materials studio建模实例
用material studio 画晶胞及参数设置

第一种情况: 从程序自带的各种晶体及有机模型中导入体系的晶胞1.打开MS,由file>import>structures>metals\>pure-metals>Fe导入Fe的晶胞。
2.由build>Surfaces>cleave Surfaces打开对话框.在对话框中输入要建立的晶面(hkl),选择position,其中depth控制晶面层数。
3.进入build>Supercell,输入A 、B 、C的值,得到想要的超晶胞。
4.到该步骤,我们已经建立了一个周期性的超晶胞。
如果要做周期性计算,则应选择build>Crystals>buil d vaccum slab,其中真空层通常选择10埃以上。
如果建立团簇模型则选择build>Symmetry>Non-periodic Structure,去掉模型的周期性,并跟据自己的实际需要删除部分原子,得到想要的团簇模型。
5.在表面插入分子时通过菜单栏上的几个小图标添加即可。
第二种情况: 手动建模,优点是可控制晶格常数。
6.首先从文献中查到晶体的晶格常数的实验值。
7.打开build>Crystals>build crystals,可见到对话框。
在对话框中选择空间群与点群,然后在Lattice Parameter中设置晶胞基矢的长度及夹角。
8.然后打开build>Add atom,从对话框中输入坐标。
这里只需输入几个有代表性的原子的坐标,不必全部输入。
在坐标输入前首先在option页面中选择coordinate system,或者分数坐标或者卡迪尔坐标。
9.以下步骤重复2-5步。
10.需要注意的是,采取什么样的团簇并不是任意的。
原因是很多模型构造出来后在优化过程中往往不收敛。
要避免这个问题的办法是查阅文献,参考文献上模型进行选取,因为它们的模型通常是经过试验证实收敛的。
3 LAMMPS软件与materials_studio软件介绍

Lammps软件的应用
应用步骤—程序安装
安装平台环境(考虑不同的操作系统,是否并行计算) 简单易行的安装
• Windows下:命令行执行方式 • Linux下:编译选择项 • 几个关键点:编译器的选择;并行库的位置,相关库的位置
应用步骤--实例学习
输入脚本格式书写:3-1节内容,积木式搭建 分块命令学习方法: 几何模型构建:atom_style, boundary, dimension,units create_atoms, create_box, lattice, read_data, read_restart, region, replicate 物理模型构建:angle_coeff, angle_style, bond_coeff, bond_style, dielectric, dihedral_coeff 过程模型构建:Fix:is any operation that is applied to the system during timestepping or minimization. Examples include updating of atom positions and velocities due to time integration, controlling temperature, applying constraint forces to atoms, enforcing boundary conditions, computing diagnostics, etc. 输出模型构建:compute过程计算量,热力学输出量(全局量),局部表征量(单 个原子、组原子)
• # initial velocities初始化速度 • compute new mobile temp • #定义温度的计算(可动区域内统计平均) • compute new2 mobile stress/atom • #定义原子应力的计算(整个区域) • Velocity mobile create 0.01 887723 temp new • #按指定的温度(0.01)计算方法,初始化原子的速度 • Velocity upper set 0.0 0.3 0.0 • #upper原子组y方向的速度为0.3 • Velocity mobile ramp vy 0.0 0.3 y 1.25 38.75 sum yes • #mobile原子的速初始度从0到0.3线性变化 • # fixes施加约束 • fix 1 all nve • #nve系综的积分算法 • fix 2 boundary setforce NULL 0.0 0.0 • #边界boundary上力条件,钢化原子,便于加载!!
