NCBI中Blast序列比对小总结
NCBI在线BLAST使用方法与结果详解

NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
没啥好说的。
6,blast结果的描述区域。
blast比对结果解读

blast比对结果解读blast比对是一种常用的生物信息学工具,它能够比较两个或多个序列之间的相似性。
通过对blast比对的结果进行解读,我们可以初步了解待比对序列与数据库中已知序列的相似性和相关信息。
在解读blast比对结果时,我们可以从几个关键方面入手,包括查询覆盖率、引物或探针匹配、E值、位点一致性和生物学含义等。
本文将一步一步回答围绕着blast比对结果解读的几个主题。
一、查询覆盖率:查询覆盖率是指待比对序列在blast比对中与数据库序列的匹配程度。
查询覆盖率可以通过比对结果中的两个指标来评估,即比对长度和查询覆盖率百分比。
比对长度表示匹配的碱基对数,查询覆盖率百分比则是指比对长度与查询序列长度的比值。
较高的查询覆盖率通常意味着待比对序列与数据库序列具有较高的相似性,而较低的查询覆盖率则可能表明待比对序列与数据库序列存在较大区别。
二、引物或探针匹配:在解读blast比对结果时,我们可以关注引物或探针的匹配情况。
引物或探针匹配的结果可以用来评估待比对序列与数据库序列之间是否存在所需的相似性。
如果比对结果中显示待比对序列与数据库序列的引物或探针完全匹配,那么说明待比对序列很可能具有与数据库序列类似的特征。
三、E值:E值是指期望值,用来评估待比对序列与数据库序列之间的匹配巧合程度。
E值越小,表示待比对序列与数据库序列之间的匹配越显著,也就是说两个序列之间的相似性越高。
在解读blast比对结果时,我们可以根据E值的大小来判断待比对序列与数据库序列之间的相似性。
四、位点一致性:位点一致性是指待比对序列与数据库序列在特定位点上的相同碱基数目。
位点一致性可以通过比对结果中的一致性百分比来评估,即一致性碱基数目与比对长度的比值。
较高的位点一致性表明待比对序列与数据库序列在特定位点上具有较高的相似性。
五、生物学含义:最后,解读blast比对结果时我们还可以关注待比对序列与数据库序列之间的生物学含义。
通过比对结果,我们可以了解待比对序列与数据库序列之间的功能和结构相似性,进而推测待比对序列可能具有的功能和结构特征。
NCBI在线BLAST使用方法与结果详解

NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool )是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST采用一种局部的算法获得两个序列中具有相似性的序列。
Blast 中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLAST)是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLAST是蛋白序列到核酸库中的一种查询。
与BLAST)相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36 种比对阵列。
NCBI的在线BLAST下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast 所有的核酸或蛋白序列。
不同的blast 程序上面已经有了介绍。
这里以常用的核酸库作为例子。
BLAST Assembled GenomesChoose 3 species genome to search, or list all qenoiriic BLAST databases.HumanMouseRatAnafeftfoosfe 册a 帰fffl Otyza sativa口Bg tavTUS D Danic思Mo 应Drosoo打ifa tnElanog^趙F□Basic BLAST以核昔醱库的曰则为例Choose a BLTXST gram torun./ --------- > nucleoliJe blast ■5;Search 3nucleotide database using a nucleotide query AfQorithms. bhstn, megablast, discontiguous megablasttintEm bl可st Search p「otein database using a 卩rateinqueryAfgoriihms' bhstp,卩si-blast,卩hi-blastblastK Search protein database using 玄 translated nucleotide querytbiastn Search transialed nucleotide database using a protein querySearch transiated nucleotide database using a 1ranslat«tl nucleotide query2,粘贴fasta格式的序列。
NCBI在线BLAST使用方法与结果详解

NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
没啥好说的。
6,blast结果的描述区域。
NCBI在线BLAST使用方法与结果详解

