手把手教你设计引物(图文并茂)
引物设计——手把手教新手

②Analyze中,第一项为Key info,点击Selected primers,会给出两条引物的概括性信息,其中包括引物的Tm值,此值Oligo是采用nearest neighbor method计算,会比Primer5中引物的Tm值略高,此窗口中还给出引物的Delta G和3’端的Delta G.3’端的Delta G过高,会在错配位点形成双链结构并引起DNA聚合反应,因此此项绝对值应该小一些,最好不要超过9。
⑤在Primer5窗口中,若觉得某一对引物合适,可以在搜索结果窗口中,点击该引物,然后在菜单栏,选择File-Print-Current pair,使用PDF虚拟打印机,即可转换为Pdf文档,里面有该引物的详细信息。
3、用Oligo验证评估引物
①在Oligo软件界面,File菜单下,选择Open,定位到目的cDNA序列(在primer中,该序列已经被保存为Seq文件),会跳出来两个窗口,分别为Internal Stability(Delta G)窗口和Tm窗口。在Tm窗口中,点击最左下角的按钮,会出来引物定位对话框,输入候选的上游引物序列位置(Primer5已经给出)即可,而引物长度可以通过点击Change-Current oligo length来改变。定位后,点击Tm窗口的Upper按钮,确定上游引物,同样方法定位下游引物位置,点击Lower按钮,确定下游引物。引物确定后,即可以充分利用Analyze菜单中各种强大的引物分析功能了。
如何根据要求自己设计PCR引物
如何根据要求自己设计PCR引物1PCR引物设计课堂笔记○PCR这个名词大家都不陌生,但实际操作时我们常说的引物设计到底是怎么回事呢?今天我就来给大家用实例演示一下哈。
首先,我们要知道引物设计的目的是为了找到一对合适的核苷酸片段,使其能有效地扩增模板DNA序列。
引物设计是PCR的关键,附上PCR的基本流程图:○引物设计的原则:1.引物长度:一般为15-30bp,常用的是18-27bp,但不能大于38,因为过长会导致其延伸温度大于74℃,即Taq酶的最适温度。
2.引物的特异性:引物与非特异扩增序列的同源性不要超过70%或有连续8个互补碱基同源。
3.序列Tm值:引物的Tm值一般控制在55-60度, 尽可能保证上下游引物的Tm值一致,一般不超过2度。
退火温度=4×(G+C)+2×(A+T)-(5~8)4.G+C含量:有效引物中(G+C)的比例为40-60%,过高或过低都不利于引发反应。
上下游引物的GC含量不能相差太大。
5. 引物的3′端:引物的延伸是从3′端开始的,不能进行任何修饰;引物3’端的碱基一般不用A,因为A在错误引发位点的引发效率相对比较高;引物间3’端的互补、二聚体或发夹结构也可能导致PCR反应失败6.引物的5′端:引物的5′端限定着PCR产物的长度,它对扩增特异性影响不大。
因此,可以被修饰而不影响扩增的特异性。
引物5′端修饰包括:加酶切位点;标记生物素、荧光、地高辛、Eu3+等;引入蛋白质结合DNA序列;引入突变位点、插入与缺失突变序列和引入一启动子序列等。
下面以实例操作演示一下加酶切位点时如何自己设计引物:2用绿色荧光蛋白(GFP)标记蛋白NR1○简单点说,就是现在我们要把Plasmid 2中GFP基因片段添加到Plasmid 1中的NR1基因片段上,但是Plasmid 2中GFP基因片段本身并没有BamHⅠ这个酶切位点,也就说我们要在引物设计中人为地把BamHⅠ这个酶切位点的序列添加给GFP基因片段,这样PCR后得到的GFP基因片段就可以通过BamHⅠ这个酶切位点进入到Plasmid 1中,然后绿色荧光蛋白(GFP)就可以来标记蛋白NR1,达到我们之后实验中来观察蛋白NR1的目的,示意图见下。
