−∂V∂q(i)∆t∆t(18)Thus,M=∂q(i+1)∂q(i)∂q(i+1)∂p(i)∂p(i+1)(i)∂q(i+1)(i)=1−1m∂2V∂q(i)∂q(i)∆t2∆tm−∂2V∂q(i)∂q(i)∆t1(19)Thus M is symplectic.Exercise:show that for M defined in Eq.(19),MωM T=ω(symplectic)and det M=1 (area preserving).Exercise:derive the Jacobian matrix M for velocity Verlet algorithm and show that it is symplectic and area preserving.3Reversibility and chaosBecause the Verlet-family algorithms(original Verlet,leap-frog and velocity Verlet)are sym-plectic,they are also time reversible.Nonetheless,it is instructive to show the time reversibil-ity explicitly.If we run the leap-frog algorithm in reverse time,then the iteration can be written as,˜q(i+1)=˜q(i)+˜p(i)m∆t(20)˜p(i+1)=˜p(i)−∂V∂q(i+1)∆t(21)(22)where i is the iteration number,˜q(i)=q(−i∆t),˜p(i)=p((−i−12)∆t).It is not difficult toshow that if˜q(i)=q(i+1)and˜p(i)=−p(i+1),then˜q(i+1)=q(i)and˜p(i+1)=−p(i).Exercise:show that the leap-frog algorithm is reversible.Exercise:show that the velocity Verlet algorithm is reversible.Exercise:show that the Forward Euler method:q(i+1)=q(i)+∆t·p(i)/m,p(i+1)=p(i)−∆t·∂V/∂q(i),is not reversible and not symplectic.Since every integration step using the Verlet algorithm is time reversible,then in principle an entire trajectory of Molecular Dynamics simulation using the Verlet algorithm should be time reversible.To be specific,let the initial condition of the simulation be q(0)and p(0). After N steps of integration,the system evolves to point q(N)and p(N).Time reversibility means that,if we run the reverse Verlet iteration starting from q(N)and−p(N),after N steps we should get exactly to point q(0)and−p(0).In practice,however,we never have perfect reversibility.This is due to the combined effect of numerical round offerror and the chaotic nature of trajectories of many Hamiltonian systems.The chaotic nature of many Hamiltonian system can be illustrated by considering the two trajectories with very close initial conditions.Let the distance between the two points in phase space at time0be d(0).At sufficiently large t,the two phase space points will diverge exponentially,i.e.d(t)∼d(0)eλt,whereλis called the Lyapunov exponent and this behavior is called Lyapunov instability.Given that we can only represent a real number on a computer withfinite precision,we are in effect introducing a small(random) perturbation at every step of the integration.Therefore,sooner or later,the numerical trajectory will deviate from