N C B I在线B L A S T使用方法与结果详解This model paper was revised by the Standardization Office on December 10, 2020N C B I在线B L A S T使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
NCBI的在线BLAST:下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
NCBI-Blast 比对方法
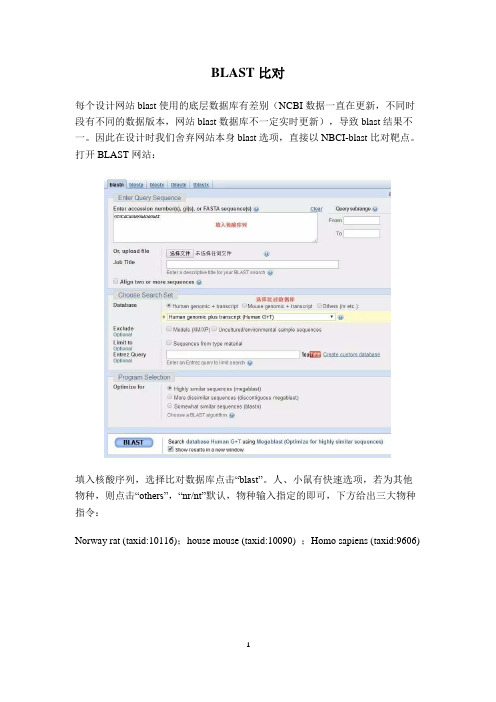
BLAST比对
每个设计网站blast使用的底层数据库有差别(NCBI数据一直在更新,不同时段有不同的数据版本,网站blast数据库不一定实时更新),导致blast结果不一。
因此在设计时我们舍弃网站本身blast选项,直接以NBCI-blast比对靶点。
打开BLAST网站:
填入核酸序列,选择比对数据库点击“blast”。
人、小鼠有快速选项,若为其他物种,则点击“others”,“nr/nt”默认,物种输入指定的即可,下方给出三大物种指令:
Norway rat (taxid:10116);house mouse (taxid:10090) ;Homo sapiens (taxid:9606)
结果界面,Max Score列数值除以2表示匹配的碱基数;完全匹配的全部都是TP53基因的15个转录本,所以,靶点位于同源区;其次,非完全靶向的Max Score最大值为30.2,也就是跟基因SIPA1L2实际结合15个碱基,错配四个,符合特异性原则,该靶点blast结果OK。
以上即靶点设计及比对的流程,该方法同样适合非编码RNA,选择多个网站设计的共有靶点以及设计2~3个靶点进行验证,更加有利于筛选出有效靶点。
如何看懂NCBIBLAST输出结果

如何看懂NCBIBLAST输出结果NCBI BLAST(Basic Local Alignment Search Tool)是一种用于比较生物序列之间的相似性的工具。
BLAST将一个查询序列与一个目标数据库中的序列进行比对,并输出比对结果。
下面将介绍如何看懂NCBI BLAST输出结果。
BLAST报告的不同部分提供了关于比对结果的详细信息。
以下是BLAST输出结果中的重要部分:1.查询信息:在输出结果的第一部分,会显示关于查询序列的信息,如查询序列的名称、长度以及描述。
这些信息可以帮助确认你是否正确提交了查询序列。
2.数据库信息:在查询信息的下方,输出结果会提供关于目标数据库的信息,包括数据库的名称、大小以及参与比对的序列数目。
这些信息可以帮助你了解比对参考的范围和样本数目。
3.参数信息:BLAST在进行比对时使用了一系列的参数,这些参数可以影响比对的灵敏度和特异性。
输出结果会显示用于比对的参数信息,包括比对算法、匹配得分、不匹配得分、开始扣分以及扩展扣分等。
这些参数提供了对比对结果的解释依据。
4.结果摘要:在参数信息的下方,会显示一个结果摘要表,提供了与查询序列最相似的多个数据库序列的信息。
这些信息包括数据库序列的名称、长度、比对得分以及比对的e值。
e值是一个表示比对结果的统计显著性的指标,越小表示比对结果越显著。
这些信息可以帮助你快速了解最相关的序列。
5.序列比对信息:在结果摘要之后,会显示每个比对的详细信息。
比对信息包括目标序列的名称和描述、比对长度、匹配得分、比对得分、e值以及比对图形。
比对图形以垂直线表示查询和目标序列之间的匹配,帮助你在比对中可视化相似区域。
6.比对统计信息:在序列比对信息之后,会显示比对的统计信息。
这些统计信息包括查询序列的覆盖率、比对序列的覆盖率以及总体比对得分。
这些信息对比对结果的解释和评估非常重要。
7.结果解释:在比对统计信息之后,会提供进一步解释和分析比对结果的信息。
NCBI在线BLAST使用方法与结果详解

N C B I在线B L A S T使用方法与结果详解BLAST Basic Local Alignment Search Tool是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具;BLAST程序能迅速与公开数据库进行相似性序列比较;BLAST结果中的得分是对一种对相似性的统计说明;BLAST 采用一种局部的算法获得两个序列中具有相似性的序列;Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询;库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对;2、BLASTX是核酸序列到蛋白库中的一种查询;先将核酸序列翻译成蛋白序列一条核酸序列会被翻译成可能的六条蛋白,再对每一条作一对一的蛋白序列比对;3、BLASTN是核酸序列到核酸库中的一种查询;库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对;4、TBLASTN是蛋白序列到核酸库中的一种查询;与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对;5、TBLASTX是核酸序列到核酸库中的一种查询;此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白每条核酸序列会产生6条可能的蛋白序列,这样每次比对会产生36种比对阵列;NCBI的在线BLAST:下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种如人,小鼠,水稻等,也可以选择blast 所有的核酸或蛋白序列;不同的blast程序上面已经有了介绍;这里以常用的核酸库作为例子;2,粘贴fasta格式的序列;选择一个要比对的数据库;关于数据库的说明请看NCBI在线blast数据库的简要说明;一般的话参数默认;3,blast参数的设置;注意显示的最大的结果数跟E值,E值是比较重要的;筛选的标准;最后会说明一下;4,注意一下你输入的序列长度;注意一下比对的数据库的说明;5,blast结果的图形显示;没啥好说的;6,blast结果的描述区域;注意分值与E值;分值越大越靠前了,E值越小也是这样;7,blast结果的详细比对结果;注意比对到的序列长度;评价一个blast结果的标准主要有三项,E值Expect,一致性Identities,缺失或插入Gaps;加上长度的话,就有四个标准了;如图中显示,比对到的序列长度为1405,看Identities这一值,才匹配到1344bp,而输入的序列长度也是为1344bp看上面的图,就说明比对到的序列要长一点;由Qurey起始1和Sbjct起始35的起始位置可知,5'端是是多了一段的;有时也要注意3'端的;附:E值Expect:表示随机匹配的可能性,E值越大,随机匹配的可能性也越大;E值接近零或为零时,具本上就是完全匹配了;一致性Identities:或相似性;匹配上的碱基数占总序列长的百分数;缺失或插入Gaps:插入或缺失;用"—"来表示;。
在NCBI使用心得2-2

