VO-Ohpic_trihydrate_DataSheet_MedChemExpress
HPLC测定奥美沙坦酯氢氯噻嗪片含量和有关物质

呵护公众健康合理用药China Licensed Pharmacist Jul.2012,Vol.9No.7奥美沙坦酯氢氯噻嗪片是治疗高血压新药,2010年在我国上市,临床用于治疗高血压、充血性心力衰竭。
奥美沙坦酯与氢氯噻嗪有互补的作用机制,两者合用既能协同降压,又可以减少氢氯噻嗪的应用剂量,具有快速起效、强效降压和持久降压的特点,适于国内众多高血压患者使用。
由于该产品上市时间短,目前文献少见对该产品有关物质测定方法的报道,本研究建立了该产品含量和关物质的HPLC测定方法,简单快速,结果准确。
1仪器与试药安捷伦1200高效液相色谱仪及其色谱工作站(安捷伦公司);奥美沙坦酯对照品(原中国药品HPLC测定奥美沙坦酯氢氯噻嗪片含量和有关物质赵建峰(北京万生药业有限责任公司,北京101113)【摘要】目的:建立高效液相色谱法(HPLC)测定奥美沙坦酯氢氯噻嗪片含量和有关物质的测定方法。
方法:采用HPLC,色谱柱:C18(4.6mm×250mm,Kromasil);流动相A:0.02mol/L磷酸二氢钠溶液(用磷酸调节pH至3.0),流动相B:甲醇-乙腈(100∶900),梯度洗脱。
结果:奥美沙坦酯和氢氯噻嗪均能与其他杂质较好分离;奥美沙坦酯浓度在0.002026~0.04052mg/mL(r= 0.999)范围内线性良好,氢氯噻嗪在0.001265~0.02530mg/mL范围内线性良好(r=1.000)。
结论:本方法灵敏、准确,可作为奥美沙坦酯氢氯噻嗪片含量和有关物质的测定方法。
【关键词】高效液相色谱法;奥美沙坦酯氢氯噻嗪片;含量测定doi:10.3969/j.issn.1672-5433.2012.07.006Determination of the Content and Related Substances of Olmesartan Medoxomil/Hydrochloroth-iazide Tablets by HPLCZhao Jianfeng(Beijing Winsunny Pharmaceutical Co.Ltd.,Beijing101113,China)ABSTRACT Objective:To establish a method by HPLC to determine the content and the related substances of olmesartan medoxomil/hydrochlorothiazide tablets.Methods:The HPLC was performed on a C18column(4.6mm×250mm,Kromasil)with mobile phase A:0.02%sodium dihydrogen phosphate solution(pH3.0adjusted with phosphoric acid)and mobile phase B:methanol-acetonitrile(100∶900). Gradient elution was conducted.Results:Olmesartan medoxomil and hydrochlorothiazide were completely separated from impurities.There was a good linear range from0.002026to0.04052mg/mL(r=0.999)for olmesartan medoxomil and from0.001265to0.02530mg/mL(r=1.000)for hydrochlorothiazide. Conclusion:The method was proved good in sensitivity and accuracy and can be applied to the determination of olmesartan medoxomil/hydrochlorothiazide tablets and its related substances.KEY WORDS HPLC;Olmesartan Medoxomil/Hydrochlorothiazide Tablets;Content Determination作者简介:赵建峰,男,工程师。
CAS号737763-37-0_360A iodide_MedBio技术参数

CAS
包装
纯度
MedBio
MED11537
CCT244747
CCT244747
1404095-34-6
5mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11470
Monomethyl auristatin E
Monomethyl auristatin E
474645-27-7
10mM (in 1mL DMSO)
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11536
ELR510444
ELR510444
1233948-35-0
25mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11426
Acetyl Angiotensinogen (1-14), porcine
5mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11516
Eribulin mesylate
Eribulin mesylate
441045-17-6
500μg
≥98%Βιβλιοθήκη 品牌货号中文名称英文名称
CAS
包装
纯度
MedBio
MED11540
AZD-5597
AZD-5597
924641-59-8
3、同类产品列表:
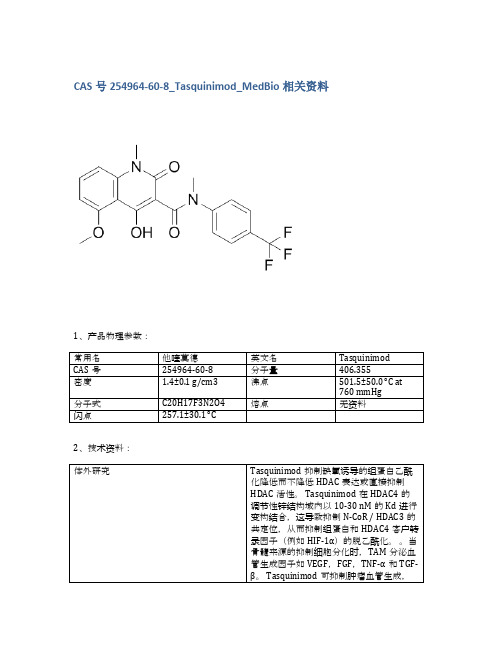
CAS号254964-60-8_Tasquinimod_MedBio相关资料
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11931
Cyclophosphamide
Cyclophosphamide
50-18-0
10mM (in 1mL DMSO)
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED12034
Mupirocin
包装
纯度
MedBio
MED11950
Resminostat hydrochloride
Resminostat hydrochloride
1187075-34-8
100mg
≥98%
品牌
CAS
包装
纯度
MedBio
MED12017
Raltitrexed
Raltitrexed
112887-68-0
10mM (in 1mL DMSO)
≥98%
体内研究
当通过管饲法或饮用水给予成年雄性小鼠(即C57B1 / 6J或无胸腺裸鼠)0.1-30mg / kg(即0.2-74μmoles/ kg)时,Tasquinimod的生物利用度和口服吸收是极好的。Tasquinimod的效力表示为抑制癌症生长50%的Tasquinimod每日口服剂量范围为0.1-1.0 mg / kg / d(即0.24-2.40μmoles/ kg /天),相对于一系列(n> 5)免疫缺陷小鼠中的人前列腺癌异种移植物。通过饮用水以5mg / kg /天的慢性剂量服用的Tasquinimod在免疫活性同系小鼠中产生> 80%的TRAMP-C2小鼠前列腺癌生长抑制(p <0.05)[2]。携带皮下LNCaP肿瘤的裸鼠用Tasquinimod治疗3周。在接种后第7天开始以1mg / kg /天和10mg / kg /天暴露于Tasquinimod。与接种后28天的未处理对照组相比,1 mg / kg /天和10 mg / kg /天的肿瘤重量均有统计学显着的剂量依赖性降低(p <0.001),说明Tasquinimod的抗肿瘤作用[3]。
奥美拉唑-欧洲8.0药典

EUROPEAN PHARMACOPOEIA 8.0Omeprazole04/2013:0942OMEPRAZOLEOmeprazolum C 17H 19N 3O 3S M r 345.4[73590-58-6]DEFINITION 5-Methoxy-2-[(RS )-[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl]-1H -benzimidazole.Content :99.0per cent to 101.0per cent (dried substance).CHARACTERSAppearance :white or almost white powder.Solubility :very slightly soluble in water,soluble in methylene chloride,sparingly soluble in ethanol (96per cent)and in methanol.It dissolves in dilute solutions of alkali hydroxides.It shows polymorphism (5.9).IDENTIFICATIONInfrared absorption spectrophotometry (2.2.24).Comparison :omeprazole CRS .If the spectra obtained in the solid state show differences,dissolve the substance to be examined and the referencesubstance separately in methanol R ,evaporate to dryness and record new spectra using the residues.TESTSSolution S .Dissolve 0.50g in methylene chloride R and dilute to 25mL with the same solvent.Appearance of solution .Solution S is clear (2.2.1).Impurities F and G :maximum 350ppm for the sum of the contents.The absorbance (2.2.25)of solution S determined at 440nm is not greater than 0.10.Related substances .Liquid chromatography (2.2.29).Prepare the solutions immediately before use .Test solution .Dissolve 3mg of the substance to be examined in the mobile phase and dilute to 25.0mL with the mobile phase.Reference solution (a).Dissolve 1mg of omeprazole CRS and 1mg of omeprazole impurity D CRS in the mobile phase and dilute to 10.0mL with the mobile phase.Reference solution (b).Dilute 1.0mL of the test solution to 100.0mL with the mobile phase.Dilute 1.0mL of this solution to 10.0mL with the mobile phase.Reference solution (c).Dissolve 3mg of omeprazole for peak identification CRS (containing impurity E)in the mobile phase and dilute to 20.0mL with the mobile phase.Column :–size :l =0.125m,Ø=4.6mm;–stationary phase :octylsilyl silica gel for chromatography R (5μm).Mobile phase :mix 27volumes of acetonitrile R and 73volumes of a 1.4g/L solution of disodium hydrogen phosphate R previously adjusted to pH 7.6with phosphoric acid R .Flow rate :1mL/min.Detection :spectrophotometer at 280nm.Injection :40μL.Run time :5times the retention time of omeprazole.Identification of impurities :use the chromatogram obtained with reference solution (a)to identify the peak due to impurity D;use the chromatogram supplied with omeprazole for peak identification CRS and the chromatogram obtainedwith reference solution (c)to identify the peak due toimpurity E.Relative retention with reference to omeprazole(retention time =about 9min):impurity E =about 0.6;impurity D =about 0.8.System suitability :reference solution (a):–resolution :minimum 3.0between the peaks due toimpurity D and omeprazole;if necessary,adjust the pH of the aqueous part of the mobile phase or the concentration of acetonitrile R ;an increase in the pH will improve theresolution.Limits :–impurities D,E :for each impurity,not more than 1.5times the area of the principal peak in the chromatogramobtained with reference solution (b)(0.15per cent);–unspecified impurities :for each