Material Studio建模

铁基块体非晶合金-纳米晶转变的动力学模拟过程Discover模块1 原子力场的分配在使用Discover模块建立基于力场的计算中,涉及几个步骤。
主要有:选择力场、指定原子类型、计算或指定电荷、选择non-bond cutoffs。
在这些步骤中,指定原子类型和计算电荷一般是自动执行的。
然而,在某些情形下需要手动指定原子类型。
原子定型使用预定义的规则对结构中的每个原子指定原子类型。
在为特定的系统确定能量和力时,定型原子使工作者能使用正确的力场参数。
通常,原子定型由Discover使用定型引擎的基本规则来自动执行,所以不需要手动原子定型。
然而,在特殊情形下,人们不得不手动的定型原子,以确保它们被正确地设置。
图 3-11)计算并显示原子类型:点击Edit→Atom Selection,如图所示弹出对话框,如图所示从右边的…的元素周期表中选择Fe,再点Select,此时所建晶胞中所有Fe原子都将被选中,原子被红色线圈住即表示原子被选中。
再编辑集合,点击Edit→Edit Sets,如图所示弹出对话框见图,点击New...,给原子集合设定一个名字。
这里设置为Fe,则3D视图中会显示“Fe”字样,再分配力场:在工具栏上点击Discover按钮,从下拉列表中选择Setup,显示Discover Setup对话框,选择Typing选项卡。
图3-2 Discover Setup对话框Typing选项卡在Forcefield types里选择相应原子力场,再点Assign(分配)按钮进行原子力场分配。
注意原子力场中的价态要与Properties Project里的原子价态(Formalcharge)一致。
2力场的选择1)Energy力场的选择:力场是经典模拟计算的核心,因为它代表着结构中每种类型的原子与围绕着它的原子是如何相互作用的。
对系统中的每个原子,力场类型都被指定了,它描述了原子的局部环境。
力场包括描述属性的不同的信息,如平衡键长度和力场类型对之间的电子相互作用。
Materials+Studio中的建模

Nanostructure Builders: Clusters
• Constructed by cutting the specified shape out of an infinite crystal. Shape options are:
– – – – – – Simple box Sphere Cylinder Cone Frustum Tetrahedron
单击此处编辑母版副标题样式
Bead type editor
单击此处编辑母版标题样式
• • • Create new bead types Set properties Share between projects
单击此处编辑母版副标题样式
Mesoscale molecule builder
单击此处编辑母版标题样式
Meso Structure Builder 层状模型 真实模型
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
Meso Structure Builder 管状模型 真实模型
单击此处编辑母版副标题样式
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
Meso Structure Builder 液滴状模型 真实模型
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
Meso Structure Builder 棒状模型 真实模型
Materials Studio整体介绍及应用案例(高分子、含能材料、化学反应)

分享与交流单击此处编辑母版标题样式单击此处编辑母版副标题样式第一名:10000元奖学金第二三名:5000元第四五六名:2000元创腾科技特为大家提供一个快速获得和阅读文献的交流学习平台,我们将收集MS软件使用者发表的SCI英文文章的中文精炼版,并将其分类整理汇总展示在创腾科技网站上凡参与活动的您都将收到创腾科技邮寄的奖品及定期的文献投送总,展示在创腾科技网站上,提供阅读和下载功能参与方式:登陆创腾科技网站奖学金页面/service/jiang.aspx ,下载word文档,将您2010-2014发表的SCI文章的中文精炼版填入word文档,发送到prize@ 单击此处编辑母版标题样式单击此处编辑母版文本样式第二级第三级第四级第五级M aterials S tudio多功能多尺度分子模拟软件Copyright ©2014, Neotrident Technology Ltd. All rights reserved.