食品中砷比色法测定标准曲线绘制
实验十五二乙基二硫代氨基甲酸银比色法测砷

实验二十一二乙基二硫代氨基甲酸银比色法测砷一、实验目的1.掌握土壤的消化方法。
2.学习比色法测定土壤中砷的含量。
二、实验原理土壤中的砷在硝酸﹑硫酸的作用下,进行消化溶解,砷(五价)在酸性溶液中经碘化钾和氯化亚锡还原为三价,与新生态氢生成砷化氢气体,通过浸过醋酸铅的棉花除去硫化物后,吸收于二乙基二硫代氨基甲酸银—三乙胺—氯仿溶液中,生成红色络合物,比色测定。
三、实验仪器1.分光光度计2.砷化氢发生器四、实验试剂1.砷标准储备液:准确称取As2O30.1320g,置于100mL烧杯中,加5mL 20%氢氧化钠溶液,温热至As2O3全部溶解后,以酚酞为指示剂,用1mol/L硫酸中和至溶液无色,再过量10mL,转入1000mL容量瓶中,用水稀释至标线,此溶液浓度为每毫升含100微克砷。
2.砷标准使用液:吸取10.00mL砷标准储备液,置于1000mL容量瓶中,用水稀释至标线,此溶液浓度为每毫升含1.00微克砷。
3.40%氯化亚锡溶液:称取40g氯化亚锡(SnCl2·2H20),溶于100mL浓盐酸中,如保存可加几粒锡粒。
此溶液现用现配。
4.15%碘化钾溶液:储于棕色瓶中。
5.浓硝酸6.浓硫酸7.(1+1)硫酸溶液8.无砷锌粒9.0.1%二乙基二硫代氨基甲酸银—三乙胺—氯仿吸收液:称取1.0g二乙基二硫代氨基甲酸银,加100mL氯仿,加18mL三乙胺,摇匀,用氯仿稀释至1000mL,放置过夜,用脱脂棉过滤后使用。
保存于棕色瓶中,避光。
10.醋酸铅棉花:称取醋酸铅10g,溶于20mL 6mol/L醋酸中,加水稀释至100mL,将脱脂棉在此溶液中浸泡1h,取出自然晾干,即可使用。
五、实验步骤1.标准曲线的绘制分别吸取0.00,1.00,2.00,3.00,5.00,7.00,9.00,13.00,15.00mL 砷标准使用液于砷化氢发生器的锥形瓶中,配成标准系列为0.00,1.00,2.00,3.00,5.00,7.00,9.00,13.00,15.00μg 砷。
砷实验报告

一、实验目的1. 学习银盐法测定砷含量的原理和方法;2. 掌握分光光度计的基本操作;3. 通过实验,了解砷在食品中的存在形式及其对健康的危害。
二、实验原理样品经消化处理后,高价砷被还原为三价砷,然后与锌粒和酸产生的新生态氢反应生成砷化氢。
砷化氢经银盐溶液吸收后,形成红色胶态物,与标准系列比较定量。
三、试剂与仪器1. 主要试剂:- 4:1硝酸高氯酸混合液- 浓硫酸- 盐酸- 氢氧化钠- 碘化钾- 40%酸性氯化亚锡溶液- 无砷锌细粒- 10%醋酸铅溶液- 醋酸铅试纸- 醋酸铅棉花- 二乙氨基二硫代甲酸银三乙醇胺三氯甲烷溶液- 砷标准溶液2. 主要仪器:- 721型分光光度计- 砷化氢吸收装置- 1150ml锥形瓶- 气管- 醋酸铅棉花- 10ml刻度离心管四、实验步骤1. 样品处理- 准确称取10克样品,置于瓷坩埚中,加入氧化镁粉2克,10%硝酸镁溶液10毫升;- 在水浴上蒸干,小火炭化后,移入550高温炉中灰化至白色灰烬;- 冷却,加入10毫升浓盐酸溶解残渣,然后用水移入100毫升量瓶中,并稀释至刻度,摇匀。
2. 标准曲线绘制- 准确吸取每毫升相当于1微克砷的标准溶液0、1.0、2.0、3.0、4.0、5.0 mL,分别置于三角烧瓶中;- 向三角烧瓶中各加入水60mL,50%H2SO4溶液15mL,15%碘化钾溶液5 mL;- 加入无砷锌细粒,摇匀,静置;- 将溶液转移到砷化氢吸收装置中,通过导管导入银盐溶液中;- 待反应完成后,用分光光度计测定吸光度,绘制标准曲线。
3. 样品测定- 按照标准曲线绘制步骤,对处理后的样品溶液进行测定;- 根据吸光度,从标准曲线上查出对应的砷含量。
五、实验结果与分析1. 标准曲线绘制结果:以吸光度为纵坐标,砷含量为横坐标,绘制标准曲线,线性范围为0~5微克/毫升。
2. 样品测定结果:根据标准曲线,计算出样品中砷的含量。
3. 结果分析:- 通过实验,验证了银盐法测定砷含量的可行性;- 通过对比标准曲线和样品测定结果,可知该样品中砷含量符合国家标准。
原子荧光测砷标准曲线

原子荧光测砷标准曲线原子荧光法是一种灵敏度高、选择性好的分析方法,广泛应用于环境、食品、医药、化工等领域。
在水质监测中,砷是一个重要的监测对象,因为其对人体健康有潜在的危害。
因此,建立准确可靠的砷测定方法至关重要。
本文将介绍原子荧光测砷标准曲线的建立方法和实验步骤。
1. 实验原理。
原子荧光法是利用样品中的砷原子在高温等离子体中产生特征辐射,再通过光电倍增管进行检测,从而实现对砷的定量分析。
建立砷的标准曲线是进行定量分析的前提,通过一系列标准溶液的测定,绘制出砷的标准曲线,从而实现对未知样品中砷含量的测定。
2. 实验步骤。
(1)准备工作,将所需试剂和仪器进行清洗和校准,确保实验条件的准确性。
(2)制备标准溶液,分别取适量的砷标准品,用去离子水稀释至不同浓度的标准溶液。
(3)仪器参数设置,打开原子荧光仪,设置激发波长、发射波长、积分时间等参数。
(4)测定标准溶液,依次吸取不同浓度的标准溶液,放入原子荧光仪中进行测定。
(5)绘制标准曲线,根据测定结果,绘制出砷的标准曲线。
3. 实验注意事项。
(1)标准溶液的制备应严格按照要求,避免误差的产生。
(2)在测定过程中,应保持仪器的稳定性,避免外界因素对测定结果的影响。
(3)实验操作时应注意安全,避免对人体和环境造成伤害。
4. 结果与分析。
通过实验测定得到的标准曲线,可以看出砷的浓度与荧光强度之间存在一定的线性关系。
根据标准曲线,可以准确测定未知样品中砷的含量,为环境监测和食品安全提供重要依据。
5. 结论。
本实验成功建立了砷的原子荧光测定标准曲线,为后续砷含量的测定提供了可靠的方法和依据。
同时,本实验也验证了原子荧光法在砷分析中的重要应用价值。
通过本文的介绍,相信读者对原子荧光测砷标准曲线的建立方法有了更深入的了解。
在实际应用中,需要根据具体情况进行实验设计和操作,以确保测定结果的准确性和可靠性。
希望本文对相关领域的研究和实践工作有所帮助。
粮食中重金属砷、汞的检测研究

