分子克隆及细胞培养基本实验方法
分子克隆实验流程

分子克隆实验流程一、引物的稀释1、引物干粉冻存于-20℃,用前12000rpm离心1min;2、按引物管上的nmol数稀释,nmol=4.92,加49.2µL ddH2O至100µM;3、稀释至10µM(5µL引物F+5µL引物R+40µL ddH2O)二、目的基因的扩增实验前准备:生物安全柜紫外照射30min,模板DNA、水、引物、buffer,dNTP提前10min拿出解冻,用75%酒精擦拭移液器及台面。
扩增体系:Reagent 25µL 50µL10xbuffer (含Mg2+) 2.5µL 5µLdNTP (10mM) 0.5µL 1µLrT aq酶0.25μL0.5μLprimer (10μM) 1.25μL 2.5μLTemplate DNA 2μL4μLddH2O 18.5µL 37µL反应程序:(延伸时间按目的片段大小进行调整)95℃预变性3min(95℃变性30 s,60℃退火30s,72℃延伸45s)x3572℃后延伸7min4℃保持电泳:120V,加2µLloading buffer,上样5µL,1000bp marker 5µL小胶:2%,0.6g琼脂糖,30ml 1xTAE中胶:2%,1g琼脂糖,50ml 1xTAE大胶:2%,2g琼脂糖,100ml 1xTAE三、目的产物切胶回收(试剂盒)四、连接实验前准备:SolutionI在冰上融化连接体系:Reagent 10µL胶回收DNA(50ng/μL)4µLPDM-18T载体1µLSolution I 5µL反应条件:16℃,4h(PCR仪,热盖105℃)/ 4℃过夜五、转化实验前准备:开启42℃水浴锅实验步骤:样品+阴性对照(无质粒)+阳性对照(感受态带的质粒)1、把感受态细胞TOP10从-80℃冰箱拿出并放置于冰上解冻;2、每管分装30 - 50μL感受态细胞(冰浴);3、向感受态细胞中加入5μL连接产物,冰浴30min。
分子克隆实验标准步骤

分⼦克隆实验标准步骤分⼦克隆实验标准步骤⼀、常规分⼦克隆实验流程:⼆、分⼦克隆实验标准步骤(含实验编号):1. PCR 扩增⽬的基因(编号Clone SOP-1)以本实验室常⽤酶KOD-Plus-Neo (TOYOBO )为例体系(50ul ):10×KOD buf 5uldNTP(2mM) 5ulMg 2+ 3ulPrimer1 1ulPrimer2 1ulTemplate50-200ngKOD0.5ulddH 2O up to 50ul程序:95℃2min98℃10s58℃30s 35cycle68℃2kb/min68℃7min12℃∞2.PCR产物的琼脂糖凝胶电泳琼脂糖凝胶的制备(编号Clone SOP-2)琼脂糖溶液的制备:称取琼脂糖,置于三⾓瓶中,按1%-1.5%的浓度加⼊相应体积的TBE或TAE缓液,将该三⾓瓶置于微波炉加热⾄琼脂糖溶解。
胶板的制备:①取有机玻璃内槽,洗净、晾⼲;②将有机玻璃内槽置于⼀⽔平位置模具上,安好挡板,放好梳⼦。
在距离底板上放置梳⼦,以便加⼊琼脂糖后可以形成完好的加样孔。
③将温热琼脂糖溶液倒⼊胶膜中,使胶液缓慢地展开,直到在整个有机玻璃板表⾯形成均匀的胶层。
④室温下静置30min左右,待凝固完全后,轻轻拔出梳⼦,在胶板上即形成相互隔开的上样孔。
制好胶后将铺胶的有机玻璃内槽放在含有0.5~1×TAE(Tris-⼄酸)或TBE(Tris-硼酸)⼯作液的电泳槽中使⽤,没过胶⾯1mm以上。
3.试剂盒回收DNA⽚段(编号Clone SOP-3)以本实验室常⽤DNA凝胶回收试剂盒(天根)为例使⽤前请先在漂洗液PW中加⼊⽆⽔⼄醇,加⼊体积请参照瓶上的标签。
①柱平衡步骤:向吸附柱CA2中(吸附柱放⼊收集管中)加⼊500µl平衡液BL,12,000rpm(~13,400×g)离⼼1min,倒掉收集管中的废液,将吸附柱重新放回收集管中。
(请使⽤当天处理过的柱⼦)②将单⼀的⽬的DNA条带从琼脂糖凝胶中切下(尽量切除多余部分)放⼊⼲净的离⼼管中,称取重量。
生物中的分子克隆

生物中的分子克隆克隆是指复制一个已经存在的个体或物品,分子克隆则是指复制分子。
在生物领域中,分子克隆技术极其重要,它能够让科学家们克隆出特定的蛋白质、基因和细胞等分子,不仅推动了基因工程、生物制药等领域的发展,还有助于对医学、生态学、进化论等问题的深入研究。
一、DNA的分子克隆DNA双链分子由四种核苷酸组成,克隆某个特定的DNA序列,需要寻找到该序列的特异性序列,一般采用如下方法:1.限制性内切酶切割法:将要克隆的DNA进行限制性内切酶切割,将切割后的DNA片段进行电泳分离,并用紫外线照射,使用UV灯观察DNA条带,选取符合要求的目标DNA条带作为模板,再使用电泳提取出目标DNA条带,进行下一步的操作。
2.基因库方法:将DNA切成片段后,将这些片段以随机顺序插入载体中,再将这些载体插入到宿主细胞中,寻找到目标片段所在的载体后,就可以从中将这个片段克隆出来。
通过上述方法,克隆出目标DNA后,还需要定位、测序、分析等步骤,才能够达到所需的效果。
二、基因的分子克隆基因是细胞中负责遗传物质传递的重要分子,克隆基因是基因工程活动中的一个重要环节。
1.针对已有的已知基因,可以使用上述DNA分子的克隆方法,将基因克隆出来,进行重组、改变等操作。
2.针对未知的基因,可以进行基因组测序与分析,利用反向遗传学法进行基因定位及功能分析。
3.对于人类常见疾病,例如乳腺癌、某些遗传性疾病等,深入研究它们的基因表达和调控,利用分子克隆技术进行基因治疗或转基因实验。
基因的克隆不仅促进了对于遗传学和基因学的深入研究,也能够产生特定的应用效果,甚至应用到生物治疗和治疗遗传性疾病等医学领域。
三、细胞的分子克隆细胞是生命活动的基本单位,克隆细胞可以使得相似的细胞在体外大量生长,提供研究的可操作性。
目前,主要有两种方法可以利用分子克隆技术克隆细胞:1.体外培养法:通过细胞培养基、激素等营养物质及生长因子,为细胞提供生长环境,使其在体外快速繁殖,而体外克隆细胞最广泛应用的领域就是生物制药,例如克隆出产生特殊蛋白质的细胞系,生产生物药品。
分子克隆操作程序-ABclonal

