203EMEA《遗传毒性杂质限度指导原则》介绍
化学药品注射剂临床前安全性评价的基本技术要求

May.2009,Vol.6No.5药品审评化学药品注射剂临床前安全性评价的基本技术要求王海学王庆利孙涛(国家食品药品监督管理局药品审评中心,北京100045)【摘要】注射剂产品在临床使用的风险性较高,技术研发单位需足够重视其安全性研究和评价。
临床前研究中需考虑进行必要的毒性实验以支持注射剂申请进入临床,并重视上市的风险评价。
【关键词】注射剂;临床前;安全性作者简介:王海学,男,博士后,副主任药师。
主要从事新药临床前药理毒理技术审评,负责抗肿瘤化学药和生物制品的技术评价工作。
Email :wanghx@国内近年出现了多起注射剂临床使用的安全性问题,这引起国家药监部门的高度重视。
为此,国家食品药品监督管理局(以下简称“国家药监局”)要求逐步加强对注射剂的风险控制要求,其中加强科学技术评价是风险控制的重要环节之一。
国家药监局近几年已经开始对注射剂的安全性评价开展了调研和技术交流,希望能够进一步完善注射剂的技术研究要求和安全性评价方法。
注射剂中的主药、辅料、杂质等通常可直接入血,其暴露量和绝对生物利用度高,因此注射剂与其他剂型比较,更需加强安全性的技术要求和风险控制。
注射剂的安全性评价包括药学、非临床和临床专业评价,其中非临床安全性评价的作用主要在于:有助于进一步加强药学质量控制,如处方组成、剂型选择、杂质限度等;预测初次进入人体试验用药的安全性;探索风险产生原因、机制和控制措施。
注射剂非临床安全性评价是在剂型选择的必要性与合理性得以满足的前提下,密切结合药学质控而采取的必要的非临床安全性评价,以达到尽可能控制产品上市的安全性风险。
1注射剂非临床安全性评价的总体考虑注射剂非临床安全性评价中需分析具体注射剂产品的风险因素,根据其风险产生原因考虑技术研究要求。
非临床安全性评价的常规关注点为评估注射剂的毒性反应性质、量效关系、暴露量、安全范围,进而为其进入临床试验或上市提供安全性支持信息。
考虑到药品风险评价的系统性,非临床安全性评价中应:①密切结合药学和临床信息考虑;②密切结合药效学、药代动力学等信息考虑;③进行规范的毒性试验研究和结果分析评价;④具体问题具体分析[1]。
基因毒性杂质

什么是基因毒性杂质对于基因毒性杂质的定义主要是指:在以DNA 反应物质为主要研究对象的体内/ 体外试验中,如果发现它们对DNA 有潜在的破坏性,那可称之为基因毒性。
对没有进行体内实验的情况下,也可以根据关联系做一些相关的体外实验去评估该物质在体内的毒性。
如果没有关联评估的,体外基因毒性物质经常被考虑为假定的体内诱变剂和致癌剂。
GUIDELINE ON THE LIMITS OF GENOTOXIC IMPURITIES ( EMEA/CHMP/QWP/251344/2006 )基因毒性杂质的风险按照目前的法规来说,(体内)基因毒性物质在任何摄入量水平上对DNA 都有潜在的破坏性,这种破坏可能导致肿瘤的产生。
因此,对于基因毒性致癌物,不能说“不存在明显的阀值,或是任何的摄入水平都具有致癌的风险”。
可接受风险的摄入量对于那些可以与DNA 进行反应的化合物,由于在较低的剂量时机体保护机制可以有效的运行,按照摄入量由高到低所造成的影响进行线性推断是很困难的。
目前,对于一个给定诱变剂,我们很难从实验方面证明它的基因毒性存在一个阀值。
特别是对某些化合物,它们可以与非DNA 靶点进行反应,或一些潜在的突变剂,在与关键靶位结合之前就迅速失去了毒性。
由于缺乏支持基因毒性阀值存在的有力证据,而使得我们很难界定一个安全的服用量。
所以有必要采取一个新观点:确定一个可接受其风险的摄入量。
可接受其风险的摄入量即毒理学阈值一般通用的被定义为Threshold of Toxicological Concern (TTC)。
具体含义为:一个“ 1.5ug/day ”的TTC 值,即相当于每天摄入1.5ug 的基因毒性杂质,被认为对于大多数药品来说是可以接受的风险(一生中致癌的风险小于100000 分之1 )。
按照这个阀值,可以根据预期的每日摄入量计算出活性药物中可接受的杂质水平。
在特定的条件下一些基因毒性杂质也可以有较高的阈值。
EMEA关于金属催化剂或金属试剂残留量限度规定的指导文件简介

EMEA关于金属催化剂或金属试剂残留量限度规定的指导文件简介2008年2月21日EMEA/CHMP颁布了金属催化剂或金属试剂残留量限度规定的指导文件(GUIDELINE ON THE SPECIFICATION LIMITS FOR RESIDUES OF METAL CATALYSTS OR METAL REAGENTS),并将于2008年9月1日在欧盟正式实施。
该指导文件从1998年6月开始起草,历经多次咨询、讨论,最后定稿。
目前国内对药物中金属残留量的控制限度还缺乏明确的技术要求。
本文对EMEA指导文件进行简要介绍,通过对14种金属的分类、分析方法和控制限度的了解,希望对药物质量控制和技术评价有所帮助。
一、该指导文件的结构框架该指导文件包括背景介绍、定义和范围、法规基础、正文、名词、参考文献、附录等七个部分。
其中正文中包括了重金属分类、暴露量限度、浓度限度的设定、分析方法、批结果和检测频率、金属残留报告水平等6个方面。
该文件有三个附录。
附录1是允许日接触量(PDE)设定的考虑因素,附录2是14种金属的单论,包括各金属简介、食物摄取情况、不同给药途径和周期的毒性数据、PDE评估结论,附录3是PDE和浓度限度的计算举例。
二、该指导文件的目的、定义和应用范围在原辅料合成中可能用到金属催化剂或金属试剂,如铂、钯、锌、铁、铬等,这些金属可能原料药中残留,它们可能以最初形式存在,也可能由于后续化学过程以其他形式存在。
原辅料中残留的金属会进一步带入到制剂中。
这些残留的金属通常不具有治疗作用,基于安全性和质控的需要进行严格控制。
该指导文件的目的是为原辅料和制剂中残留的金属催化剂或金属试剂推荐最大可接受浓度限度。
该指导原则适用于新批准和已上市的制剂,EMEA为已上市制剂设定了5年的执行过渡期。
指导文件不适用于正处于临床研究阶段的新原料药和辅料,他们可以设定更高的可接受的金属残留限度。
该指导文件不适用于原辅料中应有的金属成分(如用作成盐离子对的金属),也不适用于制剂中应有的含金属辅料(如制剂中的铁氧化物颜料)。
原料药国际注册中基因毒性杂质的法规解读

