Materials Studio软件CASTEP模块
第六讲第一原理计算方法简介及MaterialsStudio中Castep使用案例

Http Gateway Ftp
Module
parallel Windows Linux Linux IA32 IA64
Module
parallel Windows
Linux IA32
Linux IA64
Materials Visualizer Adsorption Locator Amorphous Cell Blends CASTEP and NMR CASTEP √
计算:允许选择计算选项(如基集,交换关联势和收敛判据),作业控制 和文档控制。
分析:允许处理和演示CASTEP计算结果。这一工具提供加速整体直观化以 及键结构图,态密度图形和光学性质图形。
CASTEP的任务
CASTEP计算包括单点的能量计算,几何优化或分子动 力学。可提供这些计算中的每一个以便产生特定的物理性 能。 在CASTAP计算中有很多运行步骤,可分为如下几组: 结构定义:必须规定包含所感兴趣结构的周期性的3D模型 文件,有大量方法规定一种结构:可使用构建晶体 (Build Crystal)或构建真空板(Build Vacuum Stab)来 构建,也可从已经存在的结构文档中引入,还可修正已存 在的结构。 注意: CASTEP仅能在3D周期模型文件基础上进行计算, 必须构建超单胞,以便研究分子体系。
提示: CASTAP计算所需时间随原子数平方的增加而增加。 因此,建议是用最小的初晶胞来描述体系,可使用 Build\Symmetry\Primitive Cell菜单选项来转换成初晶
CASTEP的任务
计算设置:合适的3D模型文件一旦确定,必须选择计算类型 和相关参数,例如,对于动力学计算必须确定系综和参数, 包括温度,时间步长和步数。选择运行计算的磁盘并开始 CASTEP作业。 结果分析:计算完成后,相关的CASTEP作业的文档返回用户, 在项目面板适当位置显示。这些文档进一步处理能获得所需 的观察量如光学性质。 CASTAP中选择一项任务 1 从模块面板(Module Explorer)选择CASTAP\Calculation 2 选择设置表 3 从任务列表中选择所要求的任务
第一原理计算方法简介及Materials Studio中Castep使用

提示: CASTAP计算所需时间随原子数平方的增加而增加。 因此,建议是用最小的初晶胞来描述体系,可使用 Build\Symmetry\Primitive Cell菜单选项来转换成初晶
CASTEP的任务
计算设置:合适的3D模型文件一旦确定,必须选择计算类型 和相关参数,例如,对于动力学计算必须确定系综和参数, 包括温度,时间步长和步数。选择运行计算的磁盘并开始 CASTEP作业。 结果分析:计算完成后,相关的CASTEP作业的文档返回用户, 在项目面板适当位置显示。这些文档进一步处理能获得所需 的观察量如光学性质。 CASTAP中选择一项任务 1 从模块面板(Module Explorer)选择CASTAP\Calculation 2 选择设置表 3 从任务列表中选择所要求的任务
晶胞分子晶体表面纳米结构聚合物等2458锐钛矿tio1112558pt110co2x1碳纳米管tio纳米棒2658materialstudio晶体结构模型建立建立方铅矿pbs晶体结构模型实验15步骤1查晶体结构数据如icsdpdf数据库2通过软件建模如materialstudio中模块visualizerdiamond2758castep使用2858castep是特别为固体材料学而设计的一个现代的量子力学基本程序其使用了密度泛函dft平面波赝势方法进行第一原理量子力学计算以探索如半导体陶瓷金属矿物和沸石等材料的晶体和表面性质
密度泛函理论
赝势(pseudo potential) 赝势就是把离子实的 内部势能用假想的势能 取代真实的势能,但在 求解波动方程时,不改 变能量本征值和离子实 之间区域的波函数。模 守恒赝势NCP (Norm Conserving Pseudopotential) 和 超软赝势 USPP(Ultrasoft Pseudoptential)
materialstudio中文版帮助手册

materialstudio中文版帮助手册欢迎欢迎使用Materials StudioMaterials Studio是一个采用服务器/客户机模式的软件环境,它为你的PC机带来世界最先进的材料模拟和建模技术。
Materials Studio使你能够容易地创建并研究分子模型或材料结构,使用极好的制图能力来显示结果。
与其它标准PC软件整合的工具使得容易共享这些数据。
Materials Studio的服务器/客户机结构使得你的Windows NT/2000/XP,Linux和UNIX服务器可以运行复杂的计算,并把结果直接返回你的桌面。
Materials Studio采用材料模拟中领先的十分有效并广泛应用的模拟方法。
Accelry’s的多范围的软件结合成一个集量子力学、分子力学、介观模型、分析工具模拟和统计相关为一体容易使用的建模环境。
卓越的建立结构和可视化能力和分析、显示科学数据的工具支持了这些技术。