分子克隆操作程序操作程序1 实验试剂及仪器1.1 试剂:质粒DNA小量试剂盒DNA 凝胶回收试剂盒DNA 产物回收试剂盒DNA 产物回收试剂盒Pfu,DNA marker,感受态DH5αBamHI酶,XhoI酶,EcoRI酶,T4 DNA ligase 1.2 实验仪器:PCR仪离心机凝胶成像系统:Tanon 2500,天能公司1.3 耗材:实验中所用到的PCR管,枪头2 试验步骤2.1 引物的稀释2.1.1引物一般以干粉形式保存于-20℃,临用前稀释。
12.1.2 由于引物呈很轻的干膜状附在管壁上,打开时极易散失,所以打开管子前请先离心10,000rpm,1min,然后再慢慢打开管盖。
然后在装有引物的管内根据引物说明书加入双蒸水稀释至10uM,盖上管盖,充分上下振荡30秒,再次离心,小离心机30秒(管上标注好引物名称、浓度、稀释时间)2.2 PCR扩增反应体系在0.2ml离心管中加入以下成分:(做多管的时候可以把除引物模板以外的组分混合后分装)轻弹混匀,离心收集管壁上的液滴至管底, 在PCR扩增仪上进行PCR反应,反应参数如下(不同基因的退火温度以及延伸时间略有差异,退火温度为引物Tm 上下5℃)94℃ 3 min94℃30sec58℃40sec72℃(1kb/min)2372℃ 8min4℃ ∞反应结束后,取2ulPCR 产物进行1%琼脂糖凝胶电泳,并以DNA Marker 6ul 做对照。
120V 恒压条件下电泳20min 后,于凝胶成像系统下观察并将电泳图贴到记录本中(扩增产物大约30ng/μl )2.3 PCR 产物的回收2.3.1 如果扩增产物条带单一用 Axygen AP-PCR-250纯化PCR 产物,具体操作按试剂盒说明书进行(回收产物用35ul ddH 2O 洗脱两遍)2.3.2 如果出现非特异性扩增, 目的产物用AxyPrep DNA 凝胶回收试剂盒回收(AP-GX-250),具体操作按试剂盒说明书进行 (回收产物用35ul ddH 2O 洗脱两遍) 2.4目的产物及载体酶切目的产物酶切(在1.5ML 离心管中加入以下成分, 以BamHI 和XhoI 为例)振荡混匀,短暂离心; 载体酶切(载体使用中抽质粒试剂盒)37℃温育过夜;80℃水浴20min 终止反应2.5 酶切产物回收酶切产物用DNA 产物回收试剂盒回收,具体操作按试剂盒说明书进行(目的产物酶切用35ul ddH2O 洗脱两遍,载体酶切用50ul ddH2O 洗脱两遍),将电泳图贴到记录本中。
分子克隆部分实验报告

一、实验目的1. 学习分子克隆的基本原理和方法;2. 掌握质粒的提取、纯化、线性化及目的基因的插入等实验操作;3. 熟悉DNA的纯化、鉴定及重组载体的构建等实验技术。
二、实验原理分子克隆是指将目的基因片段从基因组DNA中分离出来,并在宿主细胞中复制和扩增的过程。
实验过程中,利用限制性内切酶切割目的基因和载体,通过连接酶将二者连接形成重组载体,然后转化宿主细胞,筛选出含有目的基因的克隆。
三、实验材料1. 质粒:pET-28a2. 目的基因:EGFP3. 限制性内切酶:BamHI、EcoRI4. DNA连接酶:T4 DNA连接酶5. DNA分子量标准:DL20006. DNA纯化试剂盒7. 转化宿主细胞:大肠杆菌DH5α8. LB培养基、氨苄青霉素9. 等等四、实验步骤1. 质粒提取与纯化(1)按照试剂盒说明书提取质粒DNA;(2)用DNA纯化试剂盒纯化质粒DNA;(3)检测质粒浓度和纯度。
2. 目的基因的线性化(1)用BamHI和EcoRI双酶切目的基因片段和载体;(2)用DNA纯化试剂盒纯化酶切产物;(3)检测酶切产物浓度和纯度。
3. DNA连接(1)将纯化的目的基因片段和载体进行连接反应;(2)将连接产物转化大肠杆菌DH5α;(3)在含有氨苄青霉素的LB培养基中培养转化菌。
4. 阳性克隆的筛选(1)提取转化菌的DNA;(2)用BamHI和EcoRI双酶切提取的DNA;(3)电泳检测酶切产物,筛选出与预期大小相符的重组质粒;(4)将重组质粒进行测序验证。
五、实验结果与分析1. 质粒提取与纯化:质粒浓度约为50ng/μl,纯度大于0.8。
2. 目的基因的线性化:酶切产物浓度约为10ng/μl,纯度大于0.8。
3. DNA连接:转化菌在含有氨苄青霉素的LB培养基中生长良好。
4. 阳性克隆的筛选:电泳结果显示,重组质粒大小与预期相符。
5. 重组质粒测序验证:测序结果与预期序列一致,表明目的基因已成功插入载体。
分子克隆主要步骤

分子克隆主要步骤分子克隆是一种常用的分子生物学技术,用于复制DNA分子。
下面是分子克隆的主要步骤:1.DNA提取:首先需要从一个已知的DNA源(例如细菌、动物组织等)中提取所需的DNA。
这可以通过使用不同的提取方法(如酚/氯仿提取、自动提取仪等)来实现。
2.限制性内切酶切割:将目标DNA切割成片段。
此步骤可以通过使用限制性内切酶来实现,这些酶可以识别特定的DNA序列,并在这些序列中切割DNA,形成切割产物。
3.DNA修饰:如果需要,在第2步切割的DNA片段末端添加修饰,以便后续步骤的操作。
例如,可以在DNA片段的末端添加磷酸基团(通过激酶酶和ATP)或羟基(通过糖转移酶和dTTP)。
4.连接DNA片段:将目标DNA片段与载体DNA(通常是质粒)连接起来。
这可以通过使用DNA连接酶,如DNA连接酶I或T4DNA连接酶,将DNA片段与载体DNA的末端连接。
5.转化:将连接好的DNA导入到宿主细胞中。
这可以通过转化(常见的转化宿主细胞包括大肠杆菌和酵母)来实现。
转化可以通过热冲击法、电转化或使用化学方法来进行。
6.筛选:在经过转化的细胞中筛选出带有目标DNA的细胞。
这可以通过将转化后的细胞接种到含有适当选择标记的培养基上来实现。
只有带有目标DNA的细胞才能生长并形成克隆。
7.复制:选取带有目标DNA的细胞进行培养,并使其进行大量复制。
这可以通过将细胞培养在含有适当培养基和条件的培养皿中来实现。
8.提取:从大量复制的细胞中提取含有目标DNA的质粒。
这可以通过使用质粒提取试剂盒来实现,其中包含了一系列的化学试剂和步骤,用于纯化和提取目标DNA。
9.鉴定:验证提取的DNA是否为目标DNA。
这可以通过进行限制性内切酶切割、PCR扩增或测序等方法来实现。
分子克隆是一种重要的实验技术,可用于构建重组DNA分子、研究基因功能、制备蛋白质等。
虽然上述步骤描述了分子克隆的基本过程,但具体操作可能会因实验目的和需求而略有不同。
基因工程分子克隆实验操作流程