原料药国际注册中基因毒性杂质的法规解读摘要遗传毒素是一类极富挑战性的杂质,并已被证明即便在低浓度条件下依然具有毒性。
因此美国和欧盟的药品监管机构以及人用药品注册技术要求国际协调会(ICH)都特别指定了它们在原料药和成品药中的限量。
通过解析原料药在美国和欧盟注册中涉及到的关于基因毒性杂质控制的法规,为中国制药企业提供相关技术指导以推动中国药品出口事业的增长。
1、1. 药品中的杂质的定义及分类药品中的杂质定义为无任何疗效、且可能引起副作用的物质。
因此,必须控制杂质水平,以确保药品的安全性达到人用要求。
杂质会影响药品的安全性和研发时间,例如,药物开发中,如果必须采用多种手段进行杂质表征,并将其去除至可接受水平,所需时间将会显著增加。
根据人用药品注册技术要求国际协调会(ICH)指南,原料药相关的杂质可分为三个主要类别:有机杂质、无机(元素)杂质以及残留溶剂。
在这三类中,遗传毒性杂质是一种特例,既便在低浓度条件下也有着重大的安全风险。
这是因为它们可能具有致突变性,可能导致DNA 损伤,从而增加罹患癌症的风险。
原料药中的杂质来源包括以下几个方面:①原料及其污染物;②试剂和催化剂;③溶剂;④中间体;⑤降解产物;为保护患者,需要将药物的杂质水平降到可接受的安全限度内,因此各国都已相继制定了杂质控制指南,这些指南都专注于利用规定限度控制药品中的杂质含量,其中由ICH 与美国食品药品监督管理局(FDA)制定的指南较权威且影响大。
例如,ICH Q3A 通过制定杂质的报告、鉴定和认证阈值以监管新原料药中的杂质。
ICH Q3B 是其同等的新药杂质指南。
ICH Q3C 和 ICHQ3D 分别控制残留溶剂与重金属元素杂质的限量。
已经正式生效的ICH M7 是ICH 专家工作组制定的一本关于药物中DNA 反应性杂质的指南[5],包括如何根据结构活性分析来评估药品中杂质的潜在基因毒性,如何确定关键毒性阈值(毒性关注阈值TTC)。
FDA关于基因毒性杂志的指南

FDA关于基因毒性杂志的指南这是FDA关于基因毒性杂志的指南,或许对你有帮助FDA guidance on genotoxic impuritiesBy Nick Taylor, 16-Dec-2008Related topics: Materials & Formulation, QA/QC & validationThe FDA has issued draft guidance on how manufacturers shouldevaluate the safety of products that contain genotoxic and carcinogenic impurities.Genotoxic and carcinogenic properties can be acceptable traits of active pharmaceutical ingredients (APIs) but when these are impurities, which generally do not have a beneficial effect, their presence shouldbe minimised.To help manufacturers achieve the lowest technically feasible levelsof these impurities, or reduce them to quantities that convey no significant cancer risk, the US Food and Drug Administration (FDA) has issued guidance on the subject.By following the guidelines manufacturers should understand what the FDA requires to approve applications at various developmental stages and how these standards can be achieved.One recommendation is that manufacturers change the synthetic or purification routes to reduce impurity formation or increase its removal.The FDA regards 1.5μg per day as an acceptable level for impurities but this may not be appropriate in every case. In addition, higher levels may be allowed during clinical development.Further characterisation of the risks posed by the impurities by studying the mechanism of action or performing weight-of-evidence approaches can also add support to impurity specifications.Products released prior to the issuing of the guidance are covered by it if a specific safety signal highlighting increased risk is detected.Supplemental applications in previously approved products are also covered if they require a significant change to the labelling that suggests potential for increased carcinogenic risk.The complete guidance can be found here.本文转自诺贝尔学术资源网 ,?文献互助、学术交流和学术资源基因毒性杂质:基因毒性化合物指能直接或间接损伤细胞DNA,产生致突变和致癌作用的物质。
EMEA人用药品委员会(CHMP)《遗传毒性杂质限度指导原则》中文译稿

EMEA人用药品委员会(CHMP)《遗传毒性杂质限度指导原则》原文:European Medicines Agency: Guideline on the Limits of Genotoxic Impurities.CPMP/SWP/5199/02。
EMEA/CHMP/QWP/251344/2006。
London, 28 June 2006摘要遗传毒性杂质的毒理学评估和原料药中此类杂质的可接受限度确定是一个难题,现有ICHQ3X指南中未充分说明。
常用遗传毒性杂质数据库差异很大,而数据库是决定可接受限度评估所用方法的主要因素。
当运用已建立的风险评估方法所需资料缺乏时,包括致癌性长期试验资料或提供遗传毒性阈值机制证据的资料等,建议采用毒理学担忧阈值(TTC)所定义的普遍适用方法。
对大部分药物,遗传毒性杂质摄入量为1.5µg/天的TTC值时,认为相关的风险可接受(终身癌症风险<1/100000)。
根据该阈值,原料药中遗传毒性允许水平可根据预计每日剂量计算得到。
短期给药等特定情况下可能有理由提高限度。
1.介绍在原料药(Q3A,新原料药中的杂质)和药物制剂(Q3B,新药物制剂中的杂质)的指导原则中描述了杂质限度确定的一般概念,将限度确定定义为获得和评价特定水平下单个杂质或特定杂质谱的生物学安全性资料。
对于有潜在遗传毒性的杂质,确定可接受剂量水平通常被认为是特别重要的问题,尚未被现有专门指导原则涵盖。
2. 适用范围本指导原则阐述了如何处理新原料药中遗传毒性杂质的一般框架和实践方法。
该指导原则也适用于已有原料药的新申请,如果其合成路线、过程控制和杂质研究尚无法确保不会产生新的或更高含量的遗传毒性杂质(与EU目前批准的相同原料药相比)。
该指导原则同样适用于已上市原料药有关合成方面的变更申请。
不过,除非有特殊原因,本指导原则不适用于已批准药品。
本文中,将化合物(杂质)归类为遗传毒性物质,一般指在主要着重于检测有直接损伤DNA潜力的DNA反应物质的既定体外或体内遗传毒性试验中有阳性结果。
FDA《推荐的遗传毒性试验结果综合分析法指导原则》