第四部分如何运用BLAST 进行序列比对、检验引物特异性提到序列比对,绝大多数人都会想到BLAST,但BLAST 的使用确实又是一个很大的难题,因为他的功能比较强悍,里面涉及到的知识比较多,而且比对结束后输出的结果参数(指标)又很多。
如果把BLAST 的使用详细的都讲出来,我想我发帖发到明天也发不完,更何况我自己也不是完全懂得BLAST 的使用。
所以我在这里也就“画龙点睛”——以比对核酸序列为例来给大家介绍一下BLAST 的使用,也算是BLAST 的入门课程吧。
请大家好好体会,如果你用心看,在看帖完毕之后BLAST 的基本使用(包括其他序列的比对)应该没有问题了。
1.打开BLAST 页面,/BLAST/ 打开后如图所示:对上面这个页面进行一下必要的介绍:BLAST 的这个页面主体部分(左面)包括了三部分:BLAST Assembled Genomes、Basic BLAST、Specialized BLAST。
相信大家可以看懂这三个短语的意思,我就不多说了;我要说的是,可以认为这是三种序列比对的方法,或者说是BLAST 的三条途径。
第一部分BLAST Assembled Genomes 就是让你选择你要比对的物种,点击相应物种之后即可进入比对页面。
第二部分Basic BLAST 包含了5 个常用的BLAST,每一个都附有简短的介绍。
第三部分Specialized BLAST 是一些特殊目的的BLAST,如dart、GEO、IgBLAST、SNP 等等,这个时候你就需要在Specialized BLAST 部分做出适当的选择了。
总之,这是一个导航页面,它的目的是让你根据自己的比对目的选择相应的BLAST 途径。
下面以最基本的核酸序列比对来谈一下BLAST 的使用,期间我也会说一下其他序列比对的方法。
2.点击Basic BLAST 部分的nucleotide blast链接到一个新的页面。
打开后如图所示:介绍一下上述页面:Enter Query Sequence 部分是让我们输入序列的,你可以直接把序列粘贴进去,也可以上传序列,还可以选择你要比对的序列的范围(留空就代表要比对你要输入的整个序列)。
NCBI在线BLAST使用方法与结果详解

NCBI在线BLAST使用方法与结果详解NCBI在线BLAST(Basic Local Alignment Search Tool)是一种广泛使用的生物信息学工具,用于比对和分析DNA、RNA或蛋白质序列。
它可以对已知和未知序列进行,找到与查询序列相似的序列,并提供有关相似性和功能的信息。
使用NCBI在线BLAST可以分为四个主要步骤:选择BLAST程序,输入查询序列,选择目标数据库,解析和分析结果。
第一步:选择BLAST程序NCBI提供了多种BLAST程序可供选择,包括BLASTN(DNA对DNA的比对)、BLASTP(蛋白质对蛋白质的比对)、BLASTX(DNA对蛋白质的比对)等。
根据实际需求选择相应的BLAST程序。
第二步:输入查询序列在查询序列的文本框中输入待比对的序列。
可以输入单个序列,也可以上传包含多个序列的文件。
如果输入的序列是DNA或RNA序列,需要选择相应的序列类型。
此外,还可以选择是否使用掩码序列或低复杂性筛选来优化比对结果。
第三步:选择目标数据库用户可以选择目标数据库来与查询序列相似的序列。
NCBI提供了多个常用的数据库,如nr(非冗余蛋白质数据库)、nt(核酸数据库)等。
此外,还可以选择特定的物种数据库来限制比对范围。
第四步:解析和分析结果在BLAST运行完成后,会生成一个结果页面,其中包含了比对结果的详细信息。
结果页面包括比对统计信息、序列比对图、E值、分数等。
通过分析这些信息,可以了解查询序列与目标数据库中的序列之间的相似性和可能的功能。
此外,NCBI在线BLAST还提供了一些高级选项,例如使用特定的算法或参数来进行比对、设置比对阈值、选择比对输出格式等。
这些选项可以根据实际需求进行调整。
总结起来,使用NCBI在线BLAST可以通过选择BLAST程序、输入查询序列、选择目标数据库以及解析和分析结果来比对和分析序列。
通过权衡算法和参数选择,在特定数据库中找到与查询序列相似的序列,从而获得有关其相似性和功能的信息。
NCBIBLAST比对结果详细分析