the analytical trajectory(if the computer had infinite precision) significantly5.While the analytical trajectory is reversible,the one generated by a computer is not.This is why there exist a maximum number of iterations N,beyond which the original state cannot be recovered by running the simulation backwards.This by itself does not present too big a problem,since we seldom have the need to recover the initial condition of the simulation in this way.However,this behavior does mean that we will not be able to follow the“true”trajectory of the original system forever on a computer.Sooner or later, the“true”trajectory and the simulated one will diverge significantly.This is certainly a problem if we would like to rely entirely on simulations to tell us where is our satellite or space explorer.However,this usually does not present a problem in Molecular Simulations, in which we are usually not interested in the exact trajectories of every atom.We will return to this point in future lectures when we discuss Molecular Dynamics simulations involving many atoms.5Notice that the analytical trajectory here is not the same as the“true”trajectory of the original system. The analytical trajectory is obtained by advancing the system in discrete jumps along the time axis.。
分子模拟教案—

教案课程名称分子模拟授课题目(章、节) 第一章授课教师李慎敏授课对象化学081-2 授课时间9月2日教学方法讲授选用教具多媒体本教案以讲授一个单元(2—4学时)或一次实验(实习)为单位填写。
填写要用钢笔,字迹要清晰、工整。
按表项目逐一填写。
教案课程名称分子模拟授课题目(章、节) 第二章授课教师李慎敏授课对象化学081-2 授课时间9月9日教学方法讲授选用教具多媒体本教案以讲授一个单元(2—4学时)或一次实验(实习)为单位填写。
填写要用钢笔,字迹要清晰、工整。
按表项目逐一填写。
教案课程名称分子模拟授课题目(章、节) 第三章授课教师李慎敏授课对象化学081-2 授课时间9月16 日教学方法讲授选用教具多媒体本教案以讲授一个单元(2—4学时)或一次实验(实习)为单位填写。
填写要用钢笔,字迹要清晰、工整。
按表项目逐一填写。
教案课程名称分子模拟授课题目(章、节) 第三章授课教师李慎敏授课对象化学081-2 授课时间9月23 日教学方法讲授选用教具多媒体本教案以讲授一个单元(2—4学时)或一次实验(实习)为单位填写。
填写要用钢笔,字迹要清晰、工整。
按表项目逐一填写。
教案课程名称分子模拟授课题目(章、节) 第四章授课教师李慎敏授课对象化学081-2 授课时间10月9日教学方法讲授选用教具多媒体本教案以讲授一个单元(2—4学时)或一次实验(实习)为单位填写。
填写要用钢笔,字迹要清晰、工整。
按表项目逐一填写。
教案课程名称分子模拟授课题目(章、节) 第四章授课教师李慎敏授课对象化学081-2 授课时间10月14日教学方法讲授选用教具多媒体本教案以讲授一个单元(2—4学时)或一次实验(实习)为单位填写。
填写要用钢笔,字迹要清晰、工整。
按表项目逐一填写。
教案课程名称分子模拟授课题目(章、节) 第五章授课教师李慎敏授课对象化学081-2 授课时间10月21 日教学方法讲授选用教具多媒体本教案以讲授一个单元(2—4学时)或一次实验(实习)为单位填写。
9组_分子识别膜

分子印迹技术
使功能单体在模板分子周围进行聚合(有的再 交联),形成一个主体结构后,再将模板分子提取 出去。这样,聚合物中就形成了与模板分子的形 状和功能相对应的可进行识别的空穴,当该聚合 物用于分离由模板分子与其他物质组成的混合物 时,就能够有效地识别并分离出这些模板分子。
分子识别聚合物
采用分子印迹技术制备的、能够根据分 子的形状和特征对手性体进行有效分离的 高分子聚合物。
分子识别功能高分子膜
将制备分子识别聚合物的分子印迹技术 分子识别功能高分子膜是随着分子 应用到成膜过程中,使所制备的膜具有在 识别聚合物和分子印迹技术的发展 分子层次上对手性体进行识别的功能,它 而发展起来的一个崭新的研究领域。 是膜科学目前发展的主要方向之一。 膜