分析 检测粮食中重金属砷、汞的检测研究 刘洋 武建锋 南阳市粮油质量检测中心粮食中重金属污染是近几年来联合国粮农组织、世界卫生组织、我国食品安全组织进行食物污染检测计划中的重点项目。
粮食中的砷、汞等重金属元素的超标已经成为了威胁粮食安全的重要问题。
因此,针对粮食中的重金属砷、汞进行快速而准确的检测对于保障食品安全有着重要的意义。
粮食中重金属砷、汞对健康的危害粮食中重金属砷、汞对人体健康有着极大的危害。
微量的汞元素在人体中不会产生危害,可以经过尿液、粪便、汗液等方式排出体外,如汞含量超标则会影响人体健康。
人体吸收的汞分布在全身器官和组织,其中肝、肾、脑的含量最高,十分容易导致脑神经系统损伤。
汞的蓄积性很强,在体内的生物半衰期长达70天,在脑内更是长达250天。
汞中毒后会出现神经系统损伤症状,包括语言障碍、听力障碍、感觉障碍等,甚至导致瘫痪、肢体变形、死亡。
我国食品卫生标准GB2762-81中规定,粮食(成品粮食)中的汞含量不得超过0.02mg/kg。
长期食用砷污染的粮食会出现慢性中毒。
砷在人体能够与细胞内酶蛋白结合而丧失活性,进而影响人体的新陈代谢,从而导致死亡。
砷污染的粮食及其制品被人误食后会出现急性中毒,由于个体耐受度不同,导致中毒的剂量也有所不同。
我国食品卫生标准GB2762-81中规定粮食中的砷含量0.7mg/kg。
粮食中重金属砷、汞的检测本次研究分析微波消解结合原子荧光形态分析仪方法来测定小麦中的砷、汞含量。
材料与方法。
(1)材料与试剂:小麦样品;原子荧光形态分析仪SA5;纯水机基因型1830c;微波消解仪AntonPaar;定量取样研磨机RAS-230V;电子天平。
(2)方法:标准曲线绘制:把砷元素储备液与5%的硝酸进行逐级稀释,并配制成质量浓度为100ng/mL的标准使用液;把汞元素储备液使用5%的硝酸进行逐级稀释,并配制成为浓度为25ng/mL的标准使用液。
取砷元素、汞元素标准使用液的0、1.0ml、2.0ml、4.0ml于50ml的容量瓶当中,使用5%的硝酸定容、摇匀,等待测试。
食品中总砷的测定方法

食品中总碑的测定方法1、原理样品经消化后,以碘化钾、氯化亚锡将高价种还原为三价种,然后与锌料和酸产生的新生态氢生成神化氢,经银盐溶液吸收后,形成红色胶态物,与标准系列化比较定量。
2、试剂除特别注明外,所有试剂为分析纯,水为去离子水。
(1)硝酸一高氯酸混合液(4+1)o(2)酸性氯化亚锡溶液(40g∕IOOmD称取40g氯化亚锡(Sncl2.2H2O),加盐酸溶解并稀释到IoOm1,加入数颗金属锡粒。
(3)乙酸铅棉花用乙酸铅溶液(IOog/L浸透脱脂棉后,压除多余溶液,并使疏松,在IO(TC以下干燥,储存于玻璃瓶中。
(4)硫酸(6+94)(5)AgDDC-三乙醇胺-三氯甲烷溶液称取0.25g二乙基二硫代氨基甲酸银[(C2H5)2NCS2Ag]置于乳钵中,加少量三氯甲烷研磨,移入IoOmI量筒中,加入1.8ml三乙醇胺,再用三氯甲烷分次洗涤乳钵,洗液一并移入量筒中,再用三氯甲烷稀释至IOomI,放置过夜,滤入棕色瓶中储存。
(6)碑标准使用液(1.0μg∕ml)o3、仪器可见分光光度计4、操作步骤(1)样品处理(D湿法消化:称取样品适量,置于250~500ml定氮瓶中,先加水少许使湿润,加数粒玻璃珠、10~15ml硝酸(或硝酸-高氯酸混合液),放置片刻,小火缓缓加热,待作用缓和,放冷。
沿瓶壁加入5ml或IOmI硫酸,再加热,至瓶中液体开始变成棕色时,不断沿瓶壁滴加硝酸(或硝酸-高氯酸混合液)至有机质分解完全。
加大火力,至产生白烟,待瓶内液体再产生白烟为消化完全,该溶液应澄明无色或微带黄色,放冷。
加20ml水煮沸,除去残余的硝酸至产生白烟为止,如此处理两次,放冷。
定容到50或IOom1。
②干法消化:称取样品适量,置于用锅中,加Ig氧化镁及IOmI硝酸镁溶液,混匀,浸泡4h。
于低温或水浴上蒸干,用小火炭化至无色后移入马弗炉中加热至550℃,灼烧至完全灰化,冷却后取出。
加5ml水湿润灰分,再缓缓加热盐酸(1+1),然后将溶液移入50ml容量瓶中,用烟人盐酸(1+1),洗涤5次,洗液合并入容量瓶中,加盐酸(1+1)至刻度。
食品中砷的测定