Primer5设计引物------图文并茂一步步教你
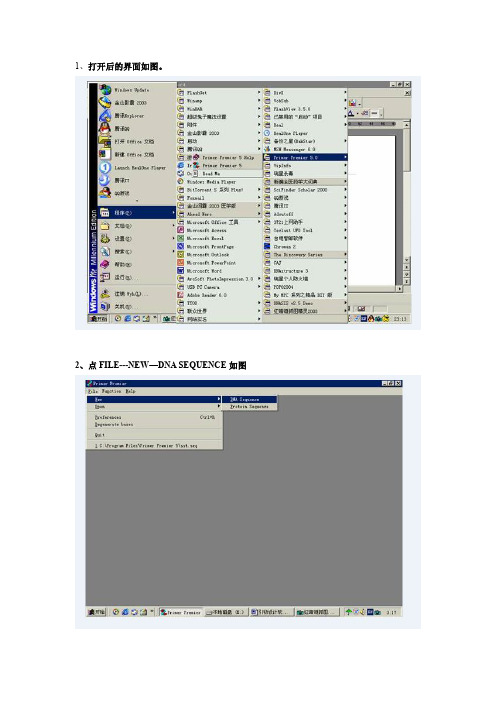
1、打开后的界面如图。
2、点FILE---NEW—DNA SEQUENCE如图3、输入目的基因片段,可以复制后用ctrl+V键拷贝到栏内,后应加数个N以备后续设计时加酶切位点及保护碱基,如图所示。
输入目的基因片段,可以复制后用ctrl+V键拷贝到栏内,后应加数个N以备后续设计时加酶切位点及保护碱基,如图所示。
4、此主题相关图片如下:选中enzyme图标,将所选质粒上的多克隆酶切位点加入左栏此主题相关图片如下:选中OK键,5、分析目的基因中所含的酶切位点,选插入位点时就应排除这些酶此主题相关图片如下:6、选中primer图标,点S图标,edit primer,开始设计正义链。
此主题相关图片如下:7、软件默认引物为二五个碱基此主题相关图片如下8、可将鼠标点在设计框的3端从右向左删除7-9个碱基,保留16-18个配对即可此主题相关图片如下:9、在引物的5端加入选好的酶切位点并在其左侧加3个保护碱基,入该图加入HIND III 酶切位点及TTA保护碱基,完成后点analyze,认为可以后点OK。
此主题相关图片如下:10、选中左上角A图标,用鼠标拉动滑块将待选引物放至目的基因末端。
11、从3端删除7-9个碱基同正义链。
此主题相关图片如下:12、将酶切位点加在5端,应将产品目录所示的酶切位点序列从右至左加入(注意不要加反)如图加入BamH I酶切位点及CGC3个保护碱基。
完成后点analyze,认为可以后点OK。
此主题相关图片如下:13、最后分析结果如图,反义链的FALSE PRIMING可以不考虑,RATING表示引物评分也可以不考虑,主要看Tm值正义链和反义链相差不应超过3度。
GC含量不应超过60%此主题相关图片如下:14、该软件有个缺点,不能保存分析结果,只能选择打印此主题相关图片如下:15、如果设计RT-PCR检测的引物就如下所示,同上输入目的基因片段,选SEARCH图标,选择参数,一般选PCR primers---both—100至250个碱基,引物长短20+/-2,search parametere 中的参数可以不选,为默认设置。
《引物设计教程》课件

适当提高退火温度有助于减少引物二 聚体的形成,因为较高的温度下二聚 体形成的概率降低。
引物特异性不高的解决策略
引物特异性验证
在引物设计完成后,应通过实验验证其特异性,确保引物只对目标序列有反应。
避免引物间的交叉反应
在设计引物时,应确保引物之间不存在交叉反应,避免与非目标序列的结合。
引物3’端的选择
Primer Premier
一款功能强大的引物设计软件,支持 多种PCR方法,可进行多参数搜索和 灵活的筛选功能。
Oligo
提供多种类型的寡核苷酸合成和设计 功能,包括引物、探针、适配体等。
GeneFisher
适用于已知序列的基因片段设计通用 引物。
BatchPrimer3
在线引物设计软件,支持多参数搜索 和灵活筛选功能。
02
引物设计的步骤
确定目标基因序列
目标基因序列的来源
可以是基因组、转录组、cDNA等。
目标基因序列的选择标准
