bcr-abl阴性非经典MPN的认识现状
存在异常染色体克隆的原发性血小板增多症转化为急性白血病1例

存在异常染色体克隆的原发性血小板增多症转化为急性白血病1例孙克;华川;王威;秦伟【期刊名称】《检验医学》【年(卷),期】2016(031)007【总页数】3页(P632-634)【作者】孙克;华川;王威;秦伟【作者单位】解放军第二五二医院检验科,河北保定071000;解放军第二五二医院检验科,河北保定071000;保定市第一中心医院东院检验科,河北保定071000;解放军第二五二医院血液科,河北保定071000【正文语种】中文【中图分类】R446.11原发性血小板增多症(essential thrombocythemia,ET)是一种少见的获得性慢性骨髓增殖性肿瘤(myeloproliferative neoplasm,MPN),以血小板持续增高为特征,临床上主要表现为出血和血栓。
ET患者进展为白血病较为少见,仅为0.6%~6.1%[1]。
与慢性粒细胞白血病(chronic myeloid leukemia,CML)不同,ET患者无特异的或独特的染色体核型异常,非特异性核型异常较为少见,仅为3.0%~5.3%[2-3]。
我们报告了1例存在异常染色体克隆的ET转化为急性髓系白血病M5的病例,并复习相关文献进行学习。
患者,女,62岁,曾于18年前因左上腹疼痛,在当地医院诊断“脾大”,并发现血小板异常增高,骨髓象诊断“血小板增多症”,予羟基脲及干扰素治疗,效果较理想。
后又于2013年8月在北京协和医院就诊,行骨髓活检发现骨髓组织较少,造血组织明显增多,脂肪组织减少,粒红比例增高,巨核细胞多见。
骨髓像:骨髓干抽,粒系以中性分叶核粒细胞为主,占81%,其余各系减少,全片可见巨核细胞39个,血小板成堆可见,考虑MPN;染色体核型分析:47,XX,+1,+9,der(1;14)(q10;q10)[5]/46,XX[12];骨髓JKA2 V617F基因阳性。
于2015年5月出现高热,腹胀明显加重,血像:白细胞59.94×109/L,血红蛋白97 g/L,血小板953×109/L,外周血涂片提示原始、幼稚细胞12%。
原发性血小板增多症

实验室检查
(一)血液 血小板(1000~3000)×109/L,涂片中血小板聚集成堆,大小不一,偶见巨核细胞碎片。 聚集试验中血小板对胶原、ADP及花生四烯酸诱导的聚集反应下降,对肾上腺素的反应消失。 白细胞增多(10~30)×109/L之间,中性粒细胞碱性磷酸酶活性增高。 半固体细胞培养有自发性巨核细胞集落形成单位(CFU-Meg)形成,有利于本病的诊断。
治疗
(二)降低血小板数 血小板大于1000×109/少血小板量,常用于妊娠、手术前准备以及骨髓抑制药不能奏效时。
谢谢
原发性血小板 增多症
定义
原发性血小板增多症(ET)为造血干细胞克隆性疾病,外周血血小板计数明显增高而功能异常,骨
髓中巨核细胞增殖旺盛,约50%~70%病人有 JAK2 V617F 基因突变。
流行病学
在发达国家及地区,约1/3的BCR-ABL-阴性MPN为ET所致。人群流行病学研
究发现,每年新发ET的发病率为1-2.5例/100,000人。由于ET患者的预期寿命似乎 与正常人群无异,所以本病的患病率要高很多,估计总患病率为9-24例/100,000 人。
发病率因种族、性别和年龄而异。在美国,黑人的发病率高于非西班牙语裔白 人,西班牙语裔白人和亚太裔的发病率较低。患者多为女性,男女发病率比约为 2:1。发病率随年龄而增长,诊断时患者的中位年龄为60岁,但多达20%的患者可 能不到40岁。
临床表现
可达半数的ET患者是在因其他原因接受全血细胞计数检查时顺带发现了血小板增多。其 他则是因疾病相关性症状(如,头痛、头晕或视觉改变)或并发症(例如,血栓形成、出血或孕早 期妊娠丢失)而就诊。与真性红细胞增多症不同,ET患者中很少见皮肤瘙痒,其发生率低于5%。 由于各研究在患者选择及对“血管舒缩症状”、“出血”或“血栓事件”的定义不同,所报道 的ET相关并发症的发生率存在较大差异。
骨髓增殖性肿瘤治疗与发病机制的研究进展

万方数据
自堕堑:鲞旦疸!Q!!生!旦箜丝鲞箜!塑!业!!堂!!堡!鲢嗵!鱼!!!坐P!!里!!』!!Y垫!!!!些!兰!塑!:!
・409・
小板计数较高。
1.1.3
罕见,但见于19名的有MPN转化的白血病[io]。IKZFl
突变可能导致JAK.STAT信号通路活化。
NFl和NRAS基因是MAPK信号转导途径的组成 成分。MAPK信号转导途径是JAK2的下游主要目标基 因之一。最近的研究显示MAPK信号转导途径的基因 如NFl和NRAS在MPN患者中较常见。NRAS基因突变 导致其蛋白活性增强,从而影响JABSTAT信号通路8。
U2AFl基因突变
NFI是RAS途径的负调控基因,NFI突变导致其失 活,减少对RAS途径的抑制。 1.2转录因子突变
1.3
1。3.1
MPL基因突变
MPL编码的蛋白是一种促血小板生成素(TPO) 的受体[州。MPL基因突变见于3%~5%的ET和8%
~1
DNA甲基化相关基因突变
TET2基因突变
0名的PMF患者,最常见的突变位于1 0号外显
TET2基因突变编码的甲基胞嘧啶双加氧酶可将
甲基胞嘧啶转化为5.羟甲基胞嘧啶,从而调控DNA甲 基化状态’s1。TET2基因突变见于16%的PV、5名的ET 及1 7%的PMF患者。
JAK2基因突变包括V61 7F突变和l 2号外显子 突变。V61 7F突变点位于JAK2基因第1 4号外显子 上,l 849位的乌嘌呤点突变为胸腺嘧啶,使得JAK2 第61 7位的缬氨酸错义编码为苯丙氨酸,该突变见于 90%以上的PV和50%左右的ET和PMF患者[1I。突变
以及免疫调节等重要的生物学过程。这条信号通路主
骨髓纤维化患者的临床特点和骨髓病理学特征分析

骨髓纤维化患者的临床特点和骨髓病理学特征分析刘亚琳;王玮;单林玲;贺鹏程;刘海波【摘要】目的:探讨骨髓微环境中纤维组织、血管系统的病理改变对部分血液系统疾病的病程及预后的影响。
方法重新评估该院诊治的骨髓纤维化患者100例骨髓病理切片,尤其是骨髓微环境中纤维组织、血管系统的病理改变,结合患者的临床症状、肝脾肿大程度、血细胞计数、骨髓细胞学检查及细胞遗传学等相关临床资料,分析骨髓纤维组织增生在血液系统疾病病程网状纤维中的意义。
结果所有骨髓活检标本(Gomori)染色均有不同程度阳性,原发性骨髓纤维化(PMF)15例,继发性骨髓纤维化( SMF)85例,SMF病因学分析显示慢性粒细胞白血病、多发性骨髓瘤、原发性血小板增多症易合并骨髓纤维化,SMF纤维化程度较轻,治疗后纤维组织增生程度有所降低。
PMF Gomori染色多为强阳性,改善微循环治疗后纤维化程度缓解不显著,差异无统计学意义。
结论 PMF较SMF纤维化程度重,SMF与疾病的病程有一定的相关性。
%Objective This study was aimed to observe the clinical and pathological feature of myelofibrosis, and to investigate the re-lationship between such pathological changes and the course and prognosis in hematological diseases.Methods One hundred patients with myelofibrosis ( MF) were retrospectively analyzed,and the pathological features especially the changes of fibrous tissue and vascu-lar system in bone marrow mircroenvironment were reanalyzed.Results There were positive inordinately in all Gomori staining,there were 15 cases of primary myelofibrosis( PMF) and 85 cases of secondarymyelofibrosis( SMF) .The etiological analysis shows that chro-nic myelocytic leukemia(CML),multiple myeloma(MM),essentialthrombocythemia(ET) and MF frequently occur in combinationg. The degree of fibrosis is mild with SMF,and the proliferative degree of fibrosis tissue will decrease after the treatment of primary dis-ease.The Gomori staining were strong positive in most of PMF ,and there were low remission after the treatment for improving microen-virenment.Conclusion The degree of fibrosis of PMF is severer than SMF.There is correlation to some degree between SMF and dis-ease course.【期刊名称】《安徽医药》【年(卷),期】2015(000)012【总页数】4页(P2335-2338)【关键词】骨髓纤维化;病理特征;骨髓活检【作者】刘亚琳;王玮;单林玲;贺鹏程;刘海波【作者单位】西安交通大学第一附属医院血液内科,陕西西安 710061;西安市第九医院血液内科,陕西西安 710054;安康市中医医院检验科,陕西安康 725000;西安交通大学第一附属医院血液内科,陕西西安 710061;西安交通大学第一附属医院血液内科,陕西西安 710061【正文语种】中文骨髓纤维化(myelofibrosis,MF)是指骨髓造血组织被纤维组织代替,以致影响造血功能所产生的病理状态,在临床上分为原发性(primary myelofibrosis,PMF)和继发性(secondary myelofibrosis,SMF)两大类,原发性骨髓纤维化(PMF)是一种以贫血、脾大、外周血中出现未成熟粒细胞、幼红细胞、泪滴状红细胞以及CD34+细胞增多,骨髓纤维化、骨硬化为特点的慢性克隆性髓系疾病。
骨髓增殖性肿瘤症状管理现状-1119-2020年华医网继续教育答案 - 副本