无论是使用高级的运算方法,还是简单地利用Materials Studio 增强你的报告或演讲,你都可以感到自己是在用的一个优秀的世界级材料科学与化学计算软件系统。
易用性与灵活性Materials Studio可以在Windows 98,Me,NT,2000和XP下运行。
用户界面符合微软标准,你可以交互控制三维图形模型、通过简单的对话框建立运算任务并分析结果,这一切对Windows用户都很熟悉。
Materials Studio的中心模块是Materials Visualizer。
它可以容易地建立和处理图形模型,包括有机无机晶体、高聚物、非晶态材料、表面和层状结构。
Materials Visualizer 也管理、显示并分析文本、图形和表格格式的数据,支持与其它字处理、电子表格和演示软件的数据交换。
Materials Studio是一个模块化的环境。
每种模块提供不同的结构确定、性质预测或模拟方法。
你可以选择符合你要求的模块与Materials Visualizer组成一个无缝的环境。
CASTEP概述

CASTAP性质任务
CASTAP性质任务允许在完成能量,几何优化或动力学运行之后求出电 子和结构性质。可以产生的性质如下:
* 态密度(DOS):利用原始模拟中产生的电荷密度和势能,非自恰计算价 带和导带的精细Monkhorst-Pack 网格上的电子本征值。
* 带结构:利用原始模拟中产生的电荷密度和势能,非自恰计算价带和导 带的布里渊区高对称性方向电子本征值。 * 光学性质:计算电子能带间转变的矩阵元素。CASTAP分析对话可用于生 成包含可以测得的光学性质的网格和图形文件。 * 布局数分析:进行Mulliken 分析。计算决定原子电荷的键总数和角动量 (以及自旋极化计算所需的磁矩)。任旋地,可产生态密度微分计算所要 求的分量。 * 应力:计算应力张量,并写入seedname.castep 文档。
CASTAP动力学任务
CASTAP动力学任务允许模拟结构中原子在计算力的影响下将如何移动。 在进行CASTAP动力学计算以前,可以选择热力学系综和相应参数,定义模拟 时间和模拟温度。
选择热力学系综
对牛顿运动定律积分允许探索体系恒值能量表面(NVE动力学)。然而,在 体系与环境进行热交换条件下发生最本质的现象。使用NVT系综(或者是确定性 的Nosé 系综或者是随机性的Langevin 系综)可模拟该条件。
在模型文档中右键单击,选择Display Styles,按下Ball and stick按钮。关闭对话框。
在3D视窗中的晶体结构是晶胞,它显示的是格子的立方对 称。如果存在的话,CASTEP使用的则是格子的全部对称. 既包含有两个原子的原胞和包含有8个原子的晶胞是相对应的. 不论单胞如何定义,电荷密度,键长,每一类原子的总体能量 都是一样的,并且由于使用了较少的原子 ,使计算时间得以减少。
实验1:Materials_Studio软件简介及基本操作要点

《计算材料学》实验讲义实验一:Materials Studio软件简介及基本操作一、前言1. 计算材料学概述随着科学技术的不断发展,科学研究的体系越来越复杂,理论研究往往不能给出复杂体系解析表达,或者即使能够给出解析表达也常常不能求解,传统的解析推导方法已不敷应用,也就失去了对实验研究的指导意义。
反之,失去了理论指导的实验研究,也只能在原有的工作基础上,根据科研人员的经验理解、分析与判断,在各种工艺条件下反复摸索,反复实验,最终造成理论研究和实验研究相互脱节。
近年来,随着计算机科学的发展和计算机运算能力的不断提高,为复杂体系的研究提供了新的手段。
在材料学领域,随着对材料性能的要求不断的提高,材料学研究对象的空间尺度在不断变小,纳米结构、原子像已成为材料研究的内容,对功能材料甚至要研究到电子层次,仅仅依靠实验室的实验来进行材料研究已难以满足现代新材料研究和发展的要求。
然而计算机模拟技术可以根据有关的基本理论,在计算机虚拟环境下从纳观、微观、介观、宏观尺度对材料进行多层次研究,进而实现材料服役性能的改善和材料设计。
因此,计算材料学应运而生,并得到迅速发展,目前已成为与实验室实验具有同样重要地位的研究手段。
计算材料学是材料科学与计算机科学的交叉学科,是一门正在快速发展的新兴学科,是关于材料组成、结构、性能、服役性能的计算机模拟与设计的学科,是材料科学研究里的“计算机实验”。
计算材料学主要包括两个方面的内容:一方面是计算模拟,即从实验数据出发,通过建立数学模型及数值计算,模拟实际过程;另一方面是材料的计算机设计,即直接通过理论模型和计算,预测或设计材料结构与性能。
计算材料科学是材料研究领域理论研究与实验研究的桥梁,不仅为理论研究提供了新途径,而且使实验研究进入了一个新的阶段。
计算材料学的发展是与计算机科学与技术的迅猛发展密切相关的。
从前,即便使用大型计算机也极为困难的一些材料计算,如材料的量子力学计算等,现在使用微机就能够完成,可以预见,将来计算材料学必将有更加迅速的发展。
【Materials_Studio】Castep说明

你的 CASTEP run 一跑完,你就可以使用 CASTEP 模組的分析工具來抽取及檢視 由 CASTEP 所產生的原始輸出資料。這原始輸出結果藉由大量的數據來描述你 模型的性質。
動態使用介面
