医疗器械安全有效基本要求清单 (2)
03医疗器械安全有效基本要求清单

03医疗器械安全有效基本要求清单医疗器械是一种特殊的产品,涉及到每个人的健康与安全,因此在其设计、生产、销售和使用过程中,需要符合一系列的安全和有效性要求。
以下是医疗器械的基本要求清单。
1.安全性要求:-材料安全性:医疗器械所使用的材料必须符合相关的安全标准,不得产生毒性、过敏或刺激等不良反应。
-电气安全性:电气医疗器械必须符合相关的电气安全标准,确保不会对患者或操作人员产生电击或火灾等危险。
-机械安全性:机械医疗器械必须符合相关的机械安全标准,确保在正常使用过程中不会造成意外伤害或危险。
-生物安全性:医疗器械必须符合相关的生物安全标准,确保不会对患者或操作人员产生传染病或其他生物危害。
-隔离性能:对于直接与患者接触的医疗器械,必须具备良好的隔离性能,防止交叉感染的发生。
-抗菌性能:一些医疗器械需要具有抗菌功能,以防止细菌滋生和交叉感染的发生。
-抗干扰能力:对于电子医疗器械,需要具备一定的抗干扰能力,以防止外界电磁干扰对器械功能的影响。
2.有效性要求:-准确性:医疗器械必须具备准确的测量和检测能力,确保对患者的诊断或治疗提供准确的结果或指导。
-稳定性:医疗器械必须具备稳定的性能,不受环境变化或运输过程中的振动等因素的影响。
-可靠性:医疗器械必须具备高度可靠的性能,可以长期稳定地工作,不断断续续或出现误差。
-一致性:同一款医疗器械在不同的生产批次中,其性能和质量应保持一致,不应出现明显的差异。
-适应性:医疗器械必须能够适应不同的使用环境和不同的患者需求。
-可操作性:医疗器械的使用操作应简单明了,易于操作和掌握。
对于依赖信息显示的医疗器械,需要具有友好的人机界面。
-治疗效果:对于治疗型医疗器械,需要具备良好的治疗效果,并有相关临床数据的支持。
3.归责要求:-生产者责任:医疗器械的生产者应对其生产的医疗器械负责,确保其符合相关的安全和有效性要求。
-进口者责任:医疗器械的进口者应对其进口的医疗器械负责,确保其符合相关的安全和有效性要求。
医疗器械ivd安全有效的基本要求清单英文

Basic Requirements Checklist for Safety and Effectiveness of In Vitro Diagnostic (IVD) Medical Devices In vitro diagnostic (IVD) medical devices play a crucial role in healthcare by providing important information for the diagnosis, treatment, and monitoring of diseases. To ensure the safety and effectiveness of these devices, various regulatory bodies have established specific requirements that manufacturers must adhere to. This checklist outlines the fundamental requirements for the safety and effectiveness of IVD medical devices.1. Design and Development•The device design should be based on scientific principles and well-established technology.•The device should be able to perform its intended function accurately and reliably.•The design should consider appropriate measures for user safety, including ergonomic factors and minimizing potential hazards.•The device should take into account the needs of the target population and end-users.2. Analytical Performance•The device should have appropriate analytical sensitivity and specificity for the intended use.•It should be able to correctly identify and quantify the target analyte or biomarker.•The device should demonstrate acceptable precision and accuracy in test results.•The device should have appropriate limits of detection and quantification.3. Clinical Performance•The device should have clinical validity, i.e., it should provide accurate and reliable information for clinical decision-making.•It should demonstrate clinical sensitivity and specificity, by accurately identifying the presence or absence of a particular condition.•The device should be able to differentiate between various conditions or disease stages, when relevant.•The device should have appropriate positive and negative predictive values.4. Quality Management System•The manufacturer should have a comprehensive quality management system in place to ensure the consistent quality of the device.•The system should comply with applicable international standards, such as ISO 13485.•It should include procedures for risk management, design control, post-market surveillance, and complaint handling.•The manufacturer should regularly perform internal audits and assessments to maintain the effectiveness of the quality management system.5. Labeling and Instructions for Use•The device labeling should be clear, accurate, and comprehensive.•It should include essential information for the safe and effective use of the device, such as intended use, limitations, and precautions.•The instructions for use should be easy to understand and follow.•The labeling should provide information on storage, handling, and disposal of the device, when applicable.6. Post-Market Surveillance•The manufacturer should have a system in place to monitor the performance and safety of the device after it is placed on the market.•The system should include mechanisms for adverse event reporting, trend analysis, and corrective actions.•The manufacturer should actively collect and analyze data on device malfunction, user-related errors,and any known risks.•The post-market surveillance system should enable the manufacturer to take appropriate actions, such asproduct recalls or modifications, if necessary.7. Clinical Evaluation and Evidence•The manufacturer should conduct a comprehensive clinical evaluation of the device to assess its safety andperformance.•The evaluation should consider available scientific literature, clinical data, and, if necessary, clinicalinvestigations.•The manufacturer should provide sufficient clinical evidence to support the claimed performance andindications for use of the device.•The clinical evaluation should be documented and available for review by regulatory authorities.ConclusionMeeting the basic requirements for safety and effectiveness is crucial for ensuring high-quality IVD medical devices.Manufacturers should carefully consider these requirements throughout the design, development, manufacturing, and post-market phases of the device’s lifecycle. Adhering to these requirements not only ensures compliance with regulatory standards but also helps protect the health and well-being of patients and users.Note: This checklist serves as a general guide, and manufacturers should refer to specific regulatory requirements and standards applicable to their region or intended market.。
医疗器械记录清单