impurity,not more than thearea of the principal peak in the chromatogram obtained with reference solution (b)(0.10per cent);–total :not more than 5times the area of the principal peak in the chromatogram obtained with reference solution (b)(0.5per cent);–disregard limit :0.5times the area of the principal peak in the chromatogram obtained with reference solution (b)(0.05per cent).Loss on drying (2.2.32):maximum 0.2per cent,determined on 1.000g by drying under high vacuum at 60°C for 4h.Sulfated ash (2.4.14):maximum 0.1per cent,determined on1.0g.ASSAYDissolve 0.250g in a mixture of 10mL of water R and 40mL of ethanol (96per cent)R .Titrate with 0.1M sodium hydroxide ,determining the end-point potentiometrically (2.2.20).1mL of 0.1M sodium hydroxide is equivalent to 34.54mg of C 17H 19N 3O 3S.STORAGEIn an airtight container,protected from light,at a temperature of 2°C to 8°C.IMPURITIESSpecified impurities:D,E,F,G .Other detectable impurities (the following substances would,if present at a sufficient level,be detected by one or other of the tests in the monograph.They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034).It is therefore not necessary to identify these impurities for demonstration of compliance.See also 5.10.Control of impurities in substances for pharmaceutical use ):A,B,C,H,I.A.5-methoxy-1H-benzimidazole-2-thiol,B.2-[(RS )-[(3,5-dimethylpyridin-2-yl)methyl]sulfinyl]-5-methoxy-1H -benzimidazole,General Notices (1)apply to all monographs and other texts2911Omeprazole magnesium EUROPEAN PHARMACOPOEIA8.0C.5-methoxy-2-[[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfanyl]-1H-benzimidazole(ufiprazole),D.5-methoxy-2-[[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfonyl]-1H-benzimidazole(omeprazolesulfone),E.4-methoxy-2-[[(RS)-(5-methoxy-1H-benzimidazol-2-yl)sulfinyl]methyl]-3,5-dimethylpyridine1-oxide,F.8-methoxy-1,3-dimethyl-12-thioxopyrido[1′,2′:3,4]-imidazo[1,2-a]benzimidazol-2(12H)-one,G.9-methoxy-1,3-dimethyl-12-thioxopyrido[1′,2′:3,4]-imidazo[1,2-a]benzimidazol-2(12H)-one,H.2-[(RS)-[(4-chloro-3,5-dimethylpyridin-2-yl)methyl]sulfinyl]-5-methoxy-1H-benzimidazole,I.4-methoxy-2-[[(5-methoxy-1H-benzimidazol-2-yl)sulfonyl]methyl]-3,5-dimethylpyridine1-oxide.01/2009:2374corrected6.7OMEPRAZOLE MAGNESIUMOmeprazolummagnesicumC34H36MgN6O6S2Mr713[95382-33-5]DEFINITIONMagnesium bis[5-methoxy-2-[(RS)-[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl]-1H-benzimidazol-1-ide].It contains a variable quantity of water.Content:97.5per cent to102.0per cent(anhydrous substance).CHARACTERSAppearance:white or almost white,hygroscopic powder.Solubility:very slightly soluble in water,sparingly soluble inmethanol,practically insoluble in heptane.IDENTIFICATIONCarry out either tests A,B,C or tests A,B,D.A.Optical rotation(2.2.7):−0.10°to+0.10°.Dissolve0.250g in methanol R and dilute to25.0mL withthe same solvent.B.Infrared absorption spectrophotometry(2.2.24).Comparison:omeprazole magnesium CRS.C.Atomic absorption spectrometry(2.2.23)as described inthe test for magnesium.The test solution shows the absorption maximum at285.2nm.D.Ignite about0.5g of the substance to be examinedaccording to the procedure for the sulfated ash test(2.4.14).Dissolve the residue in10mL of water R.2mL of thissolution gives the reaction of magnesium(2.3.1).TESTSAbsorbance(2.2.25):maximum0.10at440nm.Dissolve0.500g in methanol R and dilute to25.0mL with thesame solvent.Filter the solution through a membranefilter(nominal pore size0.45μm).Related substances.Liquid chromatography(2.2.29):use thenormalisation procedure.Prepare the solutions immediatelybefore use.Test solution.Dissolve3.5mg of the substance to be examinedin the mobile phase and dilute to25.0mL with the mobilephase.Reference solution(a).Dissolve1mg of omeprazole CRS and1mg of omeprazole impurity D CRS in the mobile phase anddilute to10.0mL with the mobile phase.Reference solution(b).Dissolve3mg of omeprazole for peakidentification CRS(containing impurity E)in the mobile phaseand dilute to20.0mL with the mobile phase.Reference solution(c).Dilute1.0mL of the test solution to100.0mL with the mobile phase.Dilute1.0mL of this solutionto10.0mL with the mobile phase.Column:–size:l=0.125m,Ø=4.6mm;–stationary phase:octylsilyl silica gel for chromatography R(5μm).2912See the information section on general monographs(cover pages)。
200种化学品理化特性表(83页)

200种化学品目录丙烯酸丁酯的理化及危险特性表 (1)丙烯酸的理化及危险特性表 (2)硝酸锌的理化及危险特性表 (3)过氧化二苯甲酰的理化及危险特性表 (4)过硫酸铵的理化及危险特性表 (5)丁酮的理化及危险特性表 (6)苯酚的主要理化和危险特性 (7)甲基叔丁基醚的主要理化及危险特性 (8)三聚氰酸的主要理化和危险特性 (9)季戊四醇的主要理化和危险特性 (10)硬脂醇的主要理化和危险特性 (11)多聚甲醛的主要理化和危险特性 (12)1,2-二甲苯的主要理化及危险特性 (13)1,3-二甲苯的主要理化及危险特性 (14)1,4-二甲苯的主要理化及危险特性 (15)环己烯的主要理化和危险特性 (16)二丁胺的主要理化和危险特性 (17)三乙胺的主要理化和危险特性 (18)甲基二氯硅烷的主要理化和危险特性 (19)异丁烯的主要理化和危险特性 (20)邻叔丁基苯酚的主要理化和危险特性 (21)对叔丁基苯酚的主要理化和危险特性 (22)环己烷的主要理化和危险特性 (23)二氯甲烷的主要理化和危险特性 (24)三氯甲烷的主要理化及危险特性 (25)硫脲的主要理化和危险特性 (26)六亚甲基四胺的主要理化和危险特性 (27)抗氧剂AT-10的主要理化和危险特性 (28)抗氧剂AT-76的主要理化和危险特性 (29)抗氧剂AT-3114的主要理化和危险特性 (30)催化剂JH-CMMS的主要理化和危险特性 (31)抗氧剂AT-168的主要理化和危险特性 (32)甲氧基钠的主要理化和危险特性 (33)氧气的理化及危险特性 (34)硝酸钾的理化及危险特性表 (35)硝酸钙的理化及危险特性表 (36)液氯的理化及危险特性 (37)盐酸的理化及危险特性 (38)丙酮理化及危险特性表 (39)次氯酸钠的理化及危险特性表 (40)二甲氧基甲烷的理化及危险特性表 (41)乙酸的理化及危险特性表 (42)香蕉水的理化及危险特性表 (43)甲醇的理化及危险特性表 (44)氰化亚铜的理化及危险特性 (45)硝酸的理化及危险特性 (46)硫酸的理化及危险特性 (47)铬酸的理化及危险特性 (48)铬酸酐的理化及危险特性 (49)氰化钠的理化及危险特性 (50)丁醇的理化及危险特性 (51)异丙醇的的理化及危险特性 (52)硝酸银的理化及危险特性表 (53)间戊二烯的理化及危险特性 (54)环戊二烯的理化及危险特性 (55)甲苯二异氰酸酯的理化及危险特性表 (56)200#溶剂油的理化及危险特性表 (57)聚氨酯漆固化剂理化及危险特性表 (58)聚氨酯漆稀释剂理化及危险特性表 (59)N,N--二甲基甲酰胺理化及危险特性 (60)丙烯腈理化及危险特性 (61)二氧化硫的特性 (62)异戊二烯的理化及危险特性 (63)醋酸丁酯的理化及危险特性 (64)醋酸乙酯的理化及危险特性 (65)甲苯的理化及危险特性 (66)环己酮的理化及危险特性 (67)三氯化铝(无水)的理化及危险特性 (68)氢氧化钠的理化及危险特性 (69)硝酸铅的理化及危险特性表 (70)电石理化及危险特性表 (71)乙炔理化及危险特性表 (72)氰化亚铜的理化及危险特性 (73)丙烯酸甲酯理化及危险特性................ 错误!未定义书签。