分子印迹技术
分子识别聚合物
分子识别 高分子膜
发展趋势
现状
1
技术和理论研究已取得初步成果
2
制膜方法趋于成熟
膜材料逐步趋于商业化
3
4
应用领域在较大范围内得到考察
存在问题
01
模板分子的种类还非常有限
02
分子识别膜的吸收量较小,仅限 于微量分析 应用领域的研究比较分散,不够 系统和深入 相关基质吸收的理论研究较少
03
04
虽然,到目前为止国内外已经成功合成 出了多种具有在分子层次上进行识别功能 的高分子膜,并且已经在手性化合物、氨 基酸及其衍生物以及多种药物的分离中得 到了实际应用 。但是,无论从理论方面, 如模板分子与聚合物相互作用、识别空穴 的形成等,还是制膜方法、膜材料的选择 和应用的实际方面而言,它都还处于发展 阶段,有待于向更深和更广的领域开拓。
研究历史
20世纪70年代,液相膜
分子模拟的基本原理及应用

分子模拟的基本原理及应用前言分子模拟是一种通过计算机模拟方法来研究和预测物质行为的技术。
它基于分子动力学和蒙特卡洛等模拟算法,模拟物质内部分子之间的相互作用和运动规律,以揭示宏观性质和微观机制。
本文将介绍分子模拟的基本原理和应用。
基本原理分子模拟的基本原理包括分子动力学方法和蒙特卡洛方法。
分子动力学方法分子动力学方法基于牛顿运动定律,通过模拟分子之间的相互作用力及其在时间上的演化来研究物质的行为。
分子动力学模拟首先需要确定分子位型(坐标和速度),然后通过计算力场和粒子间的相互作用力来求解其位型的演化。
常见的力场模型包括Lennard-Jones势和Coulomb势等。
分子动力学方法的优点是可以研究物质的结构动力学行为,如振动频率、扭曲和固有的化学反应等。
蒙特卡洛方法蒙特卡洛方法则是通过随机模拟分子的运动,以获得物质的统计性质。
蒙特卡洛模拟通过随机生成分子位型,然后根据一定的准则进行状态更新,最终达到平衡态,并收集数据进行统计分析。
常见的蒙特卡洛算法有Metropolis算法和Gibbs Ensemble算法等。
蒙特卡洛方法的优点是可以模拟大体系,且可以得到平衡态下的分子分布和宏观性质。
应用领域分子模拟在多个领域中被广泛应用。
材料科学分子模拟在材料科学中应用较多,可以研究材料的力学性能、热学性能、光学性质等。
1.硬质材料:通过分子模拟可以研究材料的晶体结构、点阵参数、断裂强度等力学性质。
2.聚合物材料:分子模拟可以用于研究聚合物的构象转变、玻璃化转变、熔融过程等。
3.纳米材料:通过分子模拟可以研究纳米材料的表面性质、纳米粒子的形态稳定性等。
生物医学分子模拟在生物医学领域可以用于研究药物与生物分子的相互作用、蛋白质的结构与功能、肿瘤的生长机制等。
1.药物设计:通过分子模拟可以预测药物分子与配体的结合方式,加速新药研发。
2.蛋白质结构预测:分子模拟可以进行蛋白质的二级结构和三级结构预测,帮助理解蛋白质的功能。
面向光电跟踪系统的先进运动控制(任彦著)PPT模板

1
4.3.1模型的加性分解
2
4.3.2系统控制器和辅系统补偿 器的设计
3
4.3.3光滑化的设计
4
4.3.4状态分解观测器的设计
第4章基于加性分解的鲁棒内回路控制
4.4实验验证
4.4.1实验 设置
1
4.4.2实验 结果及分析
2
06
第5章基于NNESO干扰补偿的离散滑模控制
第5章基于NNESO干扰补偿的离散滑模控制
5.1扩张状态观测器 5.2离散滑模控制 5.3基于NNESO干扰补偿的离散滑模控制 5.4本章小结
第5章基于NNESO干扰补偿的离散滑模控制
5.1扩张状态观测器
01
5.1.1线性高增益扩张状态观测 器
0 2 5.1.2非线性扩张状态观测器
0 3 5.1.3基于有限时间收敛的非线性扩 张状态观测器设计
02
5.3.2实验验证
07
第6章基于有限时间收敛的虚拟复合轴控制
第6章基于有限时间收敛的虚拟 复合轴控制
6.1虚拟复合轴控制的基本理论 6.2基于虚拟复合轴的干扰补偿 方法 6.3虚拟复合轴主、子系统控制 器的ຫໍສະໝຸດ 计 6.4实验验证 6.5本章小结
第6章基于有限时间收敛的虚拟复合轴控制
6.1虚拟复合轴控制的基本理论
6.1.1复合轴控制系 统
6.1.2虚拟复合轴控 制结构
6.1.3虚拟复合轴伺 服系统的实现
第6章基于有限时间收 敛的虚拟复合轴控制
6.2基于虚拟复合轴的干扰补偿方 法
A
6.2.1速度干扰观 测器的实现方法
6.2.2近似微分法
B
第6章基于有限时间收 敛的虚拟复合轴控制
分子动力学模拟基本原理及其应用

三一文库()〔分子动力学模拟基本原理及其应用〕李煌鲁红权陈钦煌张宏伟周叶琪童小宝【摘要】阐述了分子动力学模拟的理论基本、模拟条件、模拟应用以及钻研的重点目标。
介绍了MD计算的发展,并给出了分子动力学模拟中相关的运动方程和定理、有限差分算法、分子动力学模拟应用发展的主要过程。
并且阐述了分子动力学模拟的初始条件、周期性边界、长程作用力以及系综等。
【关键词】分子动力学;有限差分算法;模拟条件及应用中图分类号:TB383.1文献标识码:A文章编号:2095-2457(2018)05-0025-002【Abstract】Thetheoreticalbasis,simulationconditions,simulationapplicationsandkeyivesofmoleculardynamics simulationaredescribed.Inthispaper,thedevelopmentofMDcalculationisintroduced,andthemotionequationandtheorem,finitedifferencealgorithmandthemaindevelopmentproce ssofmoleculardynamicssimulationaregiven.Theinitialc onditions,periodicboundary,long-rangeforceandensembleofmoleculardynamicssimula tionarealsodiscussed.【Keywords】Moleculardynamics;Finitedifferencealgorithm;Simulationconditionsandapplication0引言分子动力模拟计算,从开始发展到现在已经有五十来年的历史。
