蛋白质结构预测在线软件
蛋白质结构预测
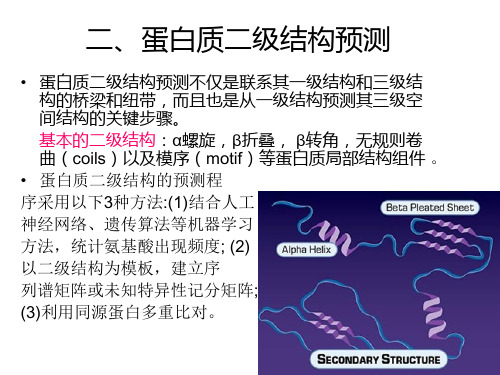
序列基序识别 二硫键识别 折叠子识别 残基接触预测 结构域预测
结构表面识别
预测蛋白质表面结构功能关键区域
5
PredictProtein Secondary Structure
PredictProtein Secondary Structure
H:螺旋 E:折叠 L:环 e:暴露表面﹥16%残基 b:其它残基
3
PredictProtein提交界面
序列提交窗口
分析方法程序详解
PROFsec(默认) PROFacc(默认) 序列预测
基于轮廓(profile)的神经网络算法预测蛋 白质二级结构 基于轮廓(profile)的神经网络算法预测残 基溶剂可及性
PHDhtm(默认)
ASP(默认) COILS(默认) PROFtmb ProSite(默认) SEG(默认) PredictNLS(默认) DISULFIND(默认) AGAPE PROFcon ProDom(默认) CHOP ConSeq
22
SWISS-MODEL
• SWISS-MODEL是一个蛋白质3D结构数据库,库中收录的蛋白质结
构都是使用SWISS-MODEL同源建模方法得来的。
– /
• 基于同源建模法与PDB数据库已知结构的蛋白质序列比对 进行预测
23
SWISS-MODEL
蛋白质三维结构预测
方法 特点 工具
同源建模法 基于序列同源比对,对于序列相似度>30% SWISS-MODEL, CPHmodels ( Homology/Comparativ 的序列模拟比较有效,最常用的方法 e modelling ) 线串法/折叠识别法 (Threading/Fold recognition) 从头预测法 ( Ab initio/De novo methods ) “串”入已知的各种蛋白质折叠骨架内,适 于对蛋白质核心结构进行预测,计算量大 基于分子动力学,寻找能量最低的构象, 计算量大,只能做小分子预测
生物学软件_大全(二)

引言概述:生物学软件在现代科学研究中扮演着重要的角色,它们为生物学家们提供了数据分析、模拟实验等功能,帮助他们更好地理解生命的复杂性。
本文将为大家介绍一系列生物学软件,帮助生物学家们在研究中更高效地工作。
正文内容:1.生物信息学软件1.1基本基因序列分析软件1.1.1BLAST:用于序列比对和相似性搜索,帮助确定生物序列的功能和结构。
1.1.2ClustalOmega:用于多序列比对的工具,帮助研究人员查找序列间的共同特征。
1.1.3EMBOSS:一套开源的生物信息学软件,包含各种工具用于序列分析、蛋白质结构分析等。
1.2基因组数据分析软件1.2.1GATK:广泛用于基因组重测序数据的分析和变异检测。
1.2.2BEDTools:用于处理基因组坐标的工具,帮助研究人员在基因组中定位感兴趣的特定区域。
1.2.3HMMER:用于比对蛋白质序列和荧光探针序列的隐马尔可夫模型工具。
2.结构生物学软件2.1Rosetta:一套用于结构预测和蛋白质构象优化的软件,帮助研究人员研究蛋白质的结构和功能。
2.2PyMOL:一种用于可视化分子结构的工具,它可以高质量的分子图像,并为研究人员提供结构分析的功能。
2.3Coot:用于蛋白质结构分析和模型建立的软件,可帮助研究人员在解析蛋白质结构时进行手动操作和调整。
2.4CCP4:一个用于蛋白质晶体学的软件套件,用于解析晶体结构和进行结构决策。
2.5SwissPdbViewer:一种用于蛋白质结构可视化和分析的软件,具有多种功能和工具。
3.蛋白质互作软件3.1STRING:综合性的蛋白质互作数据库和分析工具,帮助研究人员理解蛋白质之间的相互作用关系。
3.2Cytoscape:一个用于细胞网络分析和可视化的软件,可用于研究蛋白质之间的相互作用网络。
3.3ClusPro:一种用于蛋白质蛋白质和蛋白质配体互作的软件,可用于预测互作模型和分析互作强度。
3.4InterProSurf:一种用于预测和分析蛋白质间相互作用界面的工具,可以帮助研究人员理解蛋白质互作的机制。
15个常用生物信息在线工具

15个常用生物信息在线工具工欲善其事必先利其器小榴莲今天和大家分享16个生信网站有心的小伙伴可以收藏奥内容包括:韦恩图基因预测phylogenetic启动子区预测蛋白质一级结构分析信号肽跨膜结构域蛋白质亚细胞定位蛋白质三级结构预测短序列拼接多序列比对相似性展示绘制GO注释结果蛋白质domain基因组杂合性评估circos图韦恩图工具:Venny2.0地址:/tools/venny/index.html基因预测工具:FGENESH地址:/berry.phtml?topic=fgenesh&group=programs&subgroup=gfin dphylogenetic 工具:iTOL地址:/index.shtml启动子区预测工具:Promoter Scan地址:/services/Promoter/蛋白质一级结构分析工具:PredictProte地址:/home工具:ExPASy-ProtParam tool 地址:/protparam/信号肽工具:SignalP地址:/services/SignalP/跨膜结构域工具:TMHMM Server v. 2.0 地址:/services/TMHMM/蛋白质亚细胞定位工具:PSORT II Prediction 地址:/form2.html蛋白质三级结构预测工具:SWISS-MODEL地址:/interactive短序列拼接工具:Cap3地址:/software/cap3多序列比对相似性展示工具:SimiTriX-SimiTetra地址:/EN/tools/BioERCP/simitrix.php绘制GO注释结果工具:WEGO(Web Gene Ontology Annotation Plotting)地址:/cgi-bin/wego/index.pl蛋白质domain工具:Pfam database地址:/基因组杂合性评估工具:GenomeScope(Estimate genome heterozygosity, repeat content, and size from sequencing reads using a kmer-based statistical approach)地址:/genomescope/analysis.php?code=example2circos图工具:CIRCOS(可以用来画基因组数据的环状图,也可以用来绘制其它数据的相关环状图。
pep-fold3用法