2020年华医网继续教育答案-1119-骨髓增殖性肿瘤
症状管理现状
备注:红色选项或后方标记“[正确答案]”为正确选项
详见:医搜题
(一)骨髓增殖性肿瘤发病机制研究进展
1、MPN发病机制包括()
A、MPL突变
B、JAK2基因突变
C、C-CBL融合基因
D、以上均是[正确答案]
2、JAK2基因由()个高度保守的同源结构域组成
A、7[正确答案]
B、3
C、5
D、6
3、Bcr-abl融合基因的编码的()蛋白,具有酪氨酸激酶活性,可以调控细胞生长,参与细胞持续增殖信号传导或抑制凋亡,从而使白血病细胞大量增殖
A、TP10
B、P210[正确答案]
C、K200
D、J10
4、socs3的主要作用是()
A、适度负调细胞因子信号[正确答案]
B、介导促红细胞生成素
C、促血小板生成素
D、调节并促进细胞增殖
5、JAK2外显子()突变由Scott等于2007年在对JAK2/V617F阴性的PV和PMF 患者研究中所报道。
bcr-abl阴性MPN诊断和治疗

BCR/ABL阴性骨髓增殖性肿瘤的诊断和治疗1.概念根据2008年WHO造血与淋巴组织肿瘤分类1所提出的概念,骨髓增殖性肿瘤(MPN)包括以下8种类型:慢性髓性白血病BCR/ABL1阳性(CML),真性红细胞增多症(PV),原发性血小板增多症(ET),原发性骨髓纤维化(PMF),慢性中性粒细胞白血病(CNL),慢性嗜酸性粒细胞白血病非特质型(CEL,not otherwise specified),肥大细胞增多症(Mastocytosis),以及骨髓增殖性肿瘤未分类型(MPN-U)。
基于Dameshek2最初对于骨髓增殖性疾病的归纳,PMF、PV、ET与CML合称为经典MPN,流行病学特点、临床表现及实验室特征均有别于其他四种“非经典MPN”。
而其中CML存在特异性分子学异常BCR/ABL基因的重排,具有诊断意义。
而相应的靶向药物受体酪氨酸激酶抑制剂(TKI)伊马替尼一线应用所累积的大量临床资料和目前二代TKI的临床试验结果均证明,对大部分CML患者的治疗现在已能够达到细胞遗传学甚至分子学起效,并且能够实现预防疾病进展,延长生存期,改善生活质量,甚至治愈疾病的目标。
因此除CML之外其余各类型MPN可统称为BCR/ABL阴性MPN3。
随着对JAK2等分子标志的研究不断深入,有学者认为ET、PV、PMF可能是同一种疾病在不同时期的表现,类似于CML分三期但本质是一个连续的整体,进而提出了“连续统一体模型(continuum model)”的假设4:高危PMF和AML可能代表了疾病进展状态,而ET和PV则体现了疾病慢性期的特点;在这种由ET、PV“进展”至PMF、ET/PV后骨纤,再“进展”至AML的过程中,JAK2等位负荷、遗传不稳定性、有丝分裂重组、基因组环境、获得其他突变以及临床治疗等因素都可能起到了调控作用;与CML不同,这种“进展”速度很慢而且并非必然,且患者可能在整个病程中的任何时点“发病”。
2016年世界卫生组织(WHO)髓系肿瘤新分类
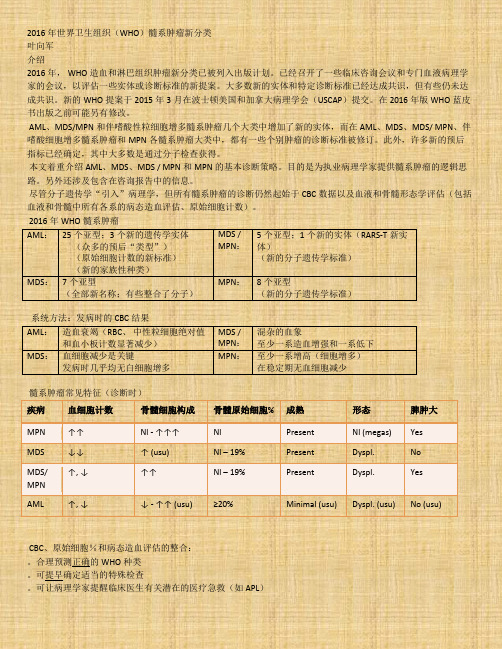
2016年世界卫生组织(WHO)髓系肿瘤新分类叶向军介绍2016年, WHO造血和淋巴组织肿瘤新分类已被列入出版计划。
已经召开了一些临床咨询会议和专门血液病理学家的会议,以评估一些实体或诊断标准的新提案。
大多数新的实体和特定诊断标准已经达成共识,但有些仍未达成共识。
新的WHO提案于2015年3月在波士顿美国和加拿大病理学会(USCAP)提交。
在2016年版WHO蓝皮书出版之前可能另有修改。
AML、MDS/MPN和伴嗜酸性粒细胞增多髓系肿瘤几个大类中增加了新的实体,而在AML、MDS、MDS/ MPN、伴嗜酸细胞增多髓系肿瘤和MPN各髓系肿瘤大类中,都有一些个别肿瘤的诊断标准被修订。
此外,许多新的预后指标已经确定,其中大多数是通过分子检查获得。
本文着重介绍AML、MDS、MDS / MPN和MPN的基本诊断策略。
目的是为执业病理学家提供髓系肿瘤的逻辑思路。
另外还涉及包含在咨询报告中的信息。
尽管分子遗传学“引入”病理学,但所有髓系肿瘤的诊断仍然起始于CBC数据以及血液和骨髓形态学评估(包括血液和骨髓中所有各系的病态造血评估、原始细胞计数)。
2016年WHO髓系肿瘤AML:25个亚型;3个新的遗传学实体(众多的预后“类型”)(原始细胞计数的新标准)(新的家族性种类)MDS /MPN:5个亚型;1个新的实体(RARS-T新实体)(新的分子遗传学标准)MDS:7个亚型(全部新名称;有些整合了分子)MPN:8个亚型(新的分子遗传学标准)系统方法:发病时的CBC结果AML:造血衰竭(RBC、中性粒细胞绝对值和血小板计数显著减少)MDS /MPN:混杂的血象至少一系造血增强和一系低下MDS:血细胞减少是关键发病时几乎均无白细胞增多MPN:至少一系增高(细胞增多)在稳定期无血细胞减少髓系肿瘤常见特征(诊断时)疾病血细胞计数骨髓细胞构成骨髓原始细胞% 成熟形态脾肿大MPN ↑↑ Nl - ↑↑↑ Nl Present Nl (megas) Yes MDS ↓↓ ↑ (usu) Nl – 19% Present Dyspl. NoMDS/MPN↑, ↓ ↑↑ Nl – 19% Present Dyspl. Yes AML ↑, ↓ ↓ - ↑↑ (usu) ≥20% Minimal (usu) Dyspl. (usu) No (usu)CBC、原始细胞%和病态造血评估的整合:。
JAK2基因突变的发现改变了骨髓增殖性肿瘤的分类和诊断

JAK2基因突变的发现改变了骨髓增殖性肿瘤的分类和诊断朱平(北京大学第一医院)从2005年报道JAK2 V617F突变发生于慢性骨髓增殖性疾病(myeloproliferative disorder,MPD)[1-3]以来,这个基因突变的发现改变了MPD的分类和诊断。
以往MPD包含慢性粒细胞白血病(CML),真性红细胞增多症(PV),原发性血小板增多症(ET),和原发性骨髓纤维化(PMF),慢性中性粒细胞白血病(CNL),慢性嗜酸细胞白血病/高嗜酸细胞综合症(CEL/HES)等多种疾病。
在修订的2008 WHO分类系统中,有无JAK2突变成为MPD主要的诊断指标[4],例如,如果JAK2突变阳性,血红蛋白增加,骨髓红系细胞明显增生可以诊断PV,即使血红蛋白低于以往WHO规定的指标值,但却持续超过正常值20g/L的PV也可以确诊。
如果JAK2突变阳性,血小板仅仅持续大于450X109/L,骨髓巨核细胞增生,可以诊断ET。
不仅MPD的诊断发生革命性进展[5],连名称都改变了。
MPD改称为骨髓增殖性肿瘤(myelo- proliferative neoplasm,MPN),MDS/MPD(myelodysplastic /myeloproliferative disorder)称为MDS/MPN。
新分类的MPN 中还包含了肥大细胞病(MCD)。
而以前的慢性嗜酸细胞白血病/高嗜酸细胞综合症(CEL/HES)分别重新划分为CEL,HES和有PDGFRA, PDGFRB或者FGFR1基因异常的嗜酸细胞增高症3种MPN,并强调这类疾病的本质是肿瘤。
现已明确,MPN具有共同的干细胞起源的克隆型遗传特征,临床出现不同表型是突变影响的蛋白、酪氨酸激酶及相关分子的不同构型和异常的信号转导所造成的。
2008年WHO修订MPN诊断标准的依据是多数患者都具有JAK2突变,主要是JAK2 V617F, 还有JAK2外显子12突变。
1例骨髓增生异常-骨髓增殖性肿瘤不能分类型的分析