FDA《推荐的遗传毒性试验结果综合分析法指导原则》介绍审评四部王庆利审评二部黄芳华审评五部彭健摘要:FDA于2006年1月正式发布了《推荐的遗传毒性试验结果综合分析法指导原则》,该指导原则介绍了目前对遗传毒性结果的综合分析方法,尤其是在遗传毒性试验结果出现阳性结果时提出了一些推荐性的建议和意见。
现在原草案翻译稿的基础上,重新翻译整理了该指导原则,以期为药物研究者提供一些参考性信息。
关键词:遗传毒性;致癌性一、简介本指导原则的目的是为了向企业和CDER的审评人员说明,CDER如何看待药物开发过程中出现的遗传毒性试验阳性结果。
当遗传毒性试验结果提示药物具有潜在的致癌性或遗传危害时,本指导原则为如何继续进行临床试验并保证受试者的安全性提供建议。
本指导原则中讨论了单次给药和多次给药临床试验相关的管理决策。
本指导原则适用于经口、静脉、局部和其他途径给药的药物。
FDA的指导原则,包括本指导原则在内,并不具有法律上的强制性,而是描述了当前FDA 对某一问题的想法,应该被认为仅是一种建议,除非是援引了特殊的管理性的或法令性的要求。
FDA指导原则中的“应该”一词是指建议的或推荐某些东西,而不是要求某些东西。
二、背景遗传毒性试验的时间安排和如何进行在ICH指导原则M3、S2A和S2B中已有描述。
我们建议参考这些指导原则,而本指导原则可认为是仅作为附件性指导原则。
通常是在啮齿类动物试验中评估致癌性风险,包括周期为2年的试验或采用替代模型而周期较短的试验。
通过ICH程序,企业和管理者接受了遗传毒性核心组合试验方案。
这些试验是用于确定化合物的遗传毒性,包括:■ 一项细菌基因突变试验■ 一项采用哺乳动物细胞进行的体外染色体损伤评估试验,或体外小鼠淋巴瘤tk+/- 试验■ 一项采用啮齿类动物造血细胞进行的体内染色体损伤试验。
以下讨论是根据当前的指导原则文件进行的。
我们建议在进行I期临床试验前完成体外遗传毒性试验。
三、遗传毒性试验结果的综合分析当遗传毒性结果为阳性结果时,对进入临床试验是否安全,FDA会考虑所有的安全性资料。
基因毒性杂质介绍及检测方法

基因毒性杂质介绍及检测⽅法1什么是基因毒性杂质基因毒性杂质(或遗传毒性杂质,Genotoxic Impurity ,GTI)是指化合物本⾝直接或间接损伤细胞DNA,产⽣基因突变或体内诱变,具有致癌可能或者倾向。
潜在基因毒性的杂质(Potential Genotoxic Impurity ,PGI)从结构上看类似基因毒性杂质,有警⽰性,但未经实验证明的黄曲霉素类、亚硝胺化合物、甲基磺酸酯等化合物均为常见的基因毒性杂质,许多化疗药物也具有⼀定的基因毒性,它们的不良反应是由化疗药物对正常细胞的基因毒性所致,如顺铂、卡铂、氟尿嘧啶等。
2为何着重研究基因毒性杂质基因毒性物质特点是在很低浓度时即可造成⼈体遗传物质的损伤,进⽽导致基因突变并可能促使肿瘤发⽣。
因其毒性较强,对⽤药的安全性产⽣了强烈的威胁,近年来也越来越多的出现因为在已上市药品中发现痕量的基因毒性杂质残留⽽发⽣⼤范围的医疗事故,被FDA强⾏召回的案例,给药⼚造成了巨⼤的经济损失。
例如某知名国际制药巨头在欧洲市场推出的HIV蛋⽩酶抑制剂维拉赛特锭(Viracept, mesylate),2007 年7⽉,EMA暂停了它在欧洲的所有市场活动,因为在其产品中发现甲基磺酸⼄酯超标,甲基磺酸⼄酯是⼀种经典的基因毒性杂质,该企业为此付出了巨⼤的代价,先内部调查残留超标的原因,因在仪器设备清洗时⼄醇未被完全清除⽽残留下来,与甲基磺酸反应形成甲基磺酸⼄酯。
在被要求解决污染问题后还被要求做毒性研究,以更好的评估对患者的风险。
同时有多达25000 名患者暴露于这个已知的遗传毒性。
直到解决了这所有问题后 EMA才恢复了它在欧洲的市场授权。
近年来各国的法规机构如ICH、FDA、EMA等都对基因毒性杂质有了更明确的要求,越来越多的药企在新药研发过程中就着重关注基因毒性杂质的控制和检测。
3哪些化合物是基因毒性杂质杂质的结构多种多样,对于绝⼤多数的杂质⽽⾔,往往没有充分的毒性或致癌研究数据,因⽽难以对其进⾏归类。
化学药品注射剂临床前安全性评价的基本技术要求.

May.2009,Vol.6No.5化学药品注射剂临床前安全性评价的基本技术要求王海学王庆利孙涛(国家食品药品监督管理局药品审评中心,北京100045)【摘要】注射剂产品在临床使用的风险性较高,技术研发单位需足够重视其安全性研究和评价。
临床前研究中需考虑进行必要的毒性实验以支持注射剂申请进入临床,并重视上市的风险评价。
【关键词】注射剂;临床前;安全性国内近年出现了多起注射剂临床使用的安全性问题,量效关系、暴露量、安全范围,进而为其进入临床试验或上这引起国家药监部门的高度重视。
为此,国家食品药品监督市提供安全性支持信息。
考虑到药品风险评价的系统性,非管理局(以下简称“国家药监局”)要求逐步加强对注射剂的临床安全性评价中应:①密切结合药学和临床信息考虑;②风险控制要求,其中加强科学技术评价是风险控制的重要密切结合药效学、药代动力学等信息考虑;③进行规范的毒环节之一。
国家药监局近几年已经开始对注射剂的安全性性试验研究和结果分析评价;④具体问题具体分析[1]。
例如评价开展了调研和技术交流,希望能够进一步完善注射剂对创新药注射剂和仿制药注射剂的非临床安全性评价中关的技术研究要求和安全性评价方法。
注的侧重点会有所不同,前者需要关注系统的毒性试验研注射剂中的主药、辅料、杂质等通常可直接入血,其暴究信息,而后者可重点关注关键性的毒性试验信息。
下面将露量和绝对生物利用度高,因此注射剂与其他剂型比较,更具体讨论几种常见的注射剂风险评价内容。
需加强安全性的技术要求和风险控制。
注射剂的安全性评价包括药学、非临床和临床专业评价,其中非临床安全性评2具体注射剂非临床安全性评价的关注要点价的作用主要在于:有助于进一步加强药学质量控制,如处2.1创新药物方组成、剂型选择、杂质限度等;预测初次进入人体试验用创新药物注射剂由于主药的安全性是未知的,制剂中药的安全性;探索风险产生原因、机制和控制措施。
注射剂的杂质或其与辅料(即与辅料的相互作用的影响)的安全性非临床安全性评价是在剂型选择的必要性与合理性得以满也可能是未知的,因此其非临床安全性评价中需全面评价足的前提下,密切结合药学质控而采取的必要的非临床安其系统的毒性试验并研究其毒性结果。
关于药物中的基因毒性杂质