NCBI BLAST比对结果详细分析
NCBI Blast结果-菜单与基本信息 1.下一步操作的菜单,你可以调整参数,重新比对、保存你的搜索条件以便下次比对、调整报告显示的参数,
以更符合你的要求、下载你比对的结果; 2.此次比对的标题,优先是你填写的,如果没有填写可能是你输入fasta序列头(大于号后面的),如果这个也没 有找到,NCBI 会自动生成一个; 3.你输入序列的信息,包括标识号、描述信息、类型、长度; 4.数据库的信息以及你选择的Blast程序; 5.查看其他报告,比如摘要、分类、距离树、结构、多重比对等。 Graphic Summary
Alignments 比对详细信息
1.比对上的序列信息;
2.比对的各种得分,这里不做一一说明,这里我最关注的是Identities,比对上(一致)的数字、一共有多少个,比 对上所占的比例。
3.具体的比对序列显示,一目了然,知道了哪些序列比对上了,哪些序列是不一样的,这里也要注意序列的位 置关系;
5.复选框,可以选择感兴趣的比对序列,在⑥处进行相应的操作;
BLAST地址: blastp&PAGE_TYPE=BlastSearch&SHOW_DEFAULTS=on&LINK_LOC=blasthome 比对用的例子:
>gi|16758036|ref|NP_445782.1| ribosomal protein L21 [Rattus norvegicus] MTNTKGKRRGTRYMFSRPFRKHGVVPLATYMRIYKKGDIVDIKGMGTVQKGMPHKCYHGKTGRVYNVTQH AVGIIVNKQVKGKILAKRINVRIEHIKHSKSRDSFLKRVKENDQKKKEAKEKGTWVQLNGQPAPPREAHF VRTNGKEPELLEPIPYEFMA 数据选择:nr 比对时间:2009年9月9日12:46:23 解读报告前需要掌握的概念
Blast分析报告

Blast分析报告1. 简介Blast(Basic Local Alignment Search Tool)是一种常用的生物信息学工具,用于比对和对比两个或多个生物序列。
它可以帮助研究人员在生物信息学研究中进行序列比对、寻找同源序列以及进行功能注释等工作。
本文将引导您详细了解和使用Blast进行分析。
2. 安装和配置Blast软件首先,您需要从NCBI(National Center for Biotechnology Information)官方网站下载并安装Blast软件。
一旦安装完成,您需要设置Blast的环境变量,以便在命令行中能够直接调用Blast命令。
3. 准备序列数据在进行Blast分析之前,您需要准备好待比对的序列数据。
这些序列可以是蛋白质序列或核酸序列,可以从NCBI数据库或其他来源获取。
确保您已经将这些序列保存在合适的文件中,并准备好进行分析。
4. 运行Blast分析接下来,您将使用命令行界面来运行Blast进行分析。
以下是一个基本的Blast命令示例:blastn -query query.fasta -db database.fasta -out result.txt在这个示例中,blastn表示您要运行的Blast程序,query.fasta是您的查询序列文件,database.fasta是您的数据库文件,result.txt是结果输出文件。
您可以根据需要调整Blast命令的参数,例如,您可以指定比对算法、设置阈值、选择输出格式等。
详细的命令选项和参数可以通过blastn -help命令来查看。
5. 解读Blast结果当Blast分析完成后,您将获得一个结果文件,其中包含了比对结果的详细信息。
您可以使用文本编辑器或其他工具打开这个结果文件,并解读其中的内容。
在结果文件中,您将看到每个查询序列和数据库序列的比对结果,包括比对得分、相似度、匹配位置等信息。
根据这些信息,您可以判断查询序列与数据库序列之间的关系,进一步分析和解释结果。
NCBI中Blast序列比对小总结

NCBI中Blast序列比对小总结NCBI中Blast可以用来进行序列比对、检验引物特异性Blast导航主页面主体包括三部分BLAST Assembled Genomes选择你要对比的物种,点击物种之后即可进入对比页面Basic BLAST包含5个常用的Blast,每一个都附有简单介绍Specialized BLAST是一些特殊目的的Blast,如Primer-BLAST、IgBLAST根据需要做出选择本学期学习了最基本的核苷酸序列的比对点击Basic BLAST部分的nucleotide链接到一个新的页面,打开后的页面特征:大体上包括三个部分Enter Query Sequence部分可以让我们输入序列,其中的Job Title部分可以为本次工作命一个名字Choose Search Set部分可以选择要与目的序列比对的物种或序列种类。
其中的Entrez Query可以对比对结果进行适当的限制。
Program Selection部分可以选择本次对比的精确度,种内种间等等。
其次Blast按钮下面有一个“Algorithm parameters”算法参数,可设置参数。
点击Blast后,出现的页面大体上包括四个部分一.所询问和比对序列的简单信息1.询问序列的简单信息——名称、描述、分子类型、序列长度2.所比对数据库的名称、描述和所用程序二.Graphic Summary——blast结果图形显示相似度颜色图(黑、蓝、绿、粉红、红,相似度由低到高)三.Descriptions——blast结果描述区1.到其他数据库的链接2.描述以表格的形式呈现(以匹配分值从大到小排序)(1)Accession下程序比对的序列名称,点击相应的可以进入更为详细的map viewer(2)Descriptions下是对所比对序列的简单描述接下来是5个结果数值:(3)Max score匹配分值,点击可进入第四部分相应序列的blast的详细比对结果(4)Total score总体分值(5)Query coverage覆盖率(6)E value——E(Expect)值,表示随机匹配的可能性。
在线blast的用法总结