基因工程分子克隆实验操作流程英文回答:Molecular Cloning Experiments Using Gene Engineering: A Comprehensive Protocol.Introduction:Molecular cloning is a fundamental technique in biotechnology that involves the isolation, manipulation, and amplification of specific DNA sequences. It plays a pivotal role in various research areas, such as gene function studies, protein production, and genetic engineering. This protocol outlines the detailed steps involved in a typical molecular cloning experiment using gene engineering techniques.Materials:Gene of interest (target DNA)。
Cloning vector (plasmid or viral vector)。
Restriction enzymes.DNA ligase.Competent cells (e.g., E. coli)。
Antibiotics.Culture media.Molecular biology reagents and equipment.Methods:1. Gene Isolation:Extract DNA from the source organism using appropriate methods.Amplify or synthesize the target gene using PCR or other techniques.2. Vector Preparation:Digest the cloning vector with appropriate restriction enzymes to create compatible ends.Dephosphorylate the vector to prevent self-ligation.3. Gene Insertion:Combine the digested gene and vector.Ligate the DNA fragments using DNA ligase.4. Transformation:Introduce the recombinant plasmid into competent cells (e.g., E. coli) by transformation.Plate the transformed cells on selective mediacontaining antibiotics.5. Colony Screening:Identify colonies that have successfully taken up the recombinant plasmid.Screen colonies for the presence of the target gene using PCR, restriction digestion, or other methods.6. Plasmid Isolation:Culture the positive clones and isolate the recombinant plasmid using plasmid extraction methods.7. Sequence Verification:Perform DNA sequencing to confirm the accuracy of the cloned gene.8. Gene Expression:If necessary, subclone the gene into an expression vector for protein production.Optimize expression conditions and purify the target protein.Applications:Molecular cloning has numerous applications in research and biotechnology, including:Gene cloning and sequencing.Protein production and characterization.Gene therapy.Genetic engineering of organisms.Development of diagnostic tools and therapeutics.Conclusion:Molecular cloning using gene engineering is a powerful technique that enables researchers to manipulate and study DNA at the molecular level. By following the outlined protocol, scientists can successfully design, execute, and analyze cloning experiments for various research and biotechnology applications.中文回答:利用基因工程进行分子克隆实验操作流程。
分子克隆的实验报告(3篇)

第1篇一、实验目的本实验旨在学习分子克隆技术的基本原理和操作步骤,掌握目的基因的扩增、克隆及表达,为后续相关研究奠定基础。
二、实验原理分子克隆技术是指将目的DNA片段从供体细胞中分离出来,通过体外重组、转化和转导等方法,将其插入到克隆载体中,再将其引入宿主细胞进行复制和扩增。
本实验采用无缝克隆技术,通过T5核酸外切酶、DNA聚合酶和DNA连接酶三种酶的共同作用,实现单片段或多片段与载体连接。
三、实验材料1. 试剂:限制性内切酶、DNA连接酶、T5核酸外切酶、DNA聚合酶、dNTPs、Taq DNA聚合酶、PCR引物、载体DNA、目的基因DNA、质粒提取试剂盒、琼脂糖凝胶电泳试剂盒等。
2. 仪器:PCR仪、凝胶成像仪、电泳仪、紫外灯、超净工作台、离心机、恒温水浴锅、移液器等。
四、实验步骤1. 目的基因扩增(1)设计引物:根据目的基因的序列设计特异性引物,引物长度一般在18-25bp,5'端添加限制酶切位点。
(2)PCR反应:配制PCR反应体系,加入引物、模板DNA、dNTPs、Taq DNA聚合酶等,进行PCR反应。
2. 载体线性化(1)酶切:使用限制性内切酶对载体DNA进行酶切,获得线性化的载体。
(2)去磷酸化:对单酶切得到的线性化载体进行去磷酸化处理。
3. 目的基因与载体连接(1)同源臂连接:将目的基因PCR产物和线性化载体进行同源臂连接,确保目的基因正确插入载体。
(2)连接反应:配制连接反应体系,加入目的基因PCR产物、线性化载体、DNA连接酶等,进行连接反应。
4. 转化与筛选(1)转化:将连接产物转化至宿主细胞中。
(2)筛选:通过抗生素筛选、酶切鉴定和测序等方法筛选出含有目的基因的克隆。
5. 目的基因表达(1)重组质粒提取:从筛选出的阳性克隆中提取重组质粒。
(2)重组质粒转化:将重组质粒转化至表达宿主细胞中。
(3)表达产物检测:通过Western blot、ELISA等方法检测目的蛋白的表达水平。
分子克隆详细步骤

分子克隆步骤:一、贴壁细胞总RNA提取:1、吸掉培养液,用PBS洗一遍?2、往培养皿中加入1ml,TRIzol,吹打几次(每10cm2面积,即3.5cm直径的培养板加1ml)3、移至1.5mlEP管,静置5分钟4、加入200ul三氯甲烷,震荡混匀,室温静置5分钟5、4度12000r/min,离心15分钟,取上清,约600ul6、加入500ul异丙醇,混匀后,静置30分钟?7、4度12000r/min,离心15分钟,弃上清8、加入1ml70%预冷酒精洗涤沉淀物9、4度7500r/min,离心5分钟10、弃上清,自然晾干11、加入50ulDEPC水溶解,测OD值*鼠尾基因组DNA粗提取:1、100ul lysis buffer for each tail,and 2ul 10mg/ml PK,55℃,overnight.2、Then,100℃ for 10min to denature the PK, use 0.5~1ul lysate as template to do PCR.Lysis buffer:(store at 4℃)KCl 0.5MTris 0.1MNP-40 1%Tween-20 1%二、RT-PCR:1、预变性体系12ul:Total RNA 2ulOligo(dT18)primer 1ulDH water 9ul65℃ 5min 速置冰上2、RT体系:20ul:预变性体系12ul5×buffer 4ulRNAase inhibiter 1ul10m dNTP 2ulMMLV 1ul42℃ 60min70℃ 5min12℃ forever3、PCR体系20ul:10×buffer 2ul10m dNTP 0.5ulPrimer(F+R) 1ul (0.5ul+0.5ul)稀释后cDNA(50ul)1ulPfu 0.2uldd water 15.3ul95℃3min、(95℃30s,55℃30s,72℃35s)×29cycle、72℃10min、12℃forever三、跑胶鉴定PCR产物:四、醇沉PCR产物:1、将PCR产物转移至1.5mlEP管中2、加入0.1倍体积预冷NaAC,3倍体积70%预冷乙醇,混匀3、—80℃静置30min4、4度14000r/min,10min离心弃上清,加1ml70%预冷乙醇洗涤沉淀5、4度14000r/min,10min离心弃上清,自然晾干6、加入25-20ul dd water 吹匀静置10-20min待溶解五、原始质粒/PCR醇沉产物双酶切体系50ul:Enzyme1 1ulEnzyme2 1ul10×Buffer 5ul (在体系中被稀释成1×)10×BSA 5ul (看需要)Template 1ugADD dd water to 50ul酶切过夜?六、单独鉴定质粒酶切产物:1、采用20ul体系:酶各0.5ul、buffer2ul、bsa0.5ul、template2ul)酶切2h2、跑胶鉴定七、电泳,切胶回收与纯化:使用DNA回收试剂盒(QIAquick Gel Extraction Kit Protocol)PCR酶切产物纯化:1.将PCR产物于需要的电压和电流下跑电泳2. 紫外灯下仔细切下含待回收DNA的凝胶,置1.5ml离心管中,称重。
分子克隆实验指南