创腾科技技术部许立芳2014年5月28日一、选择合适的方法和模块---什么是分子模拟主要内容单击此处编辑母版标题样式单击此处编辑母版副标题样式MS模拟软件简介二、MS的应用与新发展1、量子力学模块的应用2、大体系研究的新方法3、分子力学动力学介观模块的应用4、分析表征手段高分子、含能材料、催化反应、金属合金一、选择合适的方法和模块---什么是分子模拟主要内容单击此处编辑母版标题样式单击此处编辑母版副标题样式MS模拟软件简介二、MS的应用与新发展1、量子力学模块的应用2、大体系研究的新方法3、分子力学动力学介观模块的应用4、分析表征手段M aterials单击此处编辑母版标题样式单击此处编辑母版副标题样式金属材料无机非金属材料有机小分子高分子材料各种实验设备+ 各种分析设备材料性能S tudio传统试验vs. 虚拟试验C l 个候选物中找到新的共混材料但是由分子模拟的优点—降低时间和费用成本塞拉尼斯公司是全球领先的化工技术和特种材料公司单击此处编辑母版标题样式单击此处编辑母版副标题样式Celanese 公司需要从300个候选物中找到新的共混材料。
Materials Studio表面建模---根号

我以最常见的Fe (bcc) 体心立方结构为例:1. (100)-P(1x1):MS给出的基矢U和V分别为(010), (001) 显然这个面上平行于yz面2. 若想切出c(√2×√2)-45 的super cell,相应的单位向量要变为A和B,如下图,根据向量变换的基础知识就可以得到:A =U +V ;B = V – U所以A = (0 1 1) ; B = ( 0 -1 1)图中同色为同一层原子。
3. 修改MS 中U 和V 分别为(0 1 1) ; ( 0 -1 1)可以看到MS给出的最小单位变为c(√2×√2)-451 的pt(111)表面为例。
以3用MS Modeling制作slab模型结构图1,打开MS modeling,建立一个项目,如test;2,右击test项目,在下拉菜单中选择import,在弹出的菜单中选择Structures/metals/pure-metals/Pt;3, 在主菜单中的Build选择surfaces/Cleave surface,4, 在弹出的菜单中处理数据(i)在surface box界面下a)你所要做的表面,如[111]; 也就是将cleave plane [h k l]修改成[1 1 1];b) 修改深度,即将depth修改成你所需要的原子层数,如4;(ii)在options界面下将Orientation standard选项改成U矢量沿x轴方向,V在xy平面(iii)在surface Mesh的界面下修改U,V矢量。
如保持U不变,将V矢量修改成0.5 0.5 -1原因:如下图所示为Pt(1 1 1)面A则A=U+2V=(0.5 -0.5 0)+2(0 0.5 -0.5)=(0.5 0.5 -1),所以将V矢量修改为0.5 0.5 -1. 5,点击cleave,产生一个[1 1 1]的表面模型,6,在主菜单中的Build选择Crystals/Build Vaccum Slab7,在弹出的菜单中修改相应的参量,比如将真空厚度修改成14angstrom, 点击build产生一个Slab模型;8,在主菜单中的Build选择Symmetry/supercell,将单胞修改成你所需要的大小,如将A修改成2,点击Create supercell这样产生了你所要的表面在该表面,你可以非常简单的看出hcp与fcc的差别,以及top,bridge位置。
Materials studio 简单教程

.
21
此课件下载可自行编辑修改,此课件供参考! 部分内容来源于网络,如有侵权请与我联系删除!
,预览后关闭窗口。
.
11
Xi’an Jiaotong University
选择菜单栏的Build / Bonds ,打开对话框,在键和形式中勾选Monitor bonding 。然后关闭对话框。
(3)生成动画 点击 按钮,选择Bounce,然后点击
止播放。
按钮,播放动画,点击
,动画停
.
12
Xi’an Jiaotong University
Display Style中选择Ball and stick 。点击 关闭对话框。 这样在本project 中,默认显示方式被设置为 ball and stick。
.
4
Xi’an Jiaotong University
(3)绘制CH3F分子
点击工具栏中的 ,选择C原子
,并绘制单个C原子,然后点击 ,自
/ Save Project保存文件,然后选择Window / Close All,关闭所有Project Explorer窗 口。 (3)创建生成物
跟前面一样,创建新的文件,取名为SN2Product.xsd,然后复制SN2reactant.xsd 的模型到该文件。
.
8
Xi’an Jiaotong University
在SCF选项中,勾选Use smearing;在Orbital Cutoff选项中,勾选Custom,设 置Global orbital cutoff的值为5.0 Å。然后点击Assign并关闭窗口。
.