在线b l a s t的用法总结-标准化文件发布号:(9556-EUATWK-MWUB-WUNN-INNUL-DDQTY-KIIBlast(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列NCBI的在线blast:/Blast.cgi本文详细出处参考:/475/举例一:核酸序列的比对1,进入在线blast界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
(补充介绍下:1、BLASTN【 nucleotide blast】是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
2、BLASTP【protein blast】是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
3、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
)2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
实验三:利用Blast进行序列相似性比对(1)

3. 以大肠杆菌的胶原蛋白酶名称为pHK08_29的基因做为查 询序列 (1) 用Blastn能在nr/nt数据库中检索到多少条与之同源的序 列。其中大肠杆菌、弗累克斯讷氏杆菌、沙门氏菌各有多 少条序列。
(2) 换用megablast或discontiguous megablast,观察检索结 果的改变。 (3) 尝试修改Blastn的参数,观测对检索结果的影数据库中检索到多少 条与之同源的序列。 4. 用blast2分析YP_003683100与ADH70594、 YP_004926582、 YP_004925874、 YP_003273209、 YP_003646515、 YP_003514536、 ABP47302、 ADD45443、 ADW07065、 ADG78176、 ACY21316、 ABM16043、 EHP75935、 BAC74107、 YP_00407863之间的相似性.
实验三:利用Blast进行序列相似性比对
具体步骤
1.登陆blast主页 /BLAST/ 2.根据数据类型,选择合适的程序 3.填写表单信息 4.提交任务 5.查看和分析结果
1. 以大麦Mlo基因(Z83834)为查询序列 (1) 用Blastn能在nr/nt数据库中检索到多少条与之同源的序 列?有多少条是禾本科中的? (2) 换用megablast或discontiguous megablast,观察检索结 果的改变。 (3) 尝试修改Blastn的参数,观测对检索结果的影响。 (4) 找出Mlo基因的编码蛋白序列,用Blastp检索到的与 Mlo蛋白同源的序列与用PSI-Blast检索到的同源序列是否 有差别? (5) 使用BlastX预测Mlo基因的编码蛋白。 2. 用bl2seq分析大麦和小麦Mlo基因mRNA序列编码区和蛋白 质产物的同源性
使用 NCBI 查找DNA引物设计BLAST序列比对

最近看到很多战友在论坛上询问如何查询基因序列、如何进行引物设计、如何使用BLAST 进行序列比对……,这些问题在 NCBI 上都可以方便的找到答案。
现在我就结合我自己使用 NCBI的一些经历(经验)跟大家交流一下 BCBI 的使用。
希望大家都能发表自己的使用心得,让我们共同进步!我分以下几个部分说一下 NCBI 的使用:Part one 如何查找基因序列、mRNA、PromoterPart two 如何查找连续的 mRNA、cDNA、蛋白序列Part three 运用 STS 查找已经公布的引物序列Part four 如何运用 BLAST 进行序列比对、检验引物特异性特别感谢本版版主,将这个帖子置顶!从发帖到现在,很多战友对该帖给与了积极的关注,在此向给我投票的(以及想给我投票却暂时不能投票的)各位战友表示真诚的感谢,谢谢各位战友!请大家对以下我发表的内容提出自己的意见。
关于NCBI 其他方面的使用也请水平较高的战友给予补充First of all,还是让我们从查找基因序列开始。
第一部分利用Map viewer 查找基因序列、mRNA 序列、启动子(Promoter)下面以人的 IL6(白细胞介素 6)为例讲述一下具体的操作步骤1.打开Map viewer 页面,网址为:/mapview/index.html 在 search 的下拉菜单里选择物种,for 后面填写你的目的基因。
操作完毕如图所示:2.点击“GO”出现如下页面:3.在步骤二图示的右下角有一个Quick Filter,下面是让你选择的几个复选框,在Gene 前面的小方框里打勾,然后点击Filter. 出现下图:说明一下:1、染色体的红色区域即为你的目的基因所处位置。
2、下面参考序列给出了三个,是不同的部门做出来的,经我验证,序列有微小的差异,但总体来说基本相同。
尽管你分别点击后,序列代码、序列代码等有所差异,但碱基基本一致,不影响大家研究分析序列。
ncbi blast的功能和种类