分子克隆实验指南分子克隆技术是一种常用的实验手段,常用于生物学和医学领域的研究中。
这种方法可以通过将DNA分子插入到载体DNA中来制备重组DNA分子,从而达到扩增特定的DNA序列或者表达生物活性分子的目的。
分子克隆技术的原理基于DNA的重组,重组的过程通常需要以下几个步骤:1、DNA的裂解和切割:要将DNA进行克隆,首先需要将待操作的DNA裂解并用酶切成适量的小片段。
2、载体的制备:载体是与待操作的DNA进行克隆的中介物,这种载体通常采用环状DNA分子质粒,也可以采用噬菌体等其它病毒。
3、DNA的连接:将切割后的DNA与载体对应的DNA片段通过酶的帮助连接起来,形成重组的DNA分子。
4、转化:将重组的DNA分子转化到细胞中。
5、筛选:对表达成功的细胞进行筛选,得到所需要的DNA片段。
下面是一份分子克隆实验指南,供研究人员参考:1、准备实验室条件:保持实验室的清洁和安全,坚持使用一次性的实验用品,保证环境的无菌。
2、准备所需材料:重组酶、DNA、载体、培养基、试剂、菌种等。
3、DNA的制备:使用DNA分离试剂盒将所需的DNA样本从细胞中提取出来,并通过酶的作用将其切割成适当的长度。
4、制备载体:将载体放入匀质的培养基中,通过质粒扩增技术制备大量的载体。
5、连接重组:使用重组酶将切割后的DNA与载体片段连接起来。
6、转化实验:将重组的DNA分子转化到感受态细胞中,如大肠杆菌,青霉素或氨苄青霉素选择性筛选能力。
7、筛选:将所需的表达目标转移到含有感光荧光素物质的培养基中,观察感光荧光,达到筛选的目的。
8、挑选合适的细胞:将所得的高荧光表达细胞进行挑选,进行康复培养。
9、提取所需重组蛋白:采取适当的提取方法对获得的细胞进行处理,得到所需的重组蛋白。
总之,分子克隆技术是一种非常重要的实验手段,该技术的应用范围很广,能够扩大DNA等分子,开启了生物医学研究的大门,为生命科学研究做出了重要的贡献。
分子克隆及细胞培养基本实验方法

分子克隆及细胞培养基本实验方法1.载体构建实用操作技术1.1菌种的保存—20%甘油菌2体积菌液与1体积70%的甘油混合后,储存于-20℃或-70℃备用。
(甘油菌中甘油的浓度为20-30%均可)1.2甘油菌复苏、培养方法一、挑取甘油菌一环,接种在含100ug/ml Amp的LB固体培养基上(活化菌种),37℃培养过夜(约16小时);挑取一个菌落转接在含100ug/ml Amp的LB液体培养基中,37℃振荡过夜(约12~16小时)。
方法二、直接吸取10~20ul甘油菌,接种在含100ug/ml Amp的LB液体培养基中,37℃振荡过夜(约12~16小时)。
1.3小规模制备质粒DNA(QIA miniprep kit )适于从1~5ml 菌液中制备20ug高拷贝质粒⑴收集菌液,离心1000rpm,1分,弃上清⑵以250ul P1重悬细菌(P1中已加RNase)⑶加入250ul P2,颠倒4~6次轻混,约2~3分(轻混以免剪切基因组DNA,并免长时间消化)⑷加入350ul N3,迅速颠倒4~6次轻混;离心10分,13 000rpm⑸上清入QIAprep柱,离心30~60秒,滤液弃之⑹加入0.5ml PB洗,离心30~60秒⑺加入0.75ml PE洗,离心30~60秒,弃滤液,再离心1分⑻换新管,加入50ul EB,静置1分(EB 37℃预热),离心1分。
1.4酶切反应⑴体系构成(反应体系尽可能小!)pGEM3ZF-huCTLA4-Ig(ul)pAdTrack-CMV(ul)①dd.H2O 17 17②10×NEbuff 2 3 3③10×BSA 3 3④底物DNA 5 5⑤内切酶HindⅢ 1 1XbaⅠ 1 1Total : 30 ul 30ul⑵37℃水浴1~2小时,必要时延长酶切时间至12小时⑶酶切2小时后,取5-10ul 电泳观察酶解是否完全⑷65℃灭活内切酶⑸-20℃保存备用1.5回收目的片段(QIAquick Gel extraction Protocol)⑴胶,尽可能去除多余的胶,称重;⑵加入适量buff QG(300ul QG /100mg胶);>2%的胶,应加大QG用量(600ulQG /100mg);⑶水浴50℃,10min,每2-3min混匀一次,使胶完全溶解!必要时延长水浴时间,胶完全溶解后混合物颜色应为黄色,与buff QG 相似;⑷当DNA片段在<500bp或>4kb时,应加入异戊醇100ul/100mg胶,以提高产物量。
分子克隆技术实验操作手册

生物秀实验频道——专业生物实验中心 /bio101/ 分子克隆技术 实验操作手册 2005生命科学技术学院课程简介分子克隆技术是指DNA的无性繁殖技术。
分子克隆技术课程主要是针对生物技 术专业、生物科学专业、生命科学与技术基地班本科生及生物学类专业研究生而开 设的实验课。
实验内容涉及分子克隆的一些基本方法及基本操作技巧,主要包括分 子克隆和分子杂交两大部分:分子克隆技术:DNA重组技术是分子生物学的核心内容。
本实验利用质粒载体 克隆外源DNA片段,通过这个实验大家可以掌握质粒载体的抽提、外源DNA的准备、 酶切、连接及感受态细胞的制备、连接产物的转化以及阳性克隆子的鉴定和验证等。
分子杂交技术:实验室常用的分子杂交技术主要有 Southern blotting, Northern blotting,Western blotting 及 Dot blotting 等。
Southern blotting 是通过用一种或多种限制性内切酶消化基因组或其它来源的 DNA,经过琼脂糖凝胶 电泳按大小分离酶切所得的片段,随后 DNA 在原位发生变性并从凝胶转移到一固相 支持物上(硝酸纤维素膜或尼龙膜)。
DNA转移至固相支持物的过程中各DNA的相对 位置保持不变,用一定方法(如放射性同位素)标记的 DNA 探针与固着在膜上的 DNA 杂交,经X-光片自显影显现出与探针DNA互补的DNA电泳条带的位置,然后进行分 析。
本实验要求掌握植物总 DNA 的抽提、质量检测、限制性内切酶操作、DNA 的琼 脂糖凝胶电泳、转膜、探针的制备、同位素操作等方面的实验技术。
在转录水平上 研究和了解基因的表达与调控是分子生物学和基因操作的重要内容。
为让学生初步 了解有关RNA的操作过程注意事项,我们还列出Northern blotting的操作步骤以供 选择。
目 录系列一 分子克隆技术 (4)实验一 质粒的制备 (4)实验二 DNA的琼脂糖凝胶电泳 (5)实验三 外源 DNA片段在质粒载体中的克隆 (7)实验四 感受态细胞的制备 (9)CaCl2 感受态细胞的制备实验步骤 (9)电转化法制备大肠杆菌感受态细胞的实验步骤 (10)实验五 质粒的转化及转化子的鉴定 (11)热激法转化实验步骤: (11)质粒电转化大肠杆菌感受态细胞操作步骤 (12)实验六 PCR技术 (12)系列二 Southern 杂交技术 (14)实验一 植物总 DNA的快速少量抽提(CTAB 法) (14)实验二 总 DNA质量检测及酶切 (15)实验三 电泳、转膜 (16)实验四 Southern Blotting (18)系列三 Northern Blotting (24)实验一 RNA Extraction (mini prep) (24)实验二 RTPCR (Reverse transcription PCR) (26)实验三 RNA的电泳,转膜和杂交 (28)附录 试剂配方 (29)一 细菌培养试剂 (30)二 质粒抽提试剂 (30)三 DNA操作试剂 (31)四 RNA操作试剂 (34)Stock Solution: (34)Work Solution (35)系列一 分子克隆技术实验一 质粒的制备质粒是携带外源基因进入细菌中扩增或表达的主要载体,它在基因操作中具有 重要作用。
分子克隆技术