食品中砷的测定1 砷依照《GB 1002.11》规定的方法检测。
2 标准规定≤0.5mg/kg3 仪器器皿及试剂:仪器原子吸收光谱仪,万分之一天平,微波消解仪。
硝酸、高氯酸、硫酸、硫脲、抗坏血酸。
4 操作方法4.1 固体试样称取1.0~2.5g、液体试样称取5.0~10.0g(或ml)(精确至0.001g),置于50ml~100ml锥形瓶中,同时做两份试剂空白。
加硝酸20ml,高氯酸4ml,硫酸1.25ml,放置过夜。
次日置于电热板上加热消解。
若消解液处理至1ml左右时仍有未分解物质或色泽变深,取下放冷,补加硝酸5ml~10ml,再消解至2ml左右,如此反复两三次,注意避免炭化。
继续加热至消解完全后,再持续蒸发至高氯酸的白烟散尽,硫酸的白烟开始冒出。
冷却,加水25ml,再蒸发至冒硫酸白烟。
冷却,用水将内溶物转入25ml容量瓶或比色管中,加入硫脲+抗坏血酸溶液2ml,补加水至刻度,混匀,放置30min,待测。
按同一操作方法做空白试验。
4.2 仪器参考条件负高压:260V;砷空心阴极灯电流:50mA~80mA;载气:氩气;载气流速:500ml/min;屏蔽气流速800ml/min;测量方式:荧光强度;读数方式:峰面积。
4.3 标准曲线制作取25ml容量瓶或比色管6支,依次准确加入1.00μg/ml砷标准使用液0.00ml、0.10ml、0.25ml、0.50ml、1.5ml和3.0ml(分别相当于砷浓度0.0ng/ml、4.0ng/ml、10.0ng/ml、20ng/ml、60ng/ml、120ng/ml),各加硫酸溶液(1+9)12.5ml,硫脲+抗坏血酸溶液2ml,补加水至刻度,混匀后放置30min后测定。
仪器预热稳定后,将试剂空白、标准系列溶液依次引入仪器进行原子荧光强度的测定。
以原子荧光强度为纵坐标,砷浓度为横坐标绘制标准曲线,得到回归方程。
4.4 样品的测定相同条件下,将样品溶液分别引入仪器进行测定。
砷的检测实验报告

一、实验目的1. 掌握砷的检测方法。
2. 熟悉实验仪器和试剂的使用。
3. 提高分析化学实验技能。
二、实验原理砷是一种有毒的重金属元素,对人体健康具有严重的危害。
本实验采用原子荧光光谱法检测水样中的砷含量。
该方法利用砷在特定条件下能够发出特定波长的荧光,通过测定荧光强度来确定砷的含量。
三、实验仪器与试剂1. 仪器:原子荧光光谱仪、分析天平、微波消解仪、移液器、比色皿等。
2. 试剂:硝酸、盐酸、氢氧化钠、硼氢化钠、抗坏血酸、砷标准溶液等。
四、实验步骤1. 样品前处理(1)称取适量的水样,加入硝酸和盐酸,用微波消解仪消解。
(2)将消解液转移至容量瓶中,定容至刻度。
2. 标准曲线的绘制(1)分别吸取0、0.5、1.0、2.0、4.0、6.0、8.0 mL砷标准溶液于比色皿中,加入适量的硝酸和盐酸,定容至刻度。
(2)在原子荧光光谱仪上,设置好仪器参数,测定各标准溶液的荧光强度。
(3)以砷含量为横坐标,荧光强度为纵坐标,绘制标准曲线。
3. 样品测定(1)将处理好的样品溶液转移至比色皿中,加入适量的硝酸和盐酸,定容至刻度。
(2)在原子荧光光谱仪上,设置好仪器参数,测定样品溶液的荧光强度。
(3)根据标准曲线,计算样品中砷的含量。
五、实验结果与分析1. 标准曲线以砷含量为横坐标,荧光强度为纵坐标,绘制标准曲线,得到线性回归方程为:y = 0.005x + 0.002(R² = 0.998)。
2. 样品测定测定样品溶液的荧光强度,根据标准曲线计算样品中砷的含量,结果如下:样品1:砷含量为0.15 mg/L样品2:砷含量为0.20 mg/L样品3:砷含量为0.05 mg/L六、实验讨论1. 实验结果表明,原子荧光光谱法可以有效地检测水样中的砷含量。
2. 在实验过程中,需要注意以下几点:(1)样品前处理过程中,消解液要充分混合,以确保砷的充分溶解。
(2)在绘制标准曲线时,要注意标准溶液的配制和测量。
(3)在测定样品时,要严格控制实验条件,以确保实验结果的准确性。
食品中矿物质的测定