ncbi blast的功能和种类NCBI BLAST是一种广泛使用的生物信息学工具,用于比对生物序列。
BLAST是Basic Local Alignment Search Tool的缩写,它可以将一个给定的生物序列与数据库中的其他序列进行比对,以找到相似性和同源性。
NCBI BLAST提供了多种不同类型的BLAST程序,每个程序都针对不同类型的比对任务。
下面是NCBI BLAST常见的几种程序:1. BLASTN:用于比对核酸序列,如DNA和RNA。
BLASTN可以找到两个序列之间的相似性和同源性,并确定它们之间的区别。
这个程序通常用于基因组学和转录组学研究中。
2. BLASTP:用于比对蛋白质序列。
BLASTP可以找到两个蛋白质之间的相似性和同源性,并确定它们之间的区别。
这个程序通常用于蛋白质结构预测和功能注释研究中。
3. BLASTX:用于将未知核酸序列与已知蛋白质序列进行比对。
BLASTX将未知核酸序列翻译成蛋白质序列,然后与已知蛋白质序列进行比对。
这个程序通常用于寻找新基因或预测基因功能。
4. TBLASTN:用于将已知蛋白质序列与未知核酸序列进行比对。
TBLASTN将已知蛋白质序列翻译成核酸序列,然后与未知核酸序列进行比对。
这个程序通常用于在基因组中寻找新的蛋白质编码基因。
5. TBLASTX:用于将两个未知核酸序列进行比对。
TBLASTX将两个未知核酸序列翻译成蛋白质序列,然后进行比对。
这个程序通常用于寻找新基因或预测基因功能。
除了这些常见的程序之外,NCBI BLAST还提供了其他一些特殊的程序,如PSI-BLAST、RPS-BLAST和PHI-BLAST等。
PSI-BLAST是一个迭代的BLAST程序,可以在多次比对中改进结果,并生成一个更准确的蛋白质家族模型。
RPS-BLAST是一个与CDD(Conserved Domain Database)相关的程序,可以在已知域和未知蛋白质之间找到相似性和同源性。
NCBI在线BLAST使用方法与结果详解

NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool )是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST吉果中的得分是对一种对相似性的统计说明。
BLAST采用一种局部的算法获得两个序列中具有相似性的序列。
Blast 中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLAST)是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTI是蛋白序列到核酸库中的一种查询。
与BLAST)相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLAST)是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36 种比对阵列。
下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等), 也可以选择blast 所有的核酸或蛋白序列。
不同的blast 程序上面已经有了介绍。
这里以常用的核酸库作为例子。
BLAST Assembled GenomesChoose a species genome to search, list all 勺BLAST M击B词c BLAST以核昔醱库的BM为例Choose a BLAST prbgram to run.Search a nucleotide database using a nucleotide queryAlgohthm^: blastn, megablast, discontiguous megablastSearch pruteiri database using a protein queryAlgorisms bhstp, psi-blast, phi-blastSearch protein u^irg a translated nudeotide querySearch translated nucleotide database using a protein querySearch translated nucleotide database a translated nucleotide query2,粘贴fasta格式的序列。
NCBI中Blast序列比对小总结

NCBI中Blast可以用来进行序列比对、检验引物特异性Blast导航主页面主体包括三部分BLAST Assembled Genomes选择你要对比的物种,点击物种之后即可进入对比页面BasicBLAST包含5个常用的Blast,每一个都附有简单介绍SpecializedBLAST是一些特殊目的的Blast,如Primer-BLAST、IgBLAST根据需要做出选择本学期学习了最基本的核苷酸序列的比对点击BasicBLAST部分的nucleotide链接到一个新的页面,打开后的页面特征:大体上包括三个部分EnterQuerySequence部分可以让我们输入序列,其中的JobTitle部分可以为本次工作命一个名字ChooseSearchSet部分可以选择要与目的序列比对的物种或序列种类。
其中的EntrezQuery可以对比对结果进行适当的限制。
ProgramSelection部分可以选择本次对比的精确度,种内种间等等。
其次Blast按钮下面有一个“Algorithmparameters”算法参数,可设置参数。
点击Blast后,出现的页面大体上包括四个部分一."所询问和比对序列的简单信息1."询问序列的简单信息——名称、描述、分子类型、序列长度2."所比对数据库的名称、描述和所用程序二."GraphicSummary——blast结果图形显示相似度颜色图(黑、蓝、绿、粉红、红,相似度由低到高)三."Descriptions——blast结果描述区1."到其他数据库的链接2."描述以表格的形式呈现(以匹配分值从大到小排序)(1)Accession下程序比对的序列名称,点击相应的可以进入更为详细的mapviewer(2)Descriptions下是对所比对序列的简单描述接下来是5个结果数值:(3)Max score匹配分值,点击可进入第四部分相应序列的blast的详细比对结果(4)Total score总体分值(5)Query coverage覆盖率(6)E value——E(Expect)值,表示随机匹配的可能性。
ncbi序列比对方法与操作实例