分子克隆技术分子克隆技术是指利用体外的人工方法将一个DNA分子(称为目的DNA)复制到一组DNA分子(称为载体DNA)中的过程。
这项技术能够在体外精确复制和扩增DNA分子,从而可以用于研究基因功能、制备重组蛋白、基因治疗等领域。
下面是分子克隆技术的详细步骤:1.选择载体DNA:首先需要选择一个合适的载体DNA,一般会使用细菌的质粒作为载体,因为细菌质粒具有稳定、易扩增和实验操作简单的特点。
2.制备DNA片段:将目的DNA通过PCR扩增或者其他方法制备出来。
PCR扩增是指利用DNA聚合酶在体外将目的DNA的特定序列进行大规模复制的过程,一般需要利用引物引导PCR反应。
3.处理载体DNA:将载体DNA进行处理,一般需要进行酶切。
通过选择性酶切酶将载体DNA的一部分切除,形成切口,为接下来的目的DNA连接提供空位。
4.连接DNA:将目的DNA与处理后的载体DNA连接起来。
一般利用DNA连接酶进行连接,将目的DNA的末端与载体DNA的末端互补连接。
连接反应通常需要一定时间和温度来保证连接的效率和稳定性。
5.转化细胞:将连接好的DNA转化到细菌等宿主细胞中。
这一步可以通过热激转化、电转化等方法实现。
转化后,将细胞培养在含有相应抗生素的培养基上,只有携带目的DNA的细菌才能存活,从而筛选出含有目的DNA的克隆。
6.筛选克隆:通过筛选抗生素抗性或其他标记物的方法来筛选出含有目的DNA的细菌克隆。
一般需要进行筛选接种、PCR鉴定、酶切及测序等手段来确认克隆是否含有目的DNA,并进一步分析目的DNA的表达和功能。
这些步骤是分子克隆技术的基本流程,但在实际操作中可能会根据具体情况进行相应的调整和优化。
分子克隆技术的发展和应用使得我们可以对基因进行精确操作和研究,对于推动生命科学的发展和应用具有重要的意义。
分子克隆实验流程

分子克隆实验流程分子克隆是将DNA分子从一个生物体中复制并插入另一个生物体中的过程。
这种技术广泛应用于生物学研究和生物工程领域。
下面将详细介绍分子克隆的实验流程。
1.提取DNA首先,需要从源生物体中提取所需的DNA。
这可以通过不同的方法来完成,例如琼脂糖凝胶电泳、菌落PCR和基因组DNA提取试剂盒等。
提取的DNA需要是目标基因的完整、纯净的样本。
2.回收DNA片段将提取的DNA片段和载体(例如质粒)切割,以回收想要克隆的DNA 片段。
常用的酶剪切酶包括限制性内切酶和退火酶。
将酶切割后的DNA片段与载体的切割端黏合,形成重组DNA。
3.转化纯化将重组DNA转化到宿主细胞中。
通常使用大肠杆菌作为宿主细胞。
这可以通过脉冲电击、热激转化或化学转化来实现。
这一步的目的是将重组DNA导入到宿主细胞中,以便宿主细胞可以复制和表达克隆的DNA。
4.鉴定克隆细胞通常,我们通过选择性培养、荧光染色、PCR检测等方法来鉴定具有克隆DNA的细胞。
在选择性培养基上生长可以表明细胞成功地接受了重组DNA。
此外,如果重组DNA带有可观察的荧光标记,可以使用荧光显微镜进行观察。
PCR检测可以验证目标基因的存在。
5.扩增克隆细胞选取鉴定出的克隆细胞进行扩增。
这可以通过将克隆细胞培养在含有选择性抗生素的培养基上来实现。
只有带有插入DNA的细胞可以在这种培养条件下生存。
选择性培养的目的是增加克隆细胞的数量,以便后续的实验。
6.提取插入DNA从扩增的克隆细胞中提取含有插入DNA的重组质粒。
通常使用薄膜过滤法将细菌细胞去除,然后使用DNA提取试剂盒从细菌残渣中提取质粒。
提取的质粒可以通过琼脂糖凝胶电泳等方法进行纯化和鉴定。
7.鉴定插入DNA使用不同的试验方法来鉴定插入DNA的准确性和完整性。
这可能包括核酸测序、酶切鉴定、PCR扩增等。
通过这些方法可以验证克隆的DNA是否与目标基因的序列完全一致。
8.进一步应用得到插入DNA后,可以进行进一步的应用,例如重组蛋白表达、基因改良、产生转基因生物等。
分子克隆实验指南

分子克隆实验指南分子克隆是现代生物学领域的一项核心技术,也是基因研究、药物研发和农业开发过程中必不可少的手段之一。
在这篇指南中,我将会简要介绍如何进行分子克隆实验,以帮助初学者更好地理解、掌握这项技术。
同时,我也建议实验者在进行实验前,详细阅读当地的安全操作规程,并在合适的实验室环境中进行操作。
一、材料准备在进行分子克隆实验前,我们需要准备以下材料和试剂:1. DNA的扩增产物和载体DNA2. 限制性内切酶3. T4 DNA连接酶4. 细菌菌种5. 热激酶6. 磷酸缓冲液7. 离心管、PCR管、琼脂糖凝胶和电泳槽等相关实验器材。
二、实验步骤1. PCR扩增将目标DNA扩增出来,制备扩增产物。
同时,也需要提纯产物,将其溶于蒸馏水中,以备后续操作。
2. DNA限制性内切酶切割选择两种限制酶,将目标DNA和载体DNA分别切割。
切割产品应该能够被T4 DNA连接酶形成连接。
在T4 DNA连接酶形成连接的基础上,我们需要将其转化到大肠杆菌等细菌中进行培养。
3. DNA连接将切割后的DNA和载体DNA混合,加入T4DNA连接酶,进行DNA连接。
连接成功后,进行质粒的转化操作。
4. 质粒转化将DNA与转化者(例如大肠杆菌)一起培养几个小时,使其在培养基中繁殖生长。
之后,分离转化菌落,进行鉴定。
5. 鉴定正式的及相关标签元素为了确保成功的分子克隆,在选取符合需要的转化菌落后,除了进行相关的鉴定,还需要使用相关的标签元素。
这些标签元素可以用于识别有效的重组表达载体,并进行大规模的表达。
三、实验注意事项1. 选取嵌合体目标和载体的选择应按照相关配对的要求进行。
在酶切反应中,应注意酶的用量。
同时,也需要注意处理过程中不要将酶污染。
2. 进行DNA限制性内切酶切割时,应注意温度和反应时间。
3. 在进行DNA连接后,将混合物放置于水浴中,沸腾数分钟,以便DNA连接的更加稳定。
在连接DNAs之前,应该对酶进行热灭活。
4. 质粒转化后,为了鉴定高效的表达,需要进行多次筛选和鉴定。
常用分子克隆实验方法-推荐下载