(3)仪器 原子吸收分光光度计、钙空心阴极灯 (4) 操作步骤 1. 样品制备 湿样(如蔬菜,水果,鲜鱼,鲜肉等)用水清洗干净
(4)操作步骤 消化:精确称取均匀样品干样0.5~1.5g(湿样2.0~4.0g,饮料 等液体样品5.0~10.0g)于250ml高型烧杯内,加混合酸消化液 20~30ml.上盖表皿.置于电热板或电沙浴上加热消化.如未消 化好而酸液过少时,再补加几毫升混合酸消化液,继续加热消化, 直至无色透明为止.
后,要用去离子水充分洗净.干粉类样品(如面粉,奶粉等)取 样后立即装容器密封保存,防止空气中的灰尘和水分污染. 2. 样品消化 精确称取均匀样品干样0.5~1.5g(湿样2.0~ 4.0g,饮料等液体样品5.0~10.0g)于250ml高型烧杯内,加 混合酸消化液20~30ml.上盖表皿.置于电热板或电沙浴上 加热消化.如未消化好而酸液过少时,再补加几毫升混合酸 消化液,继续加热消化,直至无色透明为止.
二、铁的测定—邻二氮菲法
1,原理: 在pH为2~9的溶液中,二价铁离子与邻二氮菲生成稳 定的橙红色配合物,在510nm有最大吸收,其吸光度与铁的含量成 正比,故可比色测定.
2,试剂 ①盐酸羟胺溶液:10% ②邻二氮菲水溶液(新鲜配 制):0.12% ③醋酸钠溶液:10% ④盐酸:1mol/L ⑤铁标准溶液
③ 柠檬酸铵溶液
④ 淀粉指示液
⑤ 二硫腙-三氯甲烷溶液 ⑥ 二硫腙使用液 ⑦ 铅标准溶液 ⑧ 铅标准使用液 3.仪器 分光光度计 4操作步骤 (1)样品预处理 在采样和制备过程中,注意样品不被污染。 粮食、水果、鱼类、肉类及蛋类等水分含量高的鲜样,用食品 加工机大成匀浆存于塑料瓶中,保存备用。 (2)样品消化(灰化法)
食品中砷比色法测定标准曲线绘制
食品中砷比色法测定标准曲线的绘制在仪器分析中,如比色测定,色谱测定、原子吸收法测定等,都要用标准样品来校准仪器,即将测定所用的仪器的响应值与标准品的量(或浓度)的对应关系,制作成标准曲线。
然后,根据样品测定时所得的响应值,从标准曲线查得待测组分的量(或浓度)。
当然制作标准曲线与测定样品时,所用的方法、所处的各项条件要完全一致。
一般要同时进行。
例如:食品中砷残留量的银盐法(比色法)测定(1)砷标准贮备液:按国家标准GB/T5009.11-2003第二法,先配制成砷标准贮备液ρ(As)=1.00mg/ml。
(2)砷标准使用液:精确吸取砷标准贮备液5.00ml,用水定容至500ml,得砷标准使用液ρ(As)=1.00 μg/ml。
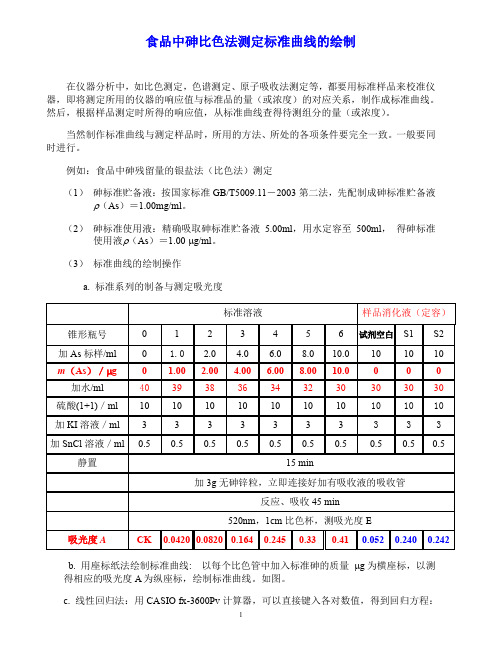
(3)标准曲线的绘制操作a. 标准系列的制备与测定吸光度b. 用座标纸法绘制标准曲线:以每个比色管中加入标准砷的质量μg为横座标,以测得相应的吸光度A为纵座标,绘制标准曲线。
如图。
c. 线性回归法:用CASIO fx-3600Pv计算器,可以直接键入各对数值,得到回归方程:Y = 24.382457 X - 0.006478; 相关系数r =0.999714 结果计算 试样中砷的含量按式(2)进行计算()()122110002/1000A A x m V V -⨯=⨯⨯()()1012 5.8050 5.80/210 5.00m m V g ml x g g V m ml gμμ-=⨯=⨯=式中:X -试样中砷的含量,mg/kgm1-测定用试样消化液中砷的质量,μg ; m0-试剂空白液中砷的质量,μg ; m -试样的质量,g ;V1-试样消化液的定容总体积, ml ;V2-用于比色时所取的试样消化液的体积, ml ;计算结果保留两位有效数字。
食品中砷的含量测量不确定度评估报告

X=
X---样品中汞的含量, mg/kg;
C---为扣除空白溶液汞后样品溶液中汞的实际浓度, μg/L;
V---样品经处理后定容溶液体积, ml;
M---样品质量, g。
M---样品质量,g。
主要不确定来源及因果图
标准溶液配制由标准曲线计算
稀释过程线性回归
标准物质
X
容量瓶允差称样量变动性
u2r
定容体积
u2r=0.00062
u3r
测定重复性
u3r=0.018
u4r
标准溶液
1000
u4r=0.010
u5r
标准曲线
1
u5r=0.0085
测量不确定度评定表(续)
合成标准不确定度
= ucr= =0.022
uc=×ucr=21.2×0. 022=0.5μg/kg
uc= ×ucr=21.2×0.022=0.5μg/kg
钦州出入境检验检疫局检验检疫综合实验室
测量不确定度评定报告
编号: 2010-03
检验项目
食品中汞的测定
试验方法
原子荧光光谱法
试验步骤、要求
称取约2 g试样(精确至0.01 g), 用微波消解仪消解后用100 mL容量瓶定容, 用原子荧光分光光度计进行测定。
仪器设备
电子天平(BS224S),原子荧光分光光度计
扩展不确定度
当置信率为95%时,包含因子k=2,则扩展不确定度
U95=k×uc=2×0.5=1.0μg/kg。
最终评定结果
合成标准不确定度
u=0.5μg/kg
扩展不确定度
U=1.0μg/kg
测量结果表示
X=(U)=(21.2±1.0)μg/kg, k=2。
食品添加剂中砷的测定方法GB8450-87

