高斯软件基础教程
高斯软件入门

G98的功能和程序结构
1. 主要功能: 基态(Ground state) •分子构型的优化 激发态(Excited state) 反应过渡态(Transition state) 基态和激发态能量
•能量计算
化学键的键能 电子亲合能和电离能 化学反应途径和势能面
IR光谱 •光谱计算 Raman光谱 电子光谱 NMR
说明: 1.根据不同的任务,某些模块需重复调用多次; 2.通常耗时较多的模块有:L5,L7,L8,L9,L10,L11等,此外, L8~L11这些模块的执行对内存和硬盘的需求较大; 3.若L9999未能正常执行完毕,则表明计算过程存在问题,需 检查之; 4.可根据各个模块的功能,对g98程序进行简化,例如如果用 户通常只用g98进行能量计算,则可只保留L1~6和L9999模块 其它模块可以删除去。 c.g98运行过程所使用的文件: 在scratch目录下有下列文件: • gxx-打头的文件为临时文件,计算结束后将自动删除,其中 对于结尾为inp的文件,记录了当前g98所执行的输入文 件内容,有时可通过该文件确定当前运行作业; • chk文件,该文件记录了g98运行的结果,包括分子结构、基 组、分子轨道、电荷密度以及偶极矩等,通常该文件在计 算结束后要保留,以便以后作补充计算之需;
电荷和电荷密度
•其它功能 偶极矩和超极矩 热力学参数 适用体系:气相和溶液
2.程序结构: a.由主引导模块(g98.exe)和 各分模块(l????.exe)组成:
b.常用模块的功能:
•L0—初始化模块; •L1—读入输入,根据所给关键词确定将要使用的模块; •L101,102,…—与构型优化和反应过渡态相关的模块; •L202—输出距离矩阵、判断化合物点群及确定新的坐标系; •L301,302…309—与基组和赝势有关模块; •L310,…319—计算单及双电子积分模块; •L401,402—SCF初始猜测模块; •L502,503,508—SCF模块; •L601,608—Mulliken布居以及自然键轨道分析模块; •L701,702…—计算能量一阶和二阶导数模块; •L8??,9??,10??,11??—与Post-SCF方法有关模块; •L9999—进程结束模块;
《高斯软件入门》课件

统计分析实例
通过实例深入理解高斯软件 在统计分析方面的应用和技 巧。
总结与展望
高斯软件的优越性
总结高斯软件的优势和特点,以及为什么它是数据分析的理想选择。
高斯软件的发展趋势
展望高斯软件未来的发展方向和趋势,激发你对技术的好奇心。
学习高斯软件的建议与方法
提供一些建议和学习方法,帮助你更高效地学习和掌握高斯软件。
高斯软件的优点和局 限性
讨论高斯软件的优点和局 限性,帮助你更好地了解 该软件的适用范围和限制。
高斯软件安装与环境配置
1 安装高斯软件的步骤
详细介绍高斯软件的安装过程,帮助你顺利完成安装。
2 配置系统环境变量
指导你如何配置系统环境变量,确保高斯软件可以在你的计算机上正常运行。
3 检查安装是否成功
提供一些简单的检查方法,确保你成功安装了高斯软件。
高斯软件基础知识
1
高斯软件的数据类型
介绍高斯软件支持的不同数据类型,如数值、字符和逻辑。
2
高斯软件的数组与向量
讲解高斯软件中数组和向量的定义、操作和应用。
3
高斯软件的函数与运算符
探索高斯软件提供的丰富函数和运算符,以及它们在数据处理中的作用。
高斯软件高级应用
高斯软件的图形绘制功能