选择基因序列时应考虑其功能、表达水平、变异程度等因素。
目标基因序列的获取方法
可以通过基因数据库、文献报道、实验测序等方法获得。
选择合适的引物序列
引物序列的设计原则
引物序列应具有特异性,避免与基因组其他序列发生非特 异性结合;长度一般在18-30bp之间;GC含量应适中, 一般在40%-60%之间。
引物长度一般在15~30碱基之间,过短可 能降低引物特异性,过长则可能导致引物 结合温度升高,不利于引物的特异性。
碱基分布均匀原则
避免二级结构原则
引物序列中的G+C含量在40%~60%之间 ,避免出现连续的4个以上的G或C。
引物自身及引物之间不能形成互补性二聚 体或发夹结构等二级结构。
引物设计知识点总结图解

引物设计知识点总结图解引物设计是分子生物学研究中至关重要的一步,它涉及到DNA/RNA的扩增、测序和定量等诸多实验。
本文将通过图解的方式,对引物设计的相关知识点进行总结。
具体涉及引物设计的基本原则、引物长度、引物序列的选择、引物的特异性、引物的GC含量以及二聚体的形成。
希望通过本文的阐述,读者能够更好地理解引物设计的要点和注意事项。
1. 引物设计的基本原则引物设计的基本原则包括:引物长度适当、引物序列具有特异性、引物的理论特性符合要求、引物的二聚体形成能力低等。
2. 引物长度的选择引物长度一般在18-30个碱基对之间,过短的引物容易出现非特异性扩增产物,而过长的引物则可能导致扩增效率降低。
因此,在设计引物时需要注意选择适当的长度。
3. 引物序列的选择引物序列的选择是引物设计过程中的核心步骤。
引物的序列应在目标区域能够满足特异性,避免与非目标区域有较高的同源性。
此外,引物序列的碱基组合应尽量避免存在重复序列、多聚碱基或者易形成二聚体的碱基组合。
4. 引物的特异性引物的特异性是评价引物设计质量的重要指标之一。
合理设计的引物应能够特异性地与目标DNA/RNA序列配对,并能够排除与非目标序列的配对。
特异性的引物可以有效地避免非特异性扩增产物的生成。
5. 引物的GC含量引物的GC含量对扩增反应的效率和特异性具有重要影响。
过高或过低的GC含量均可能导致扩增效率降低或产生非特异性扩增产物。
因此,在设计引物时需要合理调整引物的GC含量,并确保GC含量在适宜的范围内。
6. 引物的二聚体形成引物的二聚体形成是指引物之间可能发生的相互结合。
合理设计的引物应该不能形成稳定的二聚体,以避免引物在扩增反应中发生错误的配对,导致产物的异常。
通过上述的图解,我们可以清晰地了解引物设计的主要知识点和注意事项。
在实际的引物设计过程中,需要根据具体的实验目的和条件选择合适的引物设计策略,并结合生物信息学工具进行引物序列的设计和评估。
引物设计知识点归纳图解

引物设计知识点归纳图解引物设计是分子生物学和遗传学等研究领域中的重要工具,它在PCR扩增、基因克隆、基因表达分析等方面发挥着至关重要的作用。
在引物设计过程中,需要考虑引物的长度、碱基组成、熔解温度等因素,以确保引物的特异性和稳定性。
本文将围绕引物设计的基本原理、常用工具和实践技巧展开讨论,并通过图解的方式进行归纳总结。
一、引物设计的基本原理引物设计的目标是选择具有高特异性和稳定性的引物,以确保在PCR扩增等实验中的准确和可靠性。
其基本原理包括:1. 特异性:引物应与目标DNA序列的特定区域完全匹配,以避免非特异扩增。
2. 稳定性:引物应具有适当的长度和碱基组成,以确保引物在反应条件下的稳定性。
3. 熔解温度:引物的熔解温度应接近PCR扩增的反应温度,以保证引物的特异性。
