DMF以及COS和EDMF简介
CEP、COS与EDMF简介及比较

CEP、COS与EDMF简介及比较CEP/COS与EDMF简介及比较一、CEP/COS欧洲药典适用性认证(Certification of Suitability to Monograph of European Pharmacopoeia),早期简称COS,现称CEP,是由欧洲药品质量管理局(EDQM)对于已经收载到欧洲药典(EP)的原料药和辅料启动的一个独立的质量评价程序,用以证明原料药和辅料的质量是按照欧洲药典各论描述的方法严格控制的,产品质量符合欧洲药典标准。
CEP/COS证书不仅被所有欧盟成员国承认,而且被其他签订了双边协定的国家认可,如澳大利亚,加拿大等。
CEP是原料药和辅料的一种认证程序,该程序适用于生产的和提取的有机或无机物质、发酵生产的非直接基因产品及药用辅料,但前提是该物质被欧洲药典所收载。
CEP是原料药进入欧洲市场,可以被欧洲药物制剂生产厂家合法使用的不同于EDMF的另一种注册方式。
如果拟上市药品中使用的原料药获得CEP证书,上市许可申请(Marketing Authorization Application,简称MAA )可直接使用该证书,审评当局不再对原料药的质量进行评价;如果上市药品中使用的原料药取得CEP证书,就可直接用于欧洲药典协定公约成员国内的药物制剂生产厂家的药品生产。
原料药生产商或者正式授权的生产商代表可以申请CEP,而不受他们地理位置的限制(特别说明:在申请CEP认证的技术文件后面必须附加两封承诺信,一封承诺产品是按照GMP规范进行生产的,另一封承诺同意欧洲的相关审查机构进行现场检查)。
CEP由EDQM进行评估,申请成功后,EDQM将CEP证书直接颁发给原料药生产厂家。
CEP每五年需要更新一次,取得证书后,如果证书持有人或生产厂家未能达到相关要求,证书将会失效。
在更新方面,须注意以下几点:1、当发生可能影响产品质量、安全性或有效性的明显改变时,必须向欧洲认证的官方机构报告,以便重新评估或更新COS证书;2、如果没有发生任何可能影响产品质量、安全性或有效性的变化,每五年也需要更新一次与COS证书相关的认证文件;3、无论是否会影响到产品的质量,申请人对于任何行政管理方面的改变均需进行报告;EDMF则必需与使用该原料药的制剂的上市申请(MAA)同时进行,即必须由使用该原料药的欧洲终端用户申请。
各国DMF制度管理介绍

各國DMF制度管理介紹藥劑科技組盧可禎Drug Master File(簡稱DMF)為原料藥主檔案,是一套反映原料藥生產與品質管制的完整資料,包含生產廠、製程、規格、檢驗方法及安定性等方面的內容。
不同國家及地區對申請方式與DMF的編寫要求不同,以下就美國FDA、歐盟(歐洲共同體國家)、日本與韓國的DMF制度作介紹。
(一)、US FDA DMF 制度介紹簡介:根據美國的聯邦管理法(Code of Federal Regulation;CFR)第210及第211條之規定,任何進入美國市場的藥品(包括原料藥)都須向美國FDA申請註冊並遞交相關資料檔案,以便於FDA對藥物或藥品有全面的瞭解原料藥資料可與藥品查驗登記申請案之資料一併檢送,或可遞交一份DMF檔案給FDA,此由生產廠提供原料藥生產過程包括生產、加工、包裝與儲存的資料,以確定原料藥的生產過程是符合美國cGMP的要求,是為成品製劑取得核准之基礎。
但美國法律並不強制要求企業遞交DMF檔案,遞交DMF檔案與否為DMF持有者的決定。
一般準備DMF檔案可由DMF持有者以外的企業/公司於新藥臨床試驗申請(IND)、新藥申請(NDA)與學名藥申請(ANDA)時,直接引用這些資料,但這些企業/公司無法得知DMF實際內容。
申請過程:對於原料藥廠商來說,通過FDA批准主要有兩個階段:一是DMF檔案的登記,即向FDA進行DMF的備案,遞交DMF資料,經由FDA的藥品查驗及研究中心(Center for Drug Evaluation and Research, CDER)審核,初審合格後發通知函給申請人,函中會發一個DMF 登記號與說明DMF持有人的責任與義務。
二是當DMF檔案登記完成後,原料藥之使用者,即製劑藥廠於提出新藥臨床試驗申請(IND)、新藥申請(NDA)、學名藥申請(ANDA)或出口申請時,其原料藥的資料可直接引用該原料藥DMF檔案之登記號,FDA才會對DMF 技術性資料進行審查,必要時且對原料藥的生產廠進行GMP現場查核,確認原料藥生產現場與提交的DMF檔案內容一致。
COS和EDMF知识

DMF是英文Drug Master Fill的简写,中文直译为药物主文件,亦可理解为,药厂综合档案报告,其内容包括:生产、加工、包装和贮存某种药物的工厂的具体设施工艺流程、人员管理、产品分析等详细情况。
1)关于欧洲药典适用性证书(COS)与EDMF的异同欧洲药典适用性证书(COS, Certificate of Suitability), 是由欧洲药典委员会颁发的用以证明原料药品的质量是按照欧洲药典有关各论描述的方法严格控制的, 其产品质量符合欧洲药典标准的一种证书。
COS证书在欧洲药典委员会的二十八个成员国内得到承认,它与欧洲药物管理档案,即(EDMF,European Drug Master File)在程序和作用上相类似但又有不同:1.两者都是一种支持性材料,用于支持使用该原料药的制剂产品在成员国的上市申请(MAA);2.两者都是用于证明制剂产品中所使用的原料药质量的文件,是其它国家原料药品进入28个欧洲药典委员会成员国的市场必需提交的文件;3.COS和EDMF都可作为原料药进入欧洲市场的申请程序,可任选其一,没有必要重复申请;4.COS与EDMF的主要不同是COS可由原料药生产商独立申请,而EDMF则必需与使用该原料药的制剂的上市申请(MAA)同时进行。