关于药物中的基因毒性杂质众所周知,药物并⾮纯净物质,其在⽣产贮运过程中常常会引⼊或产⽣“杂质”,⽽由于杂质的存在,⼜往往会带来潜在的安全性问题,所以科研⼈员通常需要在充分研究的基础上对杂质加以有效控制。
⽽基因毒性杂质危害性⼤,需要严格控制其在药物中的限度,保障⽤药安全。
基因毒性杂质的检测⾯临杂质种类多和化学性质活泼等问题,分析⽅法复杂多样,从⽽对药物中基因毒性杂质的检测⽅法提出了很⾼的要求。
⼀、基因毒性杂质基因毒性杂质( genotoxic impurity,GTI) 定义为“经过适当遗传毒性实验模型,如细菌基因突变( Ames) 实验,证实具有遗传毒性的杂质”。
其主要包括PGLS( potentially genotoxic impurities有潜在基因毒性的杂质)和GTLs( genotoxic impurities基因毒性杂质)两种。
基因毒性杂质可能从基因突变、染⾊体畸变、DNA 损伤与修复等⼏个⽅⾯同DNA 发⽣直接或间接的相互作⽤,从⽽改变DNA 结构与构象或引起DNA 的损伤,进⽽影响DNA的功能或改变其遗传特性,最终引起突变、癌变、畸变等遗传毒性。
新药合成、原料纯化、储存运输〔与包装物接触)等过程都可能产⽣基因毒性杂质,故⽽,近年来药审机构及研发⼈员对其愈发关注!各国药品监督管理部门对药物中基因毒性杂质的控制出台了⼀系列的指导⽂件,旨在严格控制该类杂质在药物中的限度。
⼆、有关基因性杂质的参考指南1.EMEA(欧洲药品管理局)2000年,欧洲监管机构率先开始关注基因毒性杂质,Pharm Europa发表了⼀篇⽂章,提到注意在成盐⼯艺中,磺酸在⼄醇溶液中形成磺酸酯的潜在风险。
2002年,专利药物委员会(CPMP,现为⼈⽤药物委员会CHMP)发布了⼀份关于基因毒性杂质的意见书,指南中将基因毒性杂质的限度根据有⽆阈值分为两类。
2006年⾸先颁布了《基因毒性杂质限度指南》,并⾃2007年1⽉1⽇起正式实施。
遗传毒性杂质控制指导原则

遗传毒性杂质控制指导原则遗传毒性杂质控制指导原则用于指导药物遗传毒性杂质的危害评估、分类、定性和限值制定,以控制药物中遗传毒性杂质潜在的致癌风险。
为药品标准制修订,上市药品安全性再评价提供参考。
一、总则遗传毒性(Genotoxcity)是指遗传物质中任何有害变化引起的毒性,而不考虑诱发该变化的机制,又称为基因毒性。
遗传毒性杂质(Genotoxic Impurities,GTIs)是指能引起遗传毒性的杂质,包括致突变性杂质和其它类型的无致突变性杂质。
其主要来源于原料药的生产过程,如起始原料、反应物、催化剂、试剂、溶剂、中间体、副产物、降解产物等。
致突变性杂质(Mutagenic Impurities)指在较低水平时也有可能直接引起DNA损伤,导致DNA突变,从而可能引发癌症的遗传毒性杂质。
本指导原则主要关注致突变机制的遗传毒性杂质,非致突变机制的遗传毒性杂质在杂质水平的剂量下,一般可忽略其致癌风险。
药品生产、药品标准提高及上市药品再评价过程中发现杂质后,可按本指导原则进行风险评估,确定其是否为遗传毒性杂质,尤其是致突变性杂质。
如果一个杂质被鉴定为具有潜在的致癌风险,应制定相应的限值。
在制订可忽略致癌风险的杂质限值时,应进一步分析生产工艺,兼顾安全性和质量风险管理成本两方面的因素,综合考虑制定合适的限值。
本指导原则包括危害评估方法、可接受摄入量计算方法和限值制定方法。
本指导原则中描述的对杂质潜在致突变性的评估方法不适用于以下类型的原料药和制剂:生物/生物技术制品、肽类、寡核苷酸、放射性药物、发酵产品、中药和动物或植物来源的粗制品。
也不适用于已上市药物中使用的辅料、调味剂、着色剂和香料,以及与药物包材相关的可浸出物。
本指导原则中对杂质潜在致突变性的评估方法不适用于用于晚期癌症适应症的原料药和制剂,以及用于其它适应症但本身在治疗剂量下就具有遗传毒性,且预计可能与癌症风险增加有关的原料药。
在这些情况下,致突变性杂质不会显著增加原料药的致癌风险。
药物遗传毒性研究技术指导原则

药物遗传毒性研究技术指导原则药物遗传毒性研究技术指导原则一、概述遗传毒性研究(GenotoxicityStudy)是药物非临床安全性评价的重要内容,与其他研究尤其是致癌性、生殖毒性等研究有着密切的联系,是药物进入临床试验及上市的重要环节。
拟用于人体的药物,应根据受试物拟用适应症和作用特点等因素考虑进行遗传毒性试验。
遗传毒性试验是指用于检测通过不同机制直接或间接诱导遗传学损伤的受试物的体外和体内试验,这些试验能检测出DNA损伤及其损伤的固定。
以基因突变、较大范围染色体损伤或重组形式出现的DNA 损伤的固定,通常被认为是可遗传效应的基础,并且是恶性肿瘤多阶段发展过程的重要因素(恶性肿瘤发展变化是一个复杂的过程,遗传学改变可能仅在其中起部分作用)。
染色体数目的改变也与肿瘤发生有关,并可提示生殖细胞出现非整倍体的可能性。
在遗传毒性试验中呈阳性的化合物为潜在的人类致癌剂和/或致突变剂。
由于在人体中已建立了某些致突变/遗传毒性化合物的暴露与致癌性之间的相关性,而对于遗传性疾病尚难以证明有类似的相关性,因此遗传毒性试验主要用于致癌性预测。
但是,因为生殖细胞突变与人类疾病具有明确的相关性,所以也应同样重视化合物引起潜在可遗传性效应的风险。
此外,遗传毒性试验结果可能对致癌性试验的结果分析有重要作用。
因此,在药物开发的过程中,遗传毒性试验的目的是通过一系列试验来预测受试物是否有遗传毒性,在降低临床试验受试者和药品上市后使用人群的用药风险方面发挥重要作用。
本指导原则重点阐述遗传毒性试验的基本原则,介绍标准试验组合方案,阐述体内外试验的基本原则,以及对试验结果的分析评价与追加研究策略。
本指导原则适用于中药、天然药物和化学药物。
二、基本原则(一)实验管理药物遗传毒性试验必须执行《药物非临床研究质量管理规范》(GLP)。
(二)具体问题具体分析遗传毒性试验的设计,应该在对受试物认知的基础上,遵循“具体问题具体分析”的原则。
应根据受试物的结构特点、理化性质、已有的药理毒理研究信息等选择合理的试验方法,设计适宜的试验方案,并试验结果进行全面的分析与评价。
顶空气相色谱-质谱法测定依度沙班原料药中遗传毒性物质