引言:医疗器械记录清单(二)是医疗机构管理及监督医疗器械使用的重要工作。
通过建立和维护医疗器械记录清单,可以有效管理医疗器械的购买、入库、使用和报废等环节,确保医疗器械的安全性和有效性。
本文将详细介绍医疗器械记录清单的重要性,并分析其在医疗机构管理中的具体应用。
概述:医疗机构管理医疗器械需要建立一套完善的记录清单体系,以追踪每一台医疗器械的购入、维修、更新、使用和报废等环节。
只有通过清单记录,医疗机构才能确保医疗器械的有效管理和监督。
医疗器械记录清单的建立需要包含详细的信息,如医疗器械的名称、型号、生产商、购买日期、使用科室等。
医疗机构应该建立专门的部门或岗位负责医疗器械记录清单的管理和维护。
正文内容:一、清单的建立和更新1. 设立专门的医疗器械管理部门或岗位,负责医疗器械记录清单的建立和维护。
2. 对于已有的医疗器械,对其进行归档,建立详细的清单信息。
对于新增的医疗器械,应立即建立清单,并更新到总清单中。
3. 清单应包括医疗器械的基本信息、购买日期、购买渠道、使用科室等详细信息,以便后续管理和查询。
二、清单的入库管理1. 医疗器械购入后,应及时进行入库管理,将其添加到清单中。
2. 应将医疗器械放置在划定的存放区域,并进行编号或标识,方便后续管理。
3. 入库的医疗器械应进行验收,检查其是否完好,并记录入库日期和验收人员,以便后续跟踪和维护。
三、清单的使用管理1. 针对不同科室的医疗器械使用需求,建立清单使用计划,包括使用时间、使用人员等。
2. 医疗器械的使用应符合相关法规和规范要求,使用人员要对医疗器械进行正确操作,并记录使用情况。
3. 对于有特殊需求的医疗器械,应加强使用管理,定期进行维护和保养,确保其良好运行。
四、清单的维护与报废1. 定期对医疗器械进行巡检和维护,并记录维护情况和维护人员。
2. 对于维修无效或无法修复的医疗器械,应及时进行报废,并进行记录和处理,避免继续使用对患者安全造成威胁。
医疗器械安全有效基本要求清单
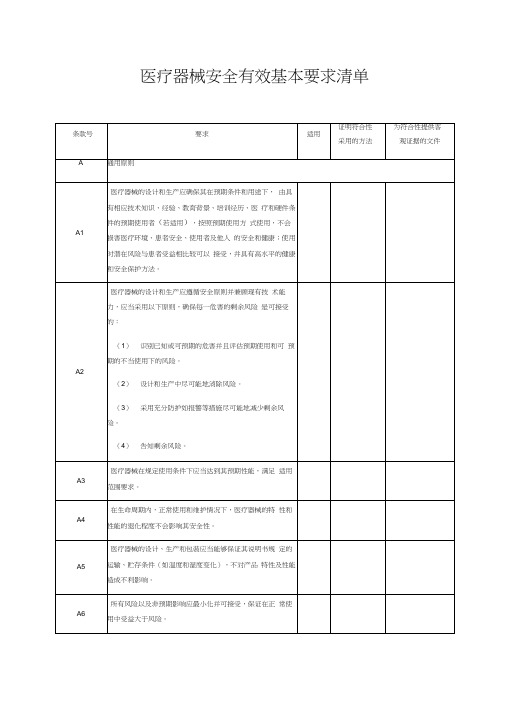
医疗器械安全有效基本要求清单
全使用产品实现其预期功能。
信息应当易于理解
1. 第3列若适用,应注明“是”。
不适用应注明“否”,并说明不适用的理由。
2. 第4列应当填写证明该医疗器械符合安全有效基本要求的方法,通常可采取下列方法证明符合基本要求:
(1) 符合已发布的医疗器械部门规章、规范性文件。
(2) 符合医疗器械相关国家标准、行业标准、国际标准
(3) 符合普遍接受的测试方法。
说明
(4) 符合企业自定的方法。
(5) 与已批准上市的同类产品的比较。
(6) 临床评价。
3. 为符合性提供的证据应标明在注册申报资料中的位置和编号。
对于包含在产品注册申报资料中的文件,应当
说明其在申报资料中的具体位置。
例如:八、注册检验报告(医用电气安全:机械风险的防护部分);说明书第
章。
对于未包含在产品注册申报资料中的文件,应当注明该证据文件名称及其在质量管理体系文件中的编号备
查。
(完整版)第二类医疗器械注册申报资料要求及说明..

附件2 :第二类医疗器械注册申报资料要求及说明(征求意见稿)注册申报资料应有所提父资料目录,包括申报资料的一级和二级标题。
每项二级标题对应的资料应单独编制页码。
一、申请表(附表1)二、证明性文件1. 企业营业执照副本复印件和组织机构代码证复印件。
2. 按照《创新医疗器械特别审批程序审批》的境内医疗器械申请注册时,应当提交创新医疗器械特别审批申请审查通知单,样品委托其他企业生产的,应当提供受托企业生产许可证和委托协议。
生产许可证生产范围应涵盖申报产品类别。
三、医疗器械安全有效基本要求清单说明产品符合《医疗器械安全有效基本要求清单》(附表2)各项适用要求所采用的方法,以及证明其符合性的文件。
对于《医疗器械安全有效基本要求清单》中不适用的各项要求,应当说明其理由。
对于包含在产品注册申报资料中的文件,应当说明其在申报资料中的具体位置;对于未包含在产品注册申报资料中的文件,应当注明该证据文件名称及其在质量管理体系文件中的编号备查。
四、综述资料(一)概述描述申报产品的管理类别、分类编码及名称的确定依据。
(二)产品描述1. 无源医疗器械描述产品工作原理、作用机理(如适用)、结构组成(含配合使用的附件)、主要原材料,以及区别于其他同类产品的特征等内容;必要时提供图示说明。
2. 有源医疗器械描述产品工作原理、作用机理(如适用)、结构组成(含配合使用的附件)、主要功能及其组成部件(关键组件和软件)的功能,以及区别于其他同类产品的特征等内容;必要时提供图示说明。
(三)型号规格对于存在多种型号规格的产品,应当明确各型号规格的区别。
应当采用对比表及带有说明性文字的图片、图表,对于各种型号规格的结构组成(或配置)、功能、产品特征和运行模式、性能指标等方面加以描述。
(四)包装说明有关产品包装的信息,以及与该产品一起销售的配件包装情况;对于无菌医疗器械,应当说明与灭菌方法相适应的最初包装的信息。
(五)适用范围和禁忌症1. 适用范围:应当明确产品所提供的治疗、诊断等符合《医疗器械监督管理条例》第七十六条定义的目的,并可描述其适用的医疗阶段(如治疗后的监测、康复等);明确目标用户及其操作该产品应当具备的技能/知识/ 培训;说明产品是一次性使用还是重复使用;说明预期与其组合使用的器械。
医疗器械安全有效基本要求清单

医疗器械安全有效基本要求清单医疗器械的安全和有效性是保障患者生命安全和健康的重要要求。
因此,制定医疗器械的安全有效基本要求清单对于确保患者的安全和健康至关重要。
下面是一份医疗器械安全有效基本要求的清单:1.符合法律法规:医疗器械应符合国家相关法律法规的要求,包括药品管理法、医疗器械管理条例等。
同时,还应符合国际质量管理体系的相关要求,如ISO9001质量管理体系。
3.先进技术:医疗器械应采用先进的技术和工艺,以满足医疗技术的发展和患者的需求。
同时,如在产品的材料选择、工艺流程等方面应采用最新的科技水平,以确保产品的安全性和有效性。
4.产品标识:医疗器械应在产品上清晰明确地标示产品名称、规格型号、生产厂家等信息,以便患者正确使用和识别产品。
5.临床试验:医疗器械上市前应进行临床试验,确保其安全、有效并具备临床价值。
同时,应符合国家相关的临床试验管理要求。
6.售后服务:医疗器械生产企业应建立健全的售后服务体系,提供及时、有效的技术支持和维修服务,确保产品在使用过程中的安全和有效性。
7.风险评估和控制:医疗器械生产企业应进行风险评估,对可能存在的风险进行识别和控制。
风险控制措施应明确并得到有效的执行,确保产品的安全和有效性。
8.产品文档:医疗器械生产企业应提供产品合格证明文件、产品技术说明书、产品使用方法说明书等相关文档,使用户能够清楚了解产品的性能、使用方法和注意事项。
9.培训和教育:医疗器械生产企业应对销售人员、使用人员进行培训和教育,使其了解产品的正确使用方法和注意事项,以确保产品的安全和有效性。
10.监管要求:医疗器械生产企业应接受监管部门的监督和检查,配合相关部门进行产品的质量监控和安全监测,确保产品的安全和有效性。
综上所述,医疗器械的安全和有效性是保障患者生命安全和健康的基本要求。
制定医疗器械的安全有效基本要求清单,能够确保医疗器械符合法律法规要求、具备质量安全、采用先进技术、进行临床试验、提供售后服务、进行风险评估和控制、提供产品文档、进行培训和教育、接受监管要求等,并最终保障患者的生命安全和健康。
医疗器械安全有效的基本要求清单英文版
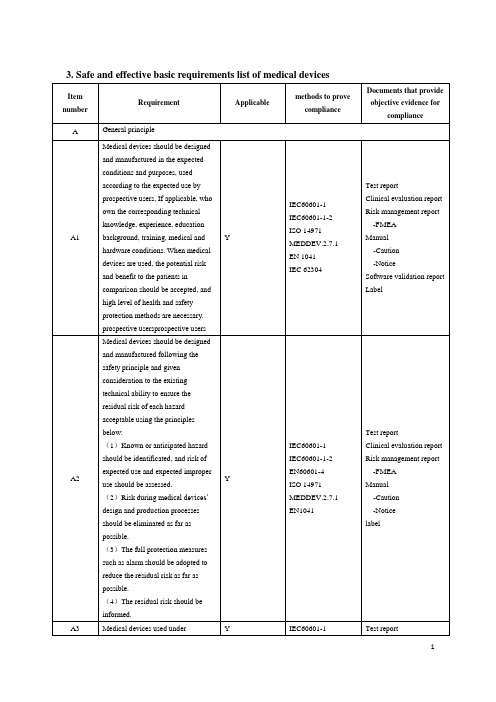
Medical devices’ design should
This product is
(1)be of easy operation
irrelevant to
B2.1
(2)reduce the microbial leakage
knowledge, experience, education
A1
background, training, medical and
Y
hardware conditions. When medical
devices are used, the potential risk
and benefit to the patients in
Test report Clinical evaluation report Risk management report
-FMEA Manual
-Caution -Notice label
Test report 1
regulations should be up to its expected performance and need its scope of application
with the relevant provisions of the with other
pharmaceutical management, and
material.
whose performance shouldn’t be
changed in the normal use
Medical devices’ design and
guarantee medical devices meet the
医疗器械注册申报资料要求及说明