也就是说,COS 的申请不需要事先找到欧洲代理商,而EDMF的申请就必需事先找到使用该原料药的欧洲代理商;5.COS 是一个证书,而EDMF只给一个参考号(Reference No.)。
申请COS的基本程序:1.按照有关当局的要求编写合格的文档(即Dossier);2.填写申请表格;3.通过银行向有关机构汇去有关费用,并递交上申请表格和所需的文档;4.有关当局接到递交后,经对资料评审合格后,颁发COS证书。
2)关于EDMF注册程序EDMF文件是欧洲药品上市申请文件或药品上市许可变更申请文件中有关活性物质(API)来源的技术支持文件,由API的生产厂家起草,经由生产制剂的上市申请厂家提交给相关成员国的药品评审机构。
DMF COS 注册介绍1

获得FDA认证的程序对于原料药来说,通过FDA批准主要有两个阶段:一是DMF文件的登记,要求递交的DMF文件对所申请的药品的生产和质量管理的全过程以及药品质量本身做一个详尽的描述。
FDA要为此文件保密,该文件是由FDA的药物评价及研究中心(Center for Drug Evaluation and Research, CDER)来审核。
二是当DMF文件的登记已经完成,而且在美国的原料药品终端用户提出了申请以后,FDA官员对原料药物的生产厂家进行GMP符合性现场检查,通过对药品生产全过程的生产管理和质量管理状况的全面考察,做出该原料药生产企业的生产和质量管理能否确保所生产药品的质量的判断。
FDA在现场检查的基础上做出是否批准该原料药品在美国市场上市的决定。
其基本程序如下所示:1.进行国际市场调研,摸清美国市场目前的销售情况,对市场发展趋势与走向做出正确的预测、分析和判断,选择好申请FDA批准的品种。
2.选择申请代理人和代理经销商,并签订委托协议书、签署委托书。
3.编写申请文件,原料药为DMF文件,由代理人完成申请文件终稿的编写并向FDA递交,取得DMF文件登记号。
4.FDA收到申请文件后,经初审合格后发通知函给申请人,并发给一个登记号,说明DMF文件持有人的责任和义务。
5.工厂按美国cGMP的要求进行厂房、设施设备的改造和并完善生产质量管理的各项软件和相关人员的强化培训。
6.应美国制剂生产厂家(即该原料药品的终端用户)的申请, FDA派官员到生产厂家按照FDA颁布的生产现场检查指南并对照已上报审核的DMF文件进行检查,FDA官员在生产现场的基础上出具书面意见给生产厂家并向FDA报告检查结果。
7.FDA审核批准后将审核结果通知生产厂家并输入美国海关的管理系统,该原料药品即获准直接进入美国市场。
8.生产厂家每年向FDA递交一份DMF修改材料,一般情况下, 每2~3年可能要接受一次复查DMF文件简介根据美国的联邦管理法规定,药品进入美国须向美国FDA申请注册并递交有关文件,化学原料药按要求提交一份药物管理档案(DMF)。
EDQM简介

一、欧洲药品质量管理局(EDQM)简介随着加入WTO和世界经济一体化进程的加快,在药品贸易方面,国内企业逐步走出国门,接受国际竞争和技术壁垒挑战。
在欧洲市场,欧洲药品质量管理局(EDQM)的COS证书是国内原料药成功进入欧洲的首选注册程序。
因此更多地了解欧洲药品质量管理局的管理系统以及COS证书的要求,成为很多企业的迫切需要。
欧洲理事会下属的EDQM是欧洲药品管理系统的核心,旨在保证在欧洲生产和销售的药品具有同等优良的品质,同时促进了资源的进一步集中和共享。
EDQM是在欧盟和欧洲理事会不断的合作中建立和发展起来的,它有效地满足了减少药品自由贸易中的技术壁垒以及合理使用公共健康资源的需要。
与欧盟相比,欧洲理事会拥有更多的成员国(现在是45个成员国,不久将变成46个),这意味着欧盟以外的欧洲国家可以与欧盟国家平等地参与药品方面的合作,同时也推动了这些国家加入欧盟的进程。
1、这种合作通常基于以下几个方面:A、国际协商会议(由欧盟和32个欧洲成员国签署的欧洲药典协定)共同体法(欧盟法规和解释)其它约定(欧洲理事会,欧盟委员会和欧洲药品评审局(EMEA)之间签署的);B、自发的技术团体:以欧洲和非欧洲国家的观察员身份,推动欧洲药品标准与各国法规之间的融合。
2、有关合作已经在以下领域开展:A、欧洲药典标准(约1850个专论和280个总论);B、欧洲药典适应性证书程序,这一程序适用于所有原材料生产商;C、官方药物实验室网络(OMCL):该网络目前包括了来自近40个国家的100多个合作实验室。
OMCL促进了欧洲国家之间药物检验结果的互认,并保证各国患者可以获得相同质量的药品。
二、欧洲药典适应性证书(COS / CEP)1、简介欧洲药典适应性证书COS——certificate of suitability to m onograph of European Pharmacopoeia。
又称CEP证书。
是由成立于1964年的欧洲药典委员会即欧洲药物质量理事会(EDQM)颁发的用以证明原料药品的质量是按照欧洲药典有关专论描述的方法严格控制的,其产品质量符合欧洲药典标准的一种证书。
DMF文件简介

21
DMF文件内容
3.2.S.3.2 Impurities 主要包括: 杂质研究工作 杂质
开发杂质研究检测方法 与发明商的制剂对比 与USP或其他官方承认的标准品对比 降解研究 杂质追踪 杂质的分离,鉴别,合成