NCBI序列比对方法与操作实例一、序列比对方法概述1. 序列比对的概念序列比对是指通过对两个或多个生物序列进行比较分析,找到它们之间的相似性和差异性。
序列比对是生物信息学中的重要工具之一,可以帮助研究人员理解DNA、RNA、蛋白质等生物分子的结构和功能,进而推动生物医药和生物科学领域的发展。
2. 序列比对的意义在生物学研究中,通过对不同生物序列进行比对分析,可以揭示它们之间的进化关系、基因结构、功能和调控机制等重要信息,有助于揭示生物系统的内在规律。
序列比对还可以在分子生物学实验设计、基因工程、疾病诊断、新药开发等方面发挥重要作用。
3. 序列比对的方法常用的序列比对方法包括全局比对、局部比对和多序列比对等,其中全局比对适用于寻找整个序列间的相似段,局部比对适用于寻找两个序列中的部分匹配段,多序列比对则适用于比较多个序列之间的相似性和差异性。
二、NCBI序列比对工具介绍1. NCBI数据库NCBI(National Center for Biotechnology Information)是美国国家生物技术信息中心,是全球生物学信息资源的重要提供者之一。
NCBI数据库中包含大量生物信息数据,包括基因组序列、蛋白质序列、原始文献、生物信息学工具等。
2. NCBI序列比对工具NCBI提供了一系列用于序列比对的工具,其中包括BLAST(Basic Local Alignment Search Tool)、BLAT(BLAST-Like Alignment Tool)、ClustalW、MAFFT等。
这些工具可以帮助研究人员进行序列比对分析,找到感兴趣的生物序列在数据库中的同源序列或相似序列。
三、NCBI序列比对操作实例以BLAST工具为例,介绍NCBI序列比对的操作步骤。
1. 打开NCBI全球信息湾打开NCBI全球信息湾(),在全球信息湾首页的搜索栏中输入“BLAST”,进入BLAST工具的页面。
2. 输入查询序列在BLAST工具的页面中,选择适当的数据库,粘贴或上传待比对的查询序列,可以选择标准蛋白数据库、EST数据库、基因组数据库等作为比对的对象。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
NCBI中Blast可以用来进行序列比对、检验引物特异性Blast导航主页面主体包括三部分BLAST Assembled Genomes选择你要对比的物种,点击物种之后即可进入对比页面Basic BLAST包含5个常用的Blast,每一个都附有简单介绍Specialized BLAST是一些特殊目的的Blast,女口Primer-BLAST、IgBLAST根据需要做出选择本学期学习了最基本的核苷酸序列的比对点击Basic BLAST部分的nucleotide链接到一个新的页面,打开后的页面特征:大体上包括三个部分Enter Query Sequenee部分可以让我们输入序列,其中的Job Title部分可以为本次工作命一个名字Choose Search Set部分可以选择要与目的序列比对的物种或序列种类。
其中的Entrez Query可以对比对结果进行适当的限制。
Program Selection部分可以选择本次对比的精确度,种内种间等等。
其次Blast按钮下面有一个“Algorithm parameters算法参数,可设置参数。
点击Blast后,出现的页面大体上包括四个部分一•所询问和比对序列的简单信息 1 •询问序列的简单信息——名称、描述、分子类型、序列长度2 .所比对数据库的名称、描述和所用程序二.Graphic Summary blast 结果图形显示相似度颜色图(黑、蓝、绿、粉红、红,相似度由低到高)三.Descriptions -------- blast结果描述区1.到其他数据库的链接2.描述以表格的形式呈现(以匹配分值从大到小排序)(l)Accession下程序比对的序列名称,点击相应的可以进入更为详细的map viewer (2)Descriptions下是对所比对序列的简单描述接下来是5个结果数值:(3)Max score匹配分值,点击可进入第四部分相应序列的blast的详细比对结果(4)Total score总体分值(5)Query coverage 覆盖率⑹E value ――E (Expect )值,表示随机匹配的可能性。
E值越大,随机匹配的可能性也越大。
E值接近零或为零时,具本上就是完全匹配了。
(7)Max ident ――匹配一致性,即匹配上的碱基数占总序列长的百分数。
(8)Links ――到其他数据库的链接。
四.各序列blast的详细比对结果数据库中不同序列比对的详细结果,每一个结果大体上包括3部分1.所比对序列的名称、简单描述、长度。
到其他数据库的链接。
2•比对结果的5个数值:(1)score打分矩阵计算出来的值,由搜索算法决定的,值越大说明询问序列跟目标序列匹配程度越大⑵Expect是输入序列被随机搜索出来的概率,该值越小越好。
(3)ldentities是相似程度,即输入序列和搜索到序列的匹配率(4)Gaps就是空白,即比对序列只有一条链上有碱基(5)stra nd二plus/mi nus 即询问序列和数据库里面序列的互补链匹配3.输入序列和库中对比到的序列每个碱基的详细对比NCBI中Blast可以用来进行序列比对、检验引物特异性Blast导航主页面主体包括三部分BLAST Assembled Genomes选择你要对比的物种,点击物种之后即可进入对比页面Basic BLAST包含5个常用的Blast,每一个都附有简单介绍Specialized BLAST 是一些特殊目的的Blast,女口Primer-BLAST、IgBLAST 根据需要做出选择本学期学习了最基本的核苷酸序列的比对点击Basic BLAST部分的nucleotide链接到一个新的页面,打开后的页面特征:大体上包括三个部分Enter Query Sequenee部分可以让我们输入序列,其中的Job Title部分可以为本次工作命一个名字Choose Search Set部分可以选择要与目的序列比对的物种或序列种类。