常⽤分⼦克隆实验⽅法-推荐下载常⽤分⼦克隆实验⽅法I⼀、植物总DNA的⼩量提取⽅法1:提取吸附法。
⽆须巯基⼄醇、氯仿等有毒物质,产物⽆须Rnase处理。
(1)充分研磨。
称取约0.2克植物组织,加⼊液氮充分研磨3-5min,稍后加约1ml溶液A,继续研磨⾄略粘稠的组织匀浆,⽤⼤⼝1ml吸头将所有溶液移⾄1.5ml离⼼管中,55℃⽔浴30min;(2) ⾼速离⼼去杂质。
10,000rpm离⼼5min,取约600ul上清⾄新1.5ml离⼼管;(3) 核酸吸附。
往上清液中加⼊1倍的异丙醇,轻轻混匀,再加⼊总体积1/4已混匀的溶液B,静置3min;(4) 低速离⼼沉淀。
5000rpm离⼼1min,轻轻倒掉上清,并⽤吸⽔纸轻吸离⼼管⼝,再⽤移液枪吸⾛⼤部分残余液体;(5) 75%⼄醇清洗。
加⼊1ml75%⼄醇,5000rpm离⼼30s,轻轻倒掉上清,⽤吸⽔纸稍吸离⼼管⼝。
重复该步骤⼀次,再5000rpm离⼼30s,然后⽤移液枪吸⾛管底的残液,晾⼲5min;(6) 核酸洗脱。
加⼊约55ul TE(PH8.0)⾄管底,轻轻重悬硅⼟,静置3min,10,000rpm离⼼1min,⽤⼩枪头轻轻吸取出50ul管底溶液,冷藏。
⽅法2:CTAB法,此为在经典⽅法基础上,经过摸索改进,提⾼了得率,减少了污染。
(1)充分研磨。
称取约0.2克植物组织,加⼊液氮充分研磨3-5min,稍后加约1mlCTAB提取液,继续研磨⾄略粘稠的组织匀浆,⽤⼤⼝1ml吸头移⾄1.5ml离⼼管,65℃⽔浴30-60min。
(2) 氯仿抽提。
10,000rpm离⼼3min,取约600ul上清。
加⼊1倍的氯仿,轻轻混匀,10,000rpm离⼼3min,取上清再抽提1遍。
(3) 核酸沉淀。
加⼊预冷的1倍异丙醇或2倍⼄醇,轻混匀,6000rpm离⼼3min,弃上清。
(4) 清洗沉淀。
轻加⼊1ml 75%⼄醇,再吸掉上清,重复⼀次,倒置于吸⽔纸或横放于离⼼管架上晾⼲5min。
分子克隆实验
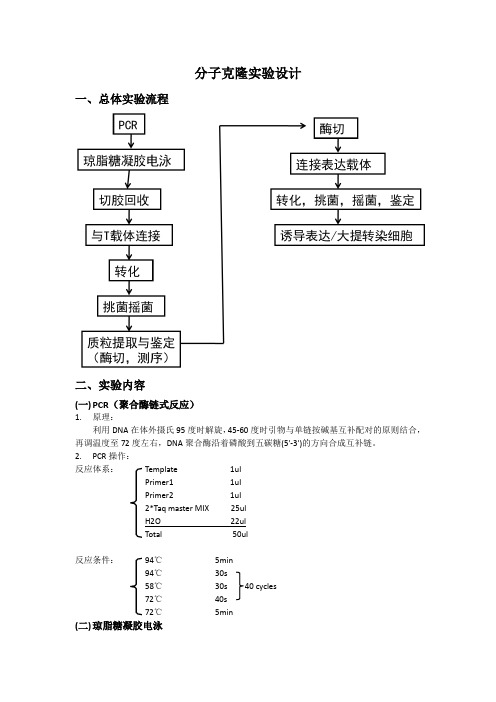
分子克隆实验设计一、总体实验流程二、实验内容(一)PCR(聚合酶链式反应)1.原理:利用DNA在体外摄氏95度时解旋,45-60度时引物与单链按碱基互补配对的原则结合,再调温度至72度左右,DNA聚合酶沿着磷酸到五碳糖(5'-3')的方向合成互补链。
2.PCR操作:反应体系:Template 1ulPrimer1 1ulPrimer2 1ul2*Taq master MIX 25ulH2O 22ulTotal 50ul反应条件:94℃5min94℃30s58℃30s 40 cycles72℃40s72℃5min(二)琼脂糖凝胶电泳1.原理:琼脂糖凝胶具有网络结构,分子通过时会受到阻力,大分子物质在涌动时受到的阻力大,因此在凝胶电泳中,带电颗粒的分离不仅取决于净电荷的性质和数量,而且还取决于分子大小。
2.凝胶电泳步骤:1)称取0.5g琼脂糖至50ml 电泳缓冲液(TAE)中,摇匀;同时装置制胶架。
2)加热至琼脂糖完全融化,期间停止加热摇动3次,即无可见漂浮物,澄清为止。
3)加入1ulEB染液,充分摇匀。
4)缓慢倒入制胶架内,冷却30-60min,至凝胶充分凝固,放入电泳槽中即可使用。
5)使用时,将样品点入相应的加样孔内,即可开始电泳,待指示剂位于整块凝胶2/3位置时,停止电泳,在紫外灯下观察样品条带。
(三)切胶回收(天根生化公司凝胶回收试剂盒)1. 将空小离心管称重。
将回收的带DNA 片段的凝胶放入其中、再次称重,确定凝胶净重。
2. 将离心管中的凝胶捣碎以助于凝胶溶解。
按胶的重量加入三倍体积的Extraction buffer (1mg凝胶加3μl Extraction buffer )。
3. 55℃恒温加热直到凝胶完全融化(5-15 分钟),每隔2-3分钟需将管内混悬液混匀一次。
4. 按照1:1比例加入异丙醇(1mg凝胶加1μl异丙醇),混匀。
5. 将混合液全部移入Spin column、6000rpm离心1分钟,弃去接液管中的液体。
分子克隆与表达实验

分子克隆与表达实验流程1.选取菌株从保存的海冰细菌中随机选取某几株,挑取其单菌落于发酵培养基(2216)活化培养,1d后将活化的菌株5ml接入70ml发酵培养基中,12℃,100rpm摇床培养,大约4~5d后对菌株在4℃下,12000rpm进行离心,收集菌体,于4℃保存。
2.提取菌株全基因组在超净工作台上取120μl离心后的菌株悬浮于400μl的1×TE缓冲液中,加入10%SDS 100μl和2mg/mL的溶菌酶20μl,混匀后置于37℃保温1h,加入5M 的NaCl 70μl充分混匀,置于65℃水浴10min,加入等体积的苯酚/氯仿/异戊醇(25:24:1)混匀,离心(12000rpm、8min),取上层水相于新离心管中并加入等体积的氯仿/异戊醇(24:1)充分抽提,取上层水相加入两倍体积的无水乙醇沉淀DNA,并用70%的乙醇淋洗两次,倾去乙醇,真空干燥后溶于25μl的1×TE缓冲液。
对全基因组进行电泳检验,其余的置-4℃冰箱保存备用。
3.根据目的蛋白设计引物本实验研究的是小分子蛋白:谷胱甘肽转移酶(GST)、硫氧还蛋白(Trx)和谷氧还蛋白(Grx)。
根据这三种蛋白的基因用Primer5设计引物。
引物名称与序列引物名称引物序列,方向5′——3′合成规格(OD)GST1(1) TTTATGGYGTMCCKCTTTCTC 2GST1(2) CA TGCTSCA TATTSATTAWCTG 2GST2(1) AGTSTCGCCGTTTAATCAACC 2GST2(2) YKCATY AAKYTGCTCRCCMSC 2GST3(1) TTTA TGGCGTACCTCTTTCTCC 2GST3(2) CGGTAA TACCTAACTGCTCAGC 2GST4(1) TCTGTTACCTCGCCTTAT 2GST4(2) AAAGCCA TTATCTTCTGC 2GST5(1) ACTTA TTTGGAGCA TTGAGCGG 2GST5(2) TCGGCGGTTCAAGAAAAG 2Trx1(1) AAGAATTCA TGAGCGAGAAAATAATCC 2Trx1(2) CGCAAGCTTTTAAAGA TTGTTTTCTA 2Trx2(1) AAGAATTCA TGAGCGAGAAAATAATC 2Trx2(2) CTCAAGCTTTTAGA TGTTGTTTTCTA 2Trx3(1) CGGAGAGCCA TA TGAGCGAGAA 2Trx3(2) TTGTTTAAATCTCGAGATTGTTTTCT 2Grx1(1) GTGTTATCTGGTGGTA TGG 2Grx1(2) TTGTGCTGCTGCGTTTTG 24.PCR扩增以菌株的全基因组为模板,对上述引物进行PCR,从中寻找含有目的基因的菌株,对含有目的基因的菌株进行验证并保种。
分子克隆技术的操作流程