EPZ015666_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:EPZ015666 is an orally available inhibitor of PRMT5 enzymatic activity in biochemical assays with IC 50 of 22 nM and broad selectivity against a panel of other histone methyltransferases.IC50 & Target: IC50: 22 nM (PRMT5)[1]In Vitro: Treatment of MCL cell lines with EPZ015666 leads to inhibition of SmD3 methylation and cell death, with IC 50 values in the nanomolar range [1]. EPZ015666, a potent peptide–competitive and SAM–cooperative inhibitor with >10,000–fold specificity againstPRMT5 relative to other methyltransferases [2].In Vivo: EPZ015666 is orally bioavailable and amenable to in vivo studies. We performed 21–d efficacy studies in severe combined immunodeficiency (SCID) mice bearing subcutaneous Z–138 and Maver–1 xenografts, with twice–daily (BID) oral dosing on four dose groups: 25, 50, 100 and 200 mg per kilogram of body weight (mg/kg). After 21 d of continuous dosing, animals areeuthanized, and blood and tissues are analyzed to determine the relationship between methyl–mark pharmacodynamics andtumor–growth inhibition (TGI). EPZ015666 showed dose–dependent exposure and TGI after 21 d in both MCL models. Tumors in all EPZ015666 dose groups measured on day 21 showed statistically significant differences in weight, volume and tumor growth compared to vehicle–treated tumors. Dosing at 200 mg/kg BID induced tumor stasis in Z–138 cells, with >93% TGI after 21 d,whereas Maver–1 cells showed >70% TGI. Additionally, a third MCL xenograft is tested using the Granta–519 cell line, afast–growing model that reached endpoint on day 18 and showed dose–dependent efficacy with 45% TGI in the 200 mg/kg group.EPZ015666 is well tolerated in all three models, with minimal bodyweight loss in the 200 mg/kg dose group and no other clinical observations [1].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[1]EPZ015666 is serially diluted threefold from 1,000 to 0.051 nM and spotted into a 384–well polypropyleneV–bottom microplate. 3H–SAM is serially diluted twofold in assay buffer for a seven–point dilution series with a top concentration of 700 nM (final assay concentration). Reactions are initiated by the addition of 4 nM enzyme and 40 nM peptide (final assayconcentrations for both). Reactions are incubated for 60 min and quenched by the addition of 10 μL per well of 600 μM unlabeled SAM in assay buffer (final assay concentration). For the peptide competition, EPZ015666 is serially diluted threefold from 1,000 to 0.051 nM and spotted into a 384–well polypropylene V–bottom microplate. Peptide is serially diluted twofold in assay buffer for a seven–point dilution series with a top concentration of 480 nM (final assay concentration). Reactions are initiated by the addition of 4 nM enzyme and 75 nM 3H–SAM (final assay concentrations for both). Reactions are incubated for 60 min, and the reactions are quenched by the addition of 10 μL per well of 600 μM unlabeled SAM in assay buffer (final assay concentration)[1].Cell Assay: EPZ015666 is dissolved in DMSO and stored, and then diluted with appropriate medium (final DMSO 0.2%)before use [1].[1]Cultured cells in linear/log–phase growth are split to a seeding density of 2×105 cells/mL in 2–20 mL of media,depending on the yield required at the end of the growth period. Compound is diluted in DMSO and added to each culture vesselProduct Name:EPZ015666Cat. No.:HY-12727CAS No.:1616391-65-1Molecular Formula:C 20H 25N 5O 3Molecular Weight:383.44Target:Histone Methyltransferase Pathway:Epigenetics Solubility:DMSOwith a final DMSO concentration of 0.2%. Cells are allowed to grow for 96 h undisturbed. At the conclusion of each treatment period, cells are harvested by centrifugation (5 min, 1,200 rpm), and cell pellets are rinsed once with PBS before being frozen on dry ice pending further processing. Long–term proliferation assays are performed on all MCL lines, with slight adjustments to initial seeding densities, depending on growth characteristics for each cell line. All assays are carried out for 12 d[1].Animal Administration: EPZ015666 is formulated in 20% N–N–dimethylacetamide in water (Mice)[1].[1]Mice[1]Male CD–1 mice (25–40 g; n=6, with 3 per time point) are treated with a single dose of EPZ015666 at 2 mg/kg by intravenoustail–vein injection and 10 mg/kg by oral gavage administration, with both doses formulated in 20% N–N–dimethylacetamide in water. Animals are fasted overnight and weighed before dose administration on the day of dosing. Approximately 30 μL ofblood are taken from animals by submandibular or retro–orbital bleeding at pre–specified time intervals (seven time points). For the last time point (24 h), samples are collected via cardiac puncture while the animals are under anesthesia (70% CO2:30% O2). Blood samples are transferred into K2–EDTA tubes and placed on wet ice before centrifugation at 4°C (3,000g, 15 min) to obtain plasma within 30 min after sample collection. Plasma samples are stored at -70±10°C before protein precipitation and LC–MS/MS analysis. We constructed standard calibration curves by analyzing a series of control plasma aliquots containing 100 ng/mL labetalol as an internal standard and 1–3,000 ng/mL EPZ015666. Four levels of quality control are also included in the analysis (3–2,400 ng/mL EPZ015666). Data are analyzed using Phoenix WinNonlin 6.2.1.References:[1]. Chan–Penebre E, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. 2015 Jun;11(6):432–7.[2]. Kryukov GV, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016 Mar 11;351(6278):1214–8.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
CAS号413611-93-5_10074-G5_MedBio_物理性质

1、产品物理参数:
常用名
10074-G5
英文名
10074-G5
CAS号
413611-93-5
分子量
332.313
密度
1.4±0.1 g/cm3
沸点
538.6±60.0 °C at 760 mmHg
分子式
C18H12N4O3
熔点
无资料
闪点
279.5±32.9 °C
2、技术资料:
体外研究
10074-G5抑制Daudi Burkitt淋巴瘤细胞的生长并破坏c-Myc / Max二聚化。针对Daudi和HL-60细胞的IC50值分别为15.6和13.5μM[1]。10074-G5在区域Arg363-Ile381中结合Myc肽Myc353-437,Kd值为2.8μM。10074-G5结合在由诱导螺旋结构域(Leu370-Arg378)的N末端的扭结(Asp379-Ile381)产生的空腔中[3]。
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11457
CDK inhibitor II
CDK inhibitor II
1269815-17-9
50mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11591
(S)-CCG-1423
(S)-CCG-1423
None
体内研究
静脉注射20 mg / kg小鼠的血浆半衰期为10074-G5,为37分钟,血药浓度峰值为58μM,比肿瘤峰值浓度高10倍[1]。
3、同类产品列表:
WHO_TRS_937__annex8_eng

© World Health OrganizationWHO Technical Report Series, No. 937, 2006Annex 8Proposal to waive in vivo bioequivalence requirements for WHO Model List of Essential Medicines immediate-release, solid oral dosage forms Introduction1. Background2. WHO revisions to the criteria for Biopharmaceutics Classifi cation Systemclassifi cation3. WHO extensions to the scope of application of the biowaiver4. WHO additional criteria for application of the biowaiver procedure5. Explanation of the tables6. Biowaiver testing procedure according to WHOIntroductionThis proposal is closely linked to the Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchange-ability (WHO Technical Report Series, No. 937, Annex 7). It aims to give national authorities suffi cient background information on the various orally administered active pharmaceutical ingredients (APIs) on the WHO Model List of Essential Medicines (EML), also taking into account local usage of the API, to enable them to make an informed decision as to whether generic formulations should be subjected to in vivo bioequivalence (B E) studies or whether a biowaiver can be granted. In light of scientifi