㊃个案分析㊃1例骨髓增生异常-骨髓增殖性肿瘤不能分类型的分析税国顺1,黄莺2,何代莉21.重庆市荣昌区妇幼保健院检验科,重庆402460;2.重庆永荣矿业有限公司总医院检验科,重庆402460关键词:骨髓增生异常-骨髓增殖性肿瘤;骨髓形态学;白血病D O I:10.3969/j.i s s n.1673-4130.2021.08.031中图法分类号:R557文章编号:1673-4130(2021)08-1019-04文献标志码:C骨髓增生异常-骨髓增殖性肿瘤(M D S-M P N)是一组临床表现㊁实验室检查和细胞形态特征上既有骨髓增生异常综合征(M D S)表现又有骨髓增殖性肿瘤(M P N)表现的髓系肿瘤[1]㊂在骨髓形态学诊断实际工作中往往接触的是不同的患者,因遗传学和生物学差异,其预后㊁治疗方案㊁分子易感性及转归可能完全不同㊂按世界卫生组织(WHO)分类,综合临床表现㊁细胞形态学㊁染色体㊁流式细胞㊁基因指标等[2]信息做出准确的诊断,对临床有较好的指导作用㊂骨髓增生异常-骨髓增殖性肿瘤不能分类型(M D S-M P N,U)是WHO在1999年提出的一个新病种,目前国内报道不多㊂经中国知网(C N K I)搜索查询,2013年至今, M D S-M P N其他类型多有报道㊂黎建云等[3]㊁葛仁英等[4]㊁吴学琼等[5]对M D S-M P N,U进行了报道㊂笔者现将1例M D S-M P N,U患者的诊治过程报道如下㊂1临床资料女,62岁,农民,头昏㊁乏力1周,晕厥1h于2019年11月13日11:52入院㊂既往史:近半个月来一直牙疼不适,在外口服药物治疗㊂查体:体温36.8ħ,脉搏78次/分,呼吸22次/分,血压131/74mm H g,发育正常,营养中等,形体正常,急性面容,面色苍白,神志欠清,呼之能应,对答稍差,抬入病房,查体合作㊂嘴唇无发绀,心㊁肺㊁腹部未见阳性体征,头颅无畸形,左前额部见约4c mˑ4c m的血肿,高出皮肤,皮肤无破溃,触压痛明显;双眼无充血㊁压痛,无上睑下垂,巩膜无黄染,双侧瞳孔等大等圆,直径约0.35c m,对光反射存在㊂听力良好,鼻腔通畅,耳鼻无出血及脑脊液漏㊂四肢肌力正常,肌张力不高㊂病理反射:双侧霍夫曼征㊁巴宾斯基征㊁查多克征㊁奥本海姆征㊁戈登征阴性㊂脑膜刺激征:颈软无强直㊂布鲁辛斯基征及双侧克尼格氏征阴性㊂入院诊断如下㊂(1)晕厥待查:①短暂性脑缺血发作②脑梗死?③脑出血?④低血糖晕厥?⑤心源性晕厥?⑥其他?(2)左额部皮下血肿㊂诊疗经过如下㊂入院后完善相关检查,头颅C T 显示:(1)双侧基底节区及半卵圆中心多发性腔隙性脑梗死;(2)脑萎缩,深部脑白质脱髓鞘改变;(3)左额部皮下血肿㊂腹部彩超显示:(1)心脏各腔室大小正常;(2)主动脉瓣局限性反流;(3)左室舒张功能减退㊂颈部血管彩超示:(1)双侧颈动脉内膜粗糙,左侧颈内动脉走行稍弯曲;(2)双侧椎动脉未见明显异常㊂经颅多普勒显示:左侧大脑中动脉㊁大脑前动脉血流速度增快㊂血液相关指标检查结果见表1㊂11月13日第1次血细胞分析手工分类200个有核细胞,中性粒细胞(N E U)58.0%,单核细胞(MO N) 2.0%,杆状核细胞6.0%,幼稚粒细胞24.0%,晚幼红细胞5.0%,网织红细胞(R e t)5.4%㊂直接抗人球蛋白试验弱阳性㊂肝功能正常,肾功能正常,血糖(G L U)9.41mm o l/L,凝血4项筛查正常,D-二聚体(D-D i m e r)2.30m g/L,乳酸脱氢酶(L D H)1084U/ L㊁α-羟丁酸脱氢酶(α-H B D H)835U/L㊁心肌肌钙蛋白I(c T n I)阴性,降钙素原(P C T)0.30n g/m L㊂入院后予头孢米诺抗感染,血塞通改善循环,奥拉西坦营养脑细胞㊂请血液科主任会诊考虑白血病可能性,于2019年11月14日转血液科进一步治疗㊂予A型R H(D)阳性红细胞悬液共4U纠正贫血㊂行骨髓形态学检查:骨髓增生明显活跃,粒细胞系统(以下简称粒系)占79.50%,红细胞系统(以下简称红系)占11.75%,粒红比例为6.77㊂粒系异常增生,原始细胞比例增高,占11%,后期细胞各阶段可见,形态可见S 颗粒㊁发育差㊁假性P-H及环形核等核畸形变现象,嗜碱性粒细胞比例增高㊂红系增生活跃,以中晚幼红细胞增生为主,形态可见芽孢㊁花瓣等核畸形变现象㊂环片一周见巨核细胞650个,产板巨核细胞254个,血小板少见㊂小巨核及多圆核巨核细胞易见㊂铁染色:细胞外铁++,细胞内铁粒幼细胞占比21%,未见环形铁粒幼细胞㊂血常规:白细胞计数增高,粒系前本文引用格式:税国顺,黄莺,何代莉.1例骨髓增生异常-骨髓增殖性肿瘤不能分类型的分析[J].国际检验医学杂志,2021,42(8):1019-1022.体细胞易见,嗜碱性粒细胞比例增高,血小板少见,无核红细胞形态大小不一㊂考虑急性髓系增殖性病变,建议到上级医院做M I GM 分型检查㊂陆军军医大学第二附属医院全军血液病中心骨髓细胞形态学检查报告显示,粒巨两系异常增生,伴三系病态造血,提示髓系肿瘤,类型倾向于M D S -M P N ,建议做相关B C R /A B L 融合基因及染色体核型分析㊂骨髓病理检查报告显示,骨髓增生极度活跃,幼稚单核细胞较易见,请结合M I GM 分型鉴别M D S -M P N 或急性髓系白血病(AM L )㊂AM L ㊁M D S ㊁M P N 免疫学分型流式报告示:淋巴细胞(L YM )占有核细胞(N C )的5.53%,为成熟T B N K 细胞;N E U 占N C 的79.45%,比例正常,C D 13/C D 16可见发育异常,C D 15减弱C D 56+㊂MO N 占N C 的0.42%,比例减低㊂红系及碎片红细胞占N C 的8.73%,比例正常㊂血型糖蛋白A +可见C D 71+㊂嗜酸性粒细胞占N C 的0.17%,嗜碱性粒细胞占N C 的0.09%,C D 38+D R-C D 11c +C D 123++C D 22+㊂前体细胞群B 占N C 的3.82%,C D 34+C D 117+D R +C D 7+C D 13+C D 38++C D 123+C D 33+C D 11c +M P O -为异常髓系体细胞㊂意见:异常髓系前体细胞增生伴粒系发育异常,考虑M D S 或M D S -M P N ㊂遗传学荧光原位杂交(F I S H )分析报告显示,B C R /A B L 融合基因阳性细胞占比0.0%㊂分子生物学血液病相关基因P C R 检测报告显示,WT 1基因数为1050,A B L 基因数为560000,WT 1/A B L 为0.19%,B C R -A B L 基因数为0,A B L 基因数为710000㊁B C R -A B L /A B L 基因数为0(B C R -A B L 基因是B C R -A B L P 210和B C R -A B L P 190混合型)㊂血液病相关基因T E L -A B L ㊁T E L -J A K 2㊁A M L 1-M D S 1/E V 11/M T G 16㊁M L L -A F 4㊁M L L -A F 9㊁M L L -A F 6/A F 10/E L L /E N L ㊁M L L -A F 17/A F 1q /A F 1q/A F X /S E P T 6㊁F 1P 1L 1-P D G F R A ㊁E T V 6-P D G F R A ㊁T E L -P D G F R B ㊁N U P 98-H o x A 13/H o x C 11/H o x D 13/H o x A 9H o x A 11/P M X 1㊁B C R -A B L ㊁AM L 1-E T O ㊁CB F β-MY H 11㊁P M L -R A R a ㊁P L Z F /S T A T 5b -R A R a ㊁F 1P 1L 1/P R K R 1A /N U M A /N P M -R A R a ㊁D E K -C A N ㊁NP M -M L F 1㊁E 2A -P B X 1㊁E 2A -H L F ㊁S I L -T A L 1㊁T E L -AM L 1全部阴性㊂按照WHO 提出的形态学㊁免疫学㊁遗传学㊁分子生物学结合的M I GM 诊断模式,本病例诊断为M D S -M P N ,U ㊂患者的骨髓象见图1㊂表1 患者不同时间的血液相关指标的检查结果时间W B C (ˑ109/L )N E U (%)L YM (%)MO N (%)E O S (%)B A S O (%)R B C (ˑ1012/L )11月13日32.3875.820.73.00.00.52.3811月13日һ28.4778.716.54.10.10.61.9911月14日21.0580.315.73.50.10.41.9011月15日*18.6077.919.12.60.00.42.31时间H B (g/L )H C T (%)M C V (f L )M C H (p g )M C H C (g /L )R DW -C V (%)R DW -S D (f L )P L T (ˑ109/L )11月13日6623.8100.027.827816.557.110411月13日һ5619.497.928.228816.255.38411月14日5318.999.528.028116.255.68111月15日*6722.798.329.129615.452.266注:W B C 为白细胞,N E U 为中性粒细胞,L YM 为淋巴细胞,MO N 为单核细胞,E O S 为嗜酸性粒细胞,B A S O 为嗜碱性粒细胞,R B C 为红细胞,H B 为血红蛋白,H C T 为血细胞比容,M C V 为红细胞平均体积,M C H 为平均血红蛋白含量,M C H C 为平均血红蛋白浓度,R DW -C V 为红细胞分布宽度变异系数,R DW -S D 为红细胞分布宽度标准差,P L T 为血小板;һ表示11月13日复查结果;*表示4U 红细胞悬液输注后的检查结果㊂图1 患者的骨髓象2治疗入院后予头孢米诺抗感染,血塞通改善循环,奥拉西坦营养脑细胞㊂输注红细胞悬液纠正贫血㊂明确诊断后,患者及家属放弃进一步治疗㊂随访后该患者5个月后病逝㊂3讨论M D S是起源于造血干细胞的一组异质性髓系恶性克隆性疾病[6],主要特征是髓系细胞发育异常,表现为无效造血㊁难治性血细胞减少,具有高危向AM L 转化的风险[7]㊂WHO在2008年提出的分型方案分为难治性血细胞减少伴单系发育异常(R C U D),包括难治性贫血(R A)㊁难治性中性粒细胞减少(R N)㊁难治性血小板减少(R T)㊁难治性贫血伴环状铁粒幼红细胞(R A R S),难治性血细胞减少伴多系发育异常(R C M D),难治性贫血伴原始细胞增多-1(R A E B-1),难治性贫血伴原始细胞增多-2(R A E B-2),M D S-末分类(M D S-U),M D S伴单纯5q-㊂M P N是一组起源于造血干细胞,一系或多系髓系细胞(包括红系㊁粒系和巨核系)增殖为主要特征的克隆性造血干细胞肿瘤,骨髓有核细胞增多,增殖的细胞可向终末分化成熟,多不伴发育异常;外周血一系或多系细胞增多,外周器官浸润,常伴有肝脾肿大㊂M P N包括慢性髓细胞白血病(C M L)㊁真性红细胞增多症(P V)㊁原发性骨髓纤维化(P M F)㊁原发性血小板增多症(E T)㊁慢性嗜酸性粒细胞白血病(C E L)㊁慢性中性粒细胞白血病(C N L)和肥大细胞增多症[8]㊂M D S-M P N患者中具有高白细胞数㊁贫血或血小板减少(也可增加),以及不同程度病态造血特征㊂骨髓和外周血原始细胞百分数通常<20%㊂尽管常见脾肿大,但患者临床常表现为M D S或M P N[9]㊂WHO在2016年提出M D S-M P N分类如下:慢性粒单细胞白血病(C MM L);不典型C MM L,B C R-A B L1阴性(a C M L);幼年型粒单核细胞白血病(J MM L); M D S-M P N伴环形铁粒幼细胞和血小板增多;M D S-M P N,U[10]㊂近年来,随着二代测序在临床的应用, M D S-M P N的基因突变谱系得以解析,从而诊断较前更为精准㊂M D S-M P N,U是WHO在1999年提出的一个新病种,由于历史尚短,在许多方面还需要认识和充分的理解㊂M D S-M P N,U为初诊时,临床㊁实验室和形态学上既有M D S也有M P N特征,符合M D S-M P N诊断标准,但又不符合C MM L㊁J MM L或a C-M L诊断条件者㊂特征为一系或一系以上血细胞增多,表现为血小板增多(ȡ450ˑ109/L),或白细胞计数增加(ȡ13ˑ109/L),伴有或不伴有明显的脾脏肿大,和未伴有M D S或M P N病史㊂在M D S特征中,骨髓一系或二系细胞异常增生,表现在外周血中一系或二系血细胞减低;在M P N特征中有一系或多系病态造血,在外周血中为一系或多系血细胞增高㊂近期没有使用细胞毒药物或生长因子治疗可以解释相关M D S-M P N特征的病史㊂无B C R-A B L1,也无P D G-F R A㊁P D G F R B或F G F R1基因重排,无t(3;3)(q21; q25)或i n v(3)(q21q25)㊂黎建云等[3]报道的2例患者中白细胞均升高(33.6ˑ109/L㊁16.8ˑ109/L),脾脏都有进行性增大㊂1例血小板正常(112ˑ109/L),年龄23岁,既往有5年苯接触史㊂1例血小板降低(59ˑ109/L)㊁年龄43岁㊂葛仁英等[4]报道的2例患者中,1例白细胞升高(24.1ˑ109/L),血小板降低(90ˑ109/L),脾肿大,年龄47岁;1例白细胞降低(1.58ˑ109/L),血小板升高(633ˑ109/L),肝脾肿大,年龄70岁㊂吴学琼等[5]报道的1例患者白细胞正常(9.5ˑ109/L),血小板异常升高(1359ˑ109/L),初诊肝脾未见肿大,年龄69岁㊂这3篇研究报道的病例其临床㊁实验室和形态学上既有M D S的特征也有M P N特征,符合M D S-M P N诊断标准,但又不符合C MM L㊁J MM L或a C M L诊断条件㊂C L A R A等[11]指出,生存率低的患者血小板减少发生的可能性较大,可能代表更具侵袭性的表型㊂本例患者发病近期无细胞毒性药物或生长因子治疗病史,肝脾无肿大㊂血细胞分析结果显示:白细胞计数增高,呈增殖性;血红蛋白㊁红细胞㊁血小板未输血前呈进行性下降,重度贫血,红细胞平均体积偏大,血小板明显减少㊂形态学上粒细胞病态造血特征明显,以粒系病态造血为主,红系和巨核系也有病态表现,符合M D S-M P N的特点;未见环形铁粒幼细胞(可与M D S-M P N伴环形铁粒幼细胞和血小板增多鉴别);免疫学方面,流式细胞学检测可见粒系异常发育,也可以见到异常表型的异常原始细胞,MO N占N C的比例为0.42%,比例减低(可与C MM L㊁a C-M L㊁J MM L鉴别)㊂遗传学方面,B C R/A B L基因阴性;分子生物学相关基因检测全部阴性㊂卢兴国等[12]指出,一些之前没有发现M P N慢性期,初诊时已经转化为M D S的患者,如果不能确定M P N病史,归类为M D S-M P N,U是适当的㊂所有这些都支持本例患者M D S-M P N,U的诊断㊂M D S-M P N,U是特异性最差的M D S-M P N亚型,发病率尚不清楚,但约占所有髓系恶性肿瘤的5%,年龄中位数为71岁,其他共同特征包括脾肿大,单核细胞计数低,20%~30%的患者J A K2V617F阳性,且目前还没有公认的M D S-M P N,U的特定细胞遗传学或分子特征,而M D S -M P N ,U 的细胞遗传学研究主要用于排除其他类似的疾病[11]㊂M D S -M P N ,U 患者少见,迄今尚无共识治疗方案㊂可依据患者个体情况选用去甲基化药物㊁免疫调节剂(如来那度胺)㊁羟基脲等降细胞药物治疗,在临床实践过程中注意个体化评估和处理㊂如有合适供体且患者自身状况允许可以考虑造血干细胞移植(H S C T )[13]㊂随着发病分子机制的不断阐释,分子靶向治疗有望改善这类患者的整体疗效㊂参考文献[1]O R A Z I A ,G E R M I N G U.T h e m y e l o d y s p l a s t i c /m ye l o -p r o l if e r a t i v e n e o p l a s m s :m y e l o pr o l -i f e r a t i v e d i s e a s e s w i t h d y s pl a s t i c f e a t u r e s [J ].L e u k e m i a ,2008,22(7):1308-1319.[2]A R B E R D A ,O R A Z I A ,HA S S E R J I A N R ,e t a l .T h e2016r e v i s i o n t o t h e W o r l d H e a l t h O r g a n i z a t i o n (WHO )c l a s s i f i c a t i o n o f m y e l o i d n e o pl a s m s a n d a c u t e l e u k e m i a [J ].B l o o d .2016,127(20):2391-2405.[3]黎建云,涂传清,唐玫琴,等.不能分类的骨髓增生异常/骨髓增殖性疾病转化为急性髓系白血病2例并文献复习[J ].临床血液学杂志,2013,26(7):474-476.[4]葛仁英,徐旭燕,毛汉文,等.骨髓增生异常/骨髓增殖性肿瘤-不能分类2例报告并文献复习[J ].内科急危重症杂志,2014,20(1):15-17.[5]吴学琼,齐小宁,李文佳,等.骨髓增生异常/骨髓增殖性肿瘤-不能分类白血病急变1例并文献复习[J ].内科急危重症杂志,2015,21(5):390-391.[6]P A N G W W ,P L U V I N A G E J V ,P R I C E E A ,e t a l .H e m -a t o p o i e t i c s t e m c e l l a n d p r o ge n i t o r c e l l m e c h a n i s m s i n m y e l o d y s p l a s t i c s yn d r o m e s [J ].P r o c N a t l A c a d S c i U S A ,2013,110(8):3011-3016.[7]T E F F E R I A ,V A R D I MA N J W.M y e l o d y s p l a s t i c s yn -d r o m e s [J ].N E n gl J M e d ,2009,361(19):1872-1885.[8]朱雨,何广胜.世界卫生组织2016年骨髓增殖性肿瘤及骨髓增生异常综合征/骨髓增殖性肿瘤分类更新解读[J ].中国实用内科杂志,2016,36(8):658-661.[9]J A F F E E S ,HA R R I S N L ,S T E I N H ,等.造血与淋巴组织肿瘤WHO 分类[M ].周小鸽,陈辉树,译.4版.北京:诊断病理学杂志社,2012.[10]叶向军,卢兴国.2016年更新版‘WHO 造血和淋巴组织肿瘤分类“之髓系肿瘤和急性白血病修订解读[J ].临床检验杂志,2016,34(9):686-689.[11]C L A R A J A ,D A V I D A S ,P A D R O N E .C l i n i c a l m a n a ge -m e n t of m y e l o d y s p l a s t i c s y n d r o m e /m y e l o pr o l i f e r a t i v e n e -o p l a s m o v e r l a p s yn d r o m e s [J ].C a n c e r B i o l M e d ,2016,13(9):360-372.[12]卢兴国,叶向军,徐根波.骨髓细胞与组织病理诊断学[M ].北京:人民卫生出版社,2020:566-580.[13]肖志坚.骨髓增生异常综合征/骨髓增殖性肿瘤的诊断和治疗[J ].中国实用内科杂志,2018,38(2):93-97.(收稿日期:2020-09-13 修回日期:2020-12-17)本文引用格式:邹自英,徐常军,黄鑫,等.圣乔治诺卡菌致老年患者肺诺卡菌病1例[J ].国际检验医学杂志,2021,42(8):1022-1024.㊃个案分析㊃圣乔治诺卡菌致老年患者肺诺卡菌病1例邹自英,徐常军,黄 鑫,许黎莉,黄 英,甘立果四川省成都市第五人民医院检验科,四川成都611130关键词:圣乔治诺卡菌; 诺卡菌病; 肺部感染D O I :10.3969/j.i s s n .1673-4130.2021.08.032中图法分类号:R 446.5文章编号:1673-4130(2021)08-1022-03文献标志码:C肺诺卡菌病影像学检查和临床表现均无特异性,磺胺类㊁碳青霉烯类㊁氨基糖苷类㊁利奈唑胺等多种抗菌药物对该病可能有效[1],体外培养生长非常缓慢,易被其他快生长定植菌覆盖,难于检出,极易被误诊为普通细菌性肺炎,使得病情迁延反复,重则危及生命㊂笔者于2020年5月连续两次成功从1例老年女性患者痰液中分离出诺卡菌,经飞行时间质谱(MA L -D I -T O F )结合形态学鉴定为少见的圣乔治诺卡菌,获得病原学诊断依据并进行目标治疗后,疗效显著,现将诊治过程报道如下㊂1 临床资料患者,女,78岁,因 反复咳嗽㊁咳痰30余年,加重伴痰中带血10余天 ,于2020年5月9日入院㊂查体:体温36.6ħ,心率76次/分,呼吸20次/分,血压121/74mm H g㊂患者自诉本次入院前30余年,因受凉后出现咳嗽㊁咳白色泡沫痰,痰不易咳出,量少,不带血,无特殊气味;咳嗽白天重于夜间,初始无胸闷㊁气紧,无畏寒㊁发热,无心悸㊁胸痛,无潮热㊁盗汗,夜间。
不典型慢性粒细胞白血病