14
Xi’an Jiaotong University
materials-studio软件介绍

计算实例 ——FTBC-C4自组装大分子单点能计算(castep计算量
较大,大模型,不建议用其做几何优化)
(1)建模,选择Castep模 块的 calculation设置参 数对话框:
1)Setup——Task: Energy-单点能 交换关联能:GGA-
PW91
计算实例 ——FTBC-C4自组装大分子单点能计算
计算实例
——GaAs光学性质计算
• 结构优化:选 择Castep模块 的 Analysis设 置参数对话框, 进行Structure 分析。
计算实例
——GaAs光学性质计算
(2)优化完成 后进行能量计 算:
选择Castep模块 的 calculation设 置参数对话框:
1)setup:Task
特点:
优点:研究分子模型或材料结构,有丰富的模型资源,建 模和制图能力。与其它标准PC软件整合,使得容易共享这 些数据。 运行平台:Windows NT/2000/XP,Linux和UNIX服务器
分析领域:多范围的软件结合成一个集量子力学、分子力 学、介观模型、分析工具模拟和统计相关为一体的建模环 境。 应用领域:材料、化工、物理等
1.MS Visualizer模块
概述: 提供了搭建分子、晶体、界面、表面及高分子材料结构模 型所需的所有工具,可以操作、观察及分析计算前后的结 构模型,处理图型、表格或文本等形式的数据,并提供软 件的基本环境和分析工具以支持Materials Studio的其 它产品。是Materials Studio产品系列的核心模块。
应用举例——建立FTBC-C4自组装大分子模型
(3)FTBC-C4自组装大分子模型的建立
同样方法 画出HOPG
衬底
Materials studio 简单教程

Xi’an Jiaotong University (2)参数设置 将Number of frames 设置为25,勾选Superimpose structures,点击Preview, 预览后关闭窗口。
Xi’an Jiaotong University 选择菜单栏的Build / Bonds ,打开对话框,在键和形式中勾选Monitor bonding。 然后关闭对话框。 (3)生成动画 点击 按钮,选择Bounce,然后点击 止播放。 按钮,播放动画,点击 ,动画停
Xi’an Jiaotong University 单击工具栏中的 按钮,选择3D Atomistic 然后点击确定。此时文件名称出现在 左侧的Project Explorer 中,名称为3D Atomistic.xsd。
பைடு நூலகம்
(2)设置球棍模型为默认显示方式 从菜单栏中选择 Modify / Default Atom Style ,打开 Default Atom Style 对话框。在 Display Style中选择Ball and stick 。点击 关闭对话框。 这样在本project 中,默认显示方式被设置为 ball and stick。
Xi’an Jiaotong University 3、创建反应物和生成物分子模型 双击打开methylfluoride DMol3 GeomOpt文件夹中的methylfluoride.xsd,利用 CTRL + A 和CTRL + C全选并复制所有原子。然后右击文件名,选择New/3D Atomistic Document,创建新文件后, CTRL + V 复制所有原子到该文件,并重命 名为SN2reactant.xsd 。 (1)添加第二个F原子 点击 按钮,选择Periodic Table中的F原子,选中已有C原子,添加一个与已 有C-F键位置相对的C-F键。然后点击 ,选择Distance,选中C原子,左键拖 拽新添加的F原子,调节C-F键的键长为3 Å。
Material_Studio建模课程论文

HUNAN UNIVERSITY《材料设计与计算机模拟》课程实验报告年 月 日学生姓名:学生学号:专业班级: 学院名称: 指导老师:一设计目的(1)熟悉Materials Studio操作界面(2)掌握Materials Studio的晶体结构建模操作(3)了解Materials Studio中CASTEP模块的基本知识(4)通过Materials Studio计算预测基本物性二设计设备Personal ComputerMS Modeling 5.5三设计内容1构建模型查阅参考文献,获得氯化钠的晶体结构数据,如错误!未找到引用源。
所示。
从表一可知,氯化钠结构所属空间群为Fm-3m,对应空间群编号为No. 225。
(verify)(what is:/?)在Materials Studio中构建氯化钠(Sodium chloride)模型,模型如错误!未找到引用源。
(a)所示,(b)为氯化钠结构的初基单胞(Primitive Cell)。