pep-fold3用法
PEP-Fold3是一款在线蛋白质二级结构预测服务器,它可以根据给定的氨基酸序列预测蛋白质的二级结构。
以下是使用PEP-Fold3进行蛋白质二级结构预测的一般步骤:
1.打开浏览器,访问PEP-Fold3的官方网站。
2.在网站首页上,找到并点击“Start”按钮,开始一个新的预测任务。
3.在打开的页面中,输入待预测的蛋白质氨基酸序列。
可以手动输入序列,也可以上传FASTA格式的序列文件。
4.选择预测参数。
在PEP-Fold3中,可以选择不同的预测参数和算法进行蛋白质二级结构预测。
选择适当的参数可以提高预测的准确性和可靠性。
5.提交预测任务。
完成输入和参数选择后,点击“Submit”按钮提交预测任务。
6.等待预测结果。
PEP-Fold3会自动进行蛋白质二级结构预测,并在完成后将结果发送到用户的邮箱中。
7.分析预测结果。
用户可以下载预测结果文件,并使用文本编辑器或生物信息学软件分析预测结果。
需要注意的是,PEP-Fold3是一种基于机器学习的蛋白质二级结构预测方法,其预测结果的准确性受到多种因素的影响,如序列长度、序列相似性、序列复杂度等。
因此,在进
行蛋白质二级结构预测时,需要综合考虑多种因素,并采用多种方法进行预测和验证。
蛋白质结构预测

6.picornaviral蛋白上的酶切位点(NetPicoRNA) 7.叶绿体传递蛋白(ChoroP) 8.跨膜螺旋(TMHMM) 9.跨膜螺旋的位臵(TOPPRED) 10.跨膜螺旋的位臵和方向(DAS) 数据库搜索: 1.基于预测的线程方法与完全基于序列的数据库搜 索(FRSVR) 2.查找远源同源蛋白序列的Markov model method (SAMT98)
评估: 1.二级结构预测准确性评估(EvalSec) 2. EVA:预测方法自动评估
PP工作原理
• PredictProtein是一项在因特网上提供氨 基酸序列分析,蛋白质结构和功能预测的服 务。通过电邮或因特网,您可以提交单一 蛋白序列或序列比对,PP将返回结果.
1.PP总流程
2. 序 列 分 析 流 程
• SWISS-MODEL反馈的第1封邮件 (Welcome to SwissModel) • SWISS-MODEL反馈的第2封邮件 (SwissModel-Fold-Recognition) • SWISS-MODEL反馈的第3封邮件 (SwissModelSecondaryStructureP...) • SWISS-MODEL反馈的第4封邮件 (SwissModel_TraceLog_AAAa06Zd K) • SWISS-MODEL反馈的第5封邮件 (SwissModel-Model-AAAa06ZdK) 中附有可用大分子结构展示软件 显示的文件AAAa06ZdK.pdb: • SWISS-MODEL反馈的第6封邮件 (SwissModel WhatCheck AAAa06ZdK...) • SWISS-MODEL反馈的第7封邮件 (PredictProtein results)
• META-PP提供单页界面,发送预测查询,到其他蛋白结 构预测服务器。在此界面提交待分析序列,选择所需要 的服务,您将以电子邮件的形式收到答复。在META-PP 提供的序列分析服务包括预测内容: 1.二级结构(JPRED) 2.模拟三维结构(automated modelling,SWISS-MODEL) 3.通过一系列方法和数据库推导三级结构(CPHmodels) 4.信号肽的剪切位点(SignalP) 5.哺乳动物体内黏蛋白的O-GalNAc(突变型)糖基化作用 位臵(NetOglyc)
蛋白二级结构(卷曲螺旋)预测软件---------COILS的使用

预测结果图: 显示沿着序 列各个部分 形成卷曲螺 旋的倾向性
二、BCL-2基因在10个物种 Homo sapiens, Xenopus laevis, Gallus gallus ,Mus musculus, Bos taurus ,Rattus norvegicus ,Capra hircus ,Pan troglodytes ,Canis lupus familiaris ,Felis catus 中ClustalW序列比对分析、MEGA 系统进化分析 、MEME进行保守位 点分析。
BCL-2基因(即B细胞淋巴瘤/白血病-2 基因)是一种原癌基因,它具有抑制凋亡 的作用,并且近年来的一些研究已开始揭 示这一作用的机制。目前已经发现的BCL2蛋白家族按功能可分为两类,一类是像 BCL-2一样具有抑制凋亡作用,而另一类 具有促进凋亡作用。
BCL-2基因在10个物种中的位置:
>gi|262050657|ref|NM_001166486.1| Bos Taurus >gi|50950156|ref|NM_001002949.1| Canis lupus familiaris >gi|548519521|ref|XM_005697325.1| PREDICTED: Capra hircus >gi|57163882|ref|NM_001009340.1| Felis catus >gi|758371883|ref|NM_205339.2| Gallus gallus >gi|72198188|ref|NM_000633.2| Homo sapiens >gi|545477919|ref|NM_009741.4| Mus musculus >gi|694971627|ref|XM_001145537.2| PREDICTED: Pan testroglody >gi|8392973|ref|NM_016993.1| Rattus norvegicus >gi|225735550|ref|NM_001146093.1| Xenopus laevis
氨基酸序列 在线转结构