附件3医疗器械注册申报资料要求及说明—\申请表应按照要求完整填写注册申请表。
二、医疗器械安全有效基本要求清单(见附件9)三、综述资料(一)概述简单描述申报产品的管理类别、分类编码及名称的确定依据、预期用途、使用环境、适用人群等信息。
(-)产品描述1.无源医疗器械应包括工作原理、结构组成(含配合使用的附件)及图示、制造材料、包装材料、型号规格及其划分依据、主要生产工艺、交付状态及作用机理等内容。
产品如有区别于其他同类产品的特征也应加以描述。
详细描述所有与人体接触部件的制成材料清单,包括规格 /化学特性。
对于含有同种异体材料、动物源性材料、药物成分或生物活性物质等的产品,应予以明确说明,包括來源、特性、使用的原因及其主要作用方式。
2 •有源医疗器械详细描述产晶结构组成(含配合使用的附件)、主耍功能、工作原理、作用机理(如适用)以及各组成部件(关键组件和软件)的功能,并详细描述关键元器件。
应足够详尽并提供图示说明,产品如有区别于其他同类产品的特征也应加以描述。
详细描述所有与人体接触部件的制成材料清单,包括规格/化学特性。
对于含有动物源性材料、药物成分或生物活性物质等的产品,应给予明确说明,包括來源、特性、使用的原因及其主要作用方式。
(三)型号规格对存在多种型号规格的产品,应详细列表说明各型号规格区别, 从结构组成(或配置)、功能、产品特征和运行模式、性能指标等方面加以描述。
应采用对比表及带有说明性文字的图片、图表对各种规格型号的不同之处加以总结。
(四)包装说明有关产品包装的信息,以及与该产品一起销售的配件包装情况; 对于无菌器械,应说明与灭菌方法相适应的最初包装的信息。
(五)预期用途和禁忌症1•预期用途:应明确产品所提供的治疗或诊断功能,并描述其适用的治疗阶段,目标用户和用户操作该产品应具备的技能/知识/培训;使用方式:一次性使用还是重复使用。
2・预期使用环境:该产品预期使用的地点如医院、医疗/临床实验室、救护车、家庭等,以及可能会影响其安全性和/或性能的环境条件(例如,温度、湿度、功率、压力、移动等);3•适用人群:目标患者人群的信息(如成人、儿童或新生儿),患者选择标准的信息及使用过程屮需要监测的相关参数;或文献资料。
医疗器械安全有效基本要求清单英文版