首先激活Build→Build Crystal,在Space Group项中选择225号空间群,在Lattice Parameters填入5.6402,应用后氯化钠结构即创建完成,通过改变3D模型的显示样式等设置使模型呈现最佳视角。
图1氯化钠晶体结构Figure1 The structure of Sodium chloride在Matrials Studio界面中,通过view→Explorer→Properties Explorer激活Properties窗口,查看构建的氯化钠模型的信息,部分信息如表2所示。
同时,注意到模型创建、计算过程中激活了Project Explorer和Job Explorer两个窗口。
表2氯化钠晶体结构数据Table 1 The structure data of Sodium chloride2结构优化2.1优化初始条件(status)type of calculation : density of statesplane wave basis set cut-off : 370.0000 eVusing functional : Perdew Burke Ernzerhof Spin= 1 of 1, K-point= 51 of 602.2优化结果表3优化后氯化钠晶体性质图1优化结果可视化电荷密度3结构计算弹性常数计算(Elastic Constants)=============================================== Elastic constants from Materials Studio: CASTEP ===============================================Summary of the calculated stresses**********************************Strain pattern: 1======================Current amplitude: 1Transformed stress tensor (GPa) :-0.576539 -0.000000 -0.000000-0.000000 -0.703165 0.041960-0.000000 0.041960 -0.703165Current amplitude: 2Transformed stress tensor (GPa) :-0.685731 -0.000000 -0.000000-0.000000 -0.728068 0.015238-0.000000 0.015238 -0.728068Current amplitude: 3Transformed stress tensor (GPa) :-0.795302 -0.000000 -0.000000-0.000000 -0.756537 -0.010879-0.000000 -0.010879 -0.756537Current amplitude: 4Transformed stress tensor (GPa) :-0.891683 -0.000000 -0.000000-0.000000 -0.776895 -0.034036-0.000000 -0.034036 -0.776895Stress corresponds to elastic coefficients (compact notation): 1 7 7 4 0 0as induced by the strain components:1 1 1 4 0 0Stress Cij value of value ofindex index stress strain1 1 -0.576539 -0.0030001 1 -0.685731 -0.0010001 1 -0.795302 0.0010001 1 -0.891683 0.003000C (gradient) : 52.750150Error on C : 1.120673Correlation coeff: 0.999549Stress intercept : -0.7373142 7 -0.703165 -0.0030002 7 -0.728068 -0.0010002 7 -0.756537 0.0010002 7 -0.776895 0.003000C (gradient) : 12.482950Error on C : 0.547309Correlation coeff: 0.998083Stress intercept : -0.7411663 7 -0.703165 -0.0030003 7 -0.728068 -0.0010003 7 -0.756537 0.0010003 7 -0.776895 0.003000C (gradient) : 12.482950Error on C : 0.547309Correlation coeff: 0.998083Stress intercept : -0.7411664 4 0.041960 -0.0030004 4 0.015238 -0.0010004 4 -0.010879 0.0010004 4 -0.034036 0.003000C (gradient) : 12.705250Error on C : 0.293880Correlation coeff: 0.999465Stress intercept : 0.003071============================Summary of elastic constants============================id i j Cij (GPa)1 1 1 52.75015 +/- 1.1214 4 4 12.70525 +/- 0.2947 1 2 12.48295 +/- 0.387=====================================Elastic Stiffness Constants Cij (GPa)=====================================52.75015 12.48295 12.48295 0.00000 0.00000 0.00000 12.48295 52.75015 12.48295 0.00000 0.00000 0.00000 12.48295 12.48295 52.75015 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 12.70525 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 12.70525 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 12.70525========================================Elastic Compliance Constants Sij (1/GPa)========================================0.0208452 -0.0039889 -0.0039889 0.0000000 0.00000000.0000000-0.0039889 0.0208452 -0.0039889 0.0000000 0.00000000.0000000-0.0039889 -0.0039889 0.0208452 0.0000000 0.00000000.00000000.0000000 0.0000000 0.0000000 0.0787076 0.00000000.00000000.0000000 0.0000000 0.0000000 0.0000000 0.07870760.00000000.0000000 0.0000000 0.0000000 0.0000000 0.00000000.0787076Bulk modulus = 25.90535 +/- 0.454 (GPa)Compressibility = 0.03860 (1/GPa)Axis Young Modulus Poisson Ratios(GPa)X 47.97270 Exy= 0.1914 Exz= 0.1914Y 47.97270 Eyx= 0.1914 Eyz= 0.1914Z 47.97270 Ezx= 0.1914 Ezy= 0.1914====================================================Elastic constants for polycrystalline material (GPa)====================================================Voigt Reuss HillBulk modulus : 25.90535 25.90535 25.90535 Shear modulus (Lame Mu) : 15.67659 14.90494 15.29076 Lame lambda : 15.45429 15.96872 15.71151光学性质计算(Optical Properties)能级、电子态密度计算(Band Structure,Density of State)。
MaterialsStudio建模操作详细步骤(本人原创)

MaterialsStudio建模操作详细步骤(本⼈原创)第2章Materials Studio建模2.1界⾯常⽤操作2.1.1 Materials Studio的启动从Windows“启动”菜单中选择“程序”Accelrys Materials Studio 4.0| Materials Studio。
如果在桌⾯上有Materials Studio图标,也可以通过双击图标来启动Materials Studio。
在启动Materials Studio时,⾸先会出现⼀个所谓的欢迎界⾯(Welcome to Materials Studio),必须创建⼀个新的项⽬或从对话框中载⼊⼀个已经存在的项⽬。