不典型慢性粒细胞白血病(aCML)aCML属于MDS/MPN,主要发病年龄在60-67岁。
发病率明显低于CML,该类病人具有和BCR/ABL阳性患者相同的症状体征,如:脾大、以粒细胞为主的白细胞升高、轻度贫血等,与CML的主要区别是:BCR/ABL阴性,有明显的粒细胞病态造血。
诊断标准为:1.外周血白细胞数增高WBC≧13×109/L.2.无ph染色体或BCR-ABL1融合基因。
3.无PDGFRA或PDGFRB重排。
4.中性粒细胞前体细胞(早、中、晚幼粒细胞)≧10%白细胞。
5.嗜碱细胞<2%。
6.单核细胞<10%。
7.骨髓粒系增生明显活跃,粒系发育异常,伴或不伴红系发育异常。
8.骨髓及外周血原始细胞<20%。
慢性粒单核细胞白血病(CMML)CMML以外周血单核细胞增多为典型表现的MDS/MPN,主要发病年龄在60岁以上,主要特征为PB单核细胞>1×109/L, PB和BM原始细胞<20%,无ph染色体和BCR-ABL1融合基因。
血液学可表现MPD为主或MDS为主。
诊断标准为:1.外周血单核细胞>1×109/L。
2.没有ph染色体或BCR-ABL1融合基因。
3.没有PDGFRA或PDGFRB重排。
4.外周血或骨髓原始细胞<20%。
5.有一系或多系髓系异常,如无发育异常或仅有轻微发育异常。
但其他条件符合者,如有以下表现者仍可诊断为CMML:1)。
骨髓细胞有获得性克隆性细胞遗传学异常;2)或单核细胞持续增高,至少3个月以上;3)除外可引起单核细胞增多的其他原因。
慢性中性粒细胞白血病(CNL)CNL是一种罕见的MPN,表现为外周血及骨髓有持续增高的中性粒细胞,并广泛浸润各组织,尤以脾肿大为甚,不伴有发热、炎症、肿瘤和其他引起白细胞增高的任何原因。
无ph染色体和BCR重排。
患者多为老年人。
诊断标准为:1.外周血白血病增多,多≧25×109/L,杆状核及分叶核粒细胞≧80%(白细胞),幼稚细胞(早、中、晚)<10%(白细胞);原始细胞<1%(白细胞)。
中高危MDS与MDS/MPN患者骨髓单个核细胞凋亡及临床特征比较