Safe and effective essential requirements listof medical devicesArticle No. RequirementApplicable(Y/NA)The adoption ways ofverification complianceDocumentationA General RequirementsA1 The medical devices must be designed and manufactured in such a way that, when be used by intended user with the corresponding technical knowledge, experience, educational background, training experience, medical and hardware condition(if applicable), under the conditions and for the purposes intended, they will not compromise the clinical condition or the safety of patients, or the safety and health of users or, where applicable, other persons, provided that any risks which may be associated with their use constitute acceptable risks when weighed against the benefits to the patient and are compatible with a high level of protection of health and safety.A2 The design and manufacture of the medical devices must conform to safety principles, and taking account of the acknowledged state of the art. The following principles shall be applied to ensure that the residual risk of each hazard is acceptable:(1)Identify known or expectable hazards, and evaluate the risk under expected use and expectable improper use.(2)Eliminate or reduce risks as far as possible in design and manufacture. (3)where appropriate take adequate protection measures including alarms if necessary, in relation to risks that cannot be eliminated.(4)Inform users of the residual risks.A3 The medical devices shall achieve itsNo. (Y/NA) verification compliance intended performance and meetapplicable scope requirement underprescribed use condition.A4 Within lifecycle, the degree of properties and performance degeneration of medical devices wouldn’t have influence on its safety under normal use and maintenance.A5 The medical devices must be designed, manufactured and packed in such a way that their characteristics and performances during their intended use will not be adversely affected (by changes such as temperature and humidity) during transport and storage taking account the instructions for use.A6 All risks and unintended effects should be minimized and accepted to ensure that the benefits outweigh the risks in normal use.B Essential requirements of safety performance for medical devices B1 Chemical, physical and biological propertiesB1.1 The materials shall ensure the medical devices meet the requirement of SectionA. Particular attention must be paid to:(1) The choice of materials used, particularly as regards toxicity and flammability (if applicable).(2) The compatibility between the materials used and biological tissues, cells and body fluids, taking account of the indications of the medical devices.(3) The choice of materials used, shall as regards hardness, abrasive resistance and fatigue strength properties( if applicable).B1.2 The medical devices must be designed, manufactured and packed in such a way as to minimize the risk posed by contaminants and residues to the persons involved in the transport, storage and useNo. (Y/NA) verification compliance of the devices and to the patients.Particular attention must be paid to thetissues exposed and the duration andfrequency of the exposure.B1.3 The medical devices must be designed and manufactured in such a way that to guarantee they can be used safely with the materials, substances and gases with which they enter into contact during their normal use or during routine procedures; if the medical devices are intended to administer medicinal products, they must be designed and manufactured in such a way as to be accordance with the regulation of drug administration, and can’t change its performance in normal use.B1.4 The medical devices must be designed and manufactured in such a way as to minimize the risk posed by leach or leakage. Particular attention must be paid to its carcinogenicity, teratogenicity and reproductive toxicity.B1.5 The medical devices must be designed and manufactured in such a way to reduce as much as possible, risk posed by the unintentional ingress of substances into the device take into account the device and nature of the environment in which it is intended to be used.B2 Infection and microbial contamination.B2.1 The medical devices must be designed and manufactured in such a way as to minimize the risk of infection with patient, user and others. Its design must be:(1) Easy handing.(2) To minimize the microbe leakage from products and/or microbe exposure in use as much as possible.(3) To prevent human contamination of medical devices and samples with microorganisms.No. (Y/NA) verification complianceB2.2 Medical devices marked with microbial requirements shall be ensured to meet microbial requirements before use.B2.3 Sterile medical devices should be ensured to meet sterility requirements before use.B2.4 Sterile medical devices or medical devices marked with microbial requirements shall be processed, manufactured or sterilized by an validated method.B2.5 Sterile medical devices shall be manufactured in appropriately controlled conditions(e.g. environment of the corresponding purification level).B2.6 Packaging for non-sterile medical devices shall keep the product without deterioration at the level of cleanliness stipulated and, if the devices are to be sterilized prior to use, minimize the risk of microbial contamination. The packaging shall be suitable the method of appropriate sterilization.B2.7 If the medical devices sold in market have sterile and non-sterile condition, its packaging or label shall be distinguished.B3 Drug and medical device combination productsB3.1 Safety, qualify and performance for the drug and medical devices combined products shall be verified.B4 Biogenic medical devicesB4.1 Tissues, cells and other substances of animal origin contained in medical devices shall comply with relevant regulations and in line with its indications requirements. The information on the geographical origin of the animals shall be retained for future reference. Processing, preservation, testing and handing of tissues, cells and substances of animalNo. (Y/NA) verification compliance origin must be carried out so as toprovide optimal security protection forpatient, user and other persons (ifapplicable). In particular viruses andother transferable agents must beaddressed by implementation ofvalidated methods of elimination orviral inactivation.B4.2 Tissues, cells and other substances of human origin contained in medical devices should be selected from appropriate sources and donors to reduce the risk of infection. Processing, preservation, testing and handing of tissues, cells and substances of human origin must be carried out so as to provide optimal security protection for patient, user and other persons (if applicable). In particular viruses and other transferable agents must be addressed by implementation of validated methods of elimination or viral inactivation.B4.3 Processing, preservation, testing and handing of cells and substances of microorganism origin contained in medical devices must be carried out so as to provide optimal security protection for patient, user and other persons (if applicable). In particular viruses and other transferable agents must be addressed by implementation of validated methods of elimination or viral inactivation.B5 Environmental propertiesB5.1 If the device is intended for use in combination with other devices or equipment, the whole combination shall be safe and shall not impair the performance of the devices or equipment. Any restrictions on use shall be indicated on the label and/or in theNo. (Y/NA) verification compliance instruction for use. Connection systemsuch as liquids, gas transmission ormechanical coupling, for example,should be designed and constructed tominimize the risk of wrong connectionsposing to the user.B5.2 B5.2.1 Medical devices shall be designed and manufactured in such a way as to remove or minimize the following risks as far as possible:The injury risk to patient, user or others, in connection with their physical or man-machine features.B5.2.2 The injury risk of incorrect handing, in connection with ergonomic features, human factors and use of the environment.B5.2.3 Risks connected with reasonably foreseeable external factors or environmental conditions, such as magnetic fields, external electrical influences, electrostatic discharge, diagnosis and treatment bring changes in radiation, pressure, humidity, temperature, as well as pressure and acceleration.B5.2.4 The risk of contact with materials, liquids and gases during normal use.B5.2.5 The risk caused by the compatibility of software and its operation environment.B5.2.6 The risk for accidental entry of substance.B5.2.7 The risks of reciprocal interference with other devices in the clinical use.B5.2.8 Risks arising where maintenance or calibration are not possible (as with implants) from ageing of the materials user or loss of accuracy of any measuring or control mechanism.B5.3 Medical devices shall be designed and manufactured in such a way as to minimize the risks of fire or explosionNo. (Y/NA) verification compliance during normal use and in single faultcondition. Particular attention must bepaid to medical devices whose intendeduse includes exposure to flammablesubstances, flammable substances orused in combination with combustibleand flammable substances.B5.4 The medical devices that need to adjusted, calibrated and maintained shall be designed and manufactured in such a way as to ensure their corresponding processes are carried out safely.B5.5 The medical devices shall be designed and manufactured in such a way as to facilitate the safe disposal of waste.B6 Medical device products with diagnostic or measuring functions.B6.1 Medical device with diagnostic or measuring functions shall be designed and manufactured in such a way as to take account of the accuracy, precision and stability. The limits of accuracy shall be indicated.B6.2 The measurement, monitoring and display scale must be designed in line with ergonomic principles.B6.3 The unit used to express the measurement value should be the standard unit in China and can be understood by users.B7 Protection against radiationB7.1 General requirements: Medical devices shall be designed, manufactured and packaged such that exposure of patient, users and other persons to radiation shall be reduced as far as possible, and wouldn’t influence its function.B7.2 Intended radiation: The medical devices that use radiation for treatment and diagnosis, its radiation should be controllable. Such devices shall be designed and manufactured to ensureNo. (Y/NA) verification compliance reproducibility and tolerance of relevantvariable parameters within theallowable range. If the medical devicesare intended to emit potentiallyhazardous radiation, they should havethe corresponding sound-light warningfunction.B7.3 Unintended radiation: Medical devices shall be designed and manufactured in such a way that exposure of patients, users and other persons to the emission of unintended, stray or scattered radiation is be reduced as far as possible..B7.4 Ionising radiation: Medical devices intended to emit ionsing radiation shall be designed and manufactured in such a way as to ensure that the quantity, geometric distribution and energy distribution (or quality) of radiation emitted can be controlled.Medical devices emitting ionsing radiation (intended for diagnostic radiology) shall be designed and manufactured in such a way as to achieve appropriate image quality for the clinical needs whilst minimizing radiation exposure of the patient and user.The medical devices shall to enable reliable monitoring and control of the delivered dose, the beam type, energy and energy distribution(if applicable).B8 Medical devices incorporate software and independent medical devices softwareB8.1 For medical devices which incorporate software or which are medical software in themselves shall be designed to ensure the repeatability, reliability and performance. In the event of a single fault condition appropriate means should be adopted to eliminate or reduce as far as possible consequentNo. (Y/NA) verification compliance risks.B8.2 For medical devices which incorporate software or which are medical software in themselves, the software must be validated according to the state of the art (need to take into account the principles of development lifecycle, risk management, validation and verification).B9 Active medical devices and the medical devices connected to active medical devicesB9.1 For active medical devices, when a single fault occurs, appropriate measures should be taken to eliminate and reduce the resulting risks as much as possible.B9.2 Medical devices where the safety of the patients depends on an internal power supply shall be equipped with a means of determining the state of the power supply.B9.3 Medical devices where the safety of the patients depends on an external power supply shall include an alarm system to signal any power failure.B9.4 Medical devices intended to monitor one or more clinical parameters of a patient shall be equipped with appropriate alarm systems to alert the user of situations which could lead to severe deterioration or death of the patient’s state of health.B9.5 Medical devices shall be designed and manufactured in such a way as to minimize the risks of creating electromagnetic fields.B9.6 Medical devices shall be designed and manufactured in such a way as to ensure the products have sufficient capacity to resist electromagnetic disturbance in order to guarantee the product can operate according to the intended use.No. (Y/NA) verification complianceB9.7 Medical devices shall be designed and manufactured in such a way as to protect the patient, user and other persons against accidental electric shocks when normal use and single failure after product is installed and maintained in according with requirements.B10 Protection against mechanical risksB10.1 Medical devices shall be designed and manufactured in such a way as to protect the patient and user against mechanical risks connected with, for example, resistance, stability and moving parts.B10.2 Medical devices shall be designed and manufactured in such a way as to reduce to the lowest possible level the risks arising from vibration generated by the devices, taking account of the means available for limiting vibrations (particularly at source),unless the vibrations are part of the specified performance.B10.3 Medical devices shall be designed and manufactured in such a way as to reduce to the lowest possible level the risks arising from the noise emitted, taking account of the means available to reduce noise (particularly at source), unless the noise emitted is part of the specified performance.B10.4 Terminals and connectors to the electricity, gas or hydraulic and pneumatic energy supplies which the user has to handle must be designed and constructed in such a way as to minimize all possible r isks.B10.5 Medical devices must be designed and manufactured in such a way as to minimize the risks of wrong connection, if the some parts of medical instrumentsNo. (Y/NA) verification compliance need to connect or reconnect use or inuse.B10.6 Accessible parts of the devices (excluding the parts or areas intended to supply beat or reach given temperatures) and their surroundings must not attain potentially dangerous temperatures under normal use.B11 Protection against the risks posed to the patient by energy supplies or substancesB11.1 Medical devices for supplying the patient with energy or substances must be designed and constructed in such a way that the flow-rate can be set and maintained accurately enough to guarantee the safety of the patient and of the user.B11.2 Medical devices should have the function of preventing and/or indicating "insufficient output" if insufficient output may cause danger. Appropriate precautions should be taken to prevent the accidental release of energy or material to a hazardous level.B11.3 The function of the controls and indicators must be clearly specified on the devices. Where a device bears instructions required for its operation or indicates operating or adjustment parameters by means of a visual system, such information must be easily understandable.B12 Protection against risks to non-professional usersB12.1 Medical devices shall be designed and manufactured in such a way as to take into account the knowledge, technology and environment of thenon-professional users, and provide adequate explanations to facilitate their understanding and use.B12.2 Medical devices shall be designed and manufactured in such a way as to minimize the risk of errors in operationNo. (Y/NA) verification compliance and understanding by non-professionalusers.B12.3 Medical devices shall, as far as possible, be provided with procedures for non-professional users to check whether the products are in normal operation during use.B13 Label and instructions for useB13.1 Taking account of the training and knowledge of the user, labels and instructions should provide the user with sufficient information to identify the manufacturer and safely use the product to achieve its intended function. The information should be easy to understand.B14 Clinical evaluationB14.1 The clinical evaluation data of medical devices shall be provided in accordance with the provisions of the current regulations.B14.2 Clinical trials should be consistent with the Helsinki Declaration. The approval of clinical trials shall be in accordance with the provisions of the current regulations.Notes 1. Column 3, where applicable, should be marked "Y". Non-application should be marked "NA" and the reasons for non-application should be stated.2. Column 4 shall fill in the method to prove that the medical device meets the essential requirements of safety and effectiveness. Generally, the following methods can be adopted to prove the essential requirements:(1) According to the issued regulations and normative documents of the medical device department.(2) According to the relevant national standards, industry standards, international standards of medical devices.(3) According to generally accepted test methods.(4) According to the enterprise's own method.(5) Comparison with similar products approved for marketing.(6) Clinical evaluation3. Evidence of conformity shall indicate the location and number in the data submitted for registration. For the documents included in the product registration declaration information, the specific location in the declaration information shall be stated. For example: viii. Registered inspection report (medical electrical safety: protective part of mechanical risk); Chapter4.2 of the instruction for use. For documents not included in the product registration declaration materials, the name of the evidence document and its serial number in the quality management system documents shall be indicated for reference.。
卷3、肌电生物反馈仪医疗器械安全有效基本要求清单