雖然機制羯大部份是明顯的,Cerius2 透過由使用者介面建立的好幾個 CASTEP 輸出輸入的資料檔來與 CASTEP 產生聯繫。你所要執行的 job 上設定的選項會被 用來產生一個檔案,此檔案會被傳送到 CASTEP 做為輸入。
膺勢
電子-離子間的交互作用可以用膺勢的觀念來描述。對於每種元素而言,CASTEP 提供了一套的的位勢:
位勢 延伸檔名
ultrasoft .usp
norm-conserving potential 使用 Lin et al.最佳化方法來產生 .recpot
norm-conserving potential 使用 Troullier-Martins 最佳化方法來產生 .pspnc
在 CASTEP 裡預設的設定是 GGA,它在很多狀況下被知道是比較好的方法。梯 度修正的方法在研究表面的過程、小分子的性質、氫鍵晶體以及有內部空間的晶 體(費時)是比較精確的。眾所皆知,LDA 會低估分子的鍵長(or 鍵能)以及 晶體的晶格參數,而 GGA 通常會補救這缺點。然而,有許多證據顯示 GGA 會 在離子晶體過度修正 LDA 結果;當 LDA 與實驗符合得非常好的時候,GGA 會 高估晶格長度。因此要推薦一個對所有系統都是最好的特定方法是很困難的。
利用materials_studio中得CASTEP模块预测锗的热力学性质

确定Ge.xsd为当前文件。
从Materials Studio 菜单栏 中选择Modules | CASTEP | Analysis 。从属性列表中选择 Thermodynamic properties。 确定Results file 选择框中显示 的是Ge_PhononDOS.castep。
勾选上Debye temperature图, 按下View按钮。 在结果文件夹中创建了两个新的图形 文档Ge Thermodynamic Properties.xcd 和Ge Debye Temperature.xcd。 Debculation 对 话框中的Setup标签,把Task 设置改为Energy。
选择Electronic标签, 按下More…按钮,显 示出CASTEP Electronic Options对话 框。选择SCF标签,勾 选上Fix occupancy。 关闭CASTEP Electronic Options对话 框。
按下View 按钮。
-100
0
100
200
300
400
创建了一个新的图形 文档Ge DOS.xcd,它 应当与下图相似。
4. 显示热力学性质
在CASTEP中,通过声子的计算,可以在准谐性近似下, 进一步计算晶体的焓、熵、自由能、晶格热容与温度的关系。 这些计算结果可以与实验数据(如,热容的测量)相比,也 可以预测结构相近的材料的稳定性或相变。 所有与能量相关的性质均画在一张图上,计算时考虑了 零点能量的修正。热容被独自画在右侧。 现在使用声子计算的结果来产生热力学性质图。
选中Job Control标签。选择你想要在其上运行工作的Gateway location。单击 Run 按钮,开始计算。 关闭 CASTEP Calculation 对话框。
第一原理计算方法及MaterialsStudio中Castep使用

第一原理常用计算软件
根据对势函数及内层电子的处理方法不同 主要分为两大类,一种是波函数中包含了 高能态和内层电子,而势函数只是原子核 的贡献,这称为全电子(all electron calculation)法,另一种处理方法是势函 数为原子核和内层电子联合产生的势,称 为离子赝势,波函数只是高能态电子的函 数,这称为赝势(pseudo-potential)法。
b. Born-Oppenheimer近似,核固定近似 中子/质子的质量是电子质量的约1835倍,即电子的运 动速率比核的运动速率要高3个数量级,因此可以实现 电子运动方程和核运动方程的近似脱耦。这样,电子可 以看作是在一组准静态原子核的平均势场下运动。
c.单电子近似 把体系中的电子运动看成是每个电子在其余电子的平均 势场作用中运动,从而把多电子的薛定谔方程简化单电 子方程。
在CASTAP计算中有很多运行步骤,可分为如下几组:
结构定义:必须规定包含所感兴趣结构的周期性的3D模型 文件,有大量方法规定一种结构:可使用构建晶体 (Build Crystal)或构建真空板(Build Vacuum Stab)来 构建,也可从已经存在的结构文档中引入,还可修正已存 在的结构。
注意: CASTEP仅能在3D周期模型文件基础上进行计算, 必须构建超单胞,以便研究分子体系。
Pseudo
Pseudo
Pseudo, PAW all-electron
操作系统
Linux
Web Site
www.abinit. org
Windows Linux
Linux
www.tcm.ph / castep/
www.pwscf.o rg/
Linux Linux
cms.mpi.un ivie.ac.at/v asp
1-Materials Studio 与 CASTEP 快速入门
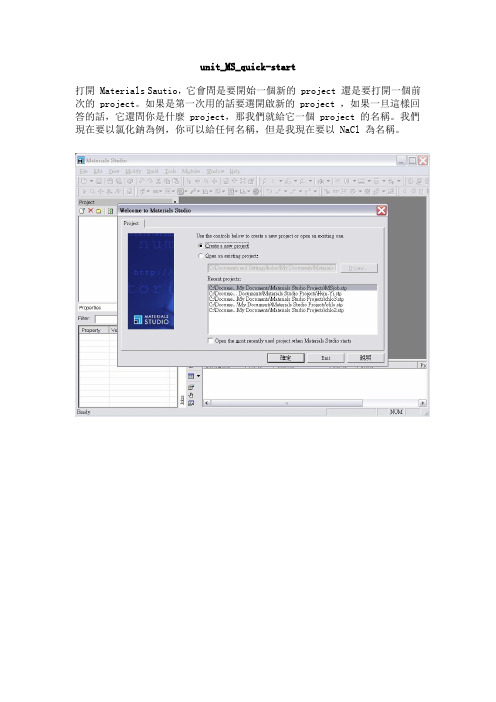
unit_MS_quick-start打開 Materials Sautio,它會問是要開始一個新的 project 還是要打開一個前次的 project。
如果是第一次用的話要選開啟新的 project ,如果一旦這樣回答的話,它還問你是什麼 project,那我們就給它一個 project 的名稱。