二、引物设计的常用工具引物设计可以借助多种在线工具和软件来完成,常用工具包括:1. Primer3:一种广泛应用的引物设计工具,可以根据给定的参数自动设计引物,并进行热力学分析。
2. NCBI Primer-BLAST:结合NCBI数据库和BLAST算法的引物设计工具,用于检验引物的特异性。
3. OligoAnalyzer:根据序列特性进行引物设计和分析的在线工具,可以计算引物的熔解温度和配对特性。
三、引物设计的实践技巧在实践过程中,为了提高引物设计的准确性和效率,可以考虑以下技巧:1. 引物长度:引物的长度通常为18-25个碱基对,较短的引物具有更高的特异性。
2. GC含量:引物的GC含量应在40-60%之间,高GC含量可以增强引物的熔解温度和特异性。
3. 引物间配对:引物之间或引物与模板之间的配对应避免,以防止引物间的二聚体形成或引物与模板的非特异结合。
4. 引物位置:引物应位于目标序列的特异区域,避免引物与非目标序列的交叉反应。
5. 引物设计的复杂性:复杂的引物设计场景,如多组引物设计、引物与探针联合设计等,可以借助专业软件进行。
引物设计实例分析(多图)
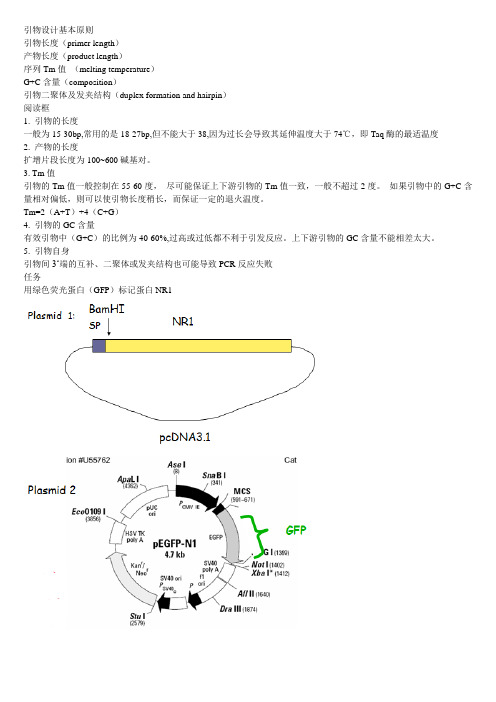
引物设计基本原则引物长度(primer length)产物长度(product length)序列Tm值(melting temperature)G+C含量(composition)引物二聚体及发夹结构(duplex formation and hairpin)阅读框1. 引物的长度一般为15-30bp,常用的是18-27bp,但不能大于38,因为过长会导致其延伸温度大于74℃,即Taq酶的最适温度2. 产物的长度扩增片段长度为100~600碱基对。
3. Tm值引物的Tm值一般控制在55-60度,尽可能保证上下游引物的Tm值一致,一般不超过2度。
如果引物中的G+C含量相对偏低,则可以使引物长度稍长,而保证一定的退火温度。
Tm=2(A+T)+4(C+G)4. 引物的GC含量有效引物中(G+C)的比例为40-60%,过高或过低都不利于引发反应。
上下游引物的GC含量不能相差太大。
5. 引物自身引物间3‘端的互补、二聚体或发夹结构也可能导致PCR反应失败任务用绿色荧光蛋白(GFP)标记蛋白NR1引物要求PCR扩增GFPGFP两边添加BamHI酶切位点保证NR1的阅读框不改变第一步:扩增GFP基本序列第二步:GC比值;Tm值第三步:酶切位点第四步:阅读框第五步保护序列Primer1: 5' GCGGggatccTATGGTGAGCAAGGGCGAGGA Primer2: 5' GCGCggatccctCTTGTACAGCTCGTCCA TGCC记得当初写本科论文,感到不知道讨论什么问题好。
愣是写了一大段的PCR条件摸索的讨论。
后来PCR成为实验最基本的一步了,但是发现在PCR中还是有许多需要注意的地方。
PCR的第一步就是引物设计了。
引物设计需要注意的地方很多,在大多数情况下,我们都是在知道已知模板序列时进行PCR扩增的。
在某些情况比如构建文库的时候也会在不知道模板序列的情况下进行设计。
这个时候随机核苷酸序列就与模板不是完全匹配。
手把手教你做PCR引物设计来自小木虫

PCR引物设计的黄金法例1.引物最幸亏模板 cDNA的守旧区内设计。
DNA序列的守旧区是经过物种间相像序列的比较确立的。