2按照质量管理体系的要求出具的各个阶段的体系文件
3.卷9《产品技术要求》
卷10《注册检验报告》
4.卷11《产品使用说明书》
卷12《最小销售单元的标签样稿》
A3
医疗器械在规定使用条件下应当达到其预期性能,满足适用范围要求。
是
1.符合YY 0607-2007、YY/T1095-2007
卷3、
肌电生物反馈仪
安全有效基本要求清单
本公司承诺:
按照国家食品药品监督管理总局《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》(2014年第43号公告)中,关于“医疗器械安全有效基本要求清单”的相关要求,提交了肌电生物反馈仪的医疗器械安全有效基本要求清单。
力康华耀生物科技(上海)有限公司
B1.4
医疗器械的设计和生产应当尽可能减少滤出物或泄漏物造成的风险,特别注意其致癌、致畸和生殖毒性。
否
产品无滤出物或泄漏物产生
/
B1.5
医疗器械的设计和生产应当考虑在预期使用条件下,产品及其使用环境的特性,尽可能减少物质意外从该产品进出所造成的风险。
是
符合 YY/T 0316-2016
卷8《风险分析报告》
B5.2.4
正常使用时可能与材料、液体和气体接触而产生的风险。
是
YY/T 0316-2016
卷8《风险分析报告》
B5.2.5
软件及其运行环境的兼容性造成的风险。
否
为烧入硬件的软件,由企业安装。
/
B5.2.6
物质意外进入的风险。
是
GB9706.1-2007
卷10《注册检验报告》
GB9706.1-2007标准条款44
是
医疗器械注册申报资料要求及说明

附件3医疗器械注册申报资料要求及说明一、申请表应按照要求完整填写注册申请表。
二、医疗器械安全有效基本要求清单(见附件9)三、综述资料(一)概述简单描述申报产品的管理类别、分类编码及名称的确定依据、预期用途、使用环境、适用人群等信息。
(二)产品描述1.无源医疗器械应包括工作原理、结构组成(含配合使用的附件)及图示、制造材料、包装材料、型号规格及其划分依据、主要生产工艺、交付状态及作用机理等内容。
产品如有区别于其他同类产品的特征也应加以描述。
详细描述所有与人体接触部件的制成材料清单,包括规格/化学特性。
对于含有同种异体材料、动物源性材料、药物成分或生物活性物质等的产品,应予以明确说明,包括来源、特性、使用的原因及其主要作用方式。
2.有源医疗器械详细描述产品结构组成(含配合使用的附件)、主要功能、工作原理、作用机理(如适用)以及各组成部件(关键组件和软件)的功能,并详细描述关键元器件。
应足够详尽并提供图示说明,产品如有区别于其他同类产品的特征也应加以描述。
详细描述所有与人体接触部件的制成材料清单,包括规格/化学特性。
对于含有动物源性材料、药物成分或生物活性物质等的产品,应给予明确说明,包括来源、特性、使用的原因及其主要作用方式。
(三)型号规格对存在多种型号规格的产品,应详细列表说明各型号规格区别,从结构组成(或配置)、功能、产品特征和运行模式、性能指标等方面加以描述。
应采用对比表及带有说明性文字的图片、图表对各种规格型号的不同之处加以总结。
(四)包装说明有关产品包装的信息,以及与该产品一起销售的配件包装情况;对于无菌器械,应说明与灭菌方法相适应的最初包装的信息。
(五)预期用途和禁忌症1.预期用途:应明确产品所提供的治疗或诊断功能,并描述其适用的治疗阶段,目标用户和用户操作该产品应具备的技能/知识/培训;使用方式:一次性使用还是重复使用。
2.预期使用环境:该产品预期使用的地点如医院、医疗/临床实验室、救护车、家庭等,以及可能会影响其安全性和/或性能的环境条件(例如,温度、湿度、功率、压力、移动等);3.适用人群:目标患者人群的信息(如成人、儿童或新生儿),患者选择标准的信息及使用过程中需要监测的相关参数;4.禁忌症:由于可能产生不良事件或风险,应明确说明该器械禁止使用的疾病或情况。
3医疗器械安全有效基本要求清单