中高危MDS与MDS/MPN患者骨髓单个核细胞凋亡及临床特征比较目的比较中高危骨髓增生异常综合征(MDS)与骨髓增生异常/骨髓增殖性肿瘤(MDS/MPN)骨髓单个核细胞(BMNCs)凋亡情况及其与患者临床特征的关系。
方法收集2013年7月~2015年7月在湖南师范大学附属长沙医院血液科就诊的8例中高危MDS及5例MDS/MPN患者的骨髓标本及临床资料,AO/EB荧光双重染色及Caspase-3活性检测BMNCs凋亡,相关性分析细胞凋亡与患者临床特征的关系。
结果凋亡在中高危MDS和MDS/MPN患者BMNCs 中的发生率分别为(10.25±1.77)%、(6.90±1.66)%,前者高于后者(P=0.032);Caspase-3检测显示,中高危MDS患者BMNCs中Caspase-3活性为(0.18±0.04)%,显著高于MDS/MPN组[(0.07±0.02)%],差异有统计学意义(P=0.028);相关性分析显示,中高危MDS患者BMNCs凋亡率与骨髓原始细胞比例呈负相关(r=-0.81,P=0.015),而MDS/MPN凋亡率与骨髓原始细胞比例呈正相关(r=0.90,P=0.037)。
结论BMNCs凋亡率、Caspase-3活性在中高危MDS和MDS/MPN 中明显不同,且BMNCs凋亡和骨髓原始细胞比例相关。
[Abstract] Objective To compare the apoptosis of bone marrow mononuclear cells (BMNCs)in myelodysplastic syndrome (MDS)and myelodysplastic/myeloproliferative neoplasms (MDS/MPN),and analyze the relationship between apoptosis and clinical characteristics of patients. Methods Bone marrow specimens and clinical data of 8 cases of intermediate/high risk MDS and 5 cases of MDS/MPN patients were collected from July 2013 to July 2015 in Department of Hematology,Changsha Hospital Affiliated to Hu’nan Normal University. Acridine orange/ethidium bromide fluorescence staining and Caspase-3 activity assay were used to detect apoptosis of BMNCs,and the correlation between apoptosis and clinical characteristics of patients was analyzed. Results The apoptotic rate in intermediate/high risk MDS and MDS/MPN patients were (10.25±1.77)%,(6.90±1.66)% respectively,the former was higher than the latter (P=0.032). Caspase-3 activity assay showed that the Caspase-3 activity of intermediate/high risk MDS BMNCs was (0.18±0.04)%,significantly higher than that of the MDS/MPN group [(0.07±0.02)%],with statistically significant difference (P=0.028). Correlation analysis showed that negative correlation was found between BMNCs apoptosis and bone marrow blast cell percentage in intermediate/high risk MDS patients (r=-0.81,P=0.015),but BMNCs apoptotic rate and the proportion of bone marrow blast cells was positively correlated in MDS/MPN patients (r=0.90,P=0.037). Conclusion The apoptotic rate and Caspase-3 activity are significantly different in intermediate/high risk MDS and MDS/MPN,and there is a correlation between apoptotic rate and bone marrow blasts.[Key words] Myelodysplastic syndrome;Myelodysplastic/myeloproliferative neoplasms;Apoptosis最新WHO將慢性髓系恶性肿瘤分为三大类:骨髓增生异常综合征(myelodysplastic syndrome,MDS)、骨髓增殖性肿瘤(myeloproliferative neoplasms,MPN)、骨髓增生异常/骨髓增殖性肿瘤(MDS/MPN)。
BCR-ABL阴性骨髓增殖性肿瘤JAK2 V617F基因突变状态及负荷的临床意义

• 38•h rfn病•淋巴瘤2021 年1月第 30卷第 1期Journal of Leukemia & Lymphoma,January 2021,Vol. 30,No. 1.论著•BCR-A B L阴性骨髓增殖性肿瘤JAK2 V617F基因突变状态及负荷的临床意义扫码阅读电子版刘辉沈杰王庆苏国宏河北省沧州市中心医院血液内二科061000通信作者:苏国宏,Email:********************【摘要】目的探讨JAK2 V617F基因突变状态及负荷对BCR-ABL阴性骨髓增殖性肿瘤(MPN)的影响。
方法回顾性分析2015年9月至2020年1月河北省沧州市中心医院199例MPN患者的临床资料。
分析■IAK2V617F突变负荷与MPN患者临床病理特征及预后评分的关系。
结果199例BCR-ABL阴性MPN患者中JAK2 V617F突变阳性138例(69.4%);其中,72例真性红细胞增多症(PV)患者中突变阳性64例(88.9%),101例原发性血小板增多症(E T)患者中突变阳性54例(53.5%), 25例骨髓纤维化(M F)患者中突变阳性20例(80.0%), 1例嗜酸粒细胞增多症(HES)患者突变阳性。
JAK2 V617F突变高负荷者占55.1%(76/138)。
突变负荷最高的类型为?¥,1^’次之,£1'最低,3组突变负荷分别为(73.9±18.3)%、(59.9±25.2)%、(25.0±16.5)%。
JAK2V617F突变负荷与PV、ET、MF患者的白细胞计数均呈正相关〇值分别为0.626、0.675、0.796,均户<0.01)。
从1^2丫6丨汗突变负荷与?¥、£1'患者的预后评分均呈正相关(r值分别为0.296、0.404,均P<0.05)。
结论BCR-ABL阴性MPN患者JAK2 V617F突变负荷与临床病理因素相关,JAK2V617F突变高负荷患者预后不良。
JAK_抑制剂在骨髓纤维化治疗中的应用进展