展示高斯软件强大的图形绘制 功能,帮助你更直观地呈现数 据。
高斯软件的数据处理功能
介绍高斯软件用于数据
介绍高斯软件在统计分析方面 的功能和应用,助你深入理解 数据潜在的规律。
高斯软件编程实例
矩阵运算实例
通过实例演示高斯软件中矩 阵运算的基本操作和应用。
数据拟合实例
展示如何使用高斯软件进行 数据拟合和曲线拟合,帮助 你更好地分析数据模型。
高斯软件-基础教程

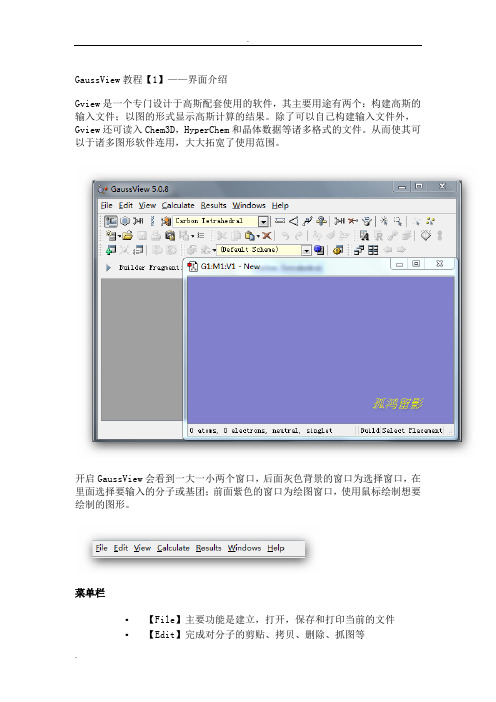
GaussView界面教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏▪【File】主要功能是建立,打开,保存和打印当前的文件▪【Edit】完成对分子的剪贴、拷贝、删除、抓图等▪【View】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后的结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
Gaussian 高斯使用指南

……………………3
2:计算原理…………………………………………………………………6
2.1 概述……………………………………………………………………6
2.2 分子力学方法…………………………………………………………6
NMR;振动圆形二色性(VCD);电子圆形二色性(ECD);旋光色散(ORD);谐性振-转耦合;
非谐性振动及振-转耦合;g 张量以及其它的超精细光谱张量。
5.模拟在反应和分子特性中溶剂的影响
在气相和在溶液之间,分子特性和化学反应经常变化很大。例如,低位构像在气相和在(不
同溶剂的)溶液中,具有完全不同的能量,构像的平衡结构也不同,化学反应具有不同的路
高斯应用指南
目
录
第一章 功能和计算原理介绍……………………………………………………2
1 Gaussian 功能介绍…………………………………………………………2
1.1 Gaussian 是一个功能强大的量子化学综合软件包……………………3
1.2 关于 Gaussian 03 的介绍…………………………
和 Parrinello 的经验,ADMP 传递电子自由度,而不是求解每个核结构的 SCF 方程。与
Car-Parrinello 不同之处在于,ADMP 传递密度矩阵而不是 MO。如果使用了原子中心基组,
2.2.3 GVIEW 使用简介…………………………………………………30
3.构建分子中的注意事项…………………………………………………45
4.怎样构建 Z-坐标………………………………………………………45
GaussView5 基础教程

GaussView教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏▪【File】主要功能是建立,打开,保存和打印当前的文件▪【Edit】完成对分子的剪贴、拷贝、删除、抓图等▪【View】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后的结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
1、开启Gview2、双击按钮,选择苯基(第一个),会看到主程序框体中出现苯环在工作窗口单击,可以看到工作窗口也出现了一个苯环。
高斯软件入门PPT课件

12
c.g98运行过程的控制:
不要随意点击!