它是一种依据经典力学的计算方法,主要是依据分子的力场来计算分子的各种性质。
分子建模与模拟导论

(10.8)
(10.9)
因为由牛顿方程可以从 x0 t t 的值算出实际加速度 x2
corrected
,可以对第二项进行修正
corrected predicted x2 x2 x2
(10.10)
其它项可以修正为
corrected predicted xn xn cn x2
Nose
(10.34)
A p, r
NVT
7
真实变量与虚变量之间的关系为
r r p p / s s s t t / s
ps2 H p , r E 2Q
1 3N 1 ps2 3N 1 3N 1 N N exp E dps exp H p, r dp dr exp N! l l 2Q l l C 3N 1 dp N dr N exp H p, r N! l
H p, r
ps2 l ln s0 E 0 2Q
(10.30)
p2 ln s0 E H p, r s 2Q l s0 exp l
另外有
ps2 H p , r E 2Q
(10.20)
3) 如果某粒子被选中, 则从温度为 T 的 Maxwell-Boltzmann 分布中随机产生一个速度赋予 该粒子,其它粒子不变。 可以证明,使用 Andersen thermostat 的体系确实是对正则系综的采样。问题在于系统 角动量不守恒,模拟的体系随时间旋转,不方便数据处理。
10.2. 正则系综的 MD 算法 基本的 MD 算法实现的是微正则系综,为了实现 MD 对正则系综的模拟,需要在牛顿 积分后引入热耦(thermostat)对系统温度进行调节。
化学软件基础-第4章 第2节-分子动力学模拟

等压等焓系综(constant-pressure, constant-enthalpy ensemble ) --NPH系综
分子动力学
12/34
系综调节
系综调节主要是指在进行分子动力学计算过 程中,对温度和压力参数的调节
调温技术: Berendsen热浴、速度标度、Gaussian热浴、
Nose-Hoover热浴
RX(I) = RXNEWI RY(I) = RYNEWI RZ(I) = RZNEWI 100 CONTINUE
分子动力学
22/34
5.1 Verlet 算法
Verlet算法的优缺点:
• 优点: 1、精确,误差O(Δ4) 2、每次积分只计算一次力 3、时间可逆 • 缺点: 1、速度有较大误差O(Δ2) 2、轨迹与速度无关,无法与热浴耦联
分子动力学
rc<L/2
15/34
5 牛顿运动方程
粒子的运动取决于经典力学 (牛顿定律(F=ma)
分子动力学
16/34
5 牛顿运动方程
原子i受力: Fi
Ei
i
x
j
y
k
z
Ei
加速度:
ai F i mi
i原子经过t时间后的位置
d2 dt 2
ri
i (rij ) 为第j个原子在i个原子处贡献的电荷密度
分子动力学
10/34
2.3 长程F-S势函数
c:正的无量纲常数
对势 ε:有能量量纲的参数
α:有长度量纲的参数
多体势 m, n:正整数
对于不同研究体系,5 个参数取值不同
分子动力学
11/34
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
方法
频率矫正因子 零点能矫正因子
HF/3-21G
0.9085
HF/6-31G(d)
0.8929
MP2(Full)/6-31G(d) 0.9427
MP2(FC)/6-31G(d) 0.9434
SVWN/6-31G(d) 0.9833
BLYP/6-31G(d)
0.9940
B3LYP/6-31G(d)
0.9613
4.2.1 Predicting the IR and Raman Spectra 预测红外和拉曼光谱
Frequencies and Intensities频率和强度
1
2
3
4
B1
B2
A1
A1
Frequencies --- 1336.0041 1383.6449 1679.5843 2028.0971
0.9409 0.9135 0.9646 0.9676 1.0079 1.0119 0.9804
4.2.3 Normal Modes简正模式
以水分子的计算结果为例:
Standard orientation:
-------------------------------------------------------------
mA mB
(2) 当 RAB 与 Req )
键的力常数
即
d 2 RAB
dt 2
ke (RAB
Req )
一个特解为:RAB Req Asin(2 t )
其中,
1 ke 2
(振动频率或特征频率)
Recap
从数学角度分析化学反应势能面
正则振动模式
i
1
2
ki
The three normal modes of H2O. The mode v2 is predominantly bending, and occurs at lower wave-number than the other two.