氨基酸序列在线转结构随着生物信息学的发展,氨基酸序列的研究在生物学领域中占据了重要地位。
氨基酸序列是指蛋白质中氨基酸的线性排列顺序,它反映了蛋白质的结构和功能。
然而,仅研究氨基酸序列并不能直接揭示蛋白质的三维结构。
为了更好地理解蛋白质的结构与功能之间的关系,科学家们需要将氨基酸序列转化为蛋白质结构。
这时,在线转结构工具就显得尤为重要。
在线转结构工具可以帮助研究人员将氨基酸序列转换为蛋白质三维结构,从而为进一步的研究提供基础。
这些工具的原理主要是基于计算机模拟和人工智能技术,通过预测氨基酸之间的相互作用和空间排列,生成蛋白质的三维结构。
接下来,我们将介绍几款常用的在线转结构工具。
首先是I-TASSER,这是一款基于模板蛋白质数据库的在线预测工具,可以根据氨基酸序列预测蛋白质结构。
其次是一站式生物信息学平台MetaPhlAn,它可以同时进行多种生物信息学分析,包括氨基酸序列转结构。
另外,MODELLER也是一款著名的在线转结构工具,以其较高的预测准确率受到研究者的青睐。
在选择在线转结构工具时,应考虑以下几点:预测准确率、服务器响应速度、用户界面以及输出结果的详细程度等。
例如,I-TASSER在预测准确率方面表现较好,但服务器响应速度相对较慢;而MetaPhlAn则具有较好的用户界面和较高的服务器响应速度。
因此,在实际应用中,可以根据自身需求和实际情况选择合适的在线转结构工具。
在线转结构在生物学研究中的应用实例众多,如结构生物学、药物设计、蛋白质互作研究等。
以药物设计为例,通过在线转结构工具预测靶点蛋白质结构,有助于研究者更好地了解药物与靶点之间的结合方式和相互作用,从而优化药物设计和筛选。
在使用在线转结构工具时,还需注意以下几点:1.确保氨基酸序列的正确性,避免使用错误或有争议的序列;2.针对不同的研究目的,选择合适的预测算法和工具;3.结合实验数据对预测结果进行验证,以提高研究可靠性。
总之,在线转结构工具为蛋白质研究提供了强大的支持。
蛋白质结构预测在线软件

蛋白质结构预测在线软件随着计算机技术的发展,越来越多的蛋白质结构预测在线软件被开发出来,并且被广泛应用于生物学研究。
本文将介绍几个常用的蛋白质结构预测在线软件,并对它们的原理和优缺点进行分析。
首先,我要介绍的是PHYRE2、PHYRE2是一款基于比较模型的蛋白质结构预测软件,它通过将待预测的蛋白质序列与已知结构库中的蛋白质序列进行比对,从而预测目标蛋白质的结构。
PHYRE2具有高度自动化的特点,可以在较短的时间内进行大量的结构预测。
但是,PHYRE2的准确性和可靠性相对较低,因为它只依赖于已知结构的信息。
其次,我要介绍的是I-TASSER。
I-TASSER是一种基于碎片装配的蛋白质结构预测软件,它通过将目标蛋白质的序列分解为小的片段,然后通过模板和螺旋转角预测来重新组装这些片段,从而得到目标蛋白质的结构。
I-TASSER具有较高的准确性和可靠性,并且在多个蛋白质结构预测比赛中表现出色。
然而,I-TASSER的计算速度较慢,需要较长的时间来进行结构预测。
另外,我要介绍的是Rosetta。
Rosetta是一种基于物理学的蛋白质结构预测软件,它通过对蛋白质的能量进行优化来确定最稳定的结构。
Rosetta具有较高的准确性和可靠性,并且可以进行全原子级别的结构预测。
然而,由于Rosetta的计算复杂性较高,需要大量的计算资源来进行结构预测。
除了以上介绍的几种蛋白质结构预测在线软件,还有许多其他的软件可供选择,如PSIPRED、HHPred等。
这些软件在原理和性能上有所差异,但都能够对蛋白质的结构进行预测,并为生物学研究提供重要的参考信息。
总结起来,蛋白质结构预测是生物信息学领域的重要课题,需要借助计算机算法来进行预测。
目前有许多蛋白质结构预测在线软件可供选择,它们在原理、准确性、可靠性和计算速度等方面有所差异。
选择合适的软件进行蛋白质结构预测,将对生物学研究产生重要的影响。
蛋白质结构预测和序列分析报告软件

蛋白质结构预测和序列分析软件2010-05-08 20:40转载自布丁布果最终编辑布丁布果4月18日蛋白质数据库及蛋白质序列分析第一节、蛋白质数据库介绍一、蛋白质一级数据库主要蛋白质序列数据库的网址SWISS-PROTt或htmlTrEMBLPIRrwwwMIPSProtein SequencesJIPIDProtein Sequence Database 已经和ExPASy三、蛋白质二级结构预测网站(数据库)4始建于SIB基于对蛋白质家族中同源序列多重序列比对得到的保守区域,这些区域通常与生物学功能相关。
数据库包括两个数据库文件:数据文件PrositeProsite/prosite5of Proteins)蛋白质二级结构构象参数数据库DSSPhttp://www.cmbi.kun.nl/gv/dssp6Proteins)蛋白质家族数据库FSSP/dall/fssp7Structure of Proteins)同源蛋白质数据库HSSPhttp://www.cmbi.kun.nl/gv/hssp在前面已经述说过了。
第二节、蛋白质序列分析方法一、多序列比对双序列比对是序列分析的基础。
基因家族的成组的序列来说,列之间的关系,征。
学模式方面起着相当重要的作用。
多序列比对有时用来区分一组序列之间的差异,但其主要用于描述一组序列之间的相似性关系,以便对一个基因家族的特征有一个简明扼要的了解。
立在某个数学或生物学模型之上。
因此,正如我们不能对双序列比对的结果得出“正确或错误”结果也没有绝对正确和绝对错误之分,为所使用的模型在多大程度上反映了序列之间的相似性关系以及它们的生物学特征。
我们称比对前序列中残基的位置为绝对位置。
序列Ⅰ的第Ⅰ相应地,位置。
而每个残基的绝对位置不同,的序列。
绝对位置是序列本身固有的属性,前的位置,而相对位置则是经过比对后的位置,也就比对过程赋予它的属性。
算法复杂性多序列比对的计算量相当可观,以下技术的复杂性。
病毒蛋白质结构预测及功能分析方法研究