JAK抑制剂在骨髓纤维化治疗中的应用进展张小东,韩孟汝,董春霞,杨林花,马艳萍,王梅芳山西医科大学第二医院血液科,太原 030001摘要:骨髓纤维化(MF)是一种源于造血干细胞的克隆性增殖的血液系统疾病,主要临床特征为血细胞减少、异常炎症细胞因子表达、进行性脾肿大等,Janus激酶—信号转导和转录活化因子通路是其主要发病机制及治疗靶点。
芦可替尼是第一个被FDA批准用于国际预后评分为中危-2及高危MF的JAK抑制剂,极大的改善了MF患者的临床症状及生存期,但安全性及耐药性问题也成为目前的主要困扰。
为克服芦可替尼的不足,菲达替尼、莫洛替尼及帕克替尼新型JAK抑制剂被研发。
菲达替尼对JAK2有较高的选择性,还可抑制溴结构域和末端外结构域通路减少核因子-kB介导的炎症反应,被批准用于初治中危-2及高危MF患者及芦可替尼治疗失败的二线选择;莫洛替尼是JAK1/2/抑制激活素A受体1型(ACVR1)抑制剂,通过抑制ACVR1介导的铁调素的产生,改善贫血症状,对合并贫血的MF患者是一种新的选择;帕克替尼由于缺乏对JAK1的抑制,骨髓抑制效应较轻,贫血及血小板减少的发生率少见,对合并贫血及血小板减少的MF患者有良好的安全性及有效性。
同时在MF的治疗进程中,为了弥补JAK抑制剂的不足,以JAK抑制剂为基础,越来越多的联合治疗被研发,大多数联合方案应用于芦可替尼单药治疗进展或反应欠佳的患者且显示了良好的临床疗效,如临床常规用药(达那唑、干扰素、免疫调节剂等)、去甲基化药物、转化生长因子β抑制剂、赖氨酸特异性去甲基酶1抑制剂等,其中联合达那唑、免疫调节剂及泼尼松+沙利度胺+达那唑的治疗方案在临床应用较多,改善了患者的贫血、血小板减少发生率及芦可替尼的剂量限制。
关键词:骨髓纤维化;JAK抑制剂;芦可替尼;达那唑;泼尼松;沙利度胺doi:10.3969/j.issn.1002-266X.2024.12.027中图分类号:R551.3 文献标志码:A 文章编号:1002-266X(2024)12-0111-04骨髓纤维化(MF)是一种BCR-ABL阴性的骨髓增殖性肿瘤(MPN),包括原发性骨髓纤维化(PMF)、真性红细胞增多症和原发性血小板增多症后骨髓纤维化,主要临床特征为血细胞减少、异常炎症细胞因子表达、进行性脾肿大、体质性症状、髓外造血、骨髓网硬蛋白/胶原蛋白纤维化及高风险向白血病转化等[1]。
自身免疫性骨髓纤维化认识现状及研究进展

自身免疫性骨髓纤维化认识现状及研究进展作者:李婷谢其冰来源:《新医学》2019年第02期【摘要】原发性骨髓纤维化是血液科骨髓增殖性肿瘤的一种亚型,90%以上合并Jak-2V617F、CALR、MPL基因突变,此外,多种疾病均可继发骨髓纤维化。
近年来,自身免疫疾病继发骨髓纤维化逐渐受到关注,但总体来说目前医学界对该病的认识仍不足,尚缺乏统一的诊断标准,其发病机制的相关研究少,治疗方式多为个案报道,尚缺乏大规模临床研究,临床医师极易忽略该病而导致误诊误治。
该文重点阐述原发性骨髓纤维化的认识现状及发病机制。
【关键词】骨髓纤维化;自身免疫疾病;認识现状;发病机制【Abstract】 Primary myelofibrosis is a subtype of bone marrow proliferative neoplasms (MPNs), 90% of which contain Jak2V617F, CALR or MPL mutations. In addition, a variety of diseases can also lead to myelofibrosis. In recent years, myelofibrosis secondary to autoimmune diseases has gradually captivated attention. Nevertheless, the pathogenesis of myelofibrosis has been poorly understood and the standard diagnostic criteria are still lacking. Large-scale clinical trials are urgently required to evaluate the clinical efficacy of different therapeutic interventions. It is highly likely to be misdiagnosed or miss diagnosis. The current understanding and pathogenesis of primary myelofibrosis were reviewed in this article.【Key words】 Myelofibrosis;Autoimmune disease;Current understanding;Pathogenesis在临床实践中,对于外周血三系减少的患者,临床医师多会进行骨髓穿刺及骨髓活组织检查(活检)进一步排除血液系统疾病。
BCR-ABL阴性骨髓增殖性肿瘤患者JAK2、CALR、MPL基因突变的临床分析

98内科急危重症杂志2019年第25卷第2期BCR-ABL阴性骨髓增殖性肿瘤患者JAK2CALR、MPL 基因突变的临床分析>汕头大学医学院第一附属医院邢学仰冯志金苏永忠•沈家欣陈芾珩陶红芳程源山林绍泽,汕头 515041摘要目的:研究BCR-ABL阴性骨髓增殖性肿瘤(M PN)患者JAK2V617F、CALR及M PL基因突变情况,并比较不同类型基因突变及突变均阴性患者部分临床参数的差异。
方法:收集227例BCR-ABL阴性M PN患者各类型基因突变情况、相关检验结果及影像学检查。
结果:JAK2V617F突变总检出率、CALR基因突变总检出率、M PL基因突变总检出率分别为71.4%,11.0%,2.6%。
真性红细胞增多症(P V)患者中未检测到CALR、M PL基因突变。
有1例原 发性血小板增多症(ET)患者有两种基因突变共存(JAK2V617F + ,CALR + )。
在P V患者中JA K2V617F突变阳性组年龄、白细胞计数、血小板计数、脾肿大发生率均高于JA K2V617F突变阴性组(均P<0.05)。
在E T患者中,JA K2V617F突变阳性组年龄、白细胞计数、血红蛋白浓度均高于CA LR突变阳性组及3种基因突变均阴性组,脾肿大及血栓发生率高于C A LR突变阳性组;而与M PL突变阳性组相比,仅表现为血红蛋白浓度增高(尸<0.05)。
在原发性骨髓纤维化(P M F)患者中,仅发现JA K2V617F突变阳性组白细胞计数高于C A L R突变阳性组。
结论:不同MPN 亚型基因突变的发生频率、分布、血栓形成风险不同,导致独特的临床表征。
C A L R阳性患者较JA K2V617F阳性患者更年轻、骨髓增殖水平更低、血栓事件风险更低,这提示危险分层更低、预后较好。
这为M PN的诊断及个体化治疗提供证据。
关键词骨髓增殖性肿瘤;JA K2V617F突变;CA LR突变;M PL突变中图分类号R551.3 文献标识码 A DOI 10. 11768/nkjwzzzz20190203Qinical characteristics of JAK2, CALR and MPL gene mutations in BCR-ABL-negative myeloproliferative neoplasms patients XINGXue-yang, FENG Zhi-jin, SU Yong-zhong, SHEN Jia-xin, CHEN Fei-heng, TAO Hong-fang, CHENG Yuan-shan, LIN Shao-ze. The First Affiliated Hospital, Shantou University Medical College, Shantou 515041, China Abstract Objective:To study the JAK2,CALR and MPL gene mutations in patients with BCR-ABL-negative myeloproliferative neoplasms (M PN) , and to compare their clinical characteristics of different types of gene mutations and negative gene mutations. Methods:All types of gene mutations in BCR-ABL-negative MPN patients were collected, including the related laboratory and imaging findings. Results:The total detection rate of JAK2V617F, CALR and MPL mutations was71.4%, 11.0%and 2.6%respectively. CALR and MPL genes were not found in polycythaemia vera (P V) patients andtwo gene mutations (JAK2V617F, CALR) coexisted in one essential thrombocythaemia (E T) patient. In PV patients, the age was older, leukocyte counts and platelet counts were greater, and the incidence of splenomegaly was higher in JAK2V617F mutation positive group than in JAK2V617F mutation negative group (all P<0.05). There was no significant difference in hemoglobin level and incidence of thrombotic events between two groups (P >0. 05). In ET patients, the age was older, white blood cell counts were greater, and the level of hemoglobin was higher in JAK2V617F mutation positive group than in CALR mutation positive group and all negative groups, and the incidence of splenomegaly and thrombotic e- vents in JAK2V617F mutation positive group was higher than in CALR mutation positive group. As compared with the MPL mutation positive group, the hemoglobin level was increased in JAK2V617F mutation positive group (P <0.05).In PMF patients, the leukocyte counts were greater in JAK2V617F mutation positive group than in CALR mutation positive group. Conclusion :Different subtypes of MPNs had different profiles of drivers gene mutations, leading to the unique clinical characteristics. CALR-positive patients were younger than JAK2V617F-positive patients, and the bone marrow proliferation level and the risk of thrombus events were also lower, which suggested that the risk stratification is lower and the prognosis is better.Keywords Myeloproliferative neoplasms;JAK2V617F mutation;CALR mutation;MPL mutation基金项目:汕头市科技计划项目(汕府科[2017]119号)通信作者:苏永忠,E-mail:suyzst@ 126. com内科急危重症杂志2019年第25卷第2期99骨髓增殖性肿瘤(myeloproliferative neoplasms,MPN)是一组发生在造血干细胞水平的异质性疾病,是由一系或多系分化相对成熟的骨髓细胞不断 克隆性增殖所导致的一组肿瘤性疾病的统称。
bcrabl is标准