c work and dis-cussion in the last decade, some of the criteria used to evaluate the API in terms of potential for a biowaiver have been revised to allow a broadened scope of application. The result is that many APIs on the EML can now be considered for the biowaiver procedure, subject to the usage and risks in the national setting.1. Background1.1Initiatives to allow biowaivers based on the BiopharmaceuticsClassifi cation SystemIn 1995 the American Department of Health and Human Services, US Food and Drug Administration (HHS-FDA) instigated the B iopharmaceutics391Classifi cation System (BCS), with the aim of granting so-called biowaiv-ers for scale-up and post-approval changes (SUPAC) (/cder/ guidance/cmc5.pdf). A biowaiver means that in vivo bioavailability and/or bioequivalence studies may be waived (i.e. not considered necessary for product approval). Instead of conducting expensive and time-consuming in vivo studies, a dissolution test could be adopted as the surrogate basis for the decision as to whether two pharmaceutical products are equivalent. At that time the biowaiver was only considered for SUPAC to pharmaceutical products.More recently, the application of the biowaiver concept has been extended to approval of certain orally administered generic products (/ cder/guidance/3618fnl.htm).Within the context of the documents cited above, only APIs with high solu-bility and high permeability and which are formulated in solid, immediate-release (IR) oral formulations can be approved on the basis of the biowaiver procedure. A major advantage of the biowaiver procedure is the simplifi ca-tion of the product approval process and the reduction of the time required, thus reducing the cost of bringing new products to market.1.2What is the Biopharmaceutics Classifi cation System?The Biopharmaceutics Classifi cation System (BCS) was proposed in 1995 by Amidon et al.1 It is a scientifi c framework which divides APIs into four groups, according to their solubility and permeability properties.1.3 Classifi cation of active pharmaceutical ingredients accordingto the Biopharmaceutics Classifi cation SystemAccording to the HHS-FDA defi nitions in the documents cited above, the four possible categories for an API according to the BCS are:•BCS class I: “high” solubility – “high” permeability•BCS class II: “low” solubility – “high” permeability•BCS class III: “high” solubility – “low” permeability•BCS class IV: “low” solubility – “low” permeability.Depending on the classifi cation, the oral availability of the API may be expected to range from being heavily dependent on the formulation and manufacturing method (e.g. Class II APIs: poorly soluble yet highly perme-able) to being mostly dependent on the API permeability properties (e.g.Class III APIs: highly soluble yet poorly permeable).1Amidon GL, Lennemas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classifi cation: the correlation of in vitro drug product dissolution and in vivo bioavailability. Phar-maceutics Research, 1995, 12:413–420.3921.4How is high or low solubility currently defi ned by the Departmentof Health and Human Services, US Food and Drug Administration?The aqueous solubility of a drug substance is considered as high according to the HHS-FDA BCS criteria when:• the ratio of the highest orally administered dose (in mg) to the solubility (mg/ml) is 250 ml or lower.—This criterion is met over the pH range 1–7.5 at 37 °C.According to HHS-FDA guidances, the determination of the equilibrium solubility should be carried out with the shake-fl ask method (other methods such as acid or base titration are permitted when their ability to predict the equilibrium solubility is justifi ed). The experiments should be carried out at a temperature of 37 ± 1°C. Further, a suffi cient number of pH conditions should be chosen to cover the pH range of 1–7.5 and each determination should be carried out at least in triplicate. The buffer solutions given in the United States Pharmacopeia (USP) are appropriate for the tests, but other buffers are also allowed for these experiments. The pH value of each buffer solution should be checked before and after each experiment. Degradation of the API due to pH or buffer composition should be reported together with other stability data.The reason for the 250-ml cut-off criterion for the dose:solubility ratio is that in pharmacokinetic bioequivalence studies, the API formulation is to be ingested with a large glass of water (8 ounces corresponds to about 250 ml). If the highest orally administered dose can be completely dissolved in this amount of water, independent of the physiological pH value (hence the determination over the pH range 1–7.5), solubility problems are not expected to hinder the uptake of the API in the small intestine.The other important parameter for the BCS is the intestinal permeability of the API.1.5How is high or low permeability currently defi ned by the Departmentof Health and Human Services, US Food and Drug Administration?According to HHS-FDA a drug is considered highly permeable, when 90 % or more of the orally administered dose is absorbed in the small intestine.Permeability can be assessed by pharmacokinetic studies (for example, mass balance studies), or intestinal permeability methods, e.g. intestinal perfusion in humans, animal models, Caco 2 cell lines or other suitable, validated cell lines. In vivo or in situ animal models or in vitro models (cell lines) are only considered appropriate by HHS-FDA for passively trans-ported drugs. It should be noted that all of these measurements assess the fraction absorbed (as opposed to the bioavailability, which can be reduced substantially by fi rst-pass metabolism).393HHS-FDA suggests use of two different methods for determining the per-meability classifi cation if results with one method are inconclusive.1.6Which pharmaceutical formulations can currently be consideredfor a biowaiver according to the Department of Health andHuman Services, US Food and Drug Administration?To be considered bioequivalent according to the HHS-FDA biowaiver pro-cedure, a pharmaceutical product:• should contain a Class I API;• should be rapidly dissolving, meaning it should release at least 85% of its content in 30 minutes in three different media (pH 1.2, pH 4.5 and pH6.8, composition see “Multisource document”)1 in a paddle (50 rpm) orbasket (100 rpm) apparatus at 37 °C and a volume of 900 ml;• should not contain excipients which could infl uence the absorption of the API;• should not contain an API with a narrow therapeutic index; and• should not be designed