病毒蛋白质结构预测及功能分析方法研究近年来,随着新型冠状病毒(COVID-19)的全球爆发,研究人员对于病毒蛋白质结构预测及功能分析方法的研究变得尤为重要。
准确预测病毒蛋白质的结构可以帮助科学家深入了解病毒的生物学特性,为疫苗和药物的设计提供重要依据。
本文将介绍几种常用的病毒蛋白质结构预测及功能分析方法。
首先,蛋白质序列分析是确定病毒蛋白质结构的关键步骤之一。
蛋白质序列包含了蛋白质构成氨基酸的排列顺序。
研究人员可以通过比对已知的蛋白质序列数据库,根据序列的相似性来推测病毒蛋白质的结构。
常用的蛋白质序列分析软件包括BLAST、FASTA和PSI-BLAST等。
这些软件可以根据已有的蛋白质序列库进行比对,找出具有相似序列的蛋白质,从而判断病毒蛋白质的可能结构。
另一种常用的病毒蛋白质结构预测方法是基于模板的建模。
这种方法利用已知结构的蛋白质作为模板,通过比对病毒蛋白质序列与已知结构的相似性,预测蛋白质的结构。
其中最常用的模板比对方法是蛋白质-蛋白质比对,通过比对序列和结构相似的蛋白质,预测目标蛋白质的结构。
另外,还有一种基于远程同源的模板比对方法,通过比对序列相似度较低但结构相似的蛋白质,预测目标蛋白质的结构。
常见的基于模板的建模软件有Phyre2、HHPred和SWISS-MODEL等。
除了基于模板的建模方法外,还有一种常用的病毒蛋白质结构预测方法是基于物理学原理的力场方法。
这种方法通过数学计算和物理学模型,模拟蛋白质分子的力学和热力学性质,预测蛋白质的结构。
著名的力场方法包括分子力学和分子动力学方法。
分子力学方法是利用经验力场,通过计算分子间的相互作用力,预测蛋白质的结构。
而分子动力学方法则是在分子力学的基础上,通过模拟蛋白质分子在时间上的演化,推测蛋白质的结构。
常用的力场方法软件包括AMBER、GROMACS和CHARMM等。
病毒蛋白质功能分析是对已预测的蛋白质结构进行功能注释和预测的过程。
功能分析可以帮助科学家了解病毒蛋白质的生物学功能和作用机制。
常用生物软件(软件及引物设计总结)

THANKS FOR WATCHING
感谢您的观看
总结词
NAMD是一款用于大规模并行计算的高 性能分子动力学模拟软件,适用于超大 型生物分子系统的模拟。
ห้องสมุดไป่ตู้VS
详细描述
NAMD采用高效的并行算法和优化的数 据结构,能够在高性能计算集群上快速运 行大规模模拟。它广泛应用于生物医学、 药物设计和材料科学等领域,尤其适用于 模拟蛋白质复合物等大型生物分子的结构 和动力学行为。
SWISS-MODEL
总结词
在线建模工具,适用于预测蛋白质三维结构 。
详细描述
SWISS-MODEL是一个在线建模工具,用于 预测蛋白质的三维结构。它基于模板建模技 术,通过搜索模板库找到与目标序列相似的 已知结构,并利用这些信息构建出目标蛋白 质的结构模型。SWISS-MODEL提供了友好 的用户界面和多种建模选项,方便用户进行
总结词
AutoDock是一款用于分子对接的软件,通过模拟分子间的 相互作用,预测小分子与大分子(如蛋白质)的结合模式。
详细描述
AutoDock使用基于网格的搜索方法,将小分子视为可旋转 和可平移的刚体,通过打分函数评估结合亲和力,并采用遗 传算法进行优化。它广泛应用于药物设计和蛋白质相互作用 研究。
Gromacs
总结词
Gromacs是一款用于分子动力学模拟的软件,通过模拟分子在真实环境中的运 动来研究其结构和功能。
详细描述
Gromacs基于势能面模型,通过求解牛顿方程模拟分子运动轨迹,可以模拟蛋 白质、核酸和脂质等生物大分子的动态行为。它广泛应用于生物物理学、药物 设计和材料科学等领域。
NAMD
Galaxy
总结词
Galaxy是一个基于Web的生物信息学分析平台,提供 了简单易用的界面和强大的数据分析能力。
蛋白质结构与功能预测网站

功能域
用户自定义区段
点击不同功能域或是以直接粘贴氨基酸序列的方式得到以下结果
氨基酸数目 相对分子质量 理论 pI 值
氨基酸组成
正/负电荷残基数
原子组成
分子式 总原子数 消光系数
半衰期
14
不稳定系数
<40 stable >40 unstable
脂肪系数 总平均亲水性
/跨膜区分析 蛋白质亲疏水性 蛋白质亲疏水性/
– Protparam 工具 /tools/protparam.html http:// /tools/protparam.html
相对分子质量 氨基酸组成 等电点(PI) 消光系数 半衰期 不稳定系数 总平均亲水性 ……
蛋白质理化性质分析工具
PeptideMass SAPS
/tools/peptide-mass.html http://www.isrec.isbsib.ch/software/SAPS_form.html
AACompIdent
PeptideMass
蛋白质理化性质分析
• Protparam 工具
/Mw http:// /tools/pi_tool.html Compute pI pI/Mw /tools/pi_tool.html
ProtParam
/tools/protparam.html http:// /tools/protparam.html
蛋白质结构与功能预测
2007年12月
DNA sequence Protein sequence
Protein structure
Protein function
蛋白质序列分析主要内容
蛋白质基本理化性质分析 蛋白质亲疏水性分析 蛋白质一级序列 跨膜区结构预测 卷曲螺旋预测 蛋白质序列分析 蛋白质二级结构 蛋白质超二级结构 蛋白质三级结构 蛋白质分类 翻译后修饰位点预测 蛋白质二级结构预测 蛋白质序列信号位点分析 蛋白质结构域分析 蛋白质三维结构模拟 蛋白质家族分析
蛋白质结构预测在线软件

蛋白质预测分析网址集锦物理性质预测:Compute PI/MW http://expaxy.hcuge.ch/ch2d/pi-tool.htmlPeptidemasshttp://expaxy.hcuge.ch/sprot/peptide-mass.htmlTGREASE ftp:///pub/fasta/SAPS http://ulrec3.unil.ch/software/SAPS_form.html基于组成的蛋白质识别预测AACompIdent http://expaxy.hcuge.ch ...htmlAACompSim http://expaxy.hcuge.ch/ch2d/aacsim.html PROPSEARCH http://www.e mbl-heidelberg.de/prs.html二级结构和折叠类预测nnpredict /~nomi/nnpredictPredictprotein http://www.embl-heidel ... protein/SOPMA http://www.ibcp.fr/predict.html SSPRED http://www.embl-heidel ... prd_info.html特殊结构或结构预测COILS http://ulrec3.unil.ch/ ... ILS_form.htmlMacStripe / ... acstripe.html与核酸序列一样,蛋白质序列的检索往往是进行相关分析的第一步,由于数据库和网络技校术的发展,蛋白序列的检索是十分方便,将蛋白质序列数据库下载到本地检索和通过国际互联网进行检索均是可行的。
由NCBI检索蛋白质序列可联网到:“http://www.ncbi.nlm.ni ... gi?db=protein”进行检索。
利用SRS系统从EMBL检索蛋白质序列联网到:/”,可利用EMBL的SRS系统进行蛋白质序列的检索。
这四个网站了解一下,让你轻松预测相互作用蛋白!