在NCBI上搜寻不一样物种的同一基因,经过序列剖析软件(比方DNAman)比对(Alignment ),各基因同样的序列就是该基因的守旧区。
2.引物长度一般在 15~30 碱基之间。
引物长度( primer length )常用的是 18-27 bp,但不该大于 38,由于过长会致使其延长温度大于 74℃,不适于 Taq DNA 聚合酶进行反响。
3.引物 GC含量在 40%~60%之间, Tm值最好靠近 72℃。
GC含量( composition )过高或过低都不利于引起反响。
上下游引物的 GC含量不可以相差太大。
此外,上下游引物的 Tm值( melting temperature )是寡核苷酸的解链温度,即在必定盐浓度条件下, 50%寡核苷酸双链解链的温度。
有效启动温度,一般高于Tm值 5~10℃。
若按公式 Tm= 4(G+C)+2(A+T)预计引物的Tm值,则有效引物的Tm为 55~80℃,其 Tm值最好靠近 72℃以使复性条件最正确。
4.引物 3′端要避开密码子的第 3 位。
如扩增编码地区,引物3′端不要停止于密码子的第 3 位,因密码子的第 3 位易发生简并,会影响扩增的特异性与效率。
5.引物 3′端不可以选择 A,最好选择 T。
引物 3′端错配时,不一样碱基引起效率存在着很大的差别,当末位的碱基为 A 时,即便在错配的状况下,也能有引起链的合成,而当末位链为 T 时,错配的引起效率大大降低,G、C错配的引起效率介于A、T 之间,所以 3′端最好选择 T。
6.碱基要随机散布。
引物序列在模板内应当没有相像性较高,特别是3’端相像性较高的序列,不然简单致使错误引起(False priming )。
降低引物与模板相像性的一种方法是,引物中四种碱基的散布最好是随机的,不要有聚嘌呤或聚嘧啶的存在。
《引物设计教程》课件

引物长度:通 常为15-30bp, 过长可能导致 非特异性扩增
引物GC含量: 尽量控制在 40-60%,避
免形成二级结 构
引 物 Tm 值 : 应接近目标序 列 的 Tm 值 , 以提高扩增效
率
引物3'端:避 免形成发卡结 构,以免影响
扩增效果
引物特异性: 确保引物与目 标序列具有高 度特异性,避 免非特异性扩
增效率和稳定性
引物3'端:避免3'端 出现连续3个或以上 的碱基,以降低错配
率
引物二级结构:使用 软件预测引物二级结 构,避免形成发卡结 构,提高扩增效率
引物特异性:通过 BLAST比对,确保引 物特异性,避免非特
异性扩增
引物浓度:优化引物 浓度,以提高扩增效
率和稳定性
引物纯度:确保引物 纯度,避免污染和降 解,提高扩增效率和
引物GC含量:尽量保持 50%左右,避免过高或 过低
引 物 Tm 值 : 确 保 引 物 Tm 值在55-65℃之间,以提 高扩增效率
引物特异性:确保引物与 目标序列具有高度特异性, 避免非特异性扩增
引物错配:避免引物内部 错配,以提高扩增效率和 准确性
引物设计软件:可使用 Primer3、OligoT等软 件辅助设计引物
引物设计不当导 致扩增失败
引物设计不当导 致非特异性扩增
引物设计不当导 致扩增效率低
引物设计不当导 致扩增产物长度 不均一
引物设计软件:选择 合适的引物设计软件, 如Primer3、Primer-
BLAST等
引物长度:确保引物 长度在18-25bp之间, 以提高特异性和扩增
效率
引物GC含量:保持 引物GC含量在4060%之间,以提高扩
手把手教你设计引物(图文并茂)