杂质研究总结报告
产品,工艺,检测方法号 目的,介绍 参考文献 检测方法 工艺杂质 降解研究结果 稳定性试验杂质检测结果
的政府药品管理部门和制药行业接受。
10
DMF文件内容
11
DMF文件内容
CTD的主要内容
模块1:行政描述信息(不属于CTD) 模块2:通用技术文件总结(专家报告) 模块3:质量 模块4:非临床研究报告
模块5:临床研究报告
12
DMF文件内容
1. REGIONAL ADMINISTRATIVE INFORMATION (区域行政信息) 主要包括:DMF持有者,总部,制造地委托检测单位(如有), 代理情况 2. COMMON TECHNICAL DOCUMENT SUMMARIES 通用技术文件总结 (专家报告) 应该由有资格的人员,对DMF文件所涉及到的药品的主要概况,包 括工艺、物料、包装、稳定性、杂质等内容的概述。 欧洲要求由欧盟承认其资格的专家撰写。
3.2.S.4.5
Justification of Specification 成品规格标准设定说明
药品国际注册基础知识

a. FDA 简介 FDA即美国食品与药物管理局的英文缩写,全称US Food and Drug Administration。FDA成立于1906年,现有职工约7500人,其中药品局约300人。FDA隶属于美国卫生教育福利部,负责全国药品、食品、生物制品、化妆品、兽药、医疗器械及诊断品等的管理。
*
FDA管制要求 FDA要求所有的外国厂商(化妆品除外)在进入美国市场以前进行生产设施及产品注册,以便于FDA对产品的全称跟踪与监控。FDA是一执法机基本流程 编写CTD (Common Technical Document,即通用技术文件) 格式DMF文件; 通过一个美国代理(商),明确其责任,并在药品注册事务上与FDA联系,向FDA递交DMF文件,递交DMF无需任何费用; FDA收到DMF后,立即进行格式审查,如果没有问题,会马上给出一个DMF号; 制剂用户上市申请引用原料药的DMF; 原料药厂家提供3批符合客户标准,商业生产的样品; 制剂用户进行制剂稳定性实验,6个月证明符合要求; 制剂用户向FDA申请对原料药厂家进行检查; FDA检查前DMF缺陷信的整改;
*
年度工作 总结汇报
药品国际注册基础知识 药品国际注册的定义: 药品国际注册是指药品出口到国外时必须获得进口国的许可,即获得许可证,按照进口国对进口药品注册等级管理办法编制相关文件,提出申请,递交资料,获得许可证的过程。
汇报时间:12月20日
Annual Work Summary Report
*
EDMF的相关内容 a. EDMF简介 欧洲药物管理档案(EDMF,即European Drug Master File),是药品制剂的制造商为取得上市许可而必须向注册当局提交的关于在制剂产品中所使用的原料药基本情况的支持性技术文件,又称原料药主文件档案(ASMF,即Active Substance Master File)。
COS DMF EDFM

一、COS(Certificate of Suitability)指的是欧洲药典适用性认证,目的是考察欧洲药典是否能够有效地控制进口药品的质量,这是中国的原料药合法地被欧盟的最终用户使用的另一种注册方式。
这种注册途径的优点是不依赖于最终用户,可以由原料药生产厂商独立地提出申请。
中国的原料药生产厂商可以向欧盟药品质量指导委员会(EDQM)提交产品的COS认证文件(COS Dossier),申请COS证书,同时生产厂商必须要承诺产品生产的质量管理严格遵循GMP标准,在文件审查和可能的现场考察通过之后,EDQM会向原料药品的生产厂商颁发COS证书。
如果作为最终用户的欧盟成员国制剂生产企业准备采用中国生产的原料时,只要在注册文件或变更文件中附上该产品的COS证书复印件即可非常容易地获得批准。
COS认证是否需要现场检查,对企业的GMP管理水平有哪些要求?随着美国、欧盟和日本三方在药品注册程序和法规上的相互协调,欧盟在进口的原料药注册中逐步接近美国FDA的偏重现场GMP检查的办法,今后有可能对每一家提出COS认证的生产厂家进行现场的GMP检查。
自1999年开始,原料药生产企业在申请COS认证的技术文件后面必须要附加两封承诺信,一封信承诺说产品是按照GMP规范进行生产的,另一封信要承诺同意欧盟的相关审查机构进行现场检查。
如果欧盟EDQM的GMP审查越来越频繁,甚至最终变成为一种必要的审查手段,生产厂家就应当对此做出充分的准备,以使自身的GMP管理状况能够适应欧盟的检查。
欧盟的GMP检查与国内的GMP认证有以下差别:首先,欧盟的GMP检查依据的ICH Q7A的指导纲要,厂家要参照此指导进行自身检查;其次,所有的质量管理文件、操作规范(SOP)和各种生产管理表格、标牌、标签和生产记录都应当具备中英文对照,能够让国外的审查官员看懂;其三,要对员工进行GMP 的全员培训,了解并适应国外检查的特点。
COS认证过程对企业是有积极意义的,会使企业的GMP管理达到国际水平,而且随着美、欧、日三方协调的进一步发展,通过欧盟的GMP检查和COS认证最终有可能直接进入美国和日本市场,至少会使美国FDA的注册变得更为容易。
DMF和EDMF的编制区别
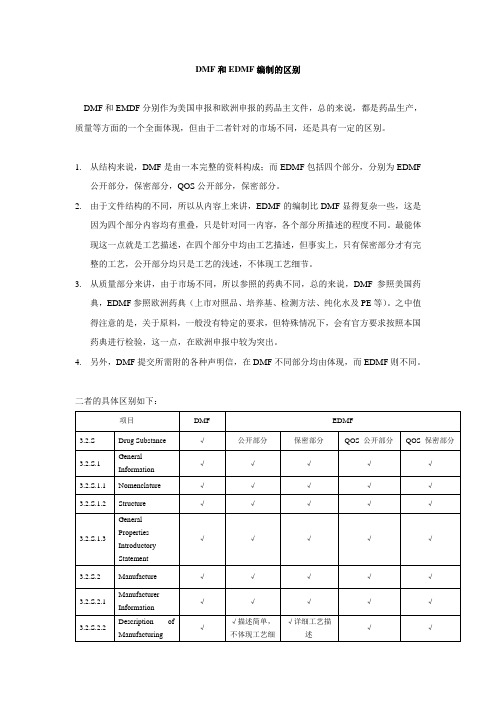
DMF和EDMF编制的区别
DMF和EMDF分别作为美国申报和欧洲申报的药品主文件,总的来说,都是药品生产,质量等方面的一个全面体现,但由于二者针对的市场不同,还是具有一定的区别。