分子克隆技术的操作流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!分子克隆技术是一种在分子水平上进行遗传操作的技术,用于复制和扩增特定的 DNA 片段。
分子克隆技术步骤

分子克隆技术步骤第一步:选取目标DNA在开始分子克隆之前,需要从一个生物体中选择一个含有所需DNA序列的样本。
这可以是任何生物体的DNA,例如人类、动物、植物或微生物。
第二步:DNA提取从所选生物体提取DNA。
这可以通过使用一系列化学和物理方法来完成,例如细胞裂解、蛋白酶处理、DNA沉淀和洗涤等。
第三步:选择一个合适的载体载体是一种DNA分子,可以容纳目标DNA序列并将其复制。
在分子克隆中最常使用的载体是质粒。
质粒是圆形的双链DNA分子,存在于许多细菌和酵母种类中,并被广泛用于分子生物学研究。
第四步:限制性内切酶切割将目标DNA和载体同时使用限制性内切酶(Restriction Enzymes)酶切。
限制性内切酶是一种可以识别和切割特定DNA序列的酶。
通过在目标DNA和载体的特定位置上切割,可以为将两者连接提供互补的末端。
第五步:DNA连接将目标DNA和载体连接在一起。
将目标DNA和载体的DNA片段混合,并在其末端形成互补碱基,然后使用DNA连接酶将两者连接在一起。
连接后的DNA分子被称为重组质粒。
第六步:转化将重组质粒引入细菌或酵母等微生物细胞中,这个过程称为转化。
这可以通过将细菌细胞暴露在低温高压胁迫下来实现,使得细胞膜变得更加渗透性,可以将质粒引入细胞内。
第七步:筛选和鉴定筛选出含有重组质粒的细菌或酵母细胞。
一种常用的筛选方法是将细菌培养在含有抗生素的培养基上,只有携带重组质粒的细菌才能在含有抗生素的环境下存活。
此外,还可以使用标记基因和特定染色剂等方法来鉴定重组质粒。
第八步:扩增和纯化用培养液扩增含有重组质粒的细菌或酵母细胞。
随着细菌或酵母细胞的生长,它们会复制重组质粒并将其传递给后代细胞。
然后使用一系列纯化步骤,如离心、洗涤和电泳等手段,将其中的重组质粒提取纯化。
总结:分子克隆技术的主要步骤包括选取目标DNA、DNA提取、选择合适的载体、限制性内切酶切割、DNA连接、转化、筛选和鉴定,以及扩增和纯化。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
分子克隆及细胞培养基本实验方法1.载体构建实用操作技术1.1菌种的保存一20%甘油菌2体积菌液与1体积70%的甘油混合后,储存于-20C或-70 C备用。
(甘油菌中甘油的浓度为20-30%均可)1.2甘油菌复苏、培养方法一、挑取甘油菌一环,接种在含100ug/ml Amp的LB固体培养基上(活化菌种),37 C培养过夜(约16小时);挑取一个菌落转接在含100ug/ml Amp的LB液体培养基中,37C振荡过夜(约12~16小时)。
方法二、直接吸取10~20ul甘油菌,接种在含100ug/ml Amp的LB液体培养基中,37 T振荡过夜(约12~16小时)。
1.3 小规模制备质粒DNA (QIA miniprep kit )适于从1~5ml菌液中制备20ug高拷贝质粒⑴收集菌液,离心1000rpm,1分,弃上清⑵以250ul P1重悬细菌(P1中已加RNase)⑶加入250ul P2,颠倒4~6次轻混,约2~3分(轻混以免剪切基因组DNA,并免长时间消化)⑷加入350ul N3,迅速颠倒4~6次轻混;离心10分,13 000rpm⑸上清入QIAprep柱,离心30~60秒,滤液弃之⑹加入0.5ml PB洗,离心30~60秒⑺加入0.75ml PE洗,离心30~60秒,弃滤液,再离心1分⑻换新管,加入50ul EB,静置1分(EB 37 C预热),离心1分。
1.4酶切反应⑴体系构成(反应体系尽可能小!)pGEM3ZF-huCTLA4-Ig (ul) pAdTrack-CMV (ul)① dd.H20 ② 10X NEbuff 2 3 ③ 10X BSA 3 ④ 底物DNA 5 ⑤ 内切酶 Hi nd 川1 Xba I1 Total : 30 ul30ul⑵37C 水浴1~2小时,必要时延长酶切时间至12小时⑶酶切2小时后,取5-10ul 电泳观察酶解是否完全 ⑷65 C 灭活内切酶 ⑸-20 C 保存备用1.5 回收目的片段(QIAquick Gel extraction Protocol )⑴胶,尽可能去除多余的胶,称重;⑵加入适量buff QG (300ul QG /100mg 胶);>2%的胶,应加大 QG 用量(600ul QG/100mg );⑶水浴50 C, 10min ,每2-3min 混匀一次,使胶完全溶解!必要时延长水浴时间, 胶完全溶解后混合物颜色应为黄色,与buff QG 相似;⑷当DNA 片段在<500bp 或>4kb 时,应加入异戊醇100ul/100mg 胶,以提高产物 量。
此步不离心。
DNA 片段在500bp~4kb 时,加入异戊醇并不能提高产量;⑸ 结合:将混合物转入 QIAquick 柱,离心13000rpm ,1min ;(柱容量 800ul/次); ⑹洗:0.75ml buff PE ,离心13000rpm ,1min ; ( DNA 用于盐敏感操作时,如平端连接、直接测序,加入 PE 后静置2-5min );弃离心液,再离心 13000rpm ,1min ,以去除剩余的乙醇;⑺将QIAquick 柱置于一清洁的 1.5ml Ep 管,加入 30~50ul buff EB 或 出0 (滴 于QIAquick 膜上!),静置 1min ,离心 15000rpm , 1min ;⑻-20 C 保存备用。
17 17 3 3 5 1 11.6连接反应反应组份数量(ul)①dd.H20 8②T4 10X buff 2③载体DNA(ul) 1目的片段(ul)8④T4DNA连接酶 1Total : 20 ul⑵16C水浴12-16小时(过夜)⑶直接用于转化或-20 C保存备用1.7感受态细胞的制备⑴ 菌种10-20ul入5ml LB液体培养基(不加Amp), 37C振摇4-6 hr,至中对数期;⑵菌液冰浴10min,分装2管,离心(4 C, 4 000-6 000rpm )收菌;⑶弃上清,将菌体悬浮于800ul 0.1M CaCb (冰预冷)中,冰浴10min,离心(4C,4000-6000rpm );⑷ 再将菌体悬浮于200ul 0.1M CaCb (冰预冷)中,冰浴,12-24hr可用。
菌体沉淀时,重悬操作要轻。