食品添加剂中砷的测定方法GB8450-87 UDC 614.3--------------------------------------------------------------------------------------- ----------------- 本标准适用于食品添加剂中砷的限量试验和定量试验。
本标准参照1983联合国粮农组织和世界卫生组织(FAO/WHO)食品添加剂联合专家委员会发布的有关砷的测定方法。
1. 二乙氨基二硫代甲酸银比色法1.1 原理在碘化钾和氯化亚锡存在下, 将样液中的高价砷还原为三价砷, 三价砷与锌粒和酸产生的新生态氢作用, 生成砷化氢气体, 经乙酸铅棉花除去硫化氢干扰后,将溶于三乙醇胺-三氯甲烷中或吡啶中的二乙氨基二硫代甲酸银溶液吸收并作用,生成紫红色络和物, 与标准比较定量。
1.2 试剂除特别注明外,寰标准所用试剂均为分析纯,水为蒸馏水或去离子水。
1.2.1 硝酸(GB 626-78)。
1.2.2硫酸(GB 625-77)。
1.2.2.1 硫酸(1+1)溶液:将1体积浓硫酸慢慢加入1体积水中,冷后叔用。
1.2.2.2 硫酸(1mol/L)溶液: 量取28ml浓硫酸,慢慢加入水中,用水稀释到500ml。
1.2.3 盐酸(GB 622-77)。
1.2.4 氢氧化钠(GB 629-77): 20%溶液。
1.2.5 氧化镁(HG 3-1294-80)。
1.2.6 硝酸镁(HG 3-1077-77): 15%溶液。
1.2.7 碘化钾(GB 1272-77):15%溶液。
贮于棕色瓶内(临用前配制)。
1.2.8 氯化亚锡(GB 638-78):40%溶液。
称取20g氯化亚锡(SnCl2.2H2O),溶于50ml盐酸。
1.2.9乙酸铅棉花:将脱脂棉浸于10%乙酸铅(HG 3-974-76)溶液中,2h后取出晾干。
1.2.10 无砷金属锌(GB 2304-80)。
1.2.11三氯甲烷(GB 682-78)。
总砷测定的规范标准操作技巧规章
1目的规范总砷测定的标准操作规程。
2范围本标准规定了食品中总砷的测定方法。
本标准第一、二法适用于各类食品中总砷的测定。
3责任质量部组织制订、化验室负责实施。
4内容4.1 依据:GB5009.11-2014 食品安全国家标准食品中总砷及无机砷的测定4.2第一法电感耦合等离子体质谱法4.2.1原理样品经酸消化处理后,消解液经过雾化由载气(氩气)导入ICP炬焰中,经过蒸发、解离、原子化、电离等过程,大部分转化为带正电荷的正离子,经离子采集系统进入质谱仪,质谱仪根据其质荷比进行分离。
对于一定的质荷比,质谱积分面积与进入质谱仪中的离子数成正比,即样品中待测物的浓度与质谱积分面积或质谱峰高成正比。
因此可通过测量质谱积分面积或质谱峰高测定样品中砷元素的浓度。
4.2.2试剂和材料除非另有说明,本方法所用试剂均为优级纯,水为GB/T 6682规定的一级水。
4.2.2.1试剂4.2.2.1.1硝酸(HNO3):MOS级(电子工业专用高纯化学品)、BV Ⅲ级。
4.2.2.1.2过氧化氢(H2O2)4.2.2.1.3质谱调谐液:Li、Y、Ce、Ti、Co,推荐使用浓度为10 ng/ml。
4.2.2.1.4内标储备液:Ge,浓度为100 Ug/mL。
4.2.2.1.5氢氧化钠(NaOH)4.2.2.2试剂配制4.2.2.2.1硝酸溶液(2+98):量取20 mL硝酸,缓缓倒入980 mL水中,混匀。
4.2.2.2.2硫酸溶液(1+9):量取硫酸100mL,缓缓倒入900 mL水中,混匀。
4.2.2.2.3内标溶液Ge 或Y(1.0 ug/mL):取1.0mL内标溶液,用硝酸溶液(2+98)稀释定容至100mL。
4.2.2.2.4 氢氧化钠溶液(100 g/L):称取10 .0g 氢氧化钠,溶于水并用水稀释至100 mL。
4.2.2.3.标准品三氧化二砷(As2O3)标准品:纯度≥99.5%。
4.2.2.4标准溶液配制4.2.2.4.1砷标准储备液(100 mg/L,按As计):准确称取于100℃干燥2h的三氧化二砷0.0132g,加1 mL氢氧化钠溶液(100 g/L)和少量水溶解,转入100 mL容量瓶中,加入适量盐酸调整其酸度接近中性,用水稀释至刻度。
食品中总砷及无机砷的测定

食品中总砷及无机砷的测定总砷的测定1 范围本标准规定了各类食品中总砷的测定方法。
本标准适用于各类食品中总砷的测定。
本方法检出限:氢化物原子荧光光度法;0.01mg/kg,线性范围为0ng/mL~200ng/mL;银盐法:0.2mg/kg;砷斑法:0.25mg/kg;硼氢化物还原比色法:0.05mg /kg。
第一法氢化物原子荧光光度法2 原理食品试样经湿消解或干灰化后,加入硫脲使五价砷预还原为三价砷,再加人硼氢化钠或硼氢化钾使还原生成砷化氢,由氩气载入石英原子化器中分解为原子态砷,在特制砷空心阴极灯的发射光激发下产生原子荧光,其荧光强度在固定条件下与被测液中的砷浓度成正比,与标准系列比较定量。
3 试剂3.1 氢氧化钠溶液(2g/L)。
3. 2 硼氢化钠(NaBH4)溶液(10g/L):称取硼氢化钠10.0g,溶于2 g/L 氢氧化钠溶液l 000mL中,混匀。
此液于冰箱可保存10天,取出后应当日使用(也可称取14 9硼氢化钾代替10g硼氢化钠)。
3.3 硫脲溶液(50g/1)3.4 硫酸溶液(1+9):量取硫酸100mL,小心倒入水900mL中,混匀。
3.5 氢氧化钠溶液(100g/L)(供配制砷标准溶液用,少量即够)。
3.6 砷标准溶液3.6.1 砷标准储备液:含砷0.1 mg/mL。
精确称取于100℃干燥 2 h以上的三氧化二砷(As203)0.132 0g,加100g/L氢氧化钠10mL溶解,用适量水转入1 000mL容量瓶中,加(1+9)硫酸25mL,用水定容至刻度。
3.6.2 砷使用标准液:含砷1μg/mL。
吸取1.00mL砷标准储备液于100mL 容量瓶中,用水稀释至刻度。
此液应当日配制使用。
3.7 湿消解试剂:硝酸、硫酸、高氯酸。
3. 8 干灰化试剂:六水硝酸镁(150g/L)、氯化镁、盐酸(1+1)。
4 仪器原于荧光光度计。
5 分析步骤5.1 试样消解5.1.1 湿消解,固体试样称样1g~2.5g,液体试样称样5g~10g(或mL)(精确至小数点后第二位),置人50mL~100mL锥形瓶中,同时做两份试剂空白。
原子荧光砷标准曲线