to be absorbed from the oral cavity.The reasoning for the above-mentioned dissolution restrictions is that whena highly soluble, highly permeable API dissolves rapidly, it behaves like asolution in the gastrointestinal tract. If this is the case, the pharmaceutical composition of the product is insignifi cant, provided that excipients which infl uence the uptake across the gut wall are excluded from the formulation.The API is not prone to precipitation after its dissolution due to its good solu-bility under all pH conditions likely to be found in the upper gastrointestinal tract. The high permeability ensures the complete uptake (> 90%) of the API during its passage through the small intestine. The rapid dissolution of the product guarantees that the API is available long enough for the uptake in the small intestine (the passage time in the small intestine is approximately four hours) and negates any slight differences between the formulations.Pharmaceutical products containing an API with a narrow therapeutic index should always be tested with in vivo methods, because the risk to the patient resulting from a possible incorrect bioequivalence decision using the bio-waiver procedure is considered too high with these kinds of APIs.As the BCS is only applicable to APIs which are absorbed from the small intestine; drugs absorbed from other sites (e.g. from the oral cavity) are not eligible for a biowaiver.It is clear that the HHS-FDA requirements for the classifi cation of APIs and eligibility criteria for the biowaiver are very strict. During the last decade,1Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability (WHO Technical Report Series, No. 937, Annex 7).394several publications and continuing scientifi c discussions have suggested that the original HHS-FDA criteria for application of the biowaiver pro-cedure could be relaxed without substantially increasing the risk to public health or to the individual patient. On the basis of these publications and dialogue, WHO has proposed revised BCS criteria and additional consid-erations for the eligibility of a pharmaceutical product for the biowaiver procedure in the “Multisource document”.12.WHO revisions to the criteria for BCS classifi cationWHO revisions to the BCS criteria are as follows:•WHO high-solubility defi nitionWhen an API shows a dose:solubility ratio of 250 ml or lower at 37 °C over a pH range of 1.2–6.8, it can be classifi ed as “highly soluble”. The decrease in pH from 7.5 in the FDA guidances to 6.8 refl ects the need to dissolve the drug before it reaches the mid-jejunum to ensure absorption from the gastrointestinal tract.• Furthermore, the dose that is to be used for the calculation is the highestdose indicated in the Model List of Essential Medicines (EML). In some countries, products may be available at doses exceeding the highest dose on the EML. In such cases, the classifi cation given in the tables at the end of this Annex may no longer be appropriate and the dose:solubil-ity ratio and the permeability will have to be reassessed at the product dose.•WHO permeability defi nitionWhen an API is absorbed to an extent of 85% or more, it is considered to be “highly permeable”. The permeability criterion was relaxed from 90% in the FDA guidance to 85% in the WHO “Multisource document”.Some examples of APIs now included in BCS Class I that were previ-ously considered to be in Class III are paracetamol, acetylsalicylic acid, allopurinol, lamivudine and promethazine.Application of these revised criteria has changed the classifi cation of some APIs in the list. Thus, the classifi cations in the tables attached to this docu-ment supersede those in previous publications. As new APIs appear on the EML, it will be necessary to classify them according to the revised BCS;so it is therefore anticipated that the tables will be revised regularly. In addition, some APIs have not yet been suffi ciently characterized to assign them a BCS classifi cation. As the tables evolve, it is anticipated that more concrete information will be generated for these APIs as well.1Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability (WHO Technical Report Series, No. 937, Annex 7).395the basket apparatus (applies to pharmaceutical products containingClass III APIs);—rapidly dissolving (release of > 85% of the labelled amount of drug in 30 minutes) in standard media at pH 1.2, 4.5 and 6.8, at a rota-tional speed of 75 rpm in the paddle apparatus or 100 rpm in the bas-ket apparatus (applies to pharmaceutical products containing Class IAPIs and/or Class II APIs which are weak acids and meet the 250 mldose:solubility requirement at pH 6.8).(4)Considerations relating to excipientsThe national authority should be aware that some excipients can infl uencemotility and/or permeability in the gastrointestinal tract. Therefore, the ex-cipients used in the multisource product formulation should be scrutinized.In this regard, the national authority can draw on the experience relat-ing to formulations which have been approved on the basis of humanbioequivalence studies in their own or in other jurisdictions.If the multisource product under consideration contains excipients thathave been used before in similar amounts in other formulations of thesame API, it can be reasonably concluded that these excipients will haveno unexpected consequences for the bioavailability of the product. If,however, the formulation contains different excipients, or amounts ofthe same excipients that are very different from usual, the national au-thority may choose to declare the biowaiver procedure inapplicable.A list of usual and acceptable excipients can be found at the following website: /cder/iig/iigfaqWEB.htm; formulations of some productscan be found on the web sites of some national drug regulatory authorities.5.Explanation of the tablesThe decision of a national authority to allow a biowaiver based on the BCS should take into consideration the solubility and permeability char-acteristics as well as the therapeutic use and therapeutic index of the API, its pharmacokinetic properties, the similarity of the dissolution profi les of the multisource and the comparator products in standard buffers with a pH of 1.2, pH 4.5 and pH 6.8 at 37 °C. Data related to the excipients compo-sition in the multisource product are also required. A systematic approach to the biowaiver decision has been established by the International Pharma-ceutical Federation (FIP) and published in the Journal of Pharmaceutical Sciences (/cgi-bin/jhome/68503813).The relevant documents can also be downloaded