手把手教你设计引物(图文并茂)不知不觉几年下来自己也快毕业了,感谢丁香园这些年来的帮助。
没有什么可回报的东西,就发个帖教教新人如何设计引物吧,尽量做到手把手的教,图文并茂。
引物设计的帖子不少,以前很多战友会推荐Oligo、PrimerPremier、DNA man等等软件。
这些软件设计完最后还是要去BLAST比对下,所以我教大家一种易懂实用的在线设计方法,觉得好的话请投个票。
就以人的PTEN基因为例,首先你要找到他的基因序列,如果你要用的是cDNA,就找它的mRNA序列。
如果你要做的是DNA,就找DNA的序列。
以cDNA为例,普遍的一种方法是上PUBMED中GENE栏搜索找到cDNA那栏,但PUBMED导出序列不太方便,我介绍个网站/index.html1. 输入目的基因,进入2.在左侧栏选择TRANSCRIPT,选择后进入3. 选择PTEN-001中的TRANSCRIPT进入,点击左侧cDNA4. 然后点击CONFIGURE THIS PAGE进入设置你要显示的内容5. 除了第一栏SHOWEXONS选择YES外,其他的都选择NO,然后取个名字保存SAVE CONFIGURATION AS6. 然后在左侧栏点击DOWNLOADVIEW AS RTF可下载你要的cDNA序列,这个文件可以用WORD打开,不同的颜色代表一个外显子间断下载后打开的WORD7. 然后根据可以根据你感兴趣的序列设计引物了,比如我在分别在第6和第7外显子分别设计上下游引物。
选取并复制第6和第7外显子序列8. 登陆/tools/primer-blast/,粘贴这段序列,设置好RANGE和PCR产物的大小,然后在下面点击GET PRIMERS,可以在线设计并比对引物9.最后选择一个比较特异性的引物,条带大小要尽量单一,其他的基因序列尽量不要比对到。
引物设计不求人!手把手教你设计RT-qPCR引物!

引物设计不求⼈!⼿把⼿教你设计RT-qPCR引物!RT-qPCR 全称 Real-time polymerase chain reaction, 即实时聚合酶链锁反应,是⼀种在DNA扩增反应中,以萤光染⾊剂检测每次聚合酶链锁反应(PCR)循环后产物总量的⽅法。
OK~这⾥对RT-qPCR的原理,应⽤及数据分析就不再赘述了,因为今天我们主要讲的是如何设计出好的PCR引物。
相信科研⼩伙伴们都已经⽤过Pubmed来设计引物了,基本就是 “傻⽠式操作”就解决了!但这⾥还需提醒⼩伙伴们两点:1. 需跨越⾄少⼀个外显⼦连接(⼀般我们⽤来做RT的RNA都是通过Trizol抽提法分离出来的,为了避免基因组的DNA也有可能被扩增出来,就要⾄少跨越⾄少⼀个exon-exon junction 了)。
2. 不要忘记选对“物种”!(这⾥以⼈种⽰例)但是Pubmed设计出来的引物⼀般不尽如⼈意,⽐如说引物之间存在极⼤的互补性,3’端的互补重叠,形成引物⼆聚体等等。
因为⽤Pubmed设计引物本⾝能设置的参数条件就很少,所以它才是最简单的。
所以,今天我要给⼤家介绍两款实⽤的软件,Primer Premier 6.0 和 Oligo 7.0。
今天主要讲讲第⼀款,⾄于第⼆款,改天再聊啦!当然,这两款软件,要想⽤全功能版本,都是需要付费的哦!不过...,我们伟⼤的祖国有强⼤的某宝,这⾥就不多说了哈。
好了,我们进⼊今天的正题……(说了这么多废话,终于要进⼊正题了!!!)⾸先,我们选⼀个基因举例来说,PPARA,⾄于为什么会选这个基因呢?因为啊,PPAR⽬前是肿瘤学研究中的⼀个“明星基因”,它的全称是Peroxisome proliferator-activated receptor, 即过氧化物酶体增殖物激活型受体,⽬前发现有三种亚型,PPAR a,PPARbeta/delta,PPAR r。