我們現在要以氯化鈉為例,你可以給任何名稱,但是我現在要以 NaCl 為名稱。
一開始進來要先介紹幾個重要的視窗,它們關係到我們進行模擬計算時所會處理及操作到的對象。
姑且可以分為這三類:一、進行計算的工作,己跑完的、正在跑的都算;二、計算工作總是有各有些不同的輸入與輸出檔案,我們經常會需要審視結果、修改輸出入的相關設定;三、材料的原子及電子結構 3D 模型帶有很多我們想要知道之關於這個材料的物性資料,例如晶體的晶胞邊長、原子的元素種類等等。
從 Veiw 的 Explorer ,它有三個 Explorer,job Exploroer、project exploroer、property explorer 。
job explorer 的開跟關是這樣按一次它就開起來。
這個是你跑什麼 job 近端遠端它都可以顯示,跑完了沒有、要不要把它移除等等,在這邊都可以操作,有很多 job 的時候會很好用。
project explorer 預設值是開著的,就是靠左邊垂直的這一塊,裡面對於跑 project 的相關物件,如文字輸出、3D結構等等都是在這裡選取,很像微軟視窗 (MS Windows) 裏頭的『檔案總管』。
要做東西總是需要選取一個 job 相關的目錄等等,所以 job explorer 在操作上來講是很重要的。
另外我也常常會打開的是 property explorer ,property explorer 在 MS 是新的東西,相對 Cerius2 而言是新的東西。
在 Cerius2 裡如果你想要知道一些 3D 物件的屬性,像是鍵長、鍵角,晶胞內原子數,就要分別去打開一些相關的表單,它才會印給你看,然而初學者還得學會這些表單藏在那裏。
material_studio_中文版帮助手册

欢迎欢迎使用Materials StudioMaterials Studio是一个采用服务器/客户机模式的软件环境,它为你的PC机带来世界最先进的材料模拟和建模技术。
Materials Studio使你能够容易地创建并研究分子模型或材料结构,使用极好的制图能力来显示结果。
与其它标准PC软件整合的工具使得容易共享这些数据。
Materials Studio的服务器/客户机结构使得你的Windows NT/2000/XP,Linux和UNIX服务器可以运行复杂的计算,并把结果直接返回你的桌面。
Materials Studio采用材料模拟中领先的十分有效并广泛应用的模拟方法。
Accelry’s的多范围的软件结合成一个集量子力学、分子力学、介观模型、分析工具模拟和统计相关为一体容易使用的建模环境。
卓越的建立结构和可视化能力和分析、显示科学数据的工具支持了这些技术。
无论是使用高级的运算方法,还是简单地利用Materials Studio增强你的报告或演讲,你都可以感到自己是在用的一个优秀的世界级材料科学与化学计算软件系统。
易用性与灵活性Materials Studio可以在Windows 98,Me,NT,2000和XP下运行。
用户界面符合微软标准,你可以交互控制三维图形模型、通过简单的对话框建立运算任务并分析结果,这一切对Windows用户都很熟悉。
Materials Studio的中心模块是Materials Visualizer。
它可以容易地建立和处理图形模型,包括有机无机晶体、高聚物、非晶态材料、表面和层状结构。
Materials Visualizer 也管理、显示并分析文本、图形和表格格式的数据,支持与其它字处理、电子表格和演示软件的数据交换。
Materials Studio是一个模块化的环境。
每种模块提供不同的结构确定、性质预测或模拟方法。
你可以选择符合你要求的模块与Materials Visualizer组成一个无缝的环境。
Materials Studio的模块

Materials Studio的模块Materials Studio是一个全尺度材料模拟平台。
平台以可视化视窗界面Materials visualizer为核心,在其上共整合了24个功能模块,囊括了量子力学方法、经典模拟方法、介观模拟方法、有限元模拟等各种常见分子模拟方法,以及晶体结构解析、晶体形貌预测、定量构效关系分析等实用工具,实现了从电子结构解析到宏观性能预测的跨尺度研究。
Materials visualizerMaterials visualizer是Materials Studio的图形化界面,也是整个平台的核心。
Materials visualizer的功能包括:●搭建、调整各类三维可视的结构模型,包括晶体、小分子、聚合物、纳米材料、团簇、表界面以及各种缺陷结构;●提供模块参数设置、结果分析的视窗界面;提供结构文件、参数文件以及结果文件的管理界面;提供计算进程的监控界面;●对模拟结果进行各种分析,可与结构模型相结合进行数据的二维、三维显示,可以给出数据的图表,可以对特定的结果进行动画演示或给出矢量图;Materials visualizer的特性包括:●支持多种结构、图形、文本文件格式的输入和输出;●支持不同功能模块间结构数据的共享;●提供Perl语言环境,以及脚本编译工具;●提供不规则多面体表面积、体积的计算工具。