顶空气相色谱-质谱法测定依度沙班原料药中遗传毒性物质徐艳梅;裴丽娟;杜高锋;宋更申【摘要】目的建立内标法测定依度沙班原料药中遗传毒性杂质甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯的含量.方法采用顶空气相色谱-质谱法,以DB-WAX毛细管柱(30 m×0.25 mm,0.25μm)为色谱柱,程序升温,高纯氦气为载气,流速为0.6 mL?min-1,进样口温度为110℃,进样方式为分流进样,分流比为20:1;顶空进样,平衡温度为60℃,平衡时间为30 min,进样体积1 mL;检测器为质谱检测器,离子源为EI源,离子源温度为200℃,接口温度为150℃,扫描方式为选择离子检测,电子能量为70 eV.结果甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯在0.05~3.0μg?mL-1(r≥0.9985)浓度范围内线性关系良好,加样回收率分别为96.4%,96.1%和96.5%,RSD分别为2.0%,1.9%,1.9%(n=6).结论该方法简便、快速、灵敏度高、专属性好,可为依度沙班原料药的质量控制提供参考依据.【期刊名称】《医药导报》【年(卷),期】2019(038)001【总页数】4页(P92-95)【关键词】依度沙班;杂质,遗传毒性;甲磺酸甲酯;甲磺酸乙酯;甲磺酸异丙酯;顶空气相色谱-质谱法【作者】徐艳梅;裴丽娟;杜高锋;宋更申【作者单位】河北省药品检验研究院,石家庄 050011;河北医科大学第一医院药剂科,石家庄 050031;河北省药品检验研究院,石家庄 050011;河北省药品检验研究院,石家庄 050011【正文语种】中文【中图分类】R973.2;R927.2依度沙班(edoxyaban)是一种新型Xa因子抑制药[1-2],具有起效快、抗凝效果可逆、具有剂量依赖的效果等特点。
2015年以来,美国食品药品管理局(FDA)、欧洲药品管理局(EMA)和瑞士药品管理局相继批准依度沙班用于非瓣膜性心房颤动患者脑卒中及系统性栓塞、急性静脉栓塞患者深静脉血栓和肺栓塞的预防及治疗[3]。
遗传毒性杂质控制指导原则
遗传毒性杂质控制指导原则遗传毒性杂质控制指导原则用于指导药物遗传毒性杂质的危害评估、分类、定性和限值制定,以控制药物中遗传毒性杂质潜在的致癌风险。
为药品标准制修订,上市药品安全性再评价提供参考。
一、总则遗传毒性(Genotoxcity)是指遗传物质中任何有害变化引起的毒性,而不考虑诱发该变化的机制,又称为基因毒性。
遗传毒性杂质(Genotoxic Impurities,GTIs)是指能引起遗传毒性的杂质,包括致突变性杂质和其它类型的无致突变性杂质。
其主要来源于原料药的生产过程,如起始原料、反应物、催化剂、试剂、溶剂、中间体、副产物、降解产物等。
致突变性杂质(Mutagenic Impurities)指在较低水平时也有可能直接引起DNA损伤,导致DNA突变,从而可能引发癌症的遗传毒性杂质。
本指导原则主要关注致突变机制的遗传毒性杂质,非致突变机制的遗传毒性杂质在杂质水平的剂量下,一般可忽略其致癌风险。
药品生产、药品标准提高及上市药品再评价过程中发现杂质后,可按本指导原则进行风险评估,确定其是否为遗传毒性杂质,尤其是致突变性杂质。
如果一个杂质被鉴定为具有潜在的致癌风险,应制定相应的限值。
在制订可忽略致癌风险的杂质限值时,应进一步分析生产工艺,兼顾安全性和质量风险管理成本两方面的因素,综合考虑制定合适的限值。
本指导原则包括危害评估方法、可接受摄入量计算方法和限值制定方法。
本指导原则中描述的对杂质潜在致突变性的评估方法不适用于以下类型的原料药和制剂:生物/生物技术制品、肽类、寡核苷酸、放射性药物、发酵产品、中药和动物或植物来源的粗制品。
也不适用于已上市药物中使用的辅料、调味剂、着色剂和香料,以及与药物包材相关的可浸出物。
本指导原则中对杂质潜在致突变性的评估方法不适用于用于晚期癌症适应症的原料药和制剂,以及用于其它适应症但本身在治疗剂量下就具有遗传毒性,且预计可能与癌症风险增加有关的原料药。
在这些情况下,致突变性杂质不会显著增加原料药的致癌风险。
遗传毒性杂质的控制

分类流程
内容
一、简介 二、ICH M7 三、遗传毒性杂质的分类 四、限度控制 五、案例 六、总结
对遗传毒性杂质的控制
遗传毒性 杂质控制
无毒理学数据, 采用TTC法
控制
控制
PDE法
PDE法
• 适用于有阈值效应的遗传毒性杂质:已有 证据表明,该类物质只有在超过一定限度 时才会产生遗传毒性。 • 杂质安全性限度的确定可以参照残留溶剂 限度的计算方法,根据相关动物的无可见 效应剂量(No-Observed Effect Level)计算 其可接受的日暴露量(Permissible Daily Exposure),再根据药品的最大日剂量计算 出杂质的接受限度。
日摄入量 (ug/天)
120
20
10
1.5
含有多个遗传毒性杂质
• 含有多个遗传毒性杂质的限度要求: • 基于TTC原理,当原料药中含有多个遗传毒 性杂质时,按照下述要求进行控制
治疗时段
日常摄入量 (ug/天)
<=1个月
120
1~12个月
60
1~10年
30
10年~终生
5
ICH M7中对时间段的划分
遗传毒性杂质的控制
王旸
2015年11月
内容
一、简介 二、ICH M7 三、遗传毒性杂质的分类 四、限度控制 五、案例 六、总结
内容
一、简介 二、ICH M7 三、遗传毒性杂质的分类 四、限度控制 五、案例 六、总结
一、 简介
• 遗传毒性杂质(genotoxic impurities): 遗传毒性泛指各种因素(物理、化学因素 )与细胞或生物体的遗传物质(主要指 DNA)发生作用而产生的毒性。 致突变性(mutagenic):与DNA相互作用 产生直接或潜在的影响,使基因突变。 致癌性(carcinogenetic):具有致癌可能性
EMEA《遗传毒性杂质限度指导原则》介绍