3医疗器械安全有效基本要求清单医疗器械的安全和有效性是医疗器械使用的基本要求,下面是一份医疗器械安全有效基本要求的清单:1.符合法规要求:医疗器械应符合国家和地区的相关法律法规要求,包括国家药监局的注册要求、地方监管部门的认证要求等。
2.设计合理性:医疗器械应具备合理的设计,确保其在使用过程中能够安全、有效地达到预期的临床效果。
设计应充分考虑人体工程学、材料选择、结构强度、功能操作等方面。
3.材料和成分的安全性:医疗器械所使用的材料和成分应符合相关国家和地区的材料安全标准,不得含有任何有害物质,不得产生副作用,不得引发过敏反应等。
4.制造工艺和质量控制:医疗器械的制造工艺应符合国家和地区的相关标准和规范,确保产品具备稳定的质量性能。
制造过程应建立严格的质量控制体系,包括材料采购、生产过程控制、产品检测等。
5.标识和包装:医疗器械的标识和包装应符合相关法规要求,对产品的使用和维护提供清晰准确的指导,确保产品的安全性和有效性。
6.临床试验和评价:医疗器械应进行临床试验和评价,确保其在实际临床应用中的安全性和有效性。
试验和评价过程应符合相关法规和标准,采用科学合理的方法和设计。
7.使用说明和操作培训:医疗器械应提供详细的使用说明书和操作培训,确保使用者能够正确并安全地操作医疗器械。
使用说明应包括产品的特性和适应症、正确的使用方法和注意事项等内容。
8.不良事件和风险管理:医疗器械制造商应建立健全的不良事件和风险管理体系,及时收集和分析产品使用过程中的不良事件和风险,并采取适当的措施进行处理和改进。
9.售后服务和维修保养:医疗器械制造商应建立健全的售后服务和维修保养体系,及时响应用户的需求,提供产品维修和维护服务,确保产品的持续有效性和安全性。
10.再次注册和监测:医疗器械应定期进行再次注册和监测,确保产品的安全性和有效性持续符合相关标准和要求。
总之,医疗器械的安全和有效性是使用者和患者使用医疗器械时最为关注的问题。
总局43号公告附件8医疗器械安全有效基本要求清单

医疗器械应当尽可能设置可供非专业用户在使用过程 中检查产品是否正常运行的程序。
B13
标签和说明书
B13.1
考虑到使用者所受的培训和所具备的知识,标签和说明 书应能让使用者获得充分的信息,以辨别生产企业,安 全使用产品实现其预期功能。信息应当易于理解。
B14
临床评价
B14.1
应当依照现行法规的规定提供医疗器械临床评价资料。
(3)防止人对医疗器械和样品的微生物污染。
B2.2
标有微生物要求的医疗器械,应当确保在使用前符合微 生物要求。
B2.3
无菌医疗器械应当确保在使用前符合无菌要求。
B2.4
无菌或标有微生物要求的医疗器械应当采用已验证的 方法对其进行加工、制造或灭菌。
B2.5
无菌医疗器械应当在相应控制状态下(如相应净化级别 的环境)生产。
B8
含软件的医疗器械和独立医疗器械软件
B8.1
含软件的医疗器械或独立医疗器械软件,其设计应当保 证重复性、可靠性和性能。当发生单一故障时,应当采 取适当的措施,尽可能地消除和减少风险。
B8.2
对于含软件的医疗器械或独立医疗器械软件,其软件必 须根据最新的技术水平进行确认(需要考虑研发周期、 风险管理要求、验证和确认要求)。
B5.3
医疗器械的设计和生产应尽可能地减少在正常使用及 单一故障状态下燃烧和爆炸的风险。尤其是在预期使用 时,暴露于可燃物、致燃物或与可燃物、致燃物联合使 用的医疗器械。
B5.4
须进行调整、校准和维护的医疗器械的设计和生产应保 证其相应过程安全进行。
B5.5
医疗器械的设计和生产应有利于废物的安全处置。
B4.2
含有人体组织、细胞和其他物质的医疗器械,应当选择 适当的来源、捐赠者,以减少感染的风险。人体组织、 细胞和其他物质的加工、保存、检测和处理等过程应当 提供患者、使用者和他人(如适用)最佳的安全保护。
二类医疗器械安全有效基本要求清单

二类医疗器械安全有效基本要求清单二类医疗器械是指在正常使用条件下,通过其自身的功能达到预防、诊断、治疗、监控或缓解人类疾病的作用,并具有一定风险的医疗器械。
为了保障二类医疗器械的安全有效性,制定了一系列基本要求清单,以下是详细介绍:1.性能要求:二类医疗器械应具有满足预期技术性能和临床性能的能力。
例如,手术器械需要具备良好的操作性能和剪切能力,体外诊断试剂需要准确可靠的测试结果等。
2.安全性要求:二类医疗器械应具备较低的风险水平。
例如,手术器械需要具备防滑、防烫等安全设计,麻醉器械需要防止氧气泄漏等。
3.有效性要求:二类医疗器械应能够在预期使用条件下提供相应的临床效果。
例如,体外诊断试剂需要准确检测出疾病标志物,医用影像设备需要清晰显示人体结构等。
4.生物相容性要求:二类医疗器械应具有与人体组织相容的能力,并且不会对人体产生明显的毒副作用。
例如,植入式医疗器械需要材料无刺激、低过敏性等。
5.标记要求:二类医疗器械应进行相应的标记,包括产品名称、规格、厂名地址、生产批号等信息。
标记应清晰易读,不易褪色等。
6.使用说明要求:二类医疗器械应提供详细的使用说明,包括产品的适应症、禁忌症、使用方法、注意事项、存储条件等。
使用说明应简明易懂,方便用户正确使用。
7.质量管理要求:二类医疗器械的生产企业应建立和实施符合相关法律法规和管理标准的质量管理体系,包括原材料采购、生产过程控制、产品检测等环节,以确保产品的质量和安全。
8.售后服务要求:二类医疗器械的生产企业应提供相应的售后服务,包括产品的维修、技术支持、不良反应的处理等。
售后服务应及时有效,确保用户的合法权益。
9.监督管理要求:二类医疗器械应具备符合国家监督管理要求的能力,接受监管部门的监督和检查,确保产品的质量和安全性。
以上是二类医疗器械安全有效基本要求的清单。
这些要求的制定是为了确保二类医疗器械在正常使用条件下具备安全有效的特性,保障患者的生命和健康安全。
二类医疗器械安全有效基本要求清单

二类医疗器械安全有效基本要求清单二类医疗器械是指根据中国国家食品药品监督管理局(CFDA)的分类管理规定,具有一定风险,需通过检验和监督才能获得销售许可的医疗器械。
医用外科口罩是二类医疗器械中的一种,其应满足一系列的安全有效基本要求。
以下是我对二类医疗器械(医用外科口罩)安全有效基本要求的详细清单:1.清晰的标识和说明书:医用外科口罩必须在包装上清晰标示产品名称、规格型号、生产批号、生产日期、有效期等重要信息,并在说明书中详尽介绍使用方法、注意事项、存储条件等内容。
2.材料安全性:口罩所使用的材料必须符合相关标准,如不含有毒、有害物质,不会对用户产生过敏反应。
常见的材料包括无纺布、熔喷布、鼻梁条等。
3.过滤效率指标:医用外科口罩用于过滤空气中的微粒,应能有效过滤颗粒物,如细菌和病毒。
通常以微米(μm)为单位的颗粒直径,如0.3μm颗粒的过滤效率为基准。
4.透气性能:医用外科口罩应保持适当的透气性能,防止用户呼吸困难。
透气性能可以通过测试透气阻力来评估,要求具体取决于使用场景和舒适性需求。
5.舒适性:医用外科口罩应采用符合人体工程学的设计,保证佩戴者的舒适性和稳定性,不会引起过多的束缚感或不适感。
6.简便易用性:医用外科口罩的佩戴和取下应简便易行,无需额外工具的辅助。
设计应具有易于调整和固定的功能,以适应不同用户需求。
7.卫生性:医用外科口罩应符合卫生要求,保证产品在生产、包装、运输等环节不受污染。
必要时,可采用单次性使用的设计,以避免交叉感染的风险。
8.性能稳定性:医用外科口罩的性能应具有一定的稳定性,如经过一定时间的使用和储存后,过滤效率不得降低,不会影响产品的基本功能。
9.生产环境要求:生产医用外科口罩的企业应具备符合相关法规要求的生产环境,如无尘车间、洁净区域等,以确保产品的质量和卫生安全。
10.监督检测要求:医用外科口罩的生产和销售应按照相关法规进行监督检测。
企业应提供符合中国国家食品药品监督管理局规定的检测报告以证明产品的安全有效性。
03医疗器械安全有效基本要求清单