1.8转化⑴新鲜感受态细胞200ul,加入2-10ul连接产物,轻轻旋转混合,冰浴30-60min ;⑵42C热休克90sec,勿摇动试管,再冰浴2min ;⑶加入液体LB培养基(无抗生素)800ul,37 C温和振摇45-60min ;⑷离心4000-6000rpm,2min,弃去900ul 上清;⑸剩余物重悬细菌,铺Amp板,平放20min,倒置培养10-14hr。
1.9鉴定重组子常规PCR法鉴定重组子(25ul反应体系,1ul菌液)或抽提质粒DNA酶切鉴定。
⑴PCR反应体系:试剂数量终浓度① dd.H2O 17.5 ul②10x PCR缓冲液 2.5 ul 1 x③ MgCI2 (25mM ) 1.5 1.5 mM④ dNTPmix(2.5mM) 1 ul 50uM⑤上游引物(25pmol/ul ) 0.5 ul 0.5uM下游引物(25pmol/ul ) 0.5 ul 0.5uM⑥TaqDNA 聚合酶(3U/ul) 0.5 ul 0.06U/ul⑦模板(菌液) 1ul终体积25ul⑵PCR反应参数:94°C, 5min ;“94C, 30sec、50 C, 30sec、72 C, 1min ” x 20-30 循环;72 C, 6min ;4 C,保存备用;⑶电泳鉴定,以挑选阳性重组子,注意阳性、阴性对照的设立。
2.大规模制备质粒DNA( QIAGEN Plasmid Maxi Protocol )适于从100~250ml菌液中制备100-200 ug高拷贝质粒⑴涂板挑取单克隆菌落,入LB选择性培养基,37 C振摇8hr后,取适量菌液(1/500-1/1000 )入100-250ml LB 选择性培养基(高拷贝100-250ml LB,低拷贝500ml LB ), 37 C 振摇12-16hr;⑵收获菌液,4 C离心6 000rpm (6 000g )1 5min,弃尽上清;注意:在此步可将菌(离心沉淀)冻存于-20 C,留待以后操作。
⑶以10ml P1重悬细菌(P1中已加RNase);⑷加入10ml P2,颠倒4~6次轻混,室温放置5min (轻混以免剪切基因组DNA , 并免长时间消化);⑸加入冰预冷10ml P3,迅速颠倒4~6次轻混,冰上放置20min,以促进沉淀形成;⑹4C离心13 OOOrpm (20 OOOg以上)30min (离心前,应再次混合样品);上清入新管,再次离心13 000rpm(20 000g 以上)15min;⑺第二次离心的同时,用10ml QBT缓冲液平衡QIAGEN柱,不需离心(重力引流);⑻结合:快速移上清(第二次离心)入QIAGEN柱,使其在重力引流下进入树脂(resin);⑼清洗:3Oml QC 洗2 次(重力引流);⑽洗脱:15ml QF 缓冲液洗脱结合的DNA ,将洗脱液收集入新管(可将洗脱液暂时存放于4 °C数小时);(11)沉淀:加入10.5ml异丙醇(室温,以免增加盐沉淀),混匀,立刻于4 C离心9 5OOrpm (15 OOOg)3Omin (离心前在管底作好标记,此步重要!),小心缓慢地倒出上清(以免弃去DNA 沉淀);注意:此步如果DNA 沉淀不可见,可以1.5ml 75%乙醇冲洗管底标记处,然后转移至1.5ml离心管,15 000 rpm (20 000g)30min,此时沉淀往往可见,可重复此步骤1 次,以收集尽可能多的质粒DNA(12)再清洗:5ml 70%乙醇(室温,以免增加盐沉淀)洗,离心9 500rpm (15000g)10min,小心缓慢地倒出上清;(13)空气中干燥沉淀5-10min,以适当体积的TE,pH8.0或10mM Tris-Cl, pH8.5 溶解DNA 沉淀(注意冲洗管壁,过度吹打会剪切DNA 应避免);(14)琼脂糖凝胶电泳分析;(15)质粒DNA浓度测定(紫外分光光度仪),OD260/OD280=1.6~1.8 。
3.细胞培养常规方法3.1 293 细胞常规复苏、培养、传代及冻存⑴培养液配制:DMEM,加入NaHCO2 3.7g/L,庆大霉素8万u,调pH至7.0-7.2(比细胞生长所需终末pH 低0.2-0.3),定容至1L,0.22um 过滤分装即可;⑵293生长要求:DMEM,含2-10%的FBS (按不同实验对细胞生长速度要求的不同而不同);⑶293细胞的复苏:取液氮中冻存的细胞1管,迅速置37C水浴1-3min融化;转移至10ml 离心管加入8ml DMEM 10% ,轻轻吹打混匀,低速离心1 000rpm ,5min ;弃上清,加入8ml DMEM 10%重悬细胞;转入25ml培养瓶补足培养液,置CO2孵箱37C、5% CO2培养;⑷293 细胞的培养:DMEM 10% ;⑸293 细胞的传代(1:2-3):弃培养液,NS 或无血清培养液洗1-2 次,加入适量(数滴,或1-3ml ;按培养的293细胞代数而定,代数低时,胰酶适当多加,反之,可能数滴即可)0.25%胰酶消化液,37 E消化1-3min,镜下观察消化情况,待细胞变圆、脱离瓶壁时,轻轻拍打瓶壁,使细胞完全脱离,加入适量培养液轻轻吹打使之分散;转入10ml 离心管,低速离心1000rpm,2-5min;弃上清,加入适量DMEM 10% 重悬细胞;转入适当大小培养瓶(1:2-3)并补足培养液,置CO2孵箱37 C、5%CO2培养;⑹293 细胞的冻存:取生长对数期的培养细胞,常规消化、离心,细胞沉淀以1-2mlDMEM 10% 重悬,计数;重悬细胞10% DMSO、40% FBS,以DMEM 10% 补足体积,细胞终浓度为1X 106/ml ;转入冻存管1.5ml/Tube,封口膜封口,置-80C冰箱24hr;次日将细胞转入液氮,长期保存。
3.2 L-O2 细胞的培养⑴L-O2细胞生长要求:DMEM/1640 (1 : 1)混合培养液,含5-10%的FBS (按不同实验对细胞生长速度要求的不同而不同);⑵L-O2细胞的传代:比例1: 3-5 ;⑶ 余基本同293 细胞。
3.3 细胞计数法⑴ 染色:常规消化、离心,弃上清,加入1 或2ml 培养液,轻轻吹打重悬细胞,吸20ul 细胞悬液,加入760ul NS ,加入20ul 台盼蓝,混匀,置2-3min ;⑵ 计数板:用96 %酒精冲洗计数板后,用擦镜纸擦净,另擦净盖玻片 1 张,把盖片覆在计数板上,使之微微移向一侧,露出计数板台面少许,以便滴加细胞悬液;⑶ 加稀释液:滴加1-2 滴已染色的细胞悬液,使之充满计数板和盖片间空隙中;⑷ 镜检:计算四大方格内细胞数,压中线者只计算左线和上线者;⑸计算公式:细胞数=4大格细胞总数/4 X 10 000 X稀释倍数;本法:细胞数=4大格细胞总数X 105;⑹ 按培养要求接种培养。