bcrabl is标准BCR-ABL是一种非常重要的蛋白质,在白血病的研究中扮演着关键的角色。
BCR-ABL是一种融合蛋白质,由BCR(Breakpoint Cluster Region)基因和ABL (Abelson leukemia virus)基因融合而成。
这种融合蛋白质的存在与慢性髓系白血病(CML)和一些急性淋巴细胞白血病(ALL)的发展密切相关。
BCR-ABL基因的突变会导致ABL激酶活性的持续增强,从而使得细胞无法正常调控增殖和凋亡,最终导致白血病的发生。
因此,BCR-ABL蛋白质的标准化研究对于白血病的治疗和预防具有重要意义。
为了确保BCR-ABL标准化研究的准确性和可靠性,科研人员们需要严格遵循一系列标准化的操作流程和实验方法。
首先,对样本的采集和处理需要严格按照标准操作程序进行,以避免外界因素对实验结果的干扰。
其次,在实验操作过程中需要严格控制各项参数,确保实验条件的一致性和可比性。
同时,对实验结果的分析和解读也需要遵循一定的标准,以保证数据的准确性和可靠性。
在BCR-ABL标准化研究中,实验方法的选择也至关重要。
常用的实验方法包括Western blot、RT-PCR、流式细胞术等,科研人员需要根据研究的具体目的和需求选择合适的实验方法,并严格按照标准操作程序进行实验操作。
只有在实验方法的选择和操作上严格遵循标准,才能确保研究结果的准确性和可靠性。
除了实验方法的选择和操作,对于BCR-ABL标准化研究来说,质控也是非常重要的环节。
科研人员需要建立健全的质控体系,对实验过程中的各个环节进行严格监控和管理,确保实验过程的可追溯性和可控性。
只有通过严格的质控措施,才能保证实验结果的可靠性和可重复性。
此外,在BCR-ABL标准化研究中,数据的管理和分析也是至关重要的。
科研人员需要建立完善的数据管理系统,对实验数据进行及时、准确的录入和整理。
在数据分析过程中,科研人员需要运用合适的统计方法,对数据进行科学、客观的分析和解读,确保研究结果的可靠性和科学性。
骨髓增殖性肿瘤患者血栓栓塞的相关因素

骨髓增殖性肿瘤患者血栓栓塞的相关因素郭慧梅;潘崚;贺建辉;化罗明;曹志新;薛蕾【期刊名称】《临床医药文献电子杂志》【年(卷),期】2013(40)10【摘要】目的研究BCR/ABL阴性骨髓增殖性肿瘤(myeloproliterative neoplasms,MPN)患者血栓栓塞的相关危险因素,用以指导临床。
方法 104例MPN患者采用等位基因特异性PCR(AS-PCR)方法检测JAK2V617F突变情况,回顾性分析104例MPN患者临床特征、实验室检查及血栓栓塞事件发生情况等资料。
先后行单因素分析和多因素分析。
结果 104例MPN患者中71例(68.3%)存在JAK2V617F突变,45例有血栓栓塞(43.3%)。
单因素分析表明,年龄、高血压病史、病种、JAK2V617F、白细胞计数(WBC)与血栓栓塞具有相关性(P<0.05);多因素分析表明,年龄(≥60岁)、JAK2V617F突变、白细胞计数升高是血栓栓塞的独立危险因素(P<0.05)。
结论高龄、JAK2V617F突变阳性或白细胞计数增高的MPN 患者血栓栓塞风险可能较高。
【总页数】3页(P958-960)【作者】郭慧梅;潘崚;贺建辉;化罗明;曹志新;薛蕾【作者单位】河北大学附属医院血液内科;河北医科大学第二医院血液内科;四川大学华西医院血液内科;保定市第一中心医院神经外科【正文语种】中文【中图分类】R733.3【相关文献】1.骨髓增殖性肿瘤患者并发急性冠脉综合征相关因素分析 [J], 张晟瑜;杨明;曾勇;韩业晨;王书杰;沈珠军;张抒扬;方全2.结直肠癌患者术后并发静脉血栓栓塞的相关因素及循证护理 [J], 李袁林3.晚期肺癌化疗患者伴静脉血栓栓塞症的相关因素分析 [J], 文静4.非霍奇金淋巴瘤合并静脉血栓栓塞症患者相关因素分析 [J], 徐燕霞;黄静;杨宇佳;张慧琪5.卵巢恶性肿瘤患者发生静脉血栓栓塞症的相关因素分析 [J], 闫华;邹存华;郝娟;张玉英;冯富忠;赵淑萍因版权原因,仅展示原文概要,查看原文内容请购买。
以门静脉高压症为主要表现的骨髓增殖性肿瘤临床特征分析