这四个网站了解一下,让你轻松预测相互作用蛋白!蛋白质互作网络是由不同蛋白通过彼此之间的相互作用构成,参与生物信号传递、基因表达调节、能量和物质代谢及细胞周期调控等生命过程的各个环节。
在进行基因机制研究中,寻找互作蛋白是一个深入探究基因功能的过程,寻找未知蛋白充满艰辛,而通过数据库预测互作蛋白能够为我们的研究拓展思路,提供方向,让我们在科研道路上更加轻松。
下面就为大家推荐四个可以用来预测相互作用蛋白的网站!一:STRING网站1.STRING数据库是比较经典的蛋白互作数据库。
数据库使用简单。
2. 输入基因名称后,可以获得蛋白互作网络图,其中圆圈节点之间的直线代表该直线连接的两个蛋白之间的相互作用关系,点击直线可查看蛋白的详细信息。
3.网站下方提示不同颜色的直线代表不同的相互作用关系证据来源。
包括蛋白质之间的直接物理相互作用,也包括蛋白质之间的间接功能相关性。
它除了包含有实验数据、从PubMed摘要中挖掘的结果和综合其他数据库数据外,还有利用生物信息学的方法预测的结果。
二:Genemania网站。
1. Genemania也是比较常用的蛋白互作网站,网站结果类似cytoscape app,但是无需下载安装软件,直接在线使用,非常方便。
2.在网站左上方可以选择查询物种以及基因名称,同时点击下方不同的图标能够设置网络图展示形式:列表式、环状等。
3 .网站右侧可以设置网络边的来源,所应用的生物信息学方法有:物理互作、基因共表达、基因共定位、基因富集分析以及网站预测等等,通过这种方法能够设定检索范围。
三:unihi 网站1. unihi是一个用于检索,分析和可视化人类分子相互作用网络的数据库。
其主要目的是为广泛的生物学和医学研究人员提供一个全面且易于使用的网络调查平台。
UniHI目前包括基因,蛋白质和药物之间近350,000个分子相互作用,以及许多其他类型的数据,如基因表达和功能注释。
2.网站支持输入单基因或者多基因名称,寻找与之互作蛋白。
蛋白质结构预测网址

蛋白质结构预测网址
下面是比较常见的蛋白质结构预测网站:
1. Protean3D:这是一个用于蛋白质结构预测的非常流行的网站,有一个易于使用的图形用户界面,可以使用免费的计算机资源来进行模拟。
它还提供了许多有用的工具,如标准化的序列比对,位点推理等。
2.I-TASSER:这是一个专为蛋白质结构预测而设计的软件包,它使用动力学模拟来模拟蛋白质的结构形态。
I-TASSER可以根据蛋白质序列生成三维结构,并可以检查构象的稳定性。
3. FoldX:这是一个蛋白质结构预测工具,准确度比较高,可以用于模拟蛋白质的结构形态,特别是用于分析和预测蛋白质突变和热稳性之间的关系。
4. RaptorX:这是一个基于深度学习(deep learning)技术的蛋白质结构预测工具,可以从蛋白质序列中推断出三维结构。
RaptorX可以预测蛋白质组装,蛋白质相互作用以及蛋白质热稳定性的变化。
5.CASTp:这是一个蛋白结构分析工具,主要用于计算蛋白质接口的热力学参数,以及蛋白质组装热力学稳定性和构象的动力学性质。
6. ProSight:这是一款用于预测蛋白质结构的软件,它可以以统计学的方式预测蛋白质结构,并得到极高的准确率。
常用分子生物学软件(一)2024

常用分子生物学软件(一)引言概述:分子生物学软件在当今生物学研究中发挥着重要的作用。
它们以其功能强大和易用性而受到科研人员的青睐。
本文将介绍常用的分子生物学软件,并对它们的主要功能和特点进行详细说明。
正文:一、序列分析软件1. 序列比对软件- BLAST: 用于快速比对蛋白质或核酸序列与已知数据库中的相似序列。
- ClustalW: 对多个序列进行比对,并生成多序列比对结果。
2. DNA/RNA序列分析软件- Primer3: 用于设计引物序列。
- M-fold: 对RNA序列进行二级结构预测。
3. 蛋白质序列分析软件- GRAVY: 计算蛋白质氨基酸序列的相对水溶性。
- ProtParam: 提供氨基酸序列的各种生化性质分析。
4. 基因表达软件- ExPASy Translate: 用于将DNA序列翻译成蛋白质序列。
- Primer-BLAST: 用于设计引物并进行特异性检验。
5. 组学数据分析软件- Galaxy: 提供了一个高度集成的平台,用于处理和分析基因组学数据。
- Cytoscape: 用于可视化和分析分子和基因网络。
二、结构生物学软件1. 分子建模软件- Swiss-PdbViewer: 用于分子可视化和蛋白质模型构建。
- Autodock: 用于模拟蛋白质与小分子之间的相互作用。
2. 蛋白质结构预测软件- Rosetta: 提供了一种高效精确的蛋白质结构预测方法。
- I-TASSER: 通过蛋白质比对和拓扑结构模板识别,预测蛋白质三维结构。
3. 蛋白质结构比对软件- Dali: 用于比对两个或多个蛋白质结构,分析它们之间的结构和功能相似性。
- TM-align: 使用局部结构比对算法,对两个蛋白质的结构进行全局比对。
4. 蛋白质模拟软件- GROMACS: 用于分子动力学模拟和能量最小化。
- NAMD: 适用于分子动力学和分子模拟的高性能软件。
5. 蛋白质结构可视化软件- PyMOL: 用于可视化和分析蛋白质结构。
sopma用于蛋白质二级结构的预测教程