原子荧光砷标准曲线原子荧光分析技术是一种高灵敏度、高选择性和高准确性的分析方法,广泛应用于环境监测、食品安全、医药卫生等领域。
而砷是一种常见的有毒元素,其含量的准确测定对于环境和食品安全具有重要意义。
因此,建立原子荧光砷标准曲线是进行砷含量测定的重要步骤之一。
标准曲线是一组实验数据点的图形表示,用于分析仪器的定量分析。
在建立原子荧光砷标准曲线时,需要准备一系列不同浓度的砷标准溶液,并通过原子荧光分析仪器对这些标准溶液进行测试,得到各个浓度下的砷信号强度。
然后,将这些数据绘制成标准曲线图,通过曲线的斜率和截距来计算未知样品中的砷含量。
在进行原子荧光砷标准曲线的建立时,需要注意以下几点:1. 标准溶液的制备。
标准溶液的制备是建立标准曲线的前提。
需要准确称取一定量的砷标准品,溶解于适量的溶剂中,制备成一系列不同浓度的标准溶液。
在制备过程中,要注意避免溶液的挥发和外界污染,确保标准溶液的浓度准确可靠。
2. 仪器的操作。
在进行原子荧光分析时,需要严格按照仪器操作手册的要求进行操作。
包括样品的进样、仪器的参数设置、分析条件的选择等。
只有确保仪器的稳定性和准确性,才能得到可靠的实验数据。
3. 数据处理。
得到砷标准溶液的各个浓度下的信号强度数据后,需要进行数据处理。
通常采用线性回归分析的方法,将信号强度与浓度之间的关系拟合成一条直线,得到标准曲线的方程。
通过这个方程,就可以计算出未知样品中砷的含量。
建立原子荧光砷标准曲线是一项繁琐而重要的工作。
只有建立了准确可靠的标准曲线,才能保证对未知样品中砷含量的准确测定。
因此,在进行实验时,需要严格按照操作规程进行,确保实验数据的可靠性和准确性。
总之,原子荧光砷标准曲线的建立是原子荧光分析技术中的关键步骤,对于砷含量的准确测定具有重要意义。
只有通过严谨的实验操作和数据处理,才能得到准确可靠的标准曲线,为后续的砷含量测定工作奠定基础。
砷含量测量不确定度评定报告

砷强化营养盐中砷的测量不确定度评定报告一、目的:1、给出砷强化营养盐中砷含量的测量结果的不确定度。
2、在测量结果处于临界状态时,用于对测量结果作出正确的判定。
3、用于评价实验室测量比对结果的质量。
二、适用范围:用分光光度计比色法测试砷强化营养盐中砷含量的不确定度。
三、依据:1、JJF1059-1999《测量不确定度评定与表示》2、依据CNAL/AG06《测量不确定度政策实施指南》3、CNAL/AR11《测量不确定度政策》分析4、GB/T 13025.13-1994《砷强化营养盐》 四、概述:1、主要仪器与试剂电子天平:AE100型,梅特勒-托利多仪器有限公司; 紫外可见分光光度计: 氯化钠溶液:100g/L ; 乙酸:1.0mol/L ;砷标准储备液:250µg/mL, ; 试验用水为去离子水。
2、原理砷溶于酸性溶液中,呈黄色,其黄色深度与砷含量成正比,光度法测定其含量。
3、标准曲线绘制吸取砷标准储备液10.00mL 于100mL 容量瓶中,加水稀释至刻度,摇匀,此溶液每毫升相当于砷25µg 。
用时新配。
吸取砷标准工作溶液(25µg/mL)0.0,2.0,4.0,6.0,10.0mL 于50mL 比色管中,加氯化钠溶液(100g/L) 10mL ,加水稀释至刻度,摇匀。
于波长440nm 处,以1.Ocm 比色池,试剂空白作对照,测定其吸光度。
以砷含量为横坐标,对应的吸光度为纵坐标,绘制标准曲线。
4、分析步骤称取均匀样品1.2533g ,置于400m L 烧杯中,加入乙酸(1.0mol/L)5 m L ,加水200m L,加热溶解。
冷却后,移入500m L 容量瓶中,加水稀释至刻度,摇匀。
吸取20.00 m L ,置于50m L 比色管中,加水稀释至刻度,摇匀。
于波长440nm 处,以1.Ocm 比色池,试剂空白作对照,测定其吸光度。
五、建立数学模型 X---样品测得的吸光度从标准曲线查出相应的砷量,mg/kg C x —从标准曲线上查得的砷量样品溶液的浓度,µg m ---样品的质量,单位为克(g) f ---稀释因子六、引入不确定度的因素分析1、样品质量m 的测量不确定度u (m );2、稀释因子f 的测量不确定度u (f )xC X fm=⨯3、砷质量的测量不确定度u (C x )七、测定结果1. 样品质量m 的测量不确定度u (m )1.1电子天平的最大误差为±0.0001g ,按均匀分布,考虑天平称量分两次进行,一次是空盘,一次是毛重,因此:1.2所用天平为数显式,其分辨率为0.01,按均匀分布,由分辨率产生的不确定度为:1.3样品质量m 的不确定度u (m )其相对合成标准不确定度为5()0.000583() 2.911020.0002rel u m gu m g m -===⨯2. 稀释因子f 的测量不确定度u (f )样品稀释过程:样品称量后,用100mL 容量瓶定容稀释,然后用10mL 移液管移取10mL 置于比色管中比色。
砷测定比色法测定标准曲线绘制
砷比色法测定标准曲线的绘制在仪器分析中,如比色测定,色谱测定、原子吸收法测定等,都要用标准样品来校准仪器,即将测定所用的仪器的响应值与标准品的量(或浓度)的对应关系,制作成标准曲线。
然后,根据样品测定时所得的响应值,从标准曲线查得待测组分的量(或浓度)。
当然制作标准曲线与测定样品时,所用的方法、所处的各项条件要完全一致。
一般要同时进行。
例如:食品中砷残留量的银盐法(比色法)测定1、砷标准贮备液:按国家标准GB/T5009.11-2003第二法,先配制成砷标准贮备液ρ(As)=1.00mg/ml。
2、砷标准使用液:精确吸取砷标准贮备液5.00ml,用水定容至500ml,得砷标准使用液ρ(As)=1.00 µg/ml。
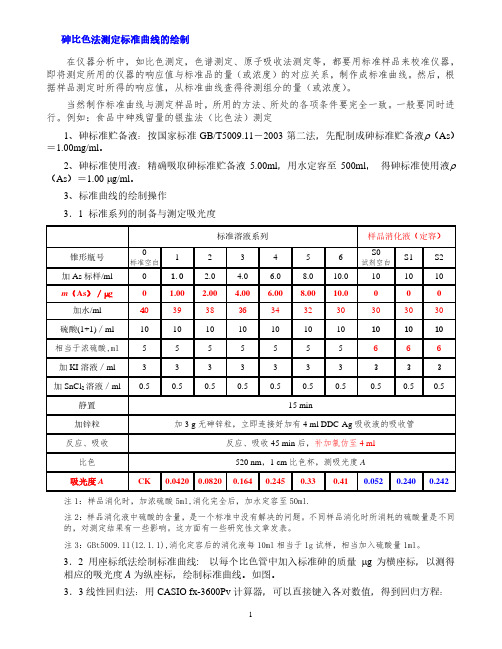
3、标准曲线的绘制操作3.1 标准系列的制备与测定吸光度标准溶液系列样品消化液(定容)锥形瓶号标准空白123456S0试剂空白S1S2加As标样/ml0 1.0 2.0 4.0 6.08.010.0101010m(As)/µg 0 1.00 2.00 4.00 6.008.0010.00 0 0 加水/ml40 393836 343230303030硫酸(1+1)/ml1010101010101010 10 10 相当于浓硫酸,ml 5 5 5 5 5 5 5 6 6 6 加KI溶液/ml 3 3 3 3 3 3 3 3 3 3 加SnCl2溶液/ml 0.5 0.5 0.5 0.5 0.5 0.5 0.5 0.5 0.5 0.5 静置15 min加锌粒加3 g无砷锌粒,立即连接好加有4 ml DDC-Ag吸收液的吸收管反应、吸收反应、吸收45 min后,补加氯仿至4 ml比色520 nm,1 cm比色杯,测吸光度A吸光度A CK 0.0420 0.0820 0.164 0.245 0.33 0.41 0.052 0.240 0.242 注1:样品消化时,加浓硫酸5ml,消化完全后,加水定容至50ml.注2:样品消化液中硫酸的含量,是一个标准中没有解决的问题。
食品中铅、镉、砷的测定(国标)【精选文档】