好吧…….听着好复杂的样⼦啊!总之,管它到底是什么呢,下⾯来说说它的引物如何设计。
PCR引物流程设计详解
PCR引物设计流程详解本文目的:复制出IL-4基因片段一、查找基因序列1、进入NCBI主页,下拉选框选择Nucleotide,在搜索栏输入要查找的目的基因,即IL-4,点击搜索2 、在搜索结果选择灵长类(Homo sapiens)2、在灵长类IL-4基因中选择需要的mRNA序列3、查看基因的相关信息外显子区域CDs区域4、点击FASTA格式,并将序列保存到文档二、使用primer premier 5.0设计引物1、建立新文件,将所得的序列复制进输入框内2、点击搜索按钮,搜索引物3、设置引物设计参数(因为在之前查找基因序列的时候获知,外显子区域分别为:1-200、201-248、249-425、426-618,又知在引物设计时引物位置最好跨越一个内含子,PCR产物长度通常为100-150bp,故设定上游引物位置为201-248,下游引物位置为249-425,产物长度为100-150bp)4、确认条件后,显示搜索结果4、双击选中得分最高的引物查看引物情况(上图为上游引物情况,下图为下游引物情况)5、将设计的上下游引物复制出来,保存到文档中三、使用oligo 6.0对设计的引物进行评价1、建立新文件,将从cnki上获得的cDNA复制进输入框,并点击accept接收2、接收后显示出该序列的相关信息3、点击edit按钮录入用primer设计的上游引物,每一次输入新数据后都需要点击accept按钮接收4、同理,录入下游引物5、分析上下游引物二聚体形成情况6、分析上下游引物发卡形成情况7、分析上下游引物GC%8、检测上下游引物与PCR模板其它位置错配情况9、分析PCR整体情况四、引物特异性检验(primer blast)1、进入NCBI主页,并选择blast2、选择primer blast3、在输入框内输入模板序列和上下游引物,并设定对比数据库,点击getprimer进行对比4、查看blast结果Blast 结果显示,尽管IL-4与其它基因有相似区,但是引物的3’端没有完全互补。
引物设计图文教程。

引物设计图⽂教程。
⼀顿操作猛如虎,⼀看战绩0-5。
尽管引物设计是PCR实验成功的前提,但是蛮多⼩伙伴的实战经验稍显不⾜。
今⼉就向⼤家介绍2种引物设计⽅法。
This is the dividing line.引物设计尽管是⽼⽣常谈了,但是⼩编还是想先把引物设计的原则放出来(其实是复制粘贴啦,哪哪都有)。
1、引物应⽤核酸系列保守区设计并具有特异性。
最好位于编码区5\'端的300-400bp区域内,可以⽤DNAMAN,Alignment软件看看结果。
2、不能形成2级结构。
3、引物长度⼀般在17-25bp之间,上下游引物不能相差太⼤。
4、G+C含量在40-60%之间,45-55%最佳。
5、碱基要随机分布,尽量均匀。
6、引物⾃⾝不能有连续4个碱基的互补。
7、引物之间不能有连续4个碱基的互补。
8、引物5‘端可以修饰。
9、3’端不可修饰,⽽且要避开AT,GC富集的区域,避开T/C,A/G连续结构(2-3个)。
10、引物3\'端要避开密码⼦的第三位。
11、引物整体设计⾃由能分布5\'端⼤于3‘端。