发布日期 20070820栏目化药药物评价>>化药质量控制标题EMEA《遗传毒性杂质限度指导原则》介绍作者史继峰部门正文内容审评四部审评七室史继峰摘要:《遗传毒性杂质限度指导原则》对遗传毒性杂质进行了分类,并介绍了相关的遗传毒性杂质限度确定的原则和方法。
关键词:遗传毒性杂质毒理学担忧阈值(TTC)1. 介绍在原料药(Q3A)和药物制剂(Q3B)的杂质指导原则中,杂质限度确定的依据包括各个杂质的生物安全性数据或杂质在某特定含量水平的研究概况。
而对于遗传毒性杂质限度的确定,通常都认为是特别关键的问题,但目前尚无相关的指导原则。
2. 适用范围本指导原则阐述了如何处理新原料药中遗传毒性杂质的一般框架和实际方法。
该指导原则也适用于已有原料药的新申请,如果其合成路线、过程控制和杂质研究尚无法确保不会产生新的或更高含量的遗传毒性杂质(与EU目前批准的相同原料药相比)。
该指导原则同样适用于已上市原料药有关合成方面的补充申请。
除非有特殊原因,本指导原则不适用于已上市的产品。
3. 毒理学背景根据目前的研究实践,具有(体内)遗传毒性的化合物在任何暴露量下都有可能对DNA产生损伤,而这种损伤可能会引发肿瘤。
因此,对于遗传毒性致癌物质,应谨慎认为不存在明确的阈值,任何暴露量下都存在风险。
然而,对于一些遗传毒性事件,其产生生物学意义的阈值效应的机理正越来越为人所了解。
对于非DNA靶点的化合物和潜在致突变剂更是如此,因为它们在与关键靶点接触前就已经去毒化了。
对于这些化合物,研究的基础可以是确定关键的未观察到影响的剂量(NOEL)和采用不确定因子。
即使对能与DNA分子发生反应的化合物,由于低剂量时有多种有效的保护机制存在,而不能将高剂量下的影响以线性方式外推到很低的(人)暴露水平。
不过,目前要用实验方法证明某诱变剂的遗传毒性阈值仍然非常困难。
所以,在缺乏恰当的证据支持遗传毒性阈值存在的情况下,确定安全剂量很困难,因此非常有必要采用一个可接受风险的暴露水平概念。
药物遗传毒性研究技术指导原则

附件四则编号:药物遗传毒性研究技术指导原则药物遗传毒性研究技术指导原则一、概述遗传毒性研究(Genotoxicity Study)是药物非临床安全性评价的重要内容,它与其他毒理学研究尤其是致癌性研究、生殖毒性研究有着密切的联系,是药物进入临床试验及上市的重要环节。
拟用于人体的药物,应根据受试物拟用适应症和作用特点等因素考虑进行遗传毒性试验。
遗传毒性试验是指用于检测通过不同机制直接或间接诱导遗传学损伤的受试物的体外和体内试验,这些试验能检出DNA损伤及其损伤的固定。
以基因突变、较大范围染色体损伤、重组和染色体数目改变形式出现的DNA损伤的固定,一般被认为是可遗传效应的基础,并且是恶性肿瘤发展过程的环节之一(这种遗传学改变仅在复杂的恶性肿瘤发展变化过程中起了部分作用)。
在检测此类损伤的试验中呈阳性的化合物为潜在致癌剂和/或致突变剂,即可能诱导癌和/或遗传性疾病。
由于在人体中已建立了某些化合物的暴露和致癌性之间的关系,而对于遗传性疾病尚难以证明有类似的关系,故遗传毒性试验主要用于致癌性预测。
但是,因为已经确定生殖细胞突变与人类疾病有关,所以对可能引起可遗传效应的化合物与可能引起癌症的化合物应引起同样的关注;此外,这些试验的结果可能还有助于解释致癌性的机制和试验结果。
因此,在药物开发的过程中,遗传毒性试验的目的是通过一系列试验来预测受试物是否有遗传毒性,在降低临床试验受试者和药品上市后使用人群的用药风险方面发挥重要作用。
本指导原则重点阐述遗传毒性试验体内外试验的基本原则,并介绍标准试验组合方案,以及对试验结果的综合分析及评价。
本指导原则适用于中药、天然药物和化学药物的遗传毒性试验研究。
二、基本原则(一)实验管理药物的遗传毒性试验属于安全性评价研究,根据《中华人民共和国药品管理法》的规定,必须执行《药物非临床研究质量管理规范》。
(二)具体问题具体分析遗传毒性试验的设计,应该在对受试物认知的基础上,遵循“具体问题具体分析”的原则。
(完整版)基因毒性杂质的评估与控制