最上行按钮的功能从左至右依次为: 开始运行g98;暂停进程;运行至下一模块(link)时暂停进程; 重新启动进程;清除进程(停止运算);编辑批作业;运行完 当前任务后,暂停批作业;停止批作业的运算;观看计算结 果;打开文本编辑器;
2021/7/24
13
(2). Linux平台:
2021/7/24
18
说明:
1.根据不同的任务,某些模块需重复调用多次; 2.通常耗时较多的模块有:L5,L7,L8,L9,L10,L11等,此外,
L8~L11这些模块的执行对内存和硬盘的需求较大; 3.若L9999未能正常执行完毕,则表明计算过程存在问题,需
检查之;
4.可根据各个模块的功能,对g98程序进行简化,例如如果用 户通常只用g98进行能量计算,则可只保留L1~6和L9999模块 其它模块可以删除去。
直接运行setup.exe,其余步骤按提示操作即可;也可将其它机 器上将已安装好的G98直接拷贝到本机,但需设置运行环境。 对Linux平台: a.若G98是经过压缩过的(文件结尾为gz),用gunzip命令解压: 例如: gunzip g98.linux.tar.gz b.若G98是打包的(文件结尾为tar),用tar命令将其释放: 例如: tar xvf g98.linux.tar c.设置环境变量,以c shell为例,在用户根目录下的.cshrc文件 添加下列内容:(也可在执行g98前逐条运行) setenv g98root /home/test/src(设置g98所在目录,根据实际情况修改)
量子化学计算方法
章永凡 福州大学化学系
2003年3月
2021/7/24
GaussView高斯软件教程

(4).双击Gview界面上的 图标。出现以下窗口点击氟的元素符号“F”,就选中氟原子。
-
(5).回到工作窗口在苯环上的任一个H上单击左键,将H置换成F。
但要注意此时仅是元素符号发生了改变,C-F间的距离仍是C-H键的距离。需要用建 长工具进行调整。单击Gview界面上 图标。 然后再点击工作窗口中的C原子和F原 子,看到被选中的两个原子于周围的原子再亮度上有差异此时会出现下面的窗口
-
(2).双击窗口中 图标,得到如下窗口里面有常用的环状官能团。选中苯环(单 击即可选中)
-
(3).在当前工作窗口(打开Gview时程序自动打开一个工作窗口,如下图)也可通过 File-new 路径 新建一个工作窗口
-
在这个窗口中点鼠标左键窗口中就会出现苯分子,见下图
将鼠标放在分子上,按左键左右或前后移动,可以调节分子的角度将鼠标放在分子上, 前后移动,可以将分子放大或缩小Shift+Alt+鼠标左键组合可以在窗口内平移分子 当工作窗口内有多个分子时[在构建大的分子时,这种情况很容易出现]这时可用以下 命令可以用Shift+Alt+鼠标左键组合移动想要移动的分子,以调节各个分子间的距离 可以用Ctrl+Alt+鼠标左键组合调节其中一个分子的角度,以调节各个分子间的角度。 Ctrl+Alt+鼠标左键这个组合常和[将鼠标左键放在分子上,左右或前后移动,可以调 节分子的角度]这个功能连用。
-
根据C-F键的长度在 0.675和2.700之间的方框内进行C-F键的调整。完毕后点击OK即可。
-
(6).双击Gview界面上的 图标,出现以下窗口这是Gview里内置的链烃库,选中乙 烷
-
Gaussian 高斯使用指南
目
录
第一章 功能和计算原理介绍……………………………………………………2
1 Gaussian 功能介绍…………………………………………………………2
1.1 Gaussian 是一个功能强大的量子化学综合软件包……………………3
1.2 关于 Gaussian 03 的介绍…………………………
1 目的 ………………………………………………………………………84
2 请参照练习了解输入格式和输出解释. …………………………………84
3 常用的研究方法的介绍和高斯自带的练习 ……………………………84
第十一章 激发态计算 …………………………………………………………93
1 目的…………………………………………………………………………93
2
1:Gaussian 功能介绍
1.1 Gaussian 是一个功能强大的量子化学综合软件包。其可执行程序可在不同型号的大型
计算机,超级计算机,工作站和个人计算机上运行,并相应有不同的版本。