from the FIP web site at: http://www.fi/. These monographs provide detailed information which should be taken into account whenever available in the biowaiver consideration.3985.1Which active pharmaceutical ingredients are included in thetables?The substances listed in the 14th WHO Model List of Essential Medicines (EML) of March 2005 have been evaluated and classifi ed according to the revised criteria given above.5.2Where do the data come from?The solubility and permeability values were found in the publicly available literature, such as Martindale’s, the Merck Index and scientifi c journals.Please note that the doses used for the calculation of the dose:solubility ratio are those stated in the EML.The indications given in the tables are reproduced directly from the EML. If the EML specifi es the dosage form (e.g. sublingual tablet) this is indicated under “comments”.5.3“Worst case” approach to the Biopharmaceutics Classifi cationSystemThe drugs listed in the EML were classifi ed according to the criteria explained above. Where no clear classifi cation could be made, the “worst case” was as-sumed. For example if a substance is highly soluble, but absolute bioavailability data were not available, the test conditions for BCS Class III substances have been proposed. The same procedure was adopted for fi xed combinations, for example amoxicillin and clavulanic acid, the testing procedure was always fi xed according to the “worst” BCS classifi cation, in this example clavulanic acid (BCS Class III/1), because amoxicillin is a BCS Class I drug. This com-bination would therefore be tested according to BCS Class III requirements.The results of the revised classifi cation can be found in Tables 1–3.5.4Why are there three Tables?Table 1 lists all APIs on the EML that are administered orally, with the excep-tion of the APIs listed as complementary. Table 2 summarizes the APIs listed as complementary in the EML and Table 3 lists the APIs for which no classifi cation had previously been assigned, or that had been introduced with the 14th EML (March 2005), together with a more detailed explanation of their classifi cation.5.5 Risk assessmentTo minimize the risks of an incorrect biowaiver decision in terms of public health and risks to individual patients, the therapeutic indications of the API, known pharmacokinetic variations, food effects, etc. should be evalu-ated based on local clinical experience, taking into account the indications399for which the API is prescribed in that country as well as specifi c pharmaco-kinetic population variations (for example CYP polymorphisms). Known potential risks are listed under “potential risks” in the tables. The absence of an entry under “potential risks” should not, however, be misconstrued as meaning that there are no risks associated with the use of the medicine. 6.Biowaiver testing procedure according to WHODepending on the BCS classifi cation of the API, based on solubility and permeability characteristics listed in the accompanying tables, the testing procedure is defi ned in section 9.2.1 of the “Multisource document”1:6.1For pharmaceutical products containing BiopharmaceuticsClassifi cation System Class I (highly soluble, highlypermeable) APIsFor rapidly dissolving (as defi ned above) pharmaceutical products contain-ing BCS Class I APIs, more than 85% dissolution of the labelled amount is required within 30 minutes in standard media at pH 1.2, 4.5 and 6.8 using the paddle apparatus at 75 rpm or the basket apparatus at 100 rpm. The dis-solution profi les of the comparator and the multisource products should be compared by an f> 50 or an equivalent statistical criterion.2If within 15 minutes more than 85% of the API are released from the compar-ator and the multisource formulation under the above-mentioned conditions the products will be considered very rapidly dissolving. In this case the prod-ucts are deemed to be equivalent and a profi le comparison is not required.6.2For pharmaceutical products containing BiopharmaceuticsClassifi cation System Class III (highly soluble, lowpermeability) APIsA biowaiver can be considered only if both the multisource and the com-parator product are very rapidly dissolving. Eighty-fi ve per cent or more dissolution of the labelled amount of the API should be achieved within15 minutes in standard media at pH 1.2, 4.5 and 6.8 using the paddle ap-paratus at 75 rpm or the basket apparatus at 100 rpm.Generally, the risks of an inappropriate biowaiver decision should be more critically reviewed (e.g. site-specifi c absorption, induction/competition at the absorption site, excipient composition and therapeutic risks) for prod-ucts containing BCS Class III APIs than for BCS Class I drugs.1Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability (WHO Technical Report Series, No. 937, Annex 7).4006.3For pharmaceutical products containing APIs with highsolubility at pH 6.8 but not at pH 1.2 or 4.5 and with highpermeability (by defi nition, BCS Class II compoundswith weak acidic properties)These are eligible for a biowaiver provided that the multisource product:• is rapidly dissolving, i.e. 85% or more dissolution of the labelled amount of the API should be achieved within 30 minutes in standard media at pH 6.8 using the paddle apparatus at 75 rpm or the basket apparatus at 100 rpm; and• the multisource product exhibits similar dissolution profiles, as deter-mined with the f2 value or equivalent statistical evaluation, to those ofthe comparator product in buffers at all three pH values (pH 1.2, 4.5 and6.8).For multisource products containing BCS Class II APIs with dose:solubility ratios of 250 ml or less, at pH 6.8, the excipients should also be critically evaluated in terms of type and amounts of surfactants in the formulation.Further details of eligibility for the biowaiver and appropriate test proce-dures can be found in sections 5 and 9 of the “Multisource document”.11Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability (WHO Technical Report Series, No. 937, Annex 7).401405c h l o r p h e n a -m i n e h yd r o ge n m a l e a t e 4 m g h i g hB A 25-59%, fi r s t p a s s 3/19.2.1.2C Y P 2D 6 p o l y -m o r p h i s m a n t i a l l e r g i ce x t e n t of fi r s t -p a s s m e t a b o l i s m u n c e r t a i nc h l o r p r o m a z i n e h yd r o c h l o r i de 100 m gh i g hl o w39.2.1.2 p s y c h o t h e r a p e u -t i c m e d i c i n e c i p r o fl o x a c i nh y d r o c h l o r i d e 250 m g h i g hB A 70–82%, p o s s i b l e fi r s t p a s s , h i g h i nC a c o -2 c e l l s3/19.2.1.2 a n t i b a c t e r i a le x t