量子力学方法量子力学方法(Quantum Mechanics)是一种能够对材料体系电子结构特点进行解析的方法,精度高且几乎不依赖于任何经验参数,因此被广泛应用在各类材料的模拟研究中。
半经验量子力学方法(Semi-empirical Quantum Mechanics)同样能够对材料体系电子结构特点进行解析,但是包含有更多的经验参数以及数学、物理近似,因此,计算效率相比于纯粹的量子力学更高,但是精度会略低。
量子力学以及半经验量子力学方法均以定态薛定谔方程为核心,计算原子核满足特定排列、堆积时,核外电子的空间、能量分布,并由此进一步得到体系的电学性质、磁学性质、光学性质、热力学性质、力学性质,所能研究的材料体系类型包括:各类晶体材料及可能的各种缺陷结构,各种维度的纳米材料,各种分子及团簇材料。
1-Materials Studio 与 CASTEP 快速入门
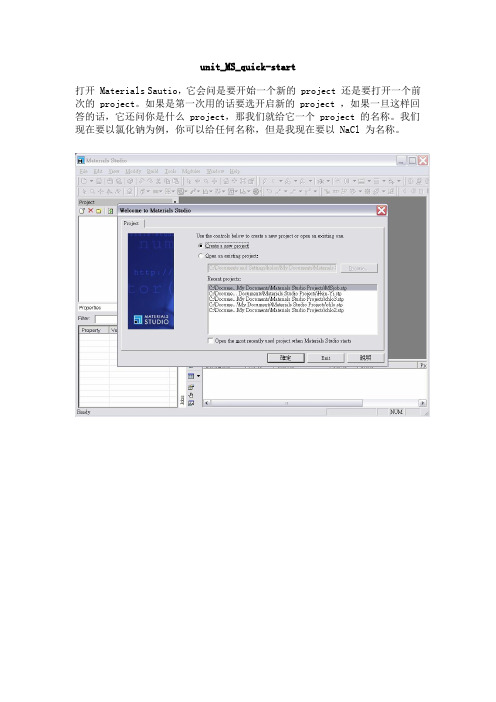
unit_MS_quick-start打开 Materials Sautio,它会问是要开始一个新的 project 还是要打开一个前次的 project。
如果是第一次用的话要选开启新的 project ,如果一旦这样回答的话,它还问你是什么 project,那我们就给它一个 project 的名称。
我们现在要以氯化钠为例,你可以给任何名称,但是我现在要以 NaCl 为名称。
一开始进来要先介绍几个重要的窗口,它们关系到我们进行模拟计算时所会处理及操作到的对象。
姑且可以分为这三类:一、进行计算的工作,己跑完的、正在跑的都算;二、计算工作总是有各有些不同的输入与输出档案,我们经常会需要审视结果、修改输出入的相关设定;三、材料的原子及电子结构 3D 模型带有很多我们想要知道之关于这个材料的物性数据,例如晶体的晶胞边长、原子的元素种类等等。
从 View(查看)的 Explorer(资源管理器),它有三个 Explorer,job Explorer、project explorer、property explorer 。
job explorer 的开跟关是这样按一次它就开起来。
这个是你跑什么 job 近端远程它都可以显示,跑完了没有、要不要把它移除等等,在这边都可以操作,有很多 job 的时候会很好用。
project explorer 默认值是开着的,就是靠左边垂直的这一块,里面对于跑 project 的相关对象,如文字输出、3D结构等等都是在这里选取,很像微软窗口 (MS Windows) 里头的『档案总管』。
要做东西总是需要选取一个 job 相关的目录等等,所以 job explorer 在操作上来讲是很重要的。
另外我也常常会打开的是 property explorer ,property explorer 在 MS 是新的东西,相对 Cerius2 而言是新的东西。
在 Cerius2 里如果你想要知道一些 3D 对象的属性,像是键长、键角,晶胞内原子数,就要分别去打开一些相关的窗体,它才会印给你看,然而初学者还得学会这些窗体藏在那里。
最新对于初学Materials-Studio-CASTEP问题整理

问题如下1、Symmetry 下的unbuild crystal, Nonperiodic, Superstructure, Make P1, Redefine options各有什么作用?答:Unbuild crystal:得到最小非对称单元的结构Nonperiodic:去掉结构的周期性,形象地说就是把盒子去掉。
Superstructure:构建超晶胞结构,也就是扩大最小重复单元(或则说晶胞)Make P1:去掉晶体结构中的所有点对称操作,只保留其平移对称性Redefine lattice:重新定义晶胞中基矢的方向2、图表的含义是什么?Atomic Populations (Mulliken)Species Ion s p d f Total Charge (e)O 1 1.91 4.99 0.00 0.00 6.90 -0.90O 2 1.91 4.99 0.00 0.00 6.90 -0.90O 3 1.91 4.99 0.00 0.00 6.90 -0.90O 4 1.91 4.99 0.00 0.00 6.90 -0.90O 5 2.01 5.08 0.00 0.00 7.08 -1.08O 6 1.84 4.87 0.00 0.00 6.71 -0.71Ca 1 2.14 6.00 0.47 0.00 8.61 1.39Ti 1 2.32 6.24 2.22 0.00 10.78 1.22Ti 2 2.32 6.24 2.22 0.00 10.78 1.22Ba 1 1.76 6.01 0.70 0.00 8.46 1.54答:以O为例子Species Ion s p d f Total Charge (e)O 1 1.87 4.79 0.00 0.00 6.65 -0.65计算以前O的电子结构是2s2 2p4,Total =6(e )计算后O的结构变为2s1.872p4.79,Total =6.65(e )-0.65 表明优化以后,O得到0.65(e )如果考虑的是纯离子,当然就是+4和-2了。
materialstudio一些基础设置问题

CASTEP的任务1. CASTEP能量任务CASTEP能量任务允许您计算指定系统的总能量,以及它的物理性质。