高斯功能:
分子能量和结构
过渡态能量和结构
键和反应能量
分子轨道
多重矩
原子电荷和电势
振动频率
红外和拉曼光谱
核磁性质
2.2.3 GVIEW 使用简介…………………………………………………30
3.构建分子中的注意事项…………………………………………………45
4.怎样构建 Z-坐标………………………………………………………45
5.关于输出的解释…………………………………………………………48
1
第四章:优化计算………………………………………………………………54
NMR;振动圆形二色性(VCD);电子圆形二色性(ECD);旋光色散(ORD);谐性振-转耦合;
GaussView基础教程

GaussView教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏▪【File】主要功能是建立,打开,保存和打印当前的文件▪【Edit】完成对分子的剪贴、拷贝、删除、抓图等▪【View】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后的结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
1、开启Gview2、双击按钮,选择苯基(第一个),会看到主程序框体中出现苯环在工作窗口单击,可以看到工作窗口也出现了一个苯环。
GaussView5基础教程
GaussView教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏▪【File】主要功能是建立,打开,保存和打印当前的文件▪【Edit】完成对分子的剪贴、拷贝、删除、抓图等▪【View】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后的结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
1、开启Gview2、双击按钮,选择苯基(第一个),会看到主程序框体中出现苯环在工作窗口单击,可以看到工作窗口也出现了一个苯环。
Gaussian 高斯使用指南
默认是 IEFPCM 模型,解析频率计算可以用于 SCRF 方法。此外改善了空穴生成技术。
模拟溶液中的很多特性。
可以对 Klamt 的 COSMO-RS 程序产生输入,通过统计力学方法,用于计算溶解能,配分系
数,蒸汽压,以及其它整体性质。
(3) 周期性边界条件(PBC)
和 Parrinello 的经验,ADMP 传递电子自由度,而不是求解每个核结构的 SCF 方程。与
Car-Parrinello 不同之处在于,ADMP 传递密度矩阵而不是 MO。如果使用了原子中心基组,
磁性质的计算。
2.通过自旋-自旋耦合常数确定构像
当没有 X-射线结构可以利用时,研究新化合物的构像是相当困难的。NMR 光谱的磁屏蔽数
据提供了分子中各原子之间的连接信息。自旋-自旋耦合常数可用来帮助识别分子的特定构
像,因为它们依赖于分子结构的扭转角。
除了以前版本提供的 NMR 屏蔽和化学位移以外,Gaussian 03 还能预测自旋-自旋耦合常数。
振动频率,IR 和 Raman 光谱,以及其它特性;极化率和超极划率;执行性能上的改善。
G03W 的界面和 G98W 相比,没有什么变化,G98W 的用户不需要重新熟悉界面。
Gaussian 03 新增加了以下内容:
新的量子化学方法
(1) ONIOM 模块做了增强
对 ONIOM(MO:MM)计算支持电子嵌入,可以在 QM 区域的计算中考虑 MM 区域的电特性。
极化率和超极化率
热力学性质
反应路径
计算可以对体系的基态或激发态执行。可以预测周期体系的能量,结构和分子轨道。因
此,Gaussian可以作为功能强大的工具,用于研究许多化学领域的课题,例如取代基的影响,
高斯计算入门 ppt课件

计算作业提交过程:
a. 用户登录网关-通过SSH远程登录软件实现
SSH软件(SSHSecureShellClient-3.2.9.exe)可从网络上免费 下载,安装过程与通常软件安装类似。安装完毕后,设置 网关外部网的IP地址以及账号名即可使用。
ppt课件
16
点击Profiles设置IP地址及用户名
(级别为0~19整数,数值越大优pp先t课件级越低) renice 19 79
21