e n t of fi r s t - p a s s m e t a b o l i s m u n c e r t a i nc l o f a z i m i n e100 m gi n s u f fi c i e n t l i t e r a t u r e l o w 4/3N o t e l i g i b l e f o r b i o w a i v e r a t p r e s e n t a n t i l e p r o s y m e d i c i n ec l o m i f e n e c i t r a t e50 m g h i g h i n s u f fi c i e n t l i t e r a t u r e 3/19.2.1.2o v u l a t i o n i n d u c e rc l o m i p r a m i n e h yd r o c h l o r i de 25 m g h i g h66% e x c r e t e d i n t h e u r i n e , t h e r e m a i n d e r b e i n g e l i m i -n a t e d i n t h e f a e c e s 3/19.2.1.2p s y c h o t h e r a p e u -t i c m e d i c i n el a c k o f a b s o l u t e b i o a v a i l a b i l i t y d a t ac l o x a c i l l i n (a s s od i u m s a l t )1000 m g h i g hl o w 39.2.1.2a n t i b a c t e r i a lc ode i n e p h o s p h a t e 30 m g h i g h l o w39.2.1.2r i s k o f a b u s eo p i o i d a n a l g e s i c ,d i a r r h oe a i n a d u l t sd a p s o n e100 m gl o w (w e a k b a s e ) h i g h 2N o t e l i g i b l e f o r b i o w a i v e rG 6P D d e fi -c i e n c ya n t i l e p r o s y m e d i c i n ed i a ze p a m 5 m g h i g hh i g h19.2.1.1p s y c h o t h e r a p e u -t i c m e d i c i n e s c o r e d t a b l e tB A , B i o a v a i l a b i l i t y ; G 6P D , g l u c o s e -6-p h o s p h a t e d e h y d r o g e n a s e .409g l y c e r y l t r i n i t r a t e 500 μgh i g hs u b l i n g u a l a p p l i c a t i o n ,p e r m e a b i l -i t y i n t h e o r a l c a v i t y m o r e i m p o r t a n t t h a n G I p e r m e a b i l i t y3/1N A hl o c a l a b s o r p t i o n a n t i a n g i n a l m e d i c i n e s u b l i n g u a l a p p l i c a t i o ng r i s e o f u l v i n 250 m gl o w (n e u t r a l ) h i g h2N o t e l i g i b l e f o r b i o w a i v e r a n t i f u n g a lh a l o p e r i d o l2 m gb o r d e r l i n e < 0.01 m g /m l 2l o w 4/3N o t e l i g i b l e f o r b i o w a i v e rp s y c h o t h e r a p e u -t i c m e d i c i n eh y d r a l a z i n e h y d r o c h l o r i d e50 m g h i g hl o w 39.2.1.2a n t i h y p e r t e n s i v e m e d i c i n eh y d r o c h l o r o -t h i a z i d e 25 m g h i g h l o w 39.2.1.2a n t i h y p e r t e n s i v e m e d i c i n e , d i u r e t i c a n d u s e d i n h e a r t f a i l u r es c o r e d t a b l e ti b u p r o f e n 400 m gl o w , w e a k a c i d (p K a 4.4,5.2)h i g h 29.2.1.3N S A I D , a n t i m i -g r a i n e m e d i c i n ei n d i n a v i r s u l f a t e 400 m g l o w l o w (?)4/2N o t e l i g i b l e f o r b i o w a i v e r C Y P 450 3A 4, f o o d e f f e c t (–)a n t i r e t r o v i r a lu n k n o w n w h e t h e r p o o r B A i s d u e t o p o o r s o l u b i l i t y o r p o o r s o l u b i l i t y a n d p o o r p e r m e a b i l i t yD :S , D o s e :s o l u b i l i t y r a t i o ; B A , b i o a v a i l a b i l i t y .426T a b l e 3C o m p o u n d s i n t r o d u c e d t o t h e W H O M o d e l L i s t o f E s s e n t i a l M e d i c i n e s s i n c e M a r c h 2005 f o r w h i c h n o c e r t a i n c l a s s i fi c a t i o n h a d b e e n p r e v i o u s l y r e p o r t e d (t h e s e c o m p o u n d s a l s o a p p e a r i n T a b l e 1 a n d T a b l e 2)M e d i c i n e aH i g h e s t o r a l s t r e n g t h a c c o r d i n g t o W H O E s s e n t i a l M e d i c i n e s L i s t a S o l u b i l i t y bP e r m e a b i l i t y c B C S c l a s s dD i s s o l u t i o n t e s t (f o r b i o w a i v e r )e P o t e n t i a l r i s k s fI n d i c a t i o n (s )a c c o r d i n g t o W H O E s s e n t i a l M e d i c i n e s L i s t (E M L )aC o m m e n t s a n d s p e c i a l d o s a g e f o r m i n d i c a t i o n s aa m l o d i p i n e 5 m gs l i g h t l y s o l u b l e (1),D :S 5 m lB A a b s60–65%,e x c r e t i o n o f d r u g m e t a b o -l i t e s i n u r i n e 90–95% (2)19.2.1.1a n t i h y p e r t e n s i v e m e d i c i n eB A a b s < 85% a s c r i b e d t o fi r s t -p a s s m e t a b o l i s ma m o d i a q u i n e(b a s e )200 m g45 m g /m l 2,D :S 4.4 m lB A > 75% (3)3/19.2.1.2C Y P 2C 8p o l y m o r p h i s m ,i n c r e a s e d r i s k f o r a g r a n u l o c y -t o s i s a n d h e p a -t o t o x i c i t y (4)a n t i m a l a r i a la m o x i c i l l i n + c l a v u l a n i c a c i d 500 m g + 125 m gf r e e l y s o l u b l e i n w a t e r (1),D :S 1.25 m la b s o r p t i o n > 73% (5)1 + 3/19.2.1.2a n t ib ac t e r i a lt e s t s b a s e d o n c l a v u l a n i c a c i d c l a s s i fi c a t i o na r t e s u n a t e 50 m gv e r ys l i g h t l y s o l u b l e (6),D :S 500 m l ;(w e a k a c i d ,p K a ~ 6.4)B A a b s 82% (1),B A a b s 88% (7),B A a b s 61% (8)4/2N o t e l i g i b l e f o r b i o w a i v e ra n t i m a l a r i a lp e r m e a b i l i t y d e p e n d s o n s e v e r i t y o f d i s e a s eD :S , D o s e : s o l u b i l i t y ; B A , B i o a v a i l a b i l i t y .427a z i t h r o m y c i n 500 m gp r a c t i c a l l y i n s o l u b l e i n w a t e r (1)< 0.01m g /m l , D :S 50 000 m lB A a b s 16% (9);B A 37%(10, 11); 4/2N o t e l i g i b l e f o r b i o w a i v e ra n t ib ac t e r i a l u n k n o w n w h e t h e r p o o r B A i sd ue t o p o o r s o l u b i l i t y o r p o o r s o l u b i l i t y a n d p o o r p e r m e a b i l i t yc a l c i u m f o l i n a t e 15 m gs p a r i n g l y s o l u b l e i n w a t e r (P h . E u r . 5.2); v e r y s o l u b l e (U S P 28); D :S 15 m l a n d 0.015 m l , r e s p e c -t i v e l yB A a b s 92% 25 m g (12, 13);B A a b s 73.4%(15 m g ) (14);f u l l y a b s o r b e d ;A UC a n d t 1/2s i m i l a r a f t e r i.v . & p .o (15)19.2.1.1 a n t i c y t o t o x i c m e d i c i n el e v o d o p a (l ) + c a r b i d o p a (c )(l ) 250 m g + (c ) 25 m g(l ) h i g h +(c ) s o l u b l e 1 i n 500 o f w a t e r , f r e e l y s o l u b l e i n 3 M H C l (1)(l ) h i g h +(c ) B A 58% (16); B A a b s88% (d o g s ) (17)(l ) 1 +(c ) 3/19.2.1.2n a r r o w t h e r a p e u t i c i n d e xa n t i p a r k i n s o n m e d i c i n et e s t s b a s e d o n c a r b i d o p a c l a s s i fi c a t i o nc e fi x i m e 400 m gs l i g h t l ys o l u b l e (2),D :S 400 m l22–54% (2)4N o t e l i g i b l e f o r b i o w a i v e ra n t ib ac t e r i a lD :S , D o s e : s o l u b i l i t y ; B A : B i o a v a i l a b i l i t y ; P h .E u r ., E u r o p e a n P h a r m a c o p o e i a ; U S P , U n i t e d S t a t e s P h a r m a c o p o e i a ; A U C , a r e a u n d e r t h e c u r v e ; i.v ., i n t r a v e n o u s .。
百普乐(培哚普利吲达帕胺片)

特殊警告 与培哚普利相关:
● 在免疫功能低下患者发生中性白细胞减少症 / 粒细胞缺乏症的危险 中性粒细胞减少症的危险与剂量及患者类型相关,并取决于患者的临床情况。没有并发症的患者极少会出现这种情况,但是与胶原血管性疾病相关的肾 功能不全的患者可能发生,如系统性红斑狼疮或硬皮病患者以及使用免疫抑制剂治疗的患者。 停止使用血管紧张素转化酶抑制剂治疗,危险性可消失。 严格遵守预先规定的剂量用药可能是防止事件发生的最好办法。但是,如果这些患者需要使用血管紧张素转化酶抑制剂,应慎重评估风险 / 效益比值。