除了总能量,原子上的力也会在计算结束时报告。
还创建了一个电荷密度文件,允许使用可视化工具直接观察电荷密度的空间分布。
还报告了在计算中使用的monkhorst - pack k点的电子能量,以便在CASTEP分析过程中生成态密度图。
能量任务对于研究可靠的结构信息体系的电子特性是非常有用的。
只要指定了应力特性,它也可以用来计算没有内部自由度的高对称系统的状态方程(即压力体积和/或能量-体积依赖)。
注意:在具有内部自由度的系统中,可以利用几何优化任务得到状态方程。
CASTEP的能量的默认单位是电子伏特(eV)。
1 eV= 0.036749308 Ha =23.0605 kcal/mole =96.4853 kJ/mole2. CASTEP几何优化任务CASTEP几何优化任务允许优化几何结构,以获得一个稳定的结构或多态性。
这是通过执行一个迭代的过程来完成的,在这个过程中,原子的坐标和可能的原胞参数被调整,从而使结构的总能量是最小的。
CASTEP几何优化是基于减小计算力和应力的大小,直到它们变得小于定义的收敛误差。
此外,还可以指定一个外部应力张量,来模拟在张力、压缩、剪切等情况下系统的行为。
在这些情况下,内部应力张量是迭代的,直到它等于施加的外部应力。
几何优化的过程一般会产生一个与实际结构相似的模型结构。
用CASTEP计算的晶格参数的准确性如图1所示(Milman等,2000)。
Figure 1. Experimental vs. CASTEP calculated lattice parameters状态方程的计算应用流体静压法的几何优化可用于确定材料的体积模量,B,压力导数、B ' = dB / dP。
这个过程包括计算状态方程(EOS),它描述了细胞体积对外部流体静压的依赖。
该方法与实际实验非常相似:在几何优化对话框中使用最小化选项键确定外部压力,通过对CASTEP进行几何优化来确定压力的单元体积。
material_studio_2016_新功能发布

material_studio_2016_新功能发布“Materials Studio 2016” 新功能发布材料科学软件技术部许立芳2016年01月11日Materials Studio 2016 新功能亮点CASTEP 加入新的结构优化算法TPSD ,提高表界面的结构优化效率CASTEP On-the-fly norm-conserving There are 38 new features or major enhancementsCASTEP 整合新的赝势,即支持利用On the fly 生成模守恒(norm conserving )赝势,特别适用于计算磁性材料和包含f 电子的元素?CASTEP 支持旋轨耦合(spin-orbit coupling )?核磁共振的J-coupling 耦合常数CASTEP TD-DFT 支持计算三维周期性材料的光谱?DMol 3中B3LYP 杂化泛函应用于大体系效率显著提升DMol 3中meta-GGA 泛函可应用于周期性结构,提高重过渡金属的计算准确性量子力学Copyright 2014 NeoTrident Technology Ltd. All rights reserved.更广泛的模拟体系:DFTB+ 模块添加多个新的slater-koster 库文件分子力学动力学添加tabulated potential ,从而支持使用自定义的函数形式用于键长、键角和范德华相互作用的模拟可编辑范德华相互作用的缩放因子,从而使Forcite Plus 支持编写OPLS 力场等CASTEP模块新功能一、新的结构优化算法:TPSD (Two-Point Steepest Descent ),适用于晶胞参数固定的体系二、整合更准确的赝势,即支持利用On-the-fly 生成模守恒(norm-conserving )赝势,提高对于磁性材料和包含 f 电子的元素的计算精度三、旋轨耦合(spin-orbital coupling )四、TD-DFT 支持计算三维周期性材料的光谱五、核磁共振的耦合常数(J-coupling )Copyright 2014 NeoTrident Technology Ltd. All rights reserved.六、包含相对论效应的赝势七、支持直接得到弹性德拜温度和平均声速八、各向异性温度因子的计算及可视化九、标记原子序号一、TPSD(Two-Point Steepest Descent )算法新的结构优化算法:推荐使用条件:对于晶胞参数含有约束条件(constraints)时,TPSD 是更有效的优化方法。
Materials Studio软件中各模块的功能简介

分子力学与分子动力学
晶体、结晶与X射线衍射 MS.Polymorph Predictor MS.Morphology MS.X-Cell MS.Reflex MS.Reflex Plus MS.Reflex QPA 高分子与介观模拟 MS.Synthia MS.Blends MS.DPD MS.MesoDyn MS.MesoPro MS.QSAR MS.QSAR Plus MS.Dmol3 Descriptor
定量结构-性质关系
Materials Studio Datasheet
-.-..... .. ......