(8) cat-显示文件内容,格式为:cat 文件名 (9) grep-一般用于从某个或多个文件中搜索某串字符,
格式为:grep “字符串” 文件名 例:grep “F=” vasp.out (10)scp-用于网关与内部网内各计算节点或外部网络之间 的文件传输
的研究:如对A1, A2, A3,先用大模型和基组对A1进行研究,
然后以该结果为参照,确定计算量适中的模型和方法并应用
于A1,A2,A3。
ppt课件
7
Gaussian03程序的使用
G03的安装和运行; G03的功能和程序结构; 输入文件的编写与主要功能的使用; 补充说明;
ppt课件
传输
ppt课件
22
(11) vi-文本编辑命令 该命令常用但较为复杂,它有2种模式:命令模式和插入 模式,二者之间关系为:
i
Esc
command mode insert mode command mode 在命令模式下,可实现以下功能及其对应按键:
delete a character: x
ppt课件
14
(2). Linux平台: 基于Linux系统的计算拓扑结构
计
计
计
GaussView高斯软件教程PPT学习教案

结算结果可视化的ቤተ መጻሕፍቲ ባይዱ现
1.几何构型 2.分子振动 3.轨道 4.电荷密度 5. 静电势
第31页/共38页
选择File,打开一个已经正常计算完的 结果,然 后选择 Results ,就可 以出现 以下选 项 见下图
主菜单
第32页/共38页
选择Vibrations选项,就可以直接查看分 子的振 动模式 (见下 图)
第26页/共38页
(10).查看分子的对称性 从Edit-Point group路径可以查看所构建分子的点群 。点击Point group后,出现如下窗口:为C1点群 ,其下 拉菜单 中的为 可能的 点群( 改变Tolerance, 也可帮 助我们 判断所 构建体 系可能 有的点 群)
第27页/共38页
第37页/共38页
第3页/共38页
Edit: 在这里可以完成对分子的剪贴,拷贝,删 除 和 抓图等。 Atom List,显示当前分子的内坐标,笛卡儿坐 标,分 数坐标 等。 Point Group 可以显示当前分子的点群及可能有的点 群。 PBC显示晶体文件(可以将CIF文件转换为图 形,在 点PBC按钮后 所给并 的对话 框中根 据选项 调节具 体显示 的格式 。 Mos用于显示分子轨道(只有检查点 文件, 此选项 才能给 出分子 轨道图 。 Sy mmetrize,对当前体系进行对称性控制。
第15页/共38页
(3).在当前工作窗口(打开Gview时程 序自动 打开一 个工作 窗口, 如下图 )也可 通过Fil e-new 路径 新建一个工作窗口
第16页/共38页
在这个窗口中点鼠标左键窗口中就会 出现苯 分子, 见下图
将鼠标放在分子上,按左键左右或前 后移动 ,可以 调节分 子的角 度将鼠 标放在 分子上 ,前后 移动, 可以将 分子放 大或缩 小Shift+ Alt+鼠 标左键 组合可 以在窗 口内平 移分子 当工作窗口内有多个分子时[在构建大 的分子 时,这 种情况 很容易 出现]这 时可用 以下命 令可以 用Shift+Alt+ 鼠标左 键组合 移动想 要移动 的分子 ,以调 节各个 分子间 的距离 可以用C trl+Alt+鼠标 左键组 合调节 其中一 个分子 的角度 ,以调 节各个 分子间 的角度 。Ctrl+ Alt+鼠 标左键 这个组 合常和[将鼠标 左键放 在分子 上,左 右或前 后移动 ,可以 调节分 子的角 度]这个 功能连 用。
Gaussian 高斯使用指南
1.1 硬件环境………………………………………………………………7
1.2 操作系统………………………………………………………………8
2.安装………………………………………………………………………8
2.1 硬盘分区方案…………………………………………………………8
系的反应机制,表面和表面反应的团簇模型,有机物光化学过程,有机和有机金属化合物的
取代影响和反应,以及均相催化作用等。
ONIOM 的其它新功能还有:定制分子力学力场;高效的 ONIOM 频率计算;ONIOM 对电、
磁性质的计算。
2.通过自旋-自旋耦合常数确定构像
当没有 X-射线结构可以利用时,研究新化合物的构像是相当困难的。NMR 光谱的磁屏蔽数