以门静脉高压症为主要表现的骨髓增殖性肿瘤临床特征分析冯丽娟;王艳;王民;张冠华;何福亮;赵新颜;王宇【期刊名称】《临床肝胆病杂志》【年(卷),期】2023(39)2【摘要】目的探讨以门静脉高压症为主要表现的骨髓增殖性肿瘤(MPN)患者的临床特征、肝脏组织学特点及诊疗方法。
方法纳入2019年1月—2022年2月在北京友谊医院肝病中心以门静脉高压症就诊,最终诊断为MPN的患者,回顾性分析其临床表现、肝脏病理特征、治疗和随访结果。
结果共纳入9例患者,所有患者均有脾大及食管胃底静脉曲张,其中8例患者有门静脉血栓。
所有患者肝功能及血常规均在正常范围或轻度异常。
6例患者行肝穿刺活检,均未见肝脏纤维间隔及假小叶形成,2例可见肝脏髓外造血现象。
所有患者均行骨髓活检及基因检测,6例为原发性血小板增多症,3例为原发性骨髓纤维化;基因检测结果显示,JAK-2V617F基因突变7例,CALR基因突变2例。
结论MPN是门静脉高压的少见病因之一,临床可表现为食管胃底静脉曲张和脾大,甚至巨脾,但无白细胞和血小板减低等脾功能亢进表现。
JAK-2V617F和CALR基因检测可提高MPN的诊断率。
【总页数】5页(P365-369)【作者】冯丽娟;王艳;王民;张冠华;何福亮;赵新颜;王宇【作者单位】首都医科大学附属北京友谊医院肝病中心【正文语种】中文【中图分类】R73【相关文献】1.以门静脉高压症为主要临床表现的骨髓纤维化合并急性胆囊炎1例报告2.BCR-ABL1阴性骨髓增殖性肿瘤骨髓病理学及临床特征分析3.JAK2阳性骨髓增殖性肿瘤患者65例临床特征分析4.云浮地区骨髓增殖性肿瘤JAK2V617F的突变率及临床表现5.费城染色体阴性骨髓增殖性肿瘤患者非驱动基因突变分布及临床特征分析因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
BCR--ABL阴性非经典骨髓增殖性肿瘤的认识现况BCR【摘要】BCR-ABL阴性非经典骨髓增殖性肿瘤包括慢性嗜酸性粒细胞白血病不另作分类(CEL-nos),慢性中性粒细胞白血病(CNL),肥大细胞增多症(Mastocytosis),骨髓增殖性肿瘤未分类型(MPN-U)。
这组疾病发病率低,多依赖排他性标准得以诊断,对病理机制、预后因素所知甚少。
近年对各亚型流行病学特点和生物学特征的不断研究已使诊断和治疗得到改进,但对一些临床变异型仍欠缺基本认识,易忽视临床症状,难于明确诊断,缺乏有效的治疗手段。
本文就CNL、肥大细胞增多症、MPN-U的相关资料进行复习,并简述对这组疾病的认识现状。
【关键词】慢性中性粒细胞白血病;肥大细胞增多症;诊断;治疗根据2008年WHO造血与淋巴组织肿瘤分类(WHO2008)1所提出的概念,骨髓增殖性肿瘤(MPN)包括以下8种类型:慢性粒细胞白血病BCR/ABL1阳性(CML),真性红细胞增多症(PV),原发性血小板增多症(ET),原发性骨髓纤维化(PMF),慢性嗜酸性粒细胞白血病不另作分类(CEL,not otherwise specified,CEL-nos),慢性中性粒细胞白血病(CNL),肥大细胞增多症(Mastocytosis),和骨髓增殖性肿瘤,未分类型(MPN-U)。
后四个类型临床称为BCR/ABL阴性非经典MPN2,其中CEL-nos 在各种嗜酸性粒细胞增高性疾病中多有讨论,本文重点对CNL、肥大细胞增多症和MPN-U的认识现况做一综述。
2.诊断与鉴别诊断2.2.11CNLCNL是一种罕见类型MPN,老年患者多见但也有青春期发病的报道,约1/5的病例伴发于其他肿瘤性疾病,浆细胞疾病尤其多发性骨髓瘤最为常见3。
诊断CNL的要点为排除各种反应性中性粒细胞(ANC)增高以及其他髓系增殖性肿瘤,并且要求证明粒细胞的单克隆性,但后者临床常难于实现。
WHO 2008提出的CNL诊断标准1主要为排他性:①外周血白细胞(WBC)>25×109/L,分叶/杆状ANC在WBC中比例>80%,不成熟粒细胞(早/中/晚幼粒细胞)<10%,髓系原始细胞<1%;②骨髓高增生表现,ANC比例/绝对值均增高,原始细胞占有核细胞比例<5%,ANC成熟正常,巨核细胞正常或核左移;③脾大;④排除生理性ANC增多,或通过细胞遗传学/分子学结果证明髓系细胞的克隆性;⑤无PH染色体/BCR-ABL1融合基因;⑥无PDGFRA,PDGFRB,FGFR1重排;⑦排除ET,PV,PMF;⑧排除骨髓增生异常综合症和MDS/MPN,无粒细胞或其他髓系形态发育异常,单核细胞<1×109/L。
需注意若骨髓中出现浆细胞异常,应予细胞遗传学/分子学检测以证明中性粒细胞的克隆性。
CNL患者可能无自觉症状,亦有部分病例出现乏力、体重下降、骨痛、盗汗、皮肤瘀斑等非特异性临床表现,重要的疾病相关症状为WBC数异常增高/浸润、脾脏肿大、消化道/粘膜出血4。
约有20% CNL患者诊断时可检测到细胞遗传学异常,多见+21、20q-、11q-、12p-;约12%的病例随访中可见克隆性演变,常见的异常染色体核型有+8、+9、、t(1;20)、-7、t(2;2)等,但这些病例大多接受了羟基脲(Hu)等细胞毒性药物的治疗5。
既往曾提出分子量为230kD的变异型BCR-ABL1融合基因产物(p230)为本病的分子学标志6,但目前并未得到认可。
其他可以提示CNL的指标还有:基于X染色体失活的克隆性检测证实粒细胞单克隆性,血清G-CSF水平正常/减低,中性粒细胞碱性磷酸酶增高,维生素B12和尿酸异常增高。
2.2.22肥大细胞增多症肥大细胞增多症是由于异常的肥大细胞(MC)克隆性增殖并于一种或多种器官中累积而引发的一组高度异质性的肿瘤性疾病。
其与肥大细胞增生/活化状态的区别在于,致病MC为肿瘤性细胞可见形态和/或分子学异常。
按病灶分布和临床表现主要分为皮肤型(CM)和系统型(SM):前者主要见于儿童,半数以上病例为出生后6个月内发病,后者多见于20岁以上的成年人且几乎均发生骨髓受累。
根据临床特征的不同,SM又包括如下变异型:惰性系统性肥大细胞增多症(ISM)、SM伴非肥大细胞系克隆性血液系统疾病(SM-AHNMD)、侵袭性系统性肥大细胞增多症(ASM)、肥大细胞白血病(MCL)、肥大细胞肉瘤(MCS)和真皮外肥大细胞瘤(ECM)。
本文对常累及血液系统的SM着重讨论。
SM患者MC广泛浸润组织器官并异常释放各种介质,临床表现种类庞杂、类型多变,但总体可分为4类1:体质性症状(乏力、体重下降、发热、多汗),皮肤表现(瘙痒、风团、荨麻疹),介质相关症状(腹痛、晕厥、低血压、心动过速等),肌肉骨骼并发症(骨痛、骨质疏松/骨量减少、肌肉疼痛、关节疼痛)。
ISM以器官浸润肿大为主,但ASM、MCL等侵袭性SM则常见器官功能的受损。
病理状态下MC异常合成/释放组胺、类胰蛋白酶、前列腺素、白细胞三烯、TNF-α及各种趋化因子、细胞因子,导致血管舒张出现一系列消化系统、神经系统、循环系统症状,临床表现轻重不一甚者可威胁生命。
多种因素可刺激SM患者引发上述病理表现,如:药物(万古霉素等多种抗生素、非甾体类抗炎药、麻醉剂/肌松剂)、物理因素(运动、皮肤摩擦)、手术、感染、酒精、蛰咬、辛辣食物、情绪压力、温度过高/低或突然改变。
SM患者中IgE介导的超敏反应性疾病的发生率同正常人群相比并无显著升高,但其在超敏反应中更易发生过敏性休克7。
需要注意,部分患者在诊断前可能已出现各种精神系统症状(沮丧、心境改变、注意力涣散、易怒、情绪不稳定),在临床上易被忽视8;而呼吸系统、内分泌系统相对较少受累,且若非合并其他血液系统疾患SM患者各种感染的发生率并无异于常人,亦未见免疫系统功能受损。
SM患者造血系统受累主要表现为贫血、白细胞增高、嗜酸性粒细胞增高、中性粒细胞减少、血小板减少。
侵袭性亚型或合并白血病的变异型可能出现骨髓衰竭,但真正以循环中出现大量MC为特征的MCL其实是十分罕见的。
约30%的SM病例在发病前或整个疾病过程中出现非肥大细胞系克隆性血液系统疾病(AHNMD),髓系或淋巴系肿瘤均可见但以前者为主,其中慢性粒单核细胞白血病最为常见。
WHO2008中特别提出了“myelomastocytic leukemia”一词,用以描述可见不成熟/不典型MC 增多但不足以诊断SM的某些髓系肿瘤病例。
WHO2008要求SM的诊断需满足主要标准+3项次要标准。
主要标准为骨髓和/或其他真皮外器官中MC多灶性、致密性浸润(≥15个MC聚集);次要标准包括:①浸润组织活检示MC中超过25%为纺锤形或不典型形态,或骨髓液涂片示MC中超过25%表现为不成熟或不典型形态;②骨髓、外周血或其他真皮外器官中可检测到KIT816发生活化性点突变;③MC表达异常免疫表型CD2和/或CD25;④血清中类胰蛋白酶(tryptase)水平持续高于20ng/mL。
需要注意的是,必须在排除了其他相关的髓系克隆性疾病之后,KIT816突变和血清类胰蛋白酶水平增高才对本病有诊断意义;并且最常见的突变类型D816V会导致非配体依赖性的KIT持续活化以及对伊马替尼的耐药9。
2.2.33MPN-U能够确诊为MPN但无法划分至任一具体类型的病例可诊断为MDS-U,主要见于如下三种情况:①PV、ET、PMF的早期阶段;②各类MPN进展至晚期或发生转化时(纤维组织增生、骨质硬化,或原始细胞增高、形态发育异常)③MPN合并了肿瘤或炎症疾病时。
在临床和实验室证据充分的情况下才能够诊断MPN-U,而且应当注意排除细胞毒药物及生长因子治疗的干扰。
对MPN-U患者至少应间隔4-6个月进行随访,以便进一步明确诊断。
临床实践中应根据每例患者所处的病程不同阶段和具体的临床表现,进行治疗选择和预后判断。
3.预后和危险因素3.1CNL的预后和危险因素大多数CNL病例表现为疾病进展缓慢、病程长,但缺乏基于大宗病例的生存分析。
病例报道提示,患者生存时间可跨越6个月至20年,中位生存期近2年,5年生存率近30%,死亡原因包括白血病转化、疾病进展(非转化)、出血、感染以及治疗相关的持续血细胞减少。
CNL起病初期有一个时长不等(数月至数年)的“慢性期”,临床症状轻、经药物治疗WBC数可控制。
疾病加速进展的征象包括:ANC进行性增多,显著/进行性脾脏肿大,贫血,血小板减少,既往治疗失效,外周血出现幼稚细胞,细胞遗传学检查示出现克隆性演变10。
约20%CNL最终出现白血病转化,病例报道示均为急性髓系白血病(AML),中位发生时间为诊断后21个月。
发生白血病转化的病例对强烈联合化疗常无效,诱导达缓解的病例亦多表现为骨髓增生程度减低或在后续治疗中发生死亡。
3.2肥大细胞增多症的预后和危险因素肥大细胞增多症各变异型之间预后差异显著。
WHO分类系统中CM预后最佳,部分儿童患者可在进入青春期后或青春期前自发缓解,但成年人则表现为非自限性,且可能进展为SM(多为ISM)。
对于仅有孤立性皮损的CM患者,应注意检查血清类胰蛋白酶水平和是否出现器官肿大以排除SM。
而伴有皮损的SM患者常表现为惰性病程,无皮损者则多为侵袭性,但孤立性骨髓肥大细胞增多(一种ISM变异型,无皮损表现)预后很好。
骨髓涂片中MC的比例和形态也是重要的独立预后因素:出现异染色性原始细胞是MCL的常见特征,并且多预示较严重的组织器官损伤;而可见双/多分叶MC 则常预示细胞具有高侵袭性增殖的特征11;此外,免疫组化标记CD117、tryptase、CD25、CD2所示形态结构特征也具重要意义。
SM中ISM和ECM预后较好,ASM较差,MCL和MCS最差,SM-AHNMD的预后则多取决于伴发的克隆性血液疾患。
2009年一项342例成人SM的临床研究结果显示12:ISM患者极少发生白血病转化,无白血病生存和预期总生存与正常人群相比无明显缩短;而ASM和MCL的中位生存分别为3.5年和2个月,该试验中SM-AHNMD患者的中位生存时间为2年。
总结提示肥大细胞增多症预后不良的因素包括:年龄>65岁,出现疾病晚期症状,无皮肤受累,血小板<10万,Hb<10g,乳酸脱氢酶升高,碱性磷酸酶升高,肝脾肿大,骨髓增生活跃,骨髓原始细胞>5%,外周血涂片可见异常形态细胞,以及骨髓中MC比例增高、形态发育异常。
4.治疗各类型BCR/ABL阴性非经典型MPN的临床特征和远期预后存在巨大差异,因此治疗的目标也有所不同,但总体策略都是根据症状特征、预后分层以及个体反应选择治疗方式。
4.4.11CNL的治疗目前尚无治疗CNL的最佳方案,主要是基于临床症状予以相应干预。