如何使用SOPMA进行蛋白质二级结构预测在蛋白质科学领域,预测蛋白质的二级结构对于理解蛋白质的功能和性质至关重要。
SOPMA(Self-Optimized Prediction Method with Alignment)是一种常用的工具,用于预测蛋白质的二级结构。
本教程将全面介绍如何使用SOPMA进行蛋白质二级结构预测,并帮助您更深入地理解这一过程。
1. SOPMA的基本原理SOPMA是一种基于序列的二级结构预测方法,它利用了蛋白质序列的保守性和物理化学性质来进行预测。
其原理基于自相似性,通过对蛋白质序列进行比对和分析,识别出其中的重复序列和保守性结构,从而预测出蛋白质的二级结构。
2. 使用SOPMA进行蛋白质二级结构预测的步骤您需要准备待预测的蛋白质序列。
在SOPMA的上线评台或使用其提供的软件工具中,输入蛋白质序列并选择相关参数,如窗口大小和权重。
接下来,点击预测按钮,SOPMA将根据其算法和模型对蛋白质的二级结构进行预测,并给出相应的结果。
3. 理解SOPMA预测结果SOPMA的预测结果通常包括了蛋白质的α-螺旋、β-折叠和无规则卷曲等二级结构元素的具体位置和可能性。
通过分析这些结果,您可以了解蛋白质的结构特征,进而推测其功能和相互作用。
总结与回顾通过本教程,我们全面介绍了如何使用SOPMA进行蛋白质二级结构预测,并帮助您更深入地理解了这一过程。
预测蛋白质二级结构是蛋白质科学研究中的重要一环,它对于理解蛋白质的结构与功能具有重要意义。
在实际应用中,您可以根据SOPMA的预测结果进一步设计实验和验证,从而推动蛋白质科学的发展。
个人观点和理解作为一名蛋白质科学研究者,我深知蛋白质二级结构预测的重要性。
SOPMA作为一种常用的预测工具,其准确性和稳定性得到了广泛认可。
我个人认为,随着技术的不断进步和算法的优化,基于序列的蛋白质二级结构预测将会更加精确和全面,为我们解读蛋白质的奥秘提供更强有力的支持。
蛋白二级结构(卷曲螺旋)预测软件---------COILS的使用
COILS数据库的构成、使 用方法和相关资源简介
• 二级结构:是指以多肽链为主链骨架盘绕折叠 而形成的结构,是多肽链的局部空间结构。 • 卷曲螺旋:是由两股或两股以上α螺旋相互缠 绕而形成超螺旋结构的总称。该结构存在于多 种天然蛋白质中,如转录因子、膜蛋白等,在 基因调分子识别等方面发挥了重要的生物学功 能。
COILS的简介
• 是目前主流预测卷曲区域工具。它基于Lupsa算 法,由Swiss EMBNet维护。用户既可以把 COILS下载到VAX/VMS操作系统上使用,也可 通过简单的Web界面使用。
• 预测方法:将序列与已知的平行双链卷曲螺
旋数据库进行比较,得到相似性得分,并据此 算出序列形成卷曲螺旋的概率。 • 网址: /software/CO ILS_form.html
BCL-2基因(即B细胞淋巴瘤/白血病-2 基因)是一种原癌基因,它具有抑制凋亡 的作用,并且近年来的一些研究已开始揭 示这一作用的机制。目前已经发现的BCL2蛋白家族按功能可分为两类,一类是像 BCL-2一样具有抑制凋亡作用,而另一类 具有促进凋亡作用。
ห้องสมุดไป่ตู้
BCL-2基因在10个物种中的位置:
>gi|262050657|ref|NM_001166486.1| Bos Taurus >gi|50950156|ref|NM_001002949.1| Canis lupus familiaris >gi|548519521|ref|XM_005697325.1| PREDICTED: Capra hircus >gi|57163882|ref|NM_001009340.1| Felis catus >gi|758371883|ref|NM_205339.2| Gallus gallus >gi|72198188|ref|NM_000633.2| Homo sapiens >gi|545477919|ref|NM_009741.4| Mus musculus >gi|694971627|ref|XM_001145537.2| PREDICTED: Pan testroglody >gi|8392973|ref|NM_016993.1| Rattus norvegicus >gi|225735550|ref|NM_001146093.1| Xenopus laevis
蛋白三维结构观察软件swisspdbviewer

蛋白三维结构观察软件Swiss PDB Viewerp Swiss PDB Viewer软件简介p示例蛋白介绍p蛋白结构分析l Swiss PDB Viewer即Spdbv,界面简明易用,可同时分析多个蛋白PDB文件。
l分析多个蛋白结构相似性,比较活性位点或其它有关位点。
l通过菜单操作,直观图形展示,可获得氢键、角度、原子距离、氨基酸突变等原始数据。
l与Swiss-Model服务器紧密关联,可直接预测蛋白三维结构。
菜单栏绘图区菜单栏File/open PDB file:打开一个PDB格式文件;File/save/save selected residues:文件保存在指定的文件夹;File/save/image:文件保存成图片;Select/group kind:寻找特定的氨基酸并在控制面板进行标识;Fit/magic fit:两个蛋白三维结构进行重叠;Color/by chain:不同二级结构用不同的颜色标注;Wind/alignment:进行序列比对。
工具栏图形在窗口中最适展示图形自由移动图形自由扩大或者缩小图形旋转测量两个原子间距测量三个原子间夹角度数测量二面角显示某位点氨基酸名称距离中心原子几埃之内其它残基或原子所选原子置于窗口中心将一个分子叠加至另一分子突变工具,可将该位点氨基酸突变为其他 氨基酸扭转工具,可以通过左右点击和移动鼠标旋转侧链原子控制面板A链二级结构氨基酸名称和位置侧链标注电子云带状展示颜色是否可移动是否可视示例蛋白介绍l乳酸脱氢酶(嗜热脂肪土芽孢杆菌)WP_033016716.1l乳酸脱氢酶:乳酸脱氢酶是一种糖酵解酶。
乳酸脱氢酶存在于机体所有组织细胞的胞质内,其中以肾脏含量较高。
乳酸脱氢酶是能催化丙酮酸生成乳酸的酶,几乎存在于所有组织中。
[1]l嗜热脂肪土芽孢杆菌:嗜热脂肪芽孢杆菌(Bacillus stearothermophilus)属嗜热性需氧芽孢杆菌,但兼有厌氧的特性,细菌繁殖体G兰氏染色阳性呈紫色,细菌芽胞孔雀绿着色。
alphafold 分子互作