注意:如果是第⼀次打开Materials Studio,会看到⼀个叫做Materials Studio ⽂件关联的对话框,如果出现这种情况,按照提⽰点击OK按钮即可。
2.1.2 创建项⽬在欢迎界⾯对话框上选择创建⼀个新的项⽬,然后点击OK。
然后会出现新建项⽬对话框,选择要存储⽂件的位置并且键⼊“tiejifeijinghejin”作为⽂件名,然后点击OK。
此时的项⽬管理器如图2-1所⽰:图2-1 Project 界⾯Materials Studio对中⽂⽀持不好,命名时最好⽤英⽂字母,可以右击点Rename,进⾏重命名。
2.1.3 输出图像可以将3D Atomistic⽂件显⽰的图像作为位图输出,输出的图像可以包含到其它⽂件中。
位图图像被存储为.bmp格式,可以使⽤简单的位图编辑器⽐如Windows的画图进⾏编辑。
从菜单栏中选择File | Export...显⽰Export对话框。
点击Export as type⽂本框右侧的选项箭头,从下拉列表中选择Structure Bitmap (*.bmp)。
⼀旦选择了位图格式,Options...按钮就被激活了。
点击Options...按钮以显⽰Bitmap Export Options对话框。
Materials Studio整体介绍及应用案例(高分子、含能材料、化学反应)
分享与交流单击此处编辑母版标题样式单击此处编辑母版副标题样式第一名:10000元奖学金第二三名:5000元第四五六名:2000元创腾科技特为大家提供一个快速获得和阅读文献的交流学习平台,我们将收集MS软件使用者发表的SCI英文文章的中文精炼版,并将其分类整理汇总展示在创腾科技网站上凡参与活动的您都将收到创腾科技邮寄的奖品及定期的文献投送总,展示在创腾科技网站上,提供阅读和下载功能参与方式:登陆创腾科技网站奖学金页面/service/jiang.aspx ,下载word文档,将您2010-2014发表的SCI文章的中文精炼版填入word文档,发送到prize@ 单击此处编辑母版标题样式单击此处编辑母版文本样式第二级第三级第四级第五级M aterials S tudio多功能多尺度分子模拟软件Copyright ©2014, Neotrident Technology Ltd. All rights reserved.创腾科技技术部许立芳2014年5月28日一、选择合适的方法和模块---什么是分子模拟主要内容单击此处编辑母版标题样式单击此处编辑母版副标题样式MS模拟软件简介二、MS的应用与新发展1、量子力学模块的应用2、大体系研究的新方法3、分子力学动力学介观模块的应用4、分析表征手段高分子、含能材料、催化反应、金属合金一、选择合适的方法和模块---什么是分子模拟主要内容单击此处编辑母版标题样式单击此处编辑母版副标题样式MS模拟软件简介二、MS的应用与新发展1、量子力学模块的应用2、大体系研究的新方法3、分子力学动力学介观模块的应用4、分析表征手段M aterials单击此处编辑母版标题样式单击此处编辑母版副标题样式金属材料无机非金属材料有机小分子高分子材料各种实验设备+ 各种分析设备材料性能S tudio传统试验vs. 虚拟试验C l 个候选物中找到新的共混材料但是由分子模拟的优点—降低时间和费用成本塞拉尼斯公司是全球领先的化工技术和特种材料公司单击此处编辑母版标题样式单击此处编辑母版副标题样式Celanese 公司需要从300个候选物中找到新的共混材料。
9 MS应用与实例

这里选择计算态密度与光 学性能。
实例2 简单性能计算
• 任务设置
开始任务
实例2 简单性能计算
任务栏
任务状态
实例2 简单性能计算
任务完成
任务结果文件 (后缀为.castep)
实例2 简单性能计算
• 分析-导入电子密度
PS: 分析性能的时候,必 须激活模型窗口。
实例2 简单性能计算
基本界面与操作
要使用 CASTEP ,首先要有晶胞,里面建立原子结构,可以通过 file ==> import 打开来 进入 structure有内建的分类,可以得到多种晶体。
实例1 构建氯化钠晶体
• 新建3D模型文件
点击鼠标右键 新建模型名称为NaCl.xsd
实例1 构建氯化钠晶体
• 点击菜单栏:Build --> Crystals --> Build Crystal...
实例3 L10-TiAl形成热计算
• TiAl晶胞的优化与总能计算
。
开始任务
实例3 L10-TiAl形成热计算
在结果文件*.castep中找到final energy
*.castep 文 件 中 可 能 有 多 个 Fina energy值,选择最后一个
实例3 L10-TiAl形成热计算
• Ti单质晶胞中单个Ti原子能量
输入晶格常数 (默认单位为Å)
实例1 构建氯化钠晶体
实例1 构建氯化钠晶体
• 添加原子
选择分数坐标
实例1 构建氯化钠晶体
• 添加原子
在 Element 选项选择Na, a: b: c: 均为0, 然后按下 Add 增加原子,如此依序 输入下列原子
国内第一MS(materialstudio)学习实例(原创)新手第一步必看materi..