Morpherials Studio 软件各模块的介绍
基本环境:MS.Materials Visualizer 量子力学 MS.Dmol3 MS.CASTEP MS.NMR CASTEP MS.VAMP MS.DISCOVER PASS MS.Amorphous Cell MS.Forcite MS.Forcite Plus MS.GULP MS.Equilibria MS.Sorption
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Logo
4
矿物晶体结构搭建
• (3)外部数据库导入结构
无机晶体学数据库ICSD
American Mineralogist Crystal Structure Database
可输入矿物名称、作者、化学元素、晶胞参数及对称性等关 键词查找,可直接获得.cif结构文件和XRD检测数据,还可 以预览3D模型。
输出结果
9
K-S求解近似处理方法-交换相关泛函
2 2
q
Zq r Rq
(r)
r r
dr VXC (r) i
i i (r)
电 动, 常数 一次迭代确定
核-电
电-电库伦作 用能
电-电交换相 关能
赝势处理
局域密度近似 LDA 广义梯度近似 GGA
决定
微观的电子结构
获 取
密度泛函
求解多粒子系统的薛定谔方程
H=E
近似求解多粒子系统薛定谔方程
Logo
7
密度泛函理论内容
[
1 2
2
Veff
(r )]
i
(r)
ii
(r
)
N
n(r) i (r) 2 i 1
Kohn-Sham方程
简化哈密顿算符
得 到
E() () Vne () Vee () Vnn
Logo
11
交换相关泛函-广义梯度近似GGA
GGA克服了LDA在描述真实体系在电子密度变化剧烈的情 况下的缺陷,提高了交换相关能计算结果的精度,也提高了密 度泛函方法计算的精度。
非定域泛函:
HF HF-LDA sX sX-LDA PBE0 B3LYP……
一般研究半导体材料采用,精度有所提高,但运算量是LDA和GGA的几十 到几百倍,在计算精度与计算时间二者中做出取舍,一般没有必要采用
Company Logo
Materials Studio软件CASTEP模块 ——架构原理及主要参数意义
主要内容
1 矿物晶体结构搭建 2 CASTEP整体架构、其中假设 3 CASTEP参数设置及其意义
Logo
2
矿物晶体结构搭建
• (1)从MS数据库中引入结构
MS数据库中为用户提供了较为常见的结构模型:
Logo
5
矿物晶体结构搭建
注意:引入结构或网上查找参数及结构文件时,注意 识别需要的结构构型或数据,有的结构是人工合成 的或者经过高温、高压处理的,不能采用,如MS 库中的ZnO
Logo
6
CASTEP整体架构、其中假设
CASTEP的整体构架就是基于密度泛函理论求解K-S方程的一种方法
基本物理性质
Logo
10
交换相关泛函-局域密度近似LDA
假设原子核外电子云均匀分布
LDA适用的情况: (1)电荷密度变化缓慢的体系(如金属) (2)电荷密度较高的体系(如过渡金属) (3)适用于大多数晶体结构(对晶胞参数描述准确)
真实情况下原子核外电子并非均匀分布
LDA不适用的情况: (1)电子分布定域性较强,电荷密度分布不均匀(化学反应中的过度态) (2)体系束缚能绝对值估计不准确 (3)低估禁带宽度的绝对值(固有缺陷)
加入原因:LDA、GGA存在对长程作用描述的缺失,导 致范德华作用行为表现不出来 适用体系:范德华力和氢键占体系总能比重较大的体系。
设置方法:
浮选吸附中一般 物理吸附最先发生, 而且需要计算氢键作 用,可视作一个必设 参数
Logo
23
Fix Occupancy、Smearing
计算的体系确定为半导体或绝缘体时,价电子较少 ,SCF收敛较容易,放开metal,可以勾选Fix Occupancy ,电子轨道固定占据,并非必选
y
x1 xj xj+1 xn
Logo
17
动能截断测试
以不同动能截断值为单一变量做收敛性测试,选取最优值
Logo
18
K点取样
K点是倒易空间的基本构成点,总 能量的计算就是对布里渊区内均匀分布 的部分特殊K点的积分后加权重求和完 成的。
y y=(x)
V
b1 b2 a1 b 3(a2
K点取样设置
以不同K点取样密度为单一变量做收敛性测试,选取最优值
Logo
21
CASTEP非必设参数
必设参数: 交换相关泛函 赝势 截断动能及测试 K点取样及测试
非必设参数:
DFT-D Fix Occupancy、Smearing
Density Mixing 自旋极化 偶极修正
Logo
22
DFT-D(Dispersion correction)色散矫正