1 目的 ………………………………………………………………………100
2 高斯自带的练习和理论模型介绍 ………………………………………100
第一章 功能和计算原理介绍
2
1:Gaussian 功能介绍
1.1 Gaussian 是一个功能强大的量子化学综合软件包。其可执行程序可在不同型号的大型
计算机,超级计算机,工作站和个人计算机上运行,并相应有不同的版本。
2.1 能量计算的格式 ………………………………………………………65
2.2 输出说明 ………………………………………………………………66
3.高斯中自带的练习 …………………………………………………………67
第七章 基组 ……………………………………………………………………69
1.基组介绍 …………………………………………………………………69
……………………3
Gaussian 高斯使用指南
像。另外,归属观测的峰值到特定的原子也比较容易。
3.研究周期性体系
Gaussian 03 扩展了化学体系的研究范围,它可以用周期性边界条件的方法(PBC)模拟周期性
体系,例如聚合物和晶体。PBC 技术把体系作为重复的单元进行模拟,以确定化合物的结
磁性质的计算。
2.通过自旋-自旋耦合常数确定构像
当没有 X-射线结构可以利用时,研究新化合物的构像是相当困难的。NMR 光谱的磁屏蔽数
据提供了分子中各原子之间的连接信息。自旋-自旋耦合常数可用来帮助识别分子的特定构
像,因为它们依赖于分子结构的扭转角。
除了以前版本提供的 NMR 屏蔽和化学位移以外,Gaussian 03 还能预测自旋-自旋耦合常数。
极化率和超极化率
热力学性质
反应路径
计算可以对体系的基态或激发态执行。可以预测周期体系的能量,结构和分子轨道。因
此,Gaussian可以作为功能强大的工具,用于研究许多化学领域的课题,例如取代基的影响,
化学反应机理,势能曲面和激发能等等。
1.2 关于 Gaussian 03 的介绍
是 Gaussian 系列电子结构程序的最新版本。它在化学、化工、生物化学、物理化学等
对正常结束的作业主要的计算结果如优化后的分子几何参数电子光谱ir和raman谱的频率和强度等都可随时从保存的检查点文件中精确提取并利用主窗口utilities中选项的功能转换为其它软件可读的格式以便进行图形显示和分析对于有gview的用户可不考虑使用utilities的功能作为和高斯配套使用的软件gview可以直接将检查点文件转换为图但检查点文件的存在是必须的
振动频率,IR 和 Raman 光谱,以及其它特性;极化率和超极划率;执行性能上的改善。
GaussView高斯软件教程
Gview是一个专门设计于高斯配套使用的软件,其 主要用途有两个构建高斯的输入文件以图的形式显示 高斯计算的结果除了可以自己构建输入文件外, Gview还可读入CHEM3D,HYPERCHEM和晶体数据 等诸多格式的文件。从而使其可以于诸多图形软件连 用,大大拓宽了使用范围(详见下图)
-
(10).查看分子的对称性 从Edit-Point group路径可以查看所构建分子的点群。点击Point group后,出现如下 窗口:为C1点群,其下拉菜单中的为可能的点群(改变Tolerance,也可帮助我们判断 所构建体系可能有的点群)
-
(11).查看分子坐标。单击Gview界面上的 图标。出现下面窗口
在构建分子前,应对分子的键长,键角和点群等有一个详细的了解。这样在构建过程 中才能更好的把握细节,如构建大分子时,各个集团间的键长,键角和二面角一定要 给的准确。相当一部分的错误(如SCF不收敛,1502报错等)都是分子建模不合理 造成的。分子模型不合理导致程序根据分子模型所得的薛定鄂方程不合理,也就导致 了对方程的解不能顺利完成,所以报错。对于大分子,不会出现分子直线排步的情况, 链烃通常为锯齿型的排列,环之间总是有一定的夹角,共面情况虽有但不多见,这些 都是在构建分子模型的过程中需要注意的。再者,分子建模是量化计算的一个难点, 特别是对金属配合物,块状体系和纳米体系的构建。只有给出合理的结构,程序才能 给出合理的解。如果不能体现出分子的对称性,则程序不能正确判断分子轨道的对称 性,这在涉计到反应机理和轨道相互作用的研究中可能会导致错误的判断。
-
(8).单击Gview界面上 图标。然后点击与乙烷正对的苯上的C原子和H原子选择 “None”,乙烷上的C-H键作类似处理然后单击Gview上的 图标,点选工作窗口中的 两个氢原子,即将它们删去