4.1 Input for Frequency Jobs 频率分析输入文件
Center Atomic
Coordinates (Angstroms)
Number Number
X
Y
Z
--------------------------------------------------------------------------
1
6
0.000000 0.000000 -0.542500
0
2E
i2
0
极小值点
2E
2 2
0
0
2E
2 3N 6
2E
2 p
0
2E
2 i
0
(i = 1, 2, ,p-1,
鞍点
p+1, 3N-6)
求解谐振子的Schrödinger方程可得其能量的量子化表达式
E振=(ν+1/2)hν特征, 振动量子数 ν=0,1,2,···
零点振动能 E0=1/2 hν特征
关键词:freq # RHF/6-31G(d) Freq Test
注意: 频率分析计算时所采用的理论方法和基组必须 与几何构型优化采用的方法和基组完全相同。
4.2 Output of Frequency Jobs 频率分析输出文件信息
●Predicting the IR and Raman Spectra ● Normal Modes ● Thermochemistry ● Characterizing Stationary Points
1.双原子分子的谐振动模型
当AB间距离RAB偏离Req时 A 和 B 都要 受到 一个力 (分子的内力)的作用
f
mA
d 2 RCA dt 2
mB
d 2 RCB dt 2
f mAmB d 2 RAB mA mB dt 2
d 2 RAB
dt 2
(1)在只有分子内的作用下,双原子分子的振动运动可用一个质量为 mAmB 的假想质点在平衡距离 Req附近的振动运动
to the force ~ the third term can be shown to be equivalent
to the force constant
Recap
对于多原子分子体系,其能量对位置的二阶 偏导数矩阵可以表示为:
Hessian Matrix
2E
R12
2E
R2R1
2E
Chapter 4 Frequency Calculations 频率分析
频率分析可以用于多种目的:
预测分子的红外和拉曼光谱(频率和强度) 几何构型优化时计算力常数矩阵 确定势能面上驻点性质 计算零点能和热力学数据,如系统的熵和焓
Molecular Vibrations
4.2 分子的振动光谱
Red. masses --- 1.3689 1.3442 1.1039 7.2497
... ---
IR Intensities --- 0.3694 23.1589 8.6240 150.1861
Raman Activities ---0.7657 4.5170 12.8594 8.1124
4.2.2 Scaling Frequencies and Zero-Point Energies 频率和零点能校正因子
R3N 6R1
2E
R1R2 2E R22 2E
R3N 6R2
2E
R1R3N 6
2E
R2R3N
2E
6
R32N 6
Recap
i
1
2
ki
通过正则变换,可以找到一组坐标i (i=1, 2, , 3N-6) 使上述Hessian Matrix对角化:
2E
12
0
0
0
坐标i称为简正坐标
泰勒级数展开
E(xi) E(xi(0))
i
(xi
xi(0)
)
E(xi(0) xi
)
1 2
i, j
(xi
xi(0) )(xj
x(j0) )
2E(xi(0), x(j0) ) xix j
~ the first term is set to zero ~ the second term can be shown to be equivalent
反应物(reactants)、生成物(products)和过渡 态(transition states)都是势能曲面的极值点。对 于N个原子的体系(3N-6维坐标),极值点的条件是 能量对位置的一阶偏导数为零:
E / Ri = 0
(i = 1, 2, , 3N-6)
其中E为能量,Ri为坐标。
Recap