Catalysts 催化剂 Ceramics 陶瓷 Glasses 玻璃 Metal-oxides 金属氧化物 Metals 金属 Minerals 矿物 Molecular-crystals 分子晶体 Nanotubes 纳米管
Organics 有机物 Polymers 聚合物 Repeat-units 重复单元 Semiconductors 半导体 Zeolites 沸石族
Logo
28
结构优化和自洽迭代过程中不收敛的处理
结构优化不收敛的处理方法: (1)观察结果文件,决定是否增加opt的循环次数; (2)从粗糙到精细,调整结构优化计算精度; (3)调整晶体结构。
Logo
29
结构优化和自洽迭代过程中不收敛的处理
自洽迭代不收敛的处理方法: (1)观察结果文件,决定是否增加SCF的迭代次数; (2)先用大的smearing值,再减小(max 0.2,文章中注明); (3)减小Density Mixing(减小新的假设的电子密度的权重); (4)增大SCF tolerance值,再降低; (5)微调构型
14
K-S求解近似处理方法-赝势
CASTEP将外层电子波函数通过平面波函数 展开,将原子实近似为新的“核”
(r)
C ei(kG)r
G i,kG
赝势
Rc
有效的减少平面波数目
pseudo wave function pseudopotential
其合理性在于,采用赝势前后: 能量本征值不发生变化 价电子波函数在Rc外的分布
Logo
26
自旋极化
体系中存在未配对电子,要考 虑自旋 假设体系中存在10个Fe3+离子, 每个离子具有5个未配对电子, 体系的Initial spin 值为50
不知自旋状态,选择Use formal spin as initial 设置方式:Modify/Electronic configuration/spin state 选择high,软件将从高自旋状 态向低自旋状态逐一考察,寻 找最低能量的自旋态组
勾选metal,可以提供空轨道,并且开放Smearing, 允许电子热占位(分数占据轨道),有助于加速自洽迭 代收敛,可以提高计算精度,不限体系,建议采用
Logo
24
Smearing
金属体系中,价电子较多,费米能级附近能量简并较多,轨道 间能量差很小,电子占据位置不确定,SCF振荡不收敛。Smearing 参数允许电子在所有轨道中按照指定的能量差ΔE 进行拖尾,类似 于物理上的热占位现象。此方法能够通过允许轨道驰豫而大大加速 SCF迭代的收敛速度。会导致虚轨道与占据轨道进行混合,因此, 会有一些轨道出现分数占位。
不变 在Rc处的电子波函数对数的
倒数不变化
只适用于周期性体系
Logo
15
K-S求解近似处理方法-赝势
赝势设置
➢ Ultrasoft (USP) 超软 推荐用,精度好,效率高
➢ Norm-conserving (NCP) 模守恒 拉曼光谱计算不支持USP,选用NCP 为了与已发表文献比较,采用NCP
Logo
3
矿物晶体结构搭建
• (2)自助搭建结构
对于结构库中没有的晶体结构,要从文献、XRD软件 标准卡片等资料中查找参数自行搭建。
如:AlAs晶体结构
空间群 F-43m(代号216) 晶格参数 a=b=c=5.6622Å α=β=γ=90o 原子占位 Al(0 0 0) As(0.25 0.25 0.25)
Logo
12
交换相关泛函的选择
虽然GGA方法弥补了LDA的缺陷,提高了计算精度,但也 很难说GGA一定优于LDA,不能说对于某一体系哪一种方法一 定适用。
实际使用过程中交换相关泛函的选择方法有两种: (1)阅读文献,根据他人已发表的成功经验直接选择, 这种方法最省时省力 (2)以交换相关泛函为单一变量做收敛性测试,与试验 结果比较,权衡计算精度与计算成本择优选用
Logo
27
偶极修正
通过剪切体相结构构建表面的时候应当尽量使上下表面的终 端原子相同,但是这种情况一般不会实现。所获得的表面为不对 称表面,两层表面之间会产生偶极场作用,影响后续的表面能、 吸附能等能量的计算。
在切得的表面不对称或者表面 有分子吸附时,能量的计算过程中 应当加入偶极修正。
推荐选用Self-consistent(自洽) 偶极修正,只有在自洽迭代不收敛 的时候才建议选用Non selfconsistent(非自洽)偶极修正。
初始的情况
Logo
采用Smearing后
初始的情况
采用Smearing后
25
Density Mixing
Density Mixing参数控制体系中如何根据特征方程来构造新的电 子密度。在计算过程中,通过加入阻尼振荡来确保整个体系的平滑 收敛。对于一个最简单的阻尼方法,遵循如下方程:
new in A out in