03医疗器械安全有效基本要求清单1.符合相关法律法规:医疗器械必须符合国家或地区的法律法规要求,包括生产、销售、使用等方面的规定。
2.注册备案:医疗器械必须按照国家的相关规定进行注册备案,确保其生产、销售和使用的合法性。
3.产品质量控制:医疗器械制造商必须建立完善的质量控制体系,包括原材料采购、生产工艺、产品检测等环节,确保产品的质量可靠。
4.安全性评价:医疗器械必须进行全面的安全性评价,包括对产品的材料、结构、功能等进行分析和测试,确保产品使用过程中不会对患者或操作人员造成伤害。
5.有效性评价:医疗器械必须进行有效性评价,即通过临床试验或其他科学方法证明其治疗或诊断效果确实存在,可以达到预期的治疗效果。
8.特殊用途产品认证:一些特殊用途的医疗器械,如放射性医疗器械、植入式医疗器械等,必须获得特殊用途产品认证,证明其安全有效性。
9.驻厂检查:国家药监局或相关部门会对医疗器械制造企业进行定期或不定期的驻厂检查,确保其生产和质量管理符合要求。
10.不良事件监测和报告:医疗器械制造商和使用单位必须建立和完善不良事件监测和报告制度,定期向药监部门汇报不良事件情况,并采取相应的措施进行处理和改进。
11.维修和召回:医疗器械制造商必须建立健全的维修和召回制度,及时回应用户投诉、维修产品,并在出现安全隐患时及时进行召回。
12.市场监管:药监部门会对市场上销售的医疗器械进行监管,并及时发布相关信息,保护消费者权益。
总结:医疗器械的安全和有效性是保障患者和操作人员的健康和安全的重要保证。
以上所述的医疗器械安全有效基本要求清单是对医疗器械的整个生命周期进行综合管理的基本要求,通过遵守这些要求,可以确保医疗器械的质量可靠,使用过程中不会对患者和操作人员造成伤害,并能达到预期的治疗效果。
医疗器械制造商和使用单位应该密切关注相关法律法规的变化,加强自身的管理和监督,为提供安全有效的医疗器械服务作出贡献。
医疗器械安全有效性基本清单

含软件的医疗器械和独立医疗器械软件
B8.1
含软件的医疗器械或独立医疗器械软件,其设计应当保证重复性、可靠性和性能。当发生单一故障时,应当采取适当的措施,尽可能地消除和减少风险。
B8.2
对于含软件的医疗器械或独立医疗器械软件,其软件必须根据最新的技术水平进行确认(需要考虑研发周期、风险管理要求、验证和确认要求)。
附件8
医疗器械安全有效基本要求清单
条款号
要求
适用
证明符合性采用的方法
为符合性提供客观证据的文件
A
通用原则
A1
医疗器械的设计和生产应确保其在预期条件和用途下,由具有相应技术知识、经验、教育背景、培训经历、医疗和硬件条件的预期使用者(若适用),按照预期使用方式使用,不会损害医疗环境、患者安全、使用者及他人的安全和健康;使用时潜在风险与患者受益相比较可以接受,并具有高水平的健康和安全保护方法。
B1.5
医疗器械的设计和生产应当考虑在预期使用条件下,产品及其使用环境的特性,尽可能减少物质意外从该产品进出所造成的风险。
B2
感染和微生物污染
B2.1
医疗器械的设计和生产应当减少患者、使用者及他人感染的风险。设计应当:
(1)易于操作。
(2)尽可能减少来自产品的微生物泄漏和/或使用中微生物暴露。
(3)防止人对医疗器械和样品的微生物污染。
B9
有源医疗器械和与其连接的器械
B9.1
对于有源医疗器械,当发生单一故障时,应当采取适当的措施,尽可能的消除和减少因此而产生的风险。
B9.2
患者安全需要通过内部电源供电的医疗器械保证的,医疗器械应当具有检测供电状态的功能。
B9.3
患者安全需要通过外部电源供电的医疗器械保证的,医疗器械应当包括显示电源故障的报警系统。
医疗器械安全有效基本要求清单(示范文本)

(1)符合已发布的医疗器械部门规章、规范性文件。
八、注册检验报告(医用电气安全:机械风险的防护部分);说明书第4.2章。
B4.3
含有微生物的细胞和其他物质的医疗器械,细胞及其他物质的加工、保存、检测和处理等过程应当提供患者、使用者和他人(如适用)最佳的安全保护。特别是病毒和其他传染原,应当采用经验证的清除或灭活方法处理。
药械组合产品
B3.1
应对该药品和药械组合产品安全、质量和性能予以验证。
是
(1)符合已发布的医疗器械部门规章、规范性文件。
八、注册检验报告(医用电气安全:机械风险的防护部分);说明书第4.2章。
生物源性医疗器械
B4.1
含有动物源性的组织、细胞和其他物质的医疗器械,该动物源性组织、细胞和物质应当符合相关法规规定,且符合其适用范围要求。动物的来源资料应当妥善保存备查。动物的组织、细胞和其他物质的加工、保存、检测和处理等过程应当提供患者、使用者和他人(如适用)最佳的安全保护。特别是病毒和其他传染原,应当采用经验证的清除或灭活方法处理。
是
(1)符合已发布的医疗器械部门规章、规范性文件。
八、注册检验报告(医用电气安全:机械风险的防护部分);说明书第4.2章。
A2
医疗器械的设计和生产应遵循安全原则并兼顾现有技术能力,应当采用以下原则,确保每一危害的剩余风险是可接受的:
(1)识别已知或可预期的危害并且评估预期使用和可预期的不当使用下的风险。
八、注册检验报告(医用电气安全:机械风险的防护部分);说明书第4.2章。
辐射防护
B7.1
一般要求:医疗器械的设计、生产和包装应当考虑尽量减少患者、使用者和他人在辐射中的暴露,同时不影响其功能。
是
(1)符合已发布的医疗器械部门规章、规范性文件。
第二类医疗器械注册申报资料要求及说明