1.从结构来说,DMF是由一本完整的资料构成;而EDMF包括四个部分,分别为EDMF
公开部分,保密部分,QOS公开部分,保密部分。
2.由于文件结构的不同,所以从内容上来讲,EDMF的编制比DMF显得复杂一些,这是
因为四个部分内容均有重叠,只是针对同一内容,各个部分所描述的程度不同。
最能体现这一点就是工艺描述,在四个部分中均由工艺描述,但事实上,只有保密部分才有完整的工艺,公开部分均只是工艺的浅述,不体现工艺细节。
3.从质量部分来讲,由于市场不同,所以参照的药典不同,总的来说,DMF参照美国药
典,EDMF参照欧洲药典(上市对照品、培养基、检测方法、纯化水及PE等)。
之中值得注意的是,关于原料,一般没有特定的要求,但特殊情况下,会有官方要求按照本国药典进行检验,这一点,在欧洲申报中较为突出。
4.另外,DMF提交所需附的各种声明信,在DMF不同部分均由体现,而EDMF则不同。
二者的具体区别如下:。
编写DMF知识培训 Drug Master File

DMF意义:
药物管理当局的要求 不注册不能获得市场准入 制剂生产商对原料药审核 无此文件制剂生产商不能对原料进行评审 贸易推销 作为提供服务的一个部分
DMF的编写格式和内容:
1. Drug Mster File Guideline(内容旧格式) 2. 按照CTD格式进行编写 格式 纸质 纸质或eCTD
药物主文件(药物主文案)
英文缩写:
FDA:Food and Drug Administration 美国食品药品管理局 EDQM:European Directorate for the Quality of Medicines (欧洲药品质量理事会) SFDA:State Food and Drug Administration(国家食品药品管 State Administration( 理局) TGA:Therapeutic Goods Administration(澳大利亚药物管 ( 理局) 理局) MCC:Medicines Control Council (南非药品管理局)
DMF的编写格式和内容:
1. Drug Master File Guideline:基本不用 2. CTD格式 Common Technical Document CTD文件是美、欧、日三方国际协调会议(International Conference of Harmonization,简称ICH)共同认可 的药品申请文件格式,由五个模块组成.
参考:
图书:SFDA培训中心图书信息 美国FDA的CGMP现场检查》 现场检查》 《美国 的 现场检查 《美国药品申报与法规管理》 美国药品申报与法规管理》 原料药质量控制系列文件及APIC“Q7如何实施”》 如何实施” 《ICH原料药质量控制系列文件及 原料药质量控制系列文件及 如何实施
EDMF与COS(CEP)的区别

EDMF与COS(CEP)的区别EDMF与COS(CEP)的区别在于:1 EDMF与药品制剂的批准有关,应当与药品注册文件同时递交到每一个相关国家;COS是一个独立的过程, 在任何时候只要提交了相关文档就可以获得证书。
2 EDMF申请过程只适用于药物活性成分,可以是已知的,也可以是新的化合物.;COS适用于药物活性成分与赋型剂,但前提是该物质被欧洲药典所收载。
3 EDMF包括两个部分:公开部分与保密部分。
COS的文档只有一个部分。
4 EDMF只被申报的国家所认可;COS被所有欧洲药典签约国接受,包括欧盟及其成员国和欧洲药典大会观察员国家。
5 CEP送交EDQM进行评估;EDMF由相关国家药审机构进行评估,如英国药品评估局。
按照欧洲共同体的相关法规,欧盟成员国以外的国家生产的原料药,要想进入欧洲市场,用于欧洲的药物制剂生产,需要提交和登记欧洲药物管理档案(EDMF,European Drug Master File)或欧洲药典适用性证书(COS ,又称CEP,Certificate of Suitability to Monographs of the European Pharmacopoeia)的复印件。
1 EDMF简介EDMF是药物制剂中所使用原料药的基本情况的支持性技术文件,适用于原料药的生产厂家不是药品上市许可证申请人的情况。
EDMF文件包含原料药生产工艺、杂质和理化性质等方面的详细技术资料和实验数据。
EDMF分为公开部分和保密部分。
如果欧洲的药品评审机构经验证,递交的EDMF申请文件是真实有效的,则给予药品上市许可证的申请人一个EDMF登记号。
这样作为原料药的生产厂家,就可以将原料药出口到欧洲,用于该药物制剂的生产。
欧盟成员国内的药物制剂生产厂家要合法地使用中国生产的原料药,就必须要向注册当局提交变更原料药的申请,并提交相应的支持性技术文件EDMF,这时通常要向原料药生产厂家或贸易中间商索取EDMF文件,得到注册当局批准之后才可以使用。
DMF和EDMF的编制区别
DMF和EDMF编制的区别
DMF和EMDF分别作为美国申报和欧洲申报的药品主文件,总的来说,都是药品生产,质量等方面的一个全面体现,但由于二者针对的市场不同,还是具有一定的区别。
1.从结构来说,DMF是由一本完整的资料构成;而EDMF包括四个部分,分别为EDMF
公开部分,保密部分,QOS公开部分,保密部分。
2.由于文件结构的不同,所以从内容上来讲,EDMF的编制比DMF显得复杂一些,这是
因为四个部分内容均有重叠,只是针对同一内容,各个部分所描述的程度不同。