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
界面教程【1】——界面介绍
是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,还可读入3D,和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏
▪【】主要功能是建立,打开,保存和打印当前的文件
▪【】完成对分子的剪贴、拷贝、删除、抓图等
▪【】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等
▪【】直接向提交计算
▪【】接收并显示计算后的结果
▪【】控制窗体,如关闭、恢复等
▪【】帮助
快速工具栏
【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;
【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等
【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸
【左面第五个】用户自定义基团,您可以将常用的基团存放到此处
这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】
这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏
这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具
教程【2】——构建分子
这里以构建一个间氟苯乙烷分子并从里递交计算为例来说明。
1、开启
2、双击按钮,选择苯基(第一个),会看到主程序框体中出现苯环
在工作窗口单击,可以看到工作窗口也出现了一个苯环。
单击左键,可以将主程序框体中的分子或基团加入到工作窗口;
按上下左右键,或者左键单击不放移动鼠标,可以调节分子的角度;
滚动鼠标滚轮,可以放大缩小分子;
按住键,左键单击不放移动鼠标,可以移动分子;
当工作窗口内有多个分子时(在构建大的分子时,这种情况很容易出现)
用鼠标左键组合,移动想要移动的分子;
用鼠标左键组合调节其中一个分子的角度;
双击图标(或者左键不放),在元素周期表中选择【F】元素,回到工作窗口在苯的任意一个【H】上单击,使之变成【F】
双击图标,从链烃库中选择【乙基】(第一个),然后点击间位上的【H】即可
至此,分子式已经构建完成,存储个人常用分子的功能,双击上的图标,在下面的对话框中键入相关项目,保存即可。
(注:我的程序此项功能不行,用户自定义基团)
查看分子坐标:单击界面的图标,查看分子的坐标。
至此,我们保存一下我们构建好的间氟苯乙烷分子,在绘图界面右键单击【】——【】然后选择保存路径即可。
教程【3】——向提交计算
上一讲我们已经构建好了一个间氟苯乙烷分子,现在打开间氟苯乙烷分子。
点击界面上的【】——【】
从对话框中我们可以选择许多参数,下面依次选择一下
【】工作类型:
【能量】|【优化】|【频率】|【优化+频率】|【扫描】|【稳定性】|【核磁】,
这里我们选择【】核磁
【】方法:每种计算模式都提供了若干种方法,这里选择默认值即可
【】题目:这项可以根据自己的项目自行命名
【 0】给检查点文件命名
其它选择默认即可
选好后点击【】提交至,并保存。
(最好存到安装文件所在磁盘)
系统会询问是否提交至,选择【】,会自动开启并计算,计算时间因硬件配置而异
计算完毕后系统会提示关闭,点击【是】
计算完毕后会生成两个文件,的是系统日志,便于查看计算结果。
这里我们选择文件【】
接下来我们又一次看到了刚才绘制的那个分子,表面上没什么不同,但这次我们可以预言它的许多性质了,在的主界面点击【】——【】可以看到刚才的计算总结,点击【】可以看到日志文件。
因为我们刚才选择计算模式是【】核磁,所以我们点击【】——【】,随即我们看到了很熟悉的核磁图谱,但似乎很乱,因为这是所有元素的核磁谱图,在左下方【】右方选择【H】,即H谱,旁边【】选择【 6-31G(d)】,这次我们就可以看到间氟苯乙烷分子理论计算的核磁谱图了。
在绘图界面右击【】——【】可以看到每个H原子对应场中的位置。