杂质 来源
降解杂质
加速试验、长期试验、光降 解、强制降解试验
环境污染
三、基因毒性杂质识别
工艺杂质
实际杂质
潜在杂质
API中实际观察到>ICH Q3A 报告限(0.05%)工艺杂质
1)合成API过程:起始物料,中间体,化学试剂 2)风险评估可能带入API中的,存在于起始物料,中间体中已识别 的杂质,以及合理机理预测产生的副产物(对于工艺早期杂质携带 入API的风险可忽略,但要提供基于风险论证的表明哪步后应该评估 杂质的潜在突变性) 3)对后工艺引入的起始物料,评价起始物料合成最后一步的潜在基 因毒性杂质
2018年
华海药业生产的缬沙坦原料药中含有微量基因毒性杂质N,N-二甲 基亚硝胺(NDMA),缬沙坦及其相关制剂从欧洲、美国和中国市 场被召回。
2018年
印度Torrent制药生产的缬沙坦片剂中也检测出含有NDMA,该公司 也从美国市场上自愿召回了14批相关药品。
二、相关概念
何为基因毒性杂质?
基因毒性杂质,也称遗传毒性杂质,通常指较低水平可直接造成DNA损伤,进而导致DNA突变, 因此可能引发癌症的DNA反应性物质。
EMA
2006年颁布《基因毒性杂质限度指南》
2010年发布《遗传毒性杂质限度指导 原则问答》 ◆为限制新活性物质中的基因毒性杂 质提供了解决问题的框架和具体方案。
◆新药必须进行基因毒性杂质分析
◆对于现有药品,不强制进行基因毒 性杂质分析评估
◆对已上市产品进行化学合成变更或 仿制药上市前,需对合成路线、过程 控制、杂质概括评价并与现有产品对 比,以确定未引入新的或更高水平的 基因毒性杂质
降解杂质
长 期 稳 定 性 试 验 , API 中 观 察 到>ICH Q3A报告限降解产物
基因毒性杂质控制相关文件及指南介绍
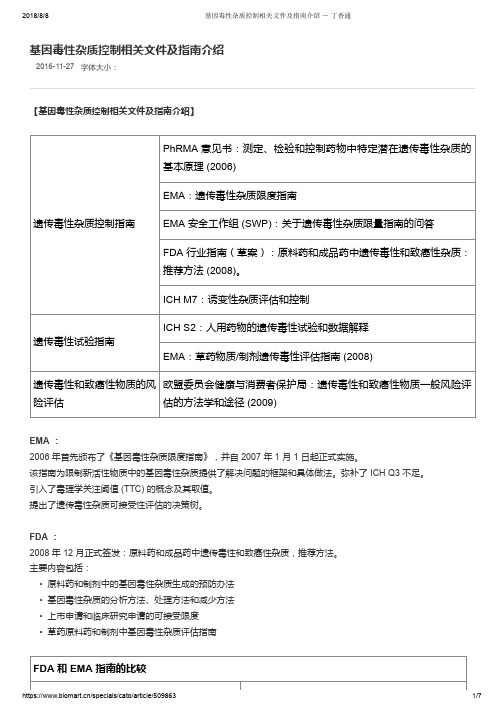
2016-11-27字体大小:基因毒性杂质控制相关文件及指南介绍【基因毒性杂质控制相关文件及指南介绍】遗传毒性杂质控制指南PhRMA 意见书:测定、检验和控制药物中特定潜在遗传毒性杂质的基本原理 (2006)EMA:遗传毒性杂质限度指南EMA 安全工作组 (SWP):关于遗传毒性杂质限量指南的问答FDA 行业指南(草案):原料药和成品药中遗传毒性和致癌性杂质:推荐方法 (2008)。
ICH M7:诱变性杂质评估和控制遗传毒性试验指南ICH S2:人用药物的遗传毒性试验和数据解释EMA:草药物质/制剂遗传毒性评估指南 (2008)遗传毒性和致癌性物质的风险评估欧盟委员会健康与消费者保护局:遗传毒性和致癌性物质一般风险评估的方法学和途径 (2009)EMA :2006 年首先颁布了《基因毒性杂质限度指南》,并自 2007 年 1 月 1 日起正式实施。
该指南为限制新活性物质中的基因毒性杂质提供了解决问题的框架和具体做法。
弥补了 ICH Q3 不足。
引入了毒理学关注阈值 (TTC) 的概念及其取值。
提出了遗传毒性杂质可接受性评估的决策树。
FDA :2008 年 12 月正式签发:原料药和成品药中遗传毒性和致癌性杂质,推荐方法。
主要内容包括:• 原料药和制剂中的基因毒性杂质生成的预防办法• 基因毒性杂质的分析方法、处理方法和减少方法• 上市申请和临床研究申请的可接受限度• 草药原料药和制剂中基因毒性杂质评估指南FDA 和 EMA 指南的比较相似点不同点推荐的鉴定和认证潜在遗传性杂质的方法相同 推荐的处理遗传毒性和致癌性杂质的方法相同FDA 指南包含致癌性杂质TTC 设定为 1.5 μg/天指南允许的 14 天内用药的 TTC 水平为 120 μg , 而非仅针对单次用药临床试验中短期暴露的 TTC 更高FDA 指南不允许根据现售药品的短期暴露情况而 提高 TTC ICH M7【基因毒性杂质的控制策略】具有阳性致癌数据的诱变杂质(第1类)---计算可接受摄入量( AI ): • M7 Addendum 中列出的 15 种化合物中有 10 个为该计算方法计算 • Carcinogenicity Potency Database (CPDB )中列明了 1574 种致癌物质的 TD50 值毒理学关注门槛---TTC 法(第 2/3 类): • ICHM7 主要讨论的方法,主要针对第 2/3 类基因毒性杂质,比如低级磺酸酯类等。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
发布日期20070820
栏目化药药物评价>>化药质量控制
标题EMEA《遗传毒性杂质限度指导原则》介绍
作者史继峰
部门
正文内容审评四部审评七室史继峰
摘要:《遗传毒性杂质限度指导原则》对遗传毒性杂质进行了分类,并介绍了相关的遗传毒性杂质限度确定的原则和方法。
关键词:遗传毒性杂质毒理学担忧阈值(TTC)
1. 介绍
在原料药(Q3A)和药物制剂(Q3B)的杂质指导原则中,杂质限度确定的依据包括各个杂质的生物安全性数据或杂质在某特定含量水平的研究概况。
而对于遗传毒性杂质限度的确定,通常都认为是特别关键的问题,但目前尚无相关的指导原则。
2. 适用范围
本指导原则阐述了如何处理新原料药中遗传毒性杂质的一般框架和实际方法。
该指导原则也适用于已有原料药的新申请,如果其合成路线、过程控制和杂质研究尚无法确保不会产生新的或更高含量的遗传毒性杂质(与EU目前批准的相同原料药相比)。
该指导原则同样适用于已上市原料药有关合成方面的补充申请。
除非有特殊原因,本指导原则不适用于已上市的产品。
3. 毒理学背景
根据目前的研究实践,具有(体内)遗传毒性的化合物在任何暴露量下都有可能对DNA产生损伤,而这种损伤可能会引发肿瘤。
因此,对于遗传毒性致癌物质,应谨慎认为不存在明确的阈值,任何暴露量下都存在风险。
然而,对于一些遗传毒性事件,其产生生物学意义的阈值效应的机理正越来越为人
所了解。
对于非DNA靶点的化合物和潜在致突变剂更是如此,因为它们在与关键靶点接触前就已经去毒化了。
对于这些化合物,研究的基础可以是确定关键的未观察到影
响的剂量(NOEL)和采用不确定因子。
即使对能与DNA分子发生反应的化合物,由于低剂量时有多种有效的保护机制存在,而不能将高剂量下的影响以线性方式外推到很低的(人)暴露水平。
不过,目前要用
实验方法证明某诱变剂的遗传毒性阈值仍然非常困难。
所以,在缺乏恰当的证据支持
遗传毒性阈值存在的情况下,确定安全剂量很困难,因此非常有必要采用一个可接受
风险的暴露水平概念。
4. 推荐
正如Q3A指导原则所述,根据合理的化学反应机理分析,在新的原料药合成、纯化和贮存过程中很有可能产生实际的和潜在的杂质。