最能体现这一点就是工艺描述,在四个部分中均由工艺描述,但事实上,只有保密部分才有完整的工艺,公开部分均只是工艺的浅述,不体现工艺细节。
3.从质量部分来讲,由于市场不同,所以参照的药典不同,总的来说,DMF参照美国药
典,EDMF参照欧洲药典(上市对照品、培养基、检测方法、纯化水及PE等)。
之中值得注意的是,关于原料,一般没有特定的要求,但特殊情况下,会有官方要求按照本国药典进行检验,这一点,在欧洲申报中较为突出。
4.另外,DMF提交所需附的各种声明信,在DMF不同部分均由体现,而EDMF则不同。
二者的具体区别如下:。
DMF基础知识

药物主档案(DMF) 药物主档案(DMF)
是不会有被通过或不通过审查之状况 最后的上市许可是针对申请者授权
DMF的种类 DMF的种类
● 美
有五种类型的DMF 国:有五种类型的DMF 有五种类型的 第一类: 制造场所与设备( 目前已废止 ) 第一类: 制造场所与设备( 第二类: 第二类:药品物质及药品中间体 第四类: 第四类:准备用材料 第五类: 第五类:补充资
欧洲DMF 欧洲DMF – 表列内容
F部分
1. 稳定度 1.1. 1.2. 1.3.湿度75% 稳定测试,在温度 30 °C/湿度60% (如果需要) 稳定测试,在温度 25 °C/湿度60% 结果的评估
美国 DMF – 表列内容
A 部份 设备与代理商之地址 B 部份 药物表列号码 C 部份 承诺事项 D 部份 公司组织及人事 E 部份 建筑物及设施 F 部份 装备 G 部份 组成及成份 H 部份 生产及制程控制
● 对DMF的任何修正均必须作确效并必须展示此改变对最终产品的品质没有改
变(如不纯物档案,药效,生物可用性及其他).
● 在许多案例中,任何修改必须先向权责单位(FDA)提出并讨论后再决定执行. ● 对DMF内容改变的管理规则,叙述在“大量活性成份在核准后之修正
1(BACPAC 1 )” 指导原则,此部份包含了在制程中早期的中间体,一直 BACPAC 到 最后的中间体.
● CoS的有效期是5年,而且必須在有改變發生時
更新(與DMF所使用的程序相同).
● ●
DMF之所有者可以授权信函(或参考函或使用许可函)使其DMF成为申请文件之一部份. DMF之所有者可以授权信函(或参考函或使用许可函)使其DMF成为申请文件之一部份. 之所有者可以授权信函 DMF成为申请文件之一部份 权责单位不会将DMF作为一份独立文件审查,它仅被作为上市许可的授权, 权责单位不会将DMF作为一份独立文件审查,它仅被作为上市许可的授权,如果没有申 DMF作为一份独立文件审查 请案用到此一DMF,则官方将不会对此DMF作审核. 请案用到此一DMF,则官方将不会对此DMF作审核. DMF DMF作审核
常见重要药用名词解释

常见重要药用名词解释创新药Innovative drug,创新药,是指具有新颖的化学结构式(NCE)或者分子式(NME)的全新药物,具有需要较长的时间投入和资金投入,成功几率小的特点。
创新药物的开发厂家一般都会申请药物专利进行保护(即专利药),获得批准上市后会申请商品名以利于推广(即品牌药)。
根据创新程度,一般在同一个“药物作用机制类别”中首个获批上市的药叫“me-first 药”,其余的称作“me-too 药”。
专利药 Patented drug,专利药,指申请了化合物专利保护药的药品。
专利药只有拥有这些药品专利权的公司才能生产,或授权其他公司生产。
专利药的研发过程包括药物发现阶段,临床前开发,新药临床申请(IND),新药I、II、III 期临床试验,新药上市申请(NDA)。
由于专利药开发通常需耗时10-15 年、耗资8-12 亿美元,通常价格昂贵。
原研药,是指专利过期后,由原专利药开发厂商生产的药品。
品牌药 Brand drug,品牌药,又叫商品名药,是指药品开发生产厂家为创新药上市申请的商品名称,以利于向医生进行学术推广,药品专利到期后可继续使用。
仿制药Generic drug,仿制药,是指与品牌药在剂量、安全性和效力、质量以及适应症上相同的一种仿制品。
仿制药是品牌药的替代药品,由于研发费用低,通常价格也较低,具有降低医疗支出、提高药品可及性、提升医疗服务水平等重要经济和社会效益。
仿制药与被仿制药具有相同的活性成分、剂型、给药途径和治疗作用,可通过“简略新药申请”(ANDA)上市,但需等待品牌药专利过期后才能销售。
药物通用名International Nonproprietary Names for Pharmaceutical Substances,简称INN。
药物通用名是由各国政府规定的、国家药典或药品标准采用的法定药物名称,对某一特定的药物分子,通用名通常是唯一的。
通用名仿制药INN generic,通用名仿制药,是指欧美发达国家一般只允许新的化学结构、新的活性成分的药物药品拥有商品名,仿制药不得使用商品名称,只能使用药物的通用名。
《国际药事法规》知识点整理
《国际药事法规》一、名词解释(常见缩写)R&D Research and development药物研发药房制剂Pharmacy compoundingNDP National Drug Policy国家药物政策GPP Good Pharmacy Practice优良药房工作规范二、简答和论述1、概述FDA组织结构及职能分工答:七中心:(1)药物评价和研究中心(CDER),负责评审所有药品(包含OTC,处方药,仿制药,含氟牙膏,止汗剂,去屑洗发水,防晒霜等)(2)生物制品评价和研究中心(CBER),评审疫苗、血浆、血液制品等(3)医疗器械和放射健康中心(CDRH),评审医疗器械和放射性产品(4)食品安全和应用营养中心(CFSAN),负责管理本国和进口食品(新鲜肉禽除外)、饮料、食品补充剂和化妆品等。