食品中铅的测定:第一法石墨炉原子吸收光谱法3 原理试样经灰化或酸消解后,注入原子吸收分光光度计石墨炉中,电热原子化后吸收283。
3 nm 共振线,在一定浓度范围,其吸收值与铅含量成正比,与标准系列比较定量。
4 试剂和材料硝酸:优级纯。
4.2 过硫酸铵。
4。
3 过氧化氢(30%)。
4。
4 高氯酸:优级纯。
4.5 硝酸(1+1):取50 mL 硝酸慢慢加入50 mL 水中。
4。
6 硝酸(0.5 mol/L):取3.2 mL 硝酸加入50 mL 水中,稀释至100 mL。
4。
7 硝酸(l mo1/L):取6.4 mL 硝酸加入50 mL 水中,稀释至100 mL。
4。
8 磷酸二氢铵溶液(20 g/L):称取2。
0 g 磷酸二氢铵,以水溶解稀释至100 mL。
4。
9 混合酸:硝酸十高氯酸(9+1)。
取9 份硝酸与1 份高氯酸混合。
4.10 铅标准储备液:准确称取1。
000 g 金属铅(99.99%),分次加少量硝酸(4。
5),加热溶解,总量不超过37 mL,移入1000 mL 容量瓶,加水至刻度。
混匀。
此溶液每毫升含 1.0 mg 铅.4。
11 铅标准使用液:每次吸取铅标准储备液1。
0 mL 于100 mL 容量瓶中,加硝酸(4.6)至刻度。
如此经多次稀释成每毫升含10。
0 ng,20.0 ng,40。
0 ng,60。
0 ng,80.0 ng 铅的标准使用液。
5 仪器和设备5。
1 原子吸收光谱仪,附石墨炉及铅空心阴极灯。
5。
2 马弗炉。
5。
3 天平:感量为1 mg.5。
4 干燥恒温箱.5。
5 瓷坩埚。
5.6 压力消解器、压力消解罐或压力溶弹。
5.7 可调式电热板、可调式电炉。
6 分析步骤6.2 试样消解(可根据实验室条件选用以下任何一种方法消解)6.2.1 湿式消解法:称取试样1 g~5 g(精确到0.001 g)于锥形瓶或高脚烧杯中,放数粒玻璃珠,加10 mL 混合酸(4。
9),加盖浸泡过夜,加一小漏斗于电炉上消解,若变棕黑色,再加混合酸,直至冒白烟,消化液呈无色透明或略带黄色,放冷,用滴管将试样消化液洗入或过滤入(视消化后试样的盐分而定)10 mL~25 mL 容量瓶中,用水少量多次洗涤锥形瓶或高脚烧杯,洗液合并于容量瓶中并定容至刻度,混匀备用;同时作试剂空白.6.3 测定6.3.1 仪器条件:根据各自仪器性能调至最佳状态.参考条件为波长283。
砷钼蓝色比色法同时测定磷、砷