其中的EntrezQuery可以对比对结果进行适当的限制。
Program Selection部分可以选择本次对比的精确度,种内种间等等。
其次Blast按钮下面有一个“Algorithm parameters算法参数,可设置参数。
点击Blast后,出现的页面大体上包括四个部分一•所询问和比对序列的简单信息1 •询问序列的简单信息——名称、描述、分子类型、序列长度2 •所比对数据库的名称、描述和所用程序二.Graphic Summary ------ blast 结果图形显示相似度颜色图(黑、蓝、绿、粉红、红,相似度由低到高)三.Descriptions ------ blast 结果描述区1 .到其他数据库的链接2 .描述以表格的形式呈现(以匹配分值从大到小排序)(1) Accession下程序比对的序列名称,点击相应的可以进入更为详细的map viewer(2) Descriptions下是对所比对序列的简单描述接下来是5个结果数值:(3) Max score匹配分值,点击可进入第四部分相应序列的blast的详细比对结果(4) Total score 总体分值(5) Query coverage 覆盖率(6) E value——E (Expect)值,表示随机匹配的可能性。
E值越大,随机匹配的可能性也越大。
E值接近零或为零时,具本上就是完全匹配了。
(7) Max ide nt ---- 匹配一致性,即匹配上的碱基数占总序列长的百分数。
(8) Links ――到其他数据库的链接。
四.各序列blast的详细比对结果数据库中不同序列比对的详细结果,每一个结果大体上包括3部分1.所比对序列的名称、简单描述、长度。
到其他数据库的链接。
2•比对结果的5个数值:(1) score打分矩阵计算出来的值,由搜索算法决定的,值越大说明询问序列跟目标序列匹配程度越大(2) Expect是输入序列被随机搜索出来的概率,该值越小越好。
⑶Identities是相似程度,即输入序列和搜索到序列的匹配率(4) Gaps就是空白,即比对序列只有一条链上有碱基(5) stra nd二plus/mi nus 即询问序列和数据库里面序列的互补链匹配3 .输入序列和库中对比到的序列每个碱基的详细对比NCBI中Blast可以用来进行序列比对、检验引物特异性Blast导航主页面主体包括三部分BLAST Assembled Genomes选择你要对比的物种,点击物种之后即可进入对比页面Basic BLAST包含5个常用的Blast,每一个都附有简单介绍Specialized BLAST 是一些特殊目的的Blast,女口Primer-BLAST、IgBLAST 根据需要做出选择本人本学期学习了最基本的核苷酸序列的比对点击Basic BLAST部分的nucleotide链接到一个新的页面,打开后的页面特征:大体上包括三个部分Enter Query Sequenee部分可以让我们输入序列,其中的Job Title部分可以为本次工作命一个名字Choose Search Set部分可以选择要与目的序列比对的物种或序列种类。
其中的Entrez Query可以对比对结果进行适当的限制。
Program Selection部分可以选择本次对比的精确度,种内种间等等。
其次Blast按钮下面有一个“Algorithm parameters算法参数,可设置参数。
点击Blast后,出现的页面大体上包括四个部分一•所询问和比对序列的简单信息1 •询问序列的简单信息——名称、描述、分子类型、序列长度2 •所比对数据库的名称、描述和所用程序二.--------------------- Graphic Summary blast 结果图形显示相似度颜色图(黑、蓝、绿、粉红、红,相似度由低到高)三.Descriptions ------ blast 结果描述区1 .到其他数据库的链接2 .描述以表格的形式呈现(以匹配分值从大到小排序)(1) Accession下程序比对的序列名称,点击相应的可以进入更为详细的map viewer(2) Descriptions下是对所比对序列的简单描述接下来是5个结果数值:(3) Max score匹配分值,点击可进入第四部分相应序列的blast的详细比对结果(4) Total score 总体分值(5) Query coverage 覆盖率(6) E value——E (Expect)值,表示随机匹配的可能性。
E值越大,随机匹配的可能性也越大。
E值接近零或为零时,具本上就是完全匹配了。
(7) Max ide nt ---- 匹配一致性,即匹配上的碱基数占总序列长的百分数。
(8) Links ――到其他数据库的链接。
四.各序列blast的详细比对结果数据库中不同序列比对的详细结果,每一个结果大体上包括3部分1.所比对序列的名称、简单描述、长度。
到其他数据库的链接。
2•比对结果的5个数值:(1) score打分矩阵计算出来的值,由搜索算法决定的,值越大说明询问序列跟目标序列匹配程度越大(2) Expect是输入序列被随机搜索出来的概率,该值越小越好。
⑶Identities是相似程度,即输入序列和搜索到序列的匹配率(4) Gaps就是空白,即比对序列只有一条链上有碱基(5) stra nd二plus/mi nus 即询问序列和数据库里面序列的互补链匹配3 .输入序列和库中对比到的序列每个碱基的详细对比。