(5)兽药中心(CVM)(6)全国毒理研究中心(NCTR)(7)烟草产品中心CTP(center for Tobacco Products)两室:1、局长办公室OC2、执法办公室ORA,负责相关领域内所有活动提供指导。
2、EMA的组织机构EMA强调“科学性”由EMEA-----EMA(1)董事会(2)五大部门:2.1人用药品发展与评估中心2.2患者健康保护中心2.3兽药与产品数据管理中心2.4信息与交流技术中心2.5行政中心(3)主要委员会:3.1管理委员会(独立的咨询委员会) 3.2人用药品委员会(CHMP)3.3兽用药品委员会CVMP3.4罕见病药品委员会COMP3.5草药委员会HMPC 3.6儿科委员会PDCO 3.7前沿疗法委员会CAT 3、英国药品管理机构3.1药品和健康产品管理局(MHRA)3.2顾问委员会:3.2.1人用药品委员会(CHM)3.2.2顺势疗法产品注册顾问委员会3.2.3英国药典委员会(BPC)3.2.4草药顾问委员会3.2.5药品广告的独立审查组3.2.6边缘产品分类的独立审查组3.2.7兽药委员会(VPC)4、FDA新药管理及FDA影响新药审批的方式(1)以下四类情况作为新药管理:1.1药品含有新化学实体(NEC)作为该药的活性成分1.2药品含有已有的活性成分,但该成分在美国从未作为医学用途使用过,包括在国外上市的物质以及在自然界新发现的物质1.3药品先前已被FDA批准上市,但现在建议新的用法、适应症1.4药品先前已经被FDA批准上市,但现在建议的剂型、给药途径或其他重要条件不同于先前批准的药品,包括处方药转非处方药。
(整理)欧洲药品上市申请所需认证EDMF文件、COSCEP认证文件的简介和区别比较

欧洲市场药品上市申请所需EDMF文件、COS(CEP)认证简介和比较EDMF文件简介欧洲药物管理档案(EDMF,即European Drug Master File)是药品制剂的制造商为取得上市许可证而必须向注册当局提交的关于在制剂产品中所使用的原料药的基本情况的支持性技术文件。
它的申请必须与使用该原料药的制剂的上市许可申请同时进行。
当原料药物的生产厂家(ASM,即the active substance manufacture)不是药品制剂上市许可证的申请人时,也就是说当制剂生产厂家使用其他厂家生产的原料药物生产制剂时,为了保护原料药物的生产及质量管理等方面有价值的技术机密而由原料药物的生产厂家提交给欧洲官方机构的文件。
分为公开部分和保密部分。
与FDA的DMF涵盖药品生产的全过程CMC(Chemistry,manufacturing and control)不同,欧洲DMF则强调第一个C,即chemistry,具体来说,EDMF的主要内容是药物及其相关杂质的化学,包括化学机构及结构解析、化学性质、杂质及其限度、杂质检查等等。
COS认证简介:Cos(certificate of suitability)认证指的是欧洲药典适用性认证,目的是考察欧洲药典是否能够有效控制进口药品的质量,这是中国的原料药合法地被欧盟最终用户使用的另一种注册方式。
优点是不依赖最终用户可以由原料药生产厂家独立地提出申请。
中国的原料药生产厂家可以向欧盟药品质量指导委员会(EDQM)提交产品的COS认证文件,申请CEP证书,同时生产厂家必须要承诺生产的质量管理严格遵循GMP标准,在文件审查和可能的现场考察通过之后,EDQM会向原料药的生产厂家颁布COS认证。
1999年,EDQM制定COS提出,必须附加一封承诺其申报的原料药是按照国际GMP规范(ICH Q7A)进行生产的,另一封要求承诺同意欧洲GMP检查机构官员进行现场检查,目前检查频率逐年提高。
COS简介

COS(CEP)证书简介一、COS证书与EDMF登记的产生背景及比较1964年,欧盟成立统一的公共卫生委员会(Public Health Committee)和欧洲药典委员会(Committee of European pharmacopoeia),颁布了在所有成员国内都具有法律效力的欧洲药典,目前成员国共31个:奥地利、比利时、波黑、克罗地亚、塞浦路斯、捷克共和国、丹麦、芬兰、法国、德国、希腊、匈牙利、冰岛、爱尔兰、意大利、卢森堡、荷兰、挪威、葡萄牙、斯洛伐克共和国、斯洛文尼亚、西班牙、瑞典、瑞士、前南斯拉夫联盟、土耳其、英国等,基本上涵盖了欧洲的主要国家。
欧盟以外国家生产的原料药要进入欧洲市场,原料药生产厂家首先要向欧洲用户(制剂上市申请者)提供上市申请所需要的支持性文件,即欧洲药物档案(European Drug Master File ,EDMF),供欧洲用户上市申请时使用。
EDMF 注册申请要求原料药生产厂家必须向每一个用户提供EDMF文件,而且欧盟药管部门也不向生产商颁发任何的证明性文件。
为减少原料药生产厂家和有关评审机构的大量的重复性工作,根据1999年12月22日生效的欧洲议会公共卫生委员会AP-CSP(99)4决议,由欧洲药典委员会的27个成员国启动了一个新的证书程序,即"欧洲药典适用性证书"(Certificate of Suitability of European Pharmacopoeia, COS或CEP)。
原料药要进口到欧洲,用于生产药物制剂,其生产厂商只需提交COS证书的复印件给欧洲用户就可进行市场申请。