依据现有的“可能引起遗传毒性的
结构”数据库,潜在的遗传毒性杂质应能被确认。
如果潜在的杂质含有可引起遗传毒
性的结构单元,该杂质应考虑进行遗传毒性试验(一般是细菌回复突变试验)(Dobo 等,2006)。
虽然Q3A指导原则认为这些研究采用含有那些需控制杂质的原料药进行是可行的,但用分离出来的杂质进行这些研究更恰当,也是高度推荐的方法。
根据以上论述,遗传毒性杂质可以归纳成以下两类:
有充分阈值相关机理证据(实验)的遗传毒性化合物
无充分阈值相关机理证据(实验)的遗传毒性化合物
4.1 有充分阈值相关机理证据的遗传毒性化合物
非线性或阈值明确的剂量效应关系的遗传毒性机理包括:与细胞分化过程中纺锤体
相互作用;拓扑异构酶抑制;DNA合成抑制;过度的防御机制;代谢过度和生理性干
扰(如诱导红血球生成,高体温和低体温)。
有明确遗传毒性阈值的化合物,不产生遗传毒性风险的暴露水平可以被确定,方法
可参照Q3C“杂质指导原则”中二类溶剂的限度确定方法。
该方法可计算“每日最大允许暴露量”(PDE),数据来源于“不确定因数”动物研究中的NOEL(未观察到
效果的最低水平)或观察到效果的最低水平(LOEL)。
4.2 无充分阈值相关机理证据的遗传毒性化合物
对于此类遗传毒性杂质,研究应包括药学和毒理学评估。
总之,如果杂质无法避免,药学方面的控制应遵循“合理可行的最低限量”原则(ALARP原则)。
符合ALARP原
则的杂质水平再经毒理学方面的进一步评估,以验证其合理性(见决策树和以下章节)。
4.2.1 药学方面评估
如果涉及潜在遗传毒性杂质,那么申请材料应提供对杂质的特别讨论资料(见Q3A (R))。
还需提供处方和制备工艺研究资料。
合成工艺和杂质研究部分应分析所有的化学物质,包括用到的试剂、中间体、副产物,已知遗传毒性和/或致癌性物质(如烷化剂)。
值得关注的是,虽然有些含有“可能引起遗传毒性的结构”(alerting structure)的
反应试剂与最终活性物质并没有共同结构,但也要考虑它们的遗传毒性。
如果有可能,应该对它们进行一些替代研究。
充分的替代研究资料包括替代的合成路线和制剂处方,不同的起始原料等。
比如,
应证明具有遗传毒性和/或致癌性的结构在化学合成中(如烷化反应)是必需的。
如果遗传毒性杂质在原料中不可避免,则应该采取适当的技术(如纯化步骤)降低
该杂质的含量,以满足安全性要求,或符合“合理可行的最低限量”原则。
药学评估
还应包括反应中间体、反应试剂等的化学稳定性研究。
杂质检查和定量应该用经过验证的合理的方法进行。
4.2.2 毒理学评估
鉴于在没有明确阈值的前提下定义安全暴露水平(零风险)是不可能的,且完全排
除原料药中的遗传毒性杂质经常是很难的,所以有必要提出一个“可接受风险水平”(acceptable risk level)的概念,比如估算一个“每日最大暴露量”值,低于该暴露
量时就可以忽略其对人体健康的风险。
“可接受风险”概念源自Q3C杂质指导原则注释“I类残留溶剂”的附录3。
然而,应用这些方法必须能获得足够多的长期致癌性研究数据。
大多数情况下,遗传毒性杂质的毒理学评估只是局限于杂质的体外研究(如Ames
试验,染色体畸变试验),但这些方法并不适用于确定杂质可接受的摄入水平。
也就
是说,根据体外数据(如Ames试验)计算杂质的“安全系数(safety multiples)”、进而确定可接受的限度,是不合适的。
此外,用含有较低(ppm级)杂质水平的原料
药研究其致癌性和遗传毒性,即使得出阴性结果也不足以确保该杂质限度的合理性,
因为这种试验方法缺少必要的灵敏度。
有些具有很强致突变性和致癌性物质与原料药
一起进行试验时,因为在非常低的暴露水平情况下,很有可能因为低于检测限而无法
检出。
所以,如果认识到含量非常低的遗传毒性杂质不存在“不可接受的风险”(unacceptable risk),那么可以采取实用的方法来控制该杂质。
4.2.3 毒理学担忧阈值(TTC)应用
“毒理学担忧阈值”用于定义那些不会产生显著致癌性或其他毒性作用、但又未明确研究的化合物的“常见暴露量”(common exposure level)(Munro等,1999)。
该TTC估计值是1.5μg/人/日。
TTC概念最早来源于FDA关于食品添加物的“规定阈值”(a threshold of regulation)(Rulis 1989),该阈值根据对致癌性数据库中343种致癌物质的分析结果得出。
随后该数据库扩大到700多个致癌性物质,这种分析结果不断得到重复验证。
当致癌性物质按概率计算致癌风险达到百万分之一的上限时(“事实上的安全剂量”),估算此时的暴露水平(µg/人)。
对于含有“可能引起遗传毒性结构”的化合物,其TTC应严格10倍(0.15µg/日)(Kroes等,2004)。
然而,用TTC评估原料药中的遗传毒性杂质限度,1.5µg/日(相当于10万分之一的患癌风险)是可以接受的。
应该承认,基于TTC值控制遗传毒性杂质是非常保守的,因为这只是根据从产生50%肿瘤发生率(TD50)到百万分之一发生率的剂量推测得到的,而且TD50数据是用最敏感的动物和最敏感的部位研究得到的(几个“最坏条件”假设)(Munro等)。
有些化合物即使摄入量低于TTC,其遗传毒性也是很强的,因此应归为高致癌风险化合物(Cheeseman等,1999)。
这些高致癌性物质包括黄曲霉素类、N-亚硝基物和偶氮类化合物,不适用TTC方法。
这类化合物的风险评估需采用专门的毒性数据。
某些情况下TTC值高于1.5µg/日也是可以接受的,如短期用药;用于治疗威胁生命疾病的药物;或人的存活期少于5年;或该杂质是已知物质,人体从其他途经(如食物)获得的暴露量远远高于药物途经。
如果遗传毒性杂质本身就是重要的代谢物,那么该杂质可以根据代谢物的可接受限度进行控制。
遗传毒性杂质的浓度限度根据以下公式计算:
浓度限度(ppm)=
对于有确切毒性数据(长期毒性研究)的致癌性物质不宜使用TTC概念,应进行特定化合物风险评估。
应强调,TTC是一个实用性的风险控制方法,是按概率方法学估算的。
比如按这一概念,如果未知致癌性遗传毒性杂质的摄入量低于TTC值,那么就可以保证患癌风险控制在十万分之一之内。
当然,TTC概念并不能确保绝对无风险。
1)结构上与高致癌性物质有关的杂质(见正文)不能采用TTC法。
2)如果有致癌性数据:摄入量超过10-5患癌风险吗?
3)具体情况具体分析,包括用药时间长短、适应症、患者人群等(见正文)。
*) 缩写:NOEL/UF-未观察到效果水平/不确定因素
PDE-每日最大允许暴露量,TTC-毒理学担忧阈值
备注。