alphafold 分子互作
Alphafold是DeepMind开发的一种蛋白质结构预测软件,它利
用深度学习技术来预测蛋白质的三维结构。
蛋白质是生物体内发挥
重要功能的分子,其结构对于理解蛋白质的功能和相互作用至关重要。
分子互作是指不同分子之间的相互作用,包括蛋白质与蛋白质、蛋白质与小分子等的相互作用。
Alphafold在预测蛋白质结构方面取得了巨大的成功,这也使
得人们开始探讨Alphafold在预测蛋白质分子互作方面的潜力。
通
过预测蛋白质的结构,Alphafold可以帮助科学家们理解蛋白质是
如何与其他分子相互作用的。
这对于药物设计、疾病治疗等方面具
有重要意义。
在分子互作的研究中,科学家们可以利用Alphafold预测蛋白
质的结构,然后进一步模拟蛋白质与其他分子之间的相互作用。
这
有助于揭示蛋白质在生物体内的功能机制,为新药物的研发提供重
要线索。
另外,Alphafold的预测结果也可以为研究人员提供有关蛋白
质结构和分子互作的新思路和假设,从而推动相关领域的科学研究。
通过结合实验数据和Alphafold的预测结果,科学家们可以更全面地理解蛋白质分子互作的机制。
总的来说,Alphafold在蛋白质结构预测领域取得了突破性进展,其在预测蛋白质分子互作方面的应用潜力也备受期待。
通过结合实验验证,Alphafold有望为我们揭示更多关于蛋白质分子互作的奥秘,推动相关领域的科学研究和应用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
蛋白质结构预测在线软件Company Document number:WUUT-WUUY-WBBGB-BWYTT-1982GT蛋白质预测分析网址集锦?物理性质预测:?Compute PI/MW??SAPS?基于组成的蛋白质识别预测?AACompIdentPROPSEARCH?二级结构和折叠类预测?nnpredict?Predictprotein?SSPRED?特殊结构或结构预测?COILS?MacStripe?与核酸序列一样,蛋白质序列的检索往往是进行相关分析的第一步,由于数据库和网络技校术的发展,蛋白序列的检索是十分方便,将蛋白质序列数据库下载到本地检索和通过国际互联网进行检索均是可行的。
?由NCBI检索蛋白质序列?可联网到:“”进行检索。
?利用SRS系统从EMBL检索蛋白质序列?联网到:”,可利用EMBL的SRS系统进行蛋白质序列的检索。
?通过EMAIL进行序列检索?当网络不是很畅通时或并不急于得到较多数量的蛋白质序列时,可采用EMAIL方式进行序列检索。
?蛋白质基本性质分析?蛋白质序列的基本性质分析是蛋白质序列分析的基本方面,一般包括蛋白质的氨基酸组成,分子质量,等电点,亲水性,和疏水性、信号肽,跨膜区及结构功能域的分析等到。
蛋白质的很多功能特征可直接由分析其序列而获得。
例如,疏水性图谱可通知来预测跨膜螺旋。
同时,也有很多短片段被细胞用来将目的蛋白质向特定细胞器进行转移的靶标(其中最典型的例子是在羧基端含有KDEL序列特征的蛋白质将被引向内质网。
WEB中有很多此类资源用于帮助预测蛋白质的功能。
?疏水性分析?位于ExPASy的ProtScale程序()可被用来计算蛋白质的疏水性图谱。
该网站充许用户计算蛋白质的50余种不同属性,并为每一种氨基酸输出相应的分值。
输入的数据可为蛋白质序列或SWISSPROT数据库的序列接受号。
需要调整的只是计算窗口的大小(n)该参数用于估计每种氨基酸残基的平均显示尺度。
?进行蛋白质的亲/疏水性分析时,也可用一些windows下的软件如,bioedit,dnamana等。
?跨膜区分析?有多种预测跨膜螺旋的方法,最简单的是直接,观察以20个氨基酸为单位的疏水性氨基酸残基的分布区域,但同时还有多种更加复杂的、精确的算法能够预测跨膜螺旋的具体位置和它们的膜向性。
这些技术主要是基于对已知跨膜螺旋的研究而得到的。
自然存在的跨膜螺旋Tmbase 数据库,可通过匿名FTP获得(,参见表一?资源名称网址说明?TMPRED基于对tmpred数据库的统计分析PHDhtm... 微机版本?,蛋白质序列含有跨膜区提示它可能作为膜受体起作用,也可能是定位于膜的锚定蛋白或者离子通道蛋白等,从而,含有跨膜区的蛋白质往往和细胞的功能状态密切相关。
“或“”?前导肽与蛋白质定位?在生物内,蛋白质的合成场所与功能场所常被一层或多层细胞膜所隔开,这样就涉及到蛋白质的转运。
合成的蛋白质只有准确地定向运行才能保证生命活动的正常进行。
一般来说,蛋白质的定位的信息存在于该蛋白质自身结构中,并通过与膜上特殊的受体相互作用而得以表达。
在起始密码子之后,有一段编码疏水性氨基酸序列的RNA片段,这个氨基酸序列就这个氨基酸序列就是信号肽序列。
含有信号肽的蛋白质一般都是分泌到细胞外,可能作为重要的细胞因子起作用,从而具有潜在的应用价值。
??卷曲螺旋分析?另一个能够直接从序列中预测的功能motif是α-螺旋的卷曲排列方式。
在这种结构中,两种螺旋通过其疏水性界面相互缠在一起形成一个十分稳定的结构。
?蛋白质卷曲的相关资源?资源网址?coiled-coil?蛋白质功能预测?基于序列同源性分析的蛋白质功能预测?到少有80个氨基酸长度范围内具有25%以上序列一致性才提示可能的显着性意义。
最快的工具如BlastP能很容易地发现显着性片段,而无需使用十分耗时的BLITZ软件。
?基于NCBI/BLAST软件的蛋白序列同源性分析?类似于核酸序列同源性分析,用户直接将待分析的蛋白质序列输入NCBI/BLAST(,选择程序BLASTP就可网上分析。
?基于WU/BLAST2软件进行分析?华盛顿大学的BLAST软件()也可进行蛋白质序列的同源性分析。
?基于motif、结构位点、结构功能域数据库的蛋白质功能预测?蛋白质的磷酸化与糖基化对蛋白质的功能影响很大,所以对其的分析也是生物信息学的一个部分。