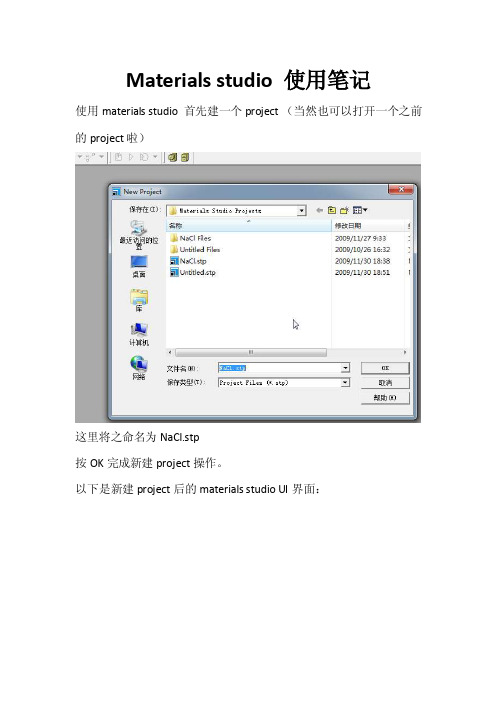
Materials studio 使用笔记使用materials studio 首先建一个project (当然也可以打开一个之前的project啦)这里将之命名为NaCl.stp按OK完成新建project操作。
以下是新建project后的materials studio UI界面:这里有几个重要的窗口,可分为这三类:一、job,己完成的、正在跑的;二、project,各种输入与输出文件,可以查看结果、修改输出入的相关设定;三、property,材料的原子及电子结构3D模型等物性数据,例如晶体晶胞边长、原子元素种类等等。
从V eiw的Explorer 可见:jobExploroer、project exploroer、property explorer。
job explorer显示运行的job,近端远程的状态都可以显示。
project explorer默认值是开着的,project的相关对象,如文字输出、3D 结构等等,job相关的目录、文件等。
property explorer,在MS相对Cerius2而言是新的东西。
只要是3D 对象有呈现的状况之下,可以直接在上面显现出各种可以看得见的特性还有可以改得了的选项。
现在,我们需要一个晶胞结构,用于演示CASTEP计算。
这个结构可以是import 的,也可以是自己手动建立。
Import方式可以通过File-import,导入structure内建的结构。
手动建立方式如下:先建立一个3D atomistic Document,方式如上但不限于以上,还有其他几种方式各位按习惯选择。
建完后:Build-crystal-Buildcrystal此时会打开Build crystal 对话框如下,根据icsd查询的NaCl晶体参数,输入进去:space group:225显示出NaCl的FM-3M结构Attice parameter 填写晶格常数,比如a,b,c及三个角度值这里,钠的a=b=c=5.64Options里基本只要用预设值即可按apply或Build即可生成晶格模型添加原子方式很多,这里也仅取一种方便的方式如下选择Na和Cl原子,钠的abc选0,Cl选0.5如下,分别按add,完成原子的添加,完成后三维图如下,是因为没有进行旋转,此时选工具旋转,即可得到从别的角度看见的三维图更改三维显示方式可以在三维图上单击右键,在弹出的菜单中选display style设置3D格式,如atom中我选择stick等就产生了形状的模型。
MaterialsStudio教程

Materials Visualizer使用一组方便的工具使我能够在3D视图中打开文档,并且调整结构的图形显示风格。
在菜单栏中选择File | Import...,显示输入文档对话框。
注意:输入文档对话框也可以通过点击标准工具栏中的输入按钮 来打开。
在输入文档对话框中定位并选择Examples/Documents/3D Model/TON.msi文件,然后点击Import。
2.2
目的:介绍Materials Studio中documents (文档)概念
模块:Materials Visualizer
时间:
先决条件:创建项目
介绍:
Materials Studio使用了各种不同的文档类型,包括:3D Atomistic、3D Mesoscale、text、chart、HTML、study table、grid、script和forcefield文档等。在本教程中,主要使用3D Atomistic文档类型。文档在在项目中进行管理,将一般的工作流记录为文档,文档可以被创建与存储。
二、计算机材料设计的专家系统
计算机材料设计专家系统是指具有相当数量的材料方面的背景知识,并能运用这些知识解决材料设计中的相关问题的计算机程序。
第2章
2.1
目的:介绍Materials Studio(MS)中project(项目)概念
模块:Materials Visualizer
时间:
先决条件:无
介绍:
到了20世纪80年代,计算机材料设计取得了重大进展。1985年,日本出版了《新材料开发与材料设计》一书,书中首次提出了“材料设计学”这一研究方向。1988年,日本科学技术厅组织功能性梯度材料的研究任务,提出将材料的设计、合成、评估这三者结合起来,按预定要求做出材料。