附件 2:第二类医疗器材注册申报资料要求及说明(征采建议稿)申报资料一级标题申报资料二级标题1.申请表2.证明性文件3.医疗器材安全有效基本要求清单4. 综述资料5. 研究资料6. 生产制造信息概括产品描绘型号规格包装说明合用范围和禁忌症参照的同类产品或前代产品的状况(若有)其余需说明的内容产品性能研究生物相容性评论研究生物安全性研究灭菌和消毒工艺研究有效期和包装研究动物研究软件研究其余无源产品 / 有源产品生产过程信息描绘生产场所7.临床评论资料8.产品风险剖析资料9.产品技术要求10.产品注册查验报告注册查验报告预评论建议11.说明书和标签样稿说明书最小销售单元的标签样稿12.切合性申明注册申报资料应有所提交资料目录,包含申报资料的一级和二级标题。
每项二级标题对应的资料应独自编制页码。
一、申请表(附表 1)二、证明性文件1.公司营业执照副本复印件和组织机构代码证复印件。
2.依照《创新医疗器材特别审批程序审批》的境内医疗器材申请注册时,应该提交创新医疗器材特别审批申请审察通知单,样品拜托其余公司生产的,应该供给受托公司生产同意证和拜托协议。
生产同意证生产范围应涵盖申报产品类型。
三、医疗器材安全有效基本要求清单说明产品切合《医疗器材安全有效基本要求清单》(附表 2)各项合用要求所采纳的方法,以及证明其切合性的文件。
对于《医疗器械安全有效基本要求清单》中不合用的各项要求,应该说明其原由。
对于包含在产品注册申报资猜中的文件,应该说明其在申报资猜中的详细地点;对于未包含在产品注册申报资猜中的文件,应该注明该凭证文件名称及其在质量管理系统文件中的编号备查。
四、综述资料(一)概括描绘申报产品的管理类型、分类编码及名称确实定依照。
(二)产品描绘1.无源医疗器材描绘产品工作原理、作用机理(如合用 )、结构构成(含配合使用的附件)、主要原资料,以及差别于其余同类产品的特点等内容;必要时供给图示说明。
2.有源医疗器材描绘产品工作原理、作用机理 (如合用 )、结构构成 (含配合使用的附件 )、主要功能及其构成零件 (重点组件和软件 )的功能,以及差别于其余同类产品的特点等内容;必需时供给图示说明。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
医疗器械的设计、生产和包装应尽可能减少污染物和残留物对从事运输、贮存、使用的人员和患者造成的风险,特别要注意与人体暴露组织接触的时间和频次。
是
YY/T0316-ห้องสมุดไป่ตู้008
《医疗器械 风险管理对医疗器械的应用》
8.产品风险分析资料
B1.3
医疗器械的设计和生产,应当能够保证产品在正常使用中接触到其他的材料、物质和气体时,仍然能够安全使用。如果医疗器械用于给药,则该产品的设计和生产需要符合药品管理的有关规定,且正常使用不改变其产品性能。
是
YY/T0316-2008
《医疗器械 风险管理对医疗器械的应用》
8.产品风险分析资料
医疗器械安全性能基本原则
化学、物理和生物学性质
B1.1
材料应当能够保证医疗器械符合A节提出的要求,特别注意:
(1) 材料的选择应特别考虑毒性、易燃性(若适用)。
(2) 依据适用范围,考虑材料与生物组织、细胞、
体液的相容性。
B2.5
无菌医疗器械应当在相应控制状态下(如相应净化级别的环境)生产。
否
非无菌医疗器械
B2.6
非无菌医疗器械的包装应当保持产品的完整性和洁净度。使用前需要灭菌的产品,其包装应当尽可能减少产品受到微生物污染的风险,且应当适合相应的灭菌方法。
是
GB/T14710-2009《医用电器环境要求及试验方法》
10.产品注册检验报告(运输和贮存试验)
是
GB/T14710-2009《医用电器环境要求及试验方法》
10.产品注册检验报告(携带污染试验)
B1.4
医疗器械的设计和生产应当尽可能减少滤出物或泄漏物造成的风险,特别注意其致癌、致畸和生殖毒性。
否
没有滤出物或泄漏物
B1.5
医疗器械的设计和生产应当考虑在预期使用条件下,产品及其使用环境的特性,尽可能减少物质意外从该产品进出所造成的风险。
是
YY/T0316-2008
《医疗器械 风险管理对医疗器械的应用》
8.产品风险分析资料
10.产品注册检验报告
A2
医疗器械的设计和生产应遵循安全原则并兼顾现有技术能力,应当采用以下原则,确保每一危害的剩余风险是可接受的:
(1)识别已知或可预期的危害并且评估预期使用和可预期的不当使用下的风险。
(2)设计和生产中尽可能地消除风险。
(3)采用充分防护如报警等措施尽可能地减少剩余风险。
(4)告知剩余风险。
是
YY/T0316-2008
《医疗器械 风险管理对医疗器械的应用》
8.产品风险分析资料
A3
医疗器械在规定使用条件下应当达到其预期性能,满足适用范围要求。
是
临床评价
7.临床评价资料
A4
在生命周期内,正常使用和维护情况下,医疗器械的特性和性能的退化程度不会影响其安全性。
医疗器械安全有效基本要求清单
适用
证明符合性采用的方法
为符合性提供客观证据的文件
A
通用原则
A1
医疗器械的设计和生产应确保其在预期条件和用途下,由具有相应技术知识、经验、教育背景、培训经历、医疗和硬件条件的预期使用者(若适用),按照预期使用方式使用,不会损害医疗环境、患者安全、使用者及他人的安全和健康;使用时潜在风险与患者受益相比较可以接受,并具有高水平的健康和安全保护方法。
《医疗器械 风险管理对医疗器械的应用》
8.产品风险分析资料
B2.2
标有微生物要求的医疗器械,应当确保在使用前符合微生物要求。
否
无微生物要求
B2.3
无菌医疗器械应当确保在使用前符合无菌要求。
否
非无菌医疗器械
B2.4
无菌或标有微生物要求的医疗器械应当采用已验证的方法对其进行加工、制造或灭菌。
否
非无菌医疗器械
是
YY/T0316-2008
《医疗器械 风险管理对医疗器械的应用》
8.产品风险分析资料
感染和微生物污染
B2.1
医疗器械的设计和生产应当减少患者、使用者及他人感染的风险。设计应当:
(1)易于操作。
(2)尽可能减少来自产品的微生物泄漏和/或使用中微生物暴露。
(3)防止人对医疗器械和样品的微生物污染。
是
YY/T0316-2008
(3)材料的选择应考虑硬度,耐磨性和疲劳强度等属性(若适用)。
是
GB4793.1-2007
《测量、控制和实验室用电气设备的安全要求 第1部分:通用要求》
YY0648-2008
《测量、控制和实验室用电气设备的安全要求 第2-101部分:体外诊断(IVD)医用设备的专用要求》
10.产品注册检验报告(耐机械冲击和撞击试验)
含有微生物的细胞和其他物质的医疗器械,细胞及其他物质的加工、保存、检测和处理等过程应当提供患者、使用者和他人(如适用)最佳的安全保护。特别是病毒和其他传染原,应当采用经验证的清除或灭活方法处理。
否
非药械组合产品
环境特性
B5.1
如医疗器械预期与其他医疗器械或设备联合使用,应当保证联合使用后的系统整体的安全性,并且不削弱各器械或设备的性能。任何联合使用上的限制应在标签和(或)说明书中载明。液体、气体传输或机械耦合等连接系统,如,应从设计和结构上尽可能减少错误连接造成对使用者的安全风险。
是
符合企业自定的方法
5.研究资料(有效期和包装研究)
A5
医疗器械的设计、生产和包装应当能够保证其说明书规定的运输、贮存条件(如温度和湿度变化),不对产品特性及性能造成不利影响。
是
GB/T14710-2009《医用电器环境要求及试验方法》
5.研究资料(有效期和包装研究)10.产品注册检验报告
A6
所有风险以及非预期影响应最小化并可接受,保证在正常使用中受益大于风险。
否
非药械组合产品
B4.2
含有人体组织、细胞和其他物质的医疗器械,应当选择适当的来源、捐赠者,以减少感染的风险。人体组织、细胞和其他物质的加工、保存、检测和处理等过程应当提供患者、使用者和他人(如适用)最佳的安全保护。特别是病毒和其他传染原,应当采用经验证的清除或灭活方法处理。
否
非药械组合产品
B4.3
B2.7
若医疗器械可以以无菌与非无菌两种状态上市,则产品的包装或标签应当加以区别。
否
非无菌医疗器械
药械组合产品
B3.1
应对该药品和药械组合产品安全、质量和性能予以验证。
否
非药械组合产品
生物源性医疗器械
B4.1
含有动物源性的组织、细胞和其他物质的医疗器械,该动物源性组织、细胞和物质应当符合相关法规规定,且符合其适用范围要求。动物的来源资料应当妥善保存备查。动物的组织、细胞和其他物质的加工、保存、检测和处理等过程应当提供患者、使用者和他人(如适用)最佳的安全保护。特别是病毒和其他传染原,应当采用经验证的清除或灭活方法处理。