?同时,分子进化方面的研究表明,蛋白质的不同区域具有不同的进化速率,一些氨基酸必须在进化过程中足够保守以实现蛋白质的功能。
在序列模式的鉴定方面有两类技术,第一类是依赖于和一致性序列(consensus sequence)或基序各残基的匹配模式,该技术可用于十分容易并快速搜索motif数据库。
?Motif数据库-PROSITE??蛋白质序列的(profile)分析??InterProScan综合分析网站???蛋白质的结构功能域分析?简单模块构架搜索工具(simple modular architecture research tool,SMART)一个较好的蛋白质结构功能域的数据,可用于蛋白质结构功能域的分析,所得到的结构域同时提供相关的资源的链接?蛋白质结构预测?PDB数据库?蛋白质基本立体结构数据库(PDB,其中有大量工具用于查看PDB数据库中的结构,如rasmol,可用于显于出蛋白质的空间结构,下载地址:)?PDBFinder 数据库?是在PDB、DSSP、HSSP基础上建立的二级库,它包含PDB序列,作者,R因子,分辨率、二级结构等,这些些信息随着PDB库每次发布新版,PDBFinder在EBI自动生成,网址为“?NRL-3D数据库?是所有已知结构蛋白质的数据库,可用于查询蛋白序列时行相似性分析以确定其结构?ISSD数据库?蛋白质序列数据库,其每个条目包含一个基因的编码序列,同相应的氨基酸序列对比,并给出相应的多肽链结构数据。
?HSSP数据库?是根据同源性导出的蛋白质二级结构数据库,每一条PDB项目都有一个对应的HSSP 文件,?蛋白质结构分类数据库?对已知蛋白质三维结构进行手工分类得到的数据库,位于剑桥的站点也提供BLAST检索服务?MMDB蛋白质分子模型数据库?是ENTREZ检索工具所使用的三维结构数据库,以ASN格式反蚋的PDB中的结构和序列数据。
NCBI同时提供一个配套的三维结构显示程序的Cn3D,?Dali/FSSP数据库?基于PDB数据库中现有的蛋白质三维结构,用自动结构对比程序Dali比较而形成的折叠单元和家庭分类库。
?蛋白质二级结构预测?基于序列进行蛋白质二级结构方面已有了大量文献描述,本质上,这些研究可被分为两大类:基于单一序列的分析和基于多重序列对齐的分析。
?文献报道PHD程序是目前此方面的最好程序,提供了从二级结构到折叠方面分析的多种资源。
其网址为?蛋白质三级结构预测?蛋白质同源家庭的分析对于确立物种之间的亲缘关系和预测新蛋白质序列的功能有重要意义,同源蛋白质(homolog)进一步划分为直系同源(ortholog)和旁系同源(paralog),前者指不同物种中具有相同功能和共同起源的基因,后者则指在同一物种内具有不同功能,但也有共同起源的基因,例如同是起源于珠蛋白的α珠蛋白、β珠蛋白和肌红蛋白。
?蛋白质分类数据库(ProtoMap)?是对SWISS-PROT数据库中的全部蛋白质由计算机自动时行层次分类,把相关者聚集分极所得到的数据库。
?蛋白质序列多重对齐分析及进化分析?如果发现一个未知蛋白质序列和较多不同和种属或同一种属的蛋白质序列具有较高的同源性(大于30%)那么提示待分析的蛋白质序列可能是相应家族的成员,从而可从分子时化的角度对蛋白质序列进行综合分析。
?常用在线蛋白工具?BCM Search Launcher??蛋白序列二级结构预测综合站点,从此出发,输入蛋白序列,可以根据需要,使用各种在线预测工具,包括Coils、nnPredict、PSSP/SSP、PSSP/NNSSP、SAPS、TMpred、SOUSI、Paircoil、Protein Hydrophilicity/Hydrophobicity Search、SOPM,使用十分方便。
?DAS?蛋白跨膜预测服务器、输入蛋白序列,预测跨膜区域。
?TopPred 2?斯德哥尔摩大学理论化学蛋白预测服务器提供的膜蛋白拓扑学预测Topology prediction of membrane proteins在线工具?SOSUI?东京农业科技大学(Tokyo University of Agriculture and Technology)提供的膜蛋白分类和二级结构预测在线工具。
?PSIpred - MEMSAT2?本蛋白结构预测服务器允许你提交一个蛋白序列,进行二级结构预测与跨膜拓朴结构预测,并将结果用EMAIL提供给您。
?HMMTOP?预测蛋白序列的跨膜螺旋与拓扑结构服务器?TMpred?预测蛋白序列跨膜区?TMHMM?预测蛋白的跨膜螺旋?The PredictProtein server?提供蛋白数据库查询,预测蛋白各种结构的服务?SMART?提供蛋白序列,在结构域数据库中查询,显示出其结构域及跨膜区等?SPLIT?膜蛋白二级结构预测服务器?PRED-TMR?提供基于SwissProt数据库统计分析的预测蛋白跨膜片段的服务?CoPreThi?基于INTERNET的JAVA程序,预测蛋白的跨膜区?TMAP?提供预测蛋白跨膜片段的服务?multalin?蛋白序列对照服务器,比较几条蛋白序列的结构。
?Protein Sequence Analysis?巴斯德研究所提供的常用蛋白序列分析在线工具,绝对精选。
?PSA?Protein Sequence Analysis 服务器为美国波士顿大学生物分子工程研究中心(the BioMolecular Engineering Research Center )开发,提交氨基酸序列,预测二级结构及折叠区域。
?PRS?EMBL提供的以未知蛋白序列的氨基酸组成而非氨基酸序列顺序进行蛋白家族及各种特性预测的服务器,并将结果通过EMAIL发给您。
?AAA?EMBL氨基酸分析服务器,与上一个服务类似,以氨基酸残基组成为蛋白分析基础数据,结合蛋白数据库进行分析。