12、定量产物长度80-150bp最好,最长是300bp。
原则忒多啦,⼩伙伴们,先别晕,看下⽅。
⽅法⼀:PrimerBank法⽹址:打开以上⽹址,进⼊PrimerBank⽹站界⾯,如下图个⼈觉得这是最便捷的引物获取⽅法,该⽹站可以⾮常迅速地检索到已经他⼈验证的引物序列及相关反应条件。
图中第⼀个⽅框,⼀般选择NCBI Gene ID;第⼆个⽅框选择你所关注的物种,常⽤的就是Mouse和Human了,如果为其它物种,则选择All Species即可;第三个⽅框中需要输⼊你关注的基因ID号码。
如何获取基因ID呢,这⾥就需要打开NCBI⽹站,选择Gene,并输⼊你的⽬的基因如p53 homo,选择第⼀个,可见其ID号为7157。
如下图所⽰。
然后将ID号7157输⼊上⽅PrimerBank⽹站的For text框中,并选择种属为⼈,点击Submit后,跳转页⾯如下。
简并引物的设计ppt课件

56Βιβλιοθήκη 78910
11
• 利用软件设计引物
Primer 5.0 支持简并引物的设计 在线设计:CODEHOP
GeneFisher2
• 对引物的修饰
降低简并度的方法: 通过密码子偏好选择合适的碱基 通过用次黄嘌呤代替简并度高的碱基
12
13
14
简并引物设计原则
• 尽量选择简并度低的氨基酸区域为引物设计区。如甲硫氨 酸和色氨酸均只有一个密码子。尽可能不用含有亮氨酸、 精氨酸、丝氨酸的肽段。
• 充分注意物种对于密码子的偏好性,选择该物种使用频率 高的密码子,以降低引物的简并性。
• 引物不要终止于简并碱基,对于大多数氨基酸残基来说, 意味着引物3’末端不要位于密码子的第三位。
2
简并引物设计方法
• 利用NCBI搜索不同物种中同一目的基 因的蛋白或cDNA编码的氨基酸序列。
因为不同物种的密码子偏好问题,不同的 核酸序列可能表达的氨基酸序列是相同的, 所以氨基酸序列才是真正保守的。
3
• 对所找到的序列进行多序列比对
将搜索到的同一基因的不同氨基酸序列进 行多序列比对
• 确定合适的保守区域
• 在简并度高的位置,可用次黄嘌呤(dI)代替简并碱基。
15
谢谢观赏
16
设计简并引物至少需要上下游各有一个保 守区域,且两个保守区域相距50~400个氨 基酸为宜,使得PCR产物在150~1200bp之间, 最重要的是每一个保守区域至少有6个氨基 酸。
4
• 利用软件设计引物
Primer 5.0 支持简并引物的设计 在线设计:CODEHOP
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
3. 选择PTEN-001中的TRANSCRIPT进入,点击左侧cDNA
4. 然后点击CONFIGURE THIS PAGE进入设置你要显示的内容
5. 除了第一栏SHOWEXONS选择YES外,其他的都选择NO,然后取个名字保存SAVE CONFIGURATION AS
6. 然后在左侧栏点击DOWNLOADVIEW AS RTF可下载你要的cDNA序列,这个文件可以用WORD打开,不同的颜色代表一个
7. 然后根据可以根据你感兴趣的序列设计引物了,比如我在分别在第6和第7外显子分别设计上下游引物。
选取并复制第6和第7
8. 登陆 /tools/primer-blast/,粘贴这段序列,设置好RANGE和PCR产物的大小,然后在下面点击GET PRIMERS,可以在线设计并比对引物
9.最后选择一个比较特异性的引物,条带大小要尽量单一,其他的基因序列尽量不要比对到。