EDMF 登记和COS证书都是原料药进入欧洲市场有效而必需的支持性材料,用于使用该原料药的制剂产品在欧洲的上市申请(MAA),它们之间的不同处是什么呢?1)评审方式上的不同:EDMF是由申请制剂上市国家的相关机构进行评审。
针对不同的制剂,不同的评审机构有不同的侧重,因而会对同一份EDMF申请文件有不同的要求。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
DMF、COS(CEP)和EDMF一、原料药申请FDA以及DMF(1)根据市场调研,摸清美国市场目前的销售情况,对市场发展趋势与走向做出正确的预测、分析,在此基础上,选好申请FDA认证的品种。
(2)选择好申请代理人(代理人)和代理经销商(经销商)并向FDA递交委托代理的证明书即委托书。
(3)编写申请文件,化学原料为DMF,交代理人修改定稿后,由代理人向FDA递交,取得DMF分配号和NDC登记号。
(4)FDA收到材料后将发函通知表示何时收到何产品的DMF资料,DMF分配号是多少,该产品由谁供货,由谁代理、谁经营,其次说明根据NDA(新药申请)程序,FDA将来检查。
(5)工厂按GMP要求进行厂房、设备和各项管理方面的准备工作。
其间,代理和代理商可能合作几次预检。
(6)FDA派员检查。
检查按他们的检查指南并对照DMF文件逐项对照,查后当场写出书面意见并由检查人员向FDA报告检查结果。
(7)FDA审核批准后通知代理商,由代理商通知外贸部签约,可知该药品已获准可直接进入美国市场。
(8)每年通过代理人向FDA递交一份DMF修改材料,2-3年接受一次复查。
根据美国的联邦管理法规定,在美国生产经营产品者须向美国食品药物管理局(FDA)申请注册并递交有关文件、化学原料药按要求提交一份新药申请(NDA)的药物管理档案:DMF。
DMF是药物管理档案(DRUGMASTERFILE)的英文名首字母缩写词,是一份文件,由生产厂提供的详细的某药品生产过程的资料,便于FDA对该厂产品有个全面了解。
DMF定为DMF系一机密参考资料,内容包括:生产、加工、包装或贮存某一物质时所用的具体厂房设施,生产方法或物件等详尽的资料。
国际上常把DMF称作“企业文化”或生产单位文化,许多国家法规要求有生产设施和监控的资料以确定药品的生产是通过GMP得到保证的。
呈报的DMF有五种类型:第一类,生产地点和厂房设施、人员;第二类,中间体、原料药和药品;第三类型,包装物料;第四类,辅料、着色剂、香料、香精及其他添家剂;第五类,非临床数据资料和临床数据资料,每一DMF应只不过含有一类资料。
上报的DMF原件在收到时经鉴定确定在格式和内容上符合规定要求,FDA就会确认收到并对其指定一个DMF编号,FDA收受DMF以及将DMF编号并不说明FDA对这一文件表示同意或不同意。
FDA为证实申请书而要查阅DMF资料前,DMF持有者必须向FDA呈报授权信件一式二份,容许FDA查阅DMF,如果FDA审查员在审查NDA(新药申请书)ANDA(新药简略申请书)等申请书时需查阅DMF,发现资料不全,则会在回复申请时把不足处指出,由申请者告知DMF持有者把DMF加以改正。
DMF持有者的责任有:改动DMF时作所需通知了授权可查阅DMF的人员名单;指定代理商;DMF所有权的转让,当DMF持有者希望终止DMF时,应向DMF部门上报一份申请书说明终止理由。
随着加入世贸组织和世界经济一体化进程的加快,我国民族医药产业就要面对一个新的课题――走出国门参与国际竞争,这既是机会也有风险存在。
既往原料药出口的经验教训很多,类似汉姆、凯华这种国外(或国外背景)公司利用我们不熟悉外部情况又急于出口的心理,以很小的代价就得到证书持有人、贸易代理权等很大价值的权益,在早期的COS证书和FDA认证通过中绝大多数权利被代理公司把持,国内生产企业只是为他人做嫁衣得到微不足道的一点利润,完全没有市场的主动权且还有技术机密无法保证的风险。
现在企业的自主意识得到很大提高,但信息不明了、理解混乱的情况仍然没得到改善,特别是后来介入的咨询公司因为出发点不一样也都说法不一,这让企业更是无法判断。
所以现在国内企业迫切需要一个权威的声音,需要与国外法规当局直接接触,鉴于此,中国药学会与FDA、COS专业咨询机构北京康利华咨询服务有限公司在11月举办了欧洲药品(COS / CEP、EDMF、GENERIC)国际研讨会,邀请EDQM官员,Q7A核心专家等参与研讨培训,国内70多家企业近百名代表参加了研讨学习。
在为期三天的会议中,专家们介绍的欧盟药品注册知识覆盖面广,内容丰富,经过一番整理,现作一简单的介绍。
二、欧洲药品质量管理局(EDQM)简介随着加入WTO和世界经济一体化进程的加快,在药品贸易方面,国内企业逐步走出国门,接受国际竞争和技术壁垒挑战。
在欧洲市场,欧洲药品质量管理局(EDQM)的COS证书是国内原料药成功进入欧洲的首选注册程序。
因此更多地了解欧洲药品质量管理局的管理系统以及COS证书的要求,成为很多企业的迫切需要。
欧洲理事会下属的EDQM是欧洲药品管理系统的核心,旨在保证在欧洲生产和销售的药品具有同等优良的品质,同时促进了资源的进一步集中和共享。
EDQM是在欧盟和欧洲理事会不断的合作中建立和发展起来的,它有效地满足了减少药品自由贸易中的技术壁垒以及合理使用公共健康资源的需要。
与欧盟相比,欧洲理事会拥有更多的成员国(现在是45个成员国,不久将变成46个),这意味着欧盟以外的欧洲国家可以与欧盟国家平等地参与药品方面的合作,同时也推动了这些国家加入欧盟的进程。