砷钼蓝色比色法同时测定磷、砷试样用NaOH-KMnO4混合熔剂半熔,以热水煮出后过滤除去Fe3+等干扰离子,在约0.3moL/L的硫酸溶液中加入酒石酸根离子、钼酸铵和抗坏血酸,加热形成钼蓝,测定磷砷含量,差减求出砷含量。
加入酒石酸根络合钼,可消除硅酸的影响,防止钼酸的还原.一、主要试剂1、抗坏血酸溶液(1.5%):称取1.5克抗坏血酸于100毫升水中充分溶解。
2、酒石酸-钼酸铵混合溶液:4%酒石酸水溶液100亳升与降1. 85%钼酸铵水溶液100毫升充分混匀。
3、砷标准溶液:称取缔0.1320克三氧化二砷,加20毫升水,0.2克氢氧化钠,加热溶解,用硫酸(1+4)中和至微酸性,移入1升容量瓶,以水定容.此溶液含砷10ug/ml.4、磷标准溶液:称取0.1917克在110度烘干的基准磷酸二氢钾(KH2PO4)溶于水中,移入1升容量瓶,以水定容.此溶液含磷10ug/ml 标准曲线的绘制:吸取5、10、15、30、40、50磷标准溶液于一组50毫升比色管中,取10、20、30、60、100ug砷标准溶液于一组50毫升比色管中,加1:2H2SO42.5毫升,酒石酸-钼酸铵混合溶液10毫升.加入0.2%KMnO40.5毫升,摇匀,稀释至约40毫升,加1.5%抗坏血酸5毫升,加水稀释至刻度,摇匀,水浴加热至沸2-3分钟,取出冷却。
用3cm 比色皿于810nm波长处测定其吸光度.二、分析步骤先取4-8克NaCO3-KMnO4混合熔剂于50毫升瓷坩埚中,准确称取0.5000-1.000克样于坩埚与熔剂混匀,上面再覆盖4克混合熔剂,放入先升至750度的马弗炉中恒温一小时.取出冷却后加水至约加毫升,捣碎熔块,于电炉上加热煮沸一分钟,趁热过滤于100毫升容量瓶中,用2%氢氧化钠溶液冲洗沉淀3-5次,定容至刻度,摇匀.抽取20毫升(视磷砷含量)溶液于50毫升比色管中,滴加酚酞指示剂2滴,边摇动边滴加(1:2)硫酸2.5毫升,酒石酸钼酸铵混合溶液10毫升,洗净管壁,摇匀,加1.5%抗坏血酸溶液5毫升,加水稀释至刻度,摇匀,水浴加热至沸2-3分钟,取出冷却后用3cm比色皿于810nm波长处测定其吸光度.另抽取一份溶液于100毫升烧杯中,滴加酚酞指示剂2滴,边摇动边滴加(1:2)硫酸至溶液从红色变无色,加(1:2)硫酸2.5毫升,碘化钾固体1克,于高温电炉上蒸至体积约5-8毫升,取下转入50毫升比色管中,加1.5%抗坏血酸溶液还原至无色或黄色,加酒石酸-钼酸铵混合溶液10毫升,摇匀,加1.5%血酸溶液还原至无色或黄色,加酒石酸-钼酸铵混合溶液10毫升,摇匀,加1.5%抗坏敌国酸溶液5毫升,显色,测量磷吸光度.查磷标准曲线,计算出磷含量.将磷砷吸光度减去磷吸光度,其差为砷吸光度,查磷标准曲线,计算出砷含量.。
砷钼酸比色法测定还原糖含量

砷钼酸比色法测定还原糖含量一、目的植物体内的还原糖主要是葡萄糖、果糖和麦芽糖。
它们在植物体内的分布,不仅反映植物体内碳水化合物的运转情况,而且也是合成其它成分碳架来源和呼吸作用的基质。
此外,水果、蔬菜中含糖量的多少,也是鉴定其品质的重要指标。
其它碳水化合物,如淀粉、蔗糖等,经水解也生成还原糖。
因此,测定还原糖的方法在研究植物体内生理生化变化和测定植物体内碳水化合物方面都是很重要的。
二,原理;还原糖是具有羰基(>C=O)的糖,能将其它物质还原而其本身被氧化。
(1) 还原糖在碱性条件及有酒石酸钾钠存在下加热,可以定量地还原二价铜离子为一价铜离子,产生砖红色的氧化亚铜沉淀,其本身被氧化。
具体反应如下:(2)氧化亚铜在酸性条件下,可将钼酸铵还原,还原型的钼酸铵再与砷酸氢二钠起作用,生成一种蓝色复合砷钼蓝,其颜色深浅在一定范围内与还原糖的含量(即被还原的Cu2O 量)成正比,用标准葡萄糖与砷钼酸作用,比色后做标准,就可测得样品还原糖含量。
三、实验材料、主要仪器和试剂1.实验材料苹果、面粉等2.主要仪器(1)分光光度计(2)水浴锅(3)具塞刻度试管:20mL×10(4)刻度吸管:1 mL×1,2 mL×4,5 mL×3(5)容量瓶:100 mL×2(6)漏斗(7)研钵3.试剂(1)铜试剂:A、4%CuSO4•5H2OB、称取24g 无水碳酸钠,用850mL 水溶于大烧杯中,加入2g 含4 分子结晶水的酒石酸钾钠,待全溶(应加热)后加入碳酸氢钠16g,再加入120g 无水硫酸钠(加热),全溶及冷却后加水至900mL,沉淀1~2d,取上清液(要求严格时过滤)备用。
使用前将A 与B 按1∶9 混匀即可使用。
(2)砷钼酸试剂:25g 钼酸铵 ( NH4)6Mo7O24·4H2O 溶于450mL 蒸馏水中(加热溶解,但温度接近150℃时易分解),待冷却后再加入21 mL 浓H2SO4 混匀。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
食品中砷比色法测定标准曲线的绘制
在仪器分析中,如比色测定,色谱测定、原子吸收法测定等,都要用标准样品来校准仪器,即将测定所用的仪器的响应值与标准品的量(或浓度)的对应关系,制作成标准曲线。
然后,根据样品测定时所得的响应值,从标准曲线查得待测组分的量(或浓度)。
当然制作标准曲线与测定样品时,所用的方法、所处的各项条件要完全一致。
一般要同时进行。
例如:食品中砷残留量的银盐法(比色法)测定
(1)砷标准贮备液:按国家标准GB/T5009.11-2003第二法,先配制成砷标准贮备液ρ(As)=1.00mg/ml。
(2)砷标准使用液:精确吸取砷标准贮备液5.00ml,用水定容至500ml,得砷标准使用液ρ(As)=1.00 μg/ml。
(3)标准曲线的绘制操作
a. 标准系列的制备与测定吸光度
b. 用座标纸法绘制标准曲线:以每个比色管中加入标准砷的质量μg为横座标,以测
得相应的吸光度A为纵座标,绘制标准曲线。
如图。
c. 线性回归法:用CASIO fx-3600Pv计算器,可以直接键入各对数值,得到回归方程:
Y = 24.382457 X - 0.006478; 相关系数r =0.9997
14 结果计算 试样中砷的含量按式(2)进行计算
()()122110002/1000
A A x m V V -⨯=⨯⨯L L L L
()()1012 5.8050 5.80/210 5.00m m V g ml x g g V m ml g
μμ-=⨯=⨯=L L L L
式中:
X -试样中砷的含量,mg/kg
m1-测定用试样消化液中砷的质量,μg ; m0-试剂空白液中砷的质量,μg ; m -试样的质量,g ;
V1-试样消化液的定容总体积, ml ;
V2-用于比色时所取的试样消化液的体积, ml ;
试样液测得的吸光度A =0.24
计算结果保留两位有效数字。