1、这种合作通常基于以下几个方面:A、国际协商会议(由欧盟和32个欧洲成员国签署的欧洲药典协定)共同体法(欧盟法规和解释)其它约定(欧洲理事会,欧盟委员会和欧洲药品评审局(EMEA)之间签署的);B、自发的技术团体:以欧洲和非欧洲国家的观察员身份,推动欧洲药品标准与各国法规之间的融合。
2、有关合作已经在以下领域开展:A、欧洲药典标准(约1850个专论和280个总论);B、欧洲药典适应性证书程序,这一程序适用于所有原材料生产商;C、官方药物实验室网络(OMCL):该网络目前包括了来自近40个国家的100多个合作实验室。
OMCL促进了欧洲国家之间药物检验结果的互认,并保证各国患者可以获得相同质量的药品。
三、欧洲药典适应性证书(COS / CEP)1、简介欧洲药典适应性证书COS——certificate of suitability to monograph of European Pharmacopoeia。
又称CEP证书。
是由成立于1964年的欧洲药典委员会即欧洲药物质量理事会(EDQM)颁发的用以证明原料药品的质量是按照欧洲药典有关专论描述的方法严格控制的,其产品质量符合欧洲药典标准的一种证书。
它是随着欧洲市场的一体化进程的逐步发展而于1992年出现的,其目的就是为了减少生产厂家和有关评审机构的大量的重复性工作,促进欧洲药品市场的一体化进程。
与美国FDA对原料药品要求的药物管理档案(DMF)相比,欧洲COS 证书申请文件更注重的是药物本身的性质,包括化学结构及结构解析、化学性质、杂质及其限度、质量控制、稳定性和安全性研究等等。
要取得此证书,生产商需要提交一份详细的档案,其中可能含有机密资料。
此证书用户表明通过应用欧洲药典有关专论,及证书上所列出的必要方法,能够检查药物原料和辅料是否适用于药品的生产,也就是说,从这一特定方法(包括原材料)生产的药物中的所有可能杂质和污染都能够由有关专论的要求完全控制。
欧洲药品质量管理局(EDQM)的COS证书程序是基于欧洲公共健康委员会部分表决通过的AP-CSP(93)4号决议而产生的。
COS证书的目的是评估和判断使用欧洲药典专论控制原料药的化学纯度、微生物品质和TSE(Transmitting animal Spongiform Encephalopathy agent 传播性动物海绵状脑病体)风险的适用性。
这一程序显示了各部门之间典型的合作关系,如CHMP(人用药委员会),CVMP(兽用药委员会)和欧洲药典委员会,这些部门在指导工业界的同时,共同努力寻找实用的解决方案,并在不使评估程序复杂化的前提下提高质量保证。
证书管理处已经明确表示:对于已有欧洲药典标准的原料药生产商,应选用COS证书程序。
由欧洲药典委员会官员、CHMP/CVMP联合工作组、CHMP生物技术工作组、CVMP免疫产品工作组、草药产品工作组和欧盟委员会代表、EMEA、EDQM 等共同组成的指导委员会,在实施COS程序时贯彻了磋商、协调和合作的原则,指导委员会每年举行一次会议,以保持有关许可、药典和适用性证书政策的连贯性。
除了负责政策方针的指导委员会,EDQM还针对化学物质和TSE风险物质分别设立了两个技术顾问团,由文件审查专家组成。
顾问团主要负责处理证书申请文件审查员提出的任何技术或科学问题。
截止到2004年3月底,EDQM共颁发COS证书1520份,其中有关化学药品的证书970份,占63.8%需做TSE风险评估的生物原料证书430件,占28.28%,两者兼有的(即需要做TSE风险评估的化学药品)43件,占2.82%,被撤销的证书75件,占4.93%,实际有效的证书总数为1445件。
中国只有24家制药企业的45件COS证书,撤销2件,实际有效的COS证书为43件,仅占全球颁发证书总额的2.97%,全部集中在化学药品。
2、COS证书的适用范围COS证书适用于已收录于欧洲药典中的以下物质:—合成或提取的有机或无机物(原料药或药用辅料);—通过发酵获得的非直接基因产物,即微生物的代谢产物,无论该微生物是否经过传统方法或r-DNA技术修饰;—具有传播动物海绵状脑病(TSE)危险的产品(注:此类产品可单独申请COS证书或进行TSE危险性评估,也可两项共同申请)。
COS证书不适用于直接基因产物(蛋白质),源于人类组织的产品、疫苗、血液制品等。
3、COS证书产生的背景、发展与现状由于有着众多的人口及发达的经济,欧洲是世界上巨大的药品消耗地区,同时也是向外输出药物制剂的最大地区。
与美国一样,出于对自身资源和环保的考虑,决大多数欧洲国家严格控制原料药的生产规模。
欧洲制药工业发达而且历史悠久,集中了很多世界主要跨国制药企业,成为世界上向外输出药品制剂最多的地区。
随着欧洲逐渐一体化,欧盟版图不断扩大,作为一个整体,它正成为世界上最大的原料药进口市场之一,也是我国众多原料药生产厂家的重要目标市场。
由于欧洲国家一般较小,经济市场一体化一直是大势所趋, 进程在不断加速。
早在1964年,欧盟就成立了统一的公共卫生委员会(Public Health Committee)和欧洲药典委员会(Committee of European Pharmacopoeia),颁布了在所有成员国内都具有法律效力的欧洲药典。
欧洲药典委员会也由最初的八个成员国发展到现在三十一个成员国,包括:奥地利、比利时、波黑、克罗地亚、塞浦路斯、捷克共和国、丹麦、芬兰、法国、德国、希腊、匈牙利、冰岛、爱尔兰、意大利、卢森堡、荷兰、挪威、葡萄牙、斯洛伐克共和国、斯洛文尼亚、西班牙、瑞典、瑞士、前南斯拉夫联盟、土耳其、英国等,基本上涵盖了欧洲的主要国家。