儿童 百普乐不能用于儿童,因为儿童单独应用或联合应用培哚普利的疗效和耐受性尚未确定。
[不良反应]
服用培哚普利可抑制肾素-血管紧张素-醛固酮轴而使吲达帕胺所致的失钾减少。服用百普乐的 2%患者出现低钾血症(钾离子水平< 3.4mmol/l)。
胃肠道 - 通常发生(> 1/100, < 1/10):便秘、口干、恶心、上腹痛、厌食、腹痛、味觉障碍。 - 极少发生(< 1/10, 000):胰腺炎。 - 在肝功能不全病例中,有引发肝性脑病的可能性(见禁忌和注意事项)。
一个月内血压即出现下降,无急速抗药反应;停药后无反弹作用。在临床试验中,同时给予培哚普利和吲达帕胺,与分别单独使用这二种药物相比,可 产生具有协同作用的抗高血压疗效。
与培哚普利相关: 培哚普利可以治疗各种程度的高血压:轻度到中度或重度。可以降低卧位和立位的收缩压和舒张压。 最大降压作用出现在服用单一剂量后 4-6 小时,降压作用可持续 24 小时以上。
培哚普利在低或正常肾素水平的患者中也可产生抗高血压作用。
培哚普利通过它的活性代谢产物—培哚普利拉产生作用。其它的代谢产物均无活性。
培哚普利可减轻心脏负荷: - 通过改变前列腺素代谢产生扩张静脉的作用:减轻前负荷, - 通过降低总外周血管阻力:减轻后负荷。
Calcipotriol_SDS_MedChemExpress

Inhibitors, Agonists, Screening LibrariesSafety Data Sheet Revision Date:Sep.-13-2017Print Date:Sep.-13-20171. PRODUCT AND COMPANY IDENTIFICATION1.1 Product identifierProduct name :CalcipotriolCatalog No. :HY-10001CAS No. :112965-21-61.2 Relevant identified uses of the substance or mixture and uses advised againstIdentified uses :Laboratory chemicals, manufacture of substances.1.3 Details of the supplier of the safety data sheetCompany:MedChemExpress USATel:609-228-6898Fax:609-228-5909E-mail:sales@1.4 Emergency telephone numberEmergency Phone #:609-228-68982. HAZARDS IDENTIFICATION2.1 Classification of the substance or mixtureNot a hazardous substance or mixture.2.2 GHS Label elements, including precautionary statementsNot a hazardous substance or mixture.2.3 Other hazardsNone.3. COMPOSITION/INFORMATION ON INGREDIENTS3.1 SubstancesSynonyms:MC 903; CalcipotrieneFormula:C27H40O3Molecular Weight:412.60CAS No. :112965-21-64. FIRST AID MEASURES4.1 Description of first aid measuresEye contactRemove any contact lenses, locate eye-wash station, and flush eyes immediately with large amounts of water. Separate eyelids with fingers to ensure adequate flushing. Promptly call a physician.Skin contactRinse skin thoroughly with large amounts of water. Remove contaminated clothing and shoes and call a physician.InhalationImmediately relocate self or casualty to fresh air. If breathing is difficult, give cardiopulmonary resuscitation (CPR). Avoid mouth-to-mouth resuscitation.IngestionWash out mouth with water; Do NOT induce vomiting; call a physician.4.2 Most important symptoms and effects, both acute and delayedThe most important known symptoms and effects are described in the labelling (see section 2.2).4.3 Indication of any immediate medical attention and special treatment neededTreat symptomatically.5. FIRE FIGHTING MEASURES5.1 Extinguishing mediaSuitable extinguishing mediaUse water spray, dry chemical, foam, and carbon dioxide fire extinguisher.5.2 Special hazards arising from the substance or mixtureDuring combustion, may emit irritant fumes.5.3 Advice for firefightersWear self-contained breathing apparatus and protective clothing.6. ACCIDENTAL RELEASE MEASURES6.1 Personal precautions, protective equipment and emergency proceduresUse full personal protective equipment. Avoid breathing vapors, mist, dust or gas. Ensure adequate ventilation. Evacuate personnel to safe areas.Refer to protective measures listed in sections 8.6.2 Environmental precautionsTry to prevent further leakage or spillage. Keep the product away from drains or water courses.6.3 Methods and materials for containment and cleaning upAbsorb solutions with finely-powdered liquid-binding material (diatomite, universal binders); Decontaminate surfaces and equipment by scrubbing with alcohol; Dispose of contaminated material according to Section 13.7. HANDLING AND STORAGE7.1 Precautions for safe handlingAvoid inhalation, contact with eyes and skin. Avoid dust and aerosol formation. Use only in areas with appropriate exhaust ventilation.7.2 Conditions for safe storage, including any incompatibilitiesKeep container tightly sealed in cool, well-ventilated area. Keep away from direct sunlight and sources of ignition.Recommended storage temperature: 4°C, protect from light, stored under nitrogenShipping at room temperature if less than 2 weeks.7.3 Specific end use(s)No data available.8. EXPOSURE CONTROLS/PERSONAL PROTECTION8.1 Control parametersComponents with workplace control parametersThis product contains no substances with occupational exposure limit values.8.2 Exposure controlsEngineering controlsEnsure adequate ventilation. Provide accessible safety shower and eye wash station.Personal protective equipmentEye protection Safety goggles with side-shields.Hand protection Protective gloves.Skin and body protection Impervious clothing.Respiratory protection Suitable respirator.Environmental exposure controls Keep the product away from drains, water courses or the soil. Cleanspillages in a safe way as soon as possible.9. PHYSICAL AND CHEMICAL PROPERTIES9.1 Information on basic physical and chemical propertiesAppearance White to off-white (Solid)Odor No data availableOdor threshold No data availablepH No data availableMelting/freezing point No data availableBoiling point/range No data availableFlash point No data availableEvaporation rate No data availableFlammability (solid, gas)No data availableUpper/lower flammability or explosive limits No data availableVapor pressure No data availableVapor density No data availableRelative density No data availableWater Solubility No data availablePartition coefficient No data availableAuto-ignition temperature No data availableDecomposition temperature No data availableViscosity No data availableExplosive properties No data availableOxidizing properties No data available9.2 Other safety informationNo data available.10. STABILITY AND REACTIVITY10.1 ReactivityNo data available.10.2 Chemical stabilityStable under recommended storage conditions.10.3 Possibility of hazardous reactionsNo data available.10.4 Conditions to avoidNo data available.10.5 Incompatible materialsStrong acids/alkalis, strong oxidising/reducing agents.10.6 Hazardous decomposition productsUnder fire conditions, may decompose and emit toxic fumes.Other decomposition products - no data available.11.TOXICOLOGICAL INFORMATION11.1 Information on toxicological effectsAcute toxicityClassified based on available data. For more details, see section 2Skin corrosion/irritationClassified based on available data. For more details, see section 2Serious eye damage/irritationClassified based on available data. For more details, see section 2Respiratory or skin sensitizationClassified based on available data. For more details, see section 2Germ cell mutagenicityClassified based on available data. For more details, see section 2CarcinogenicityIARC: No component of this product present at a level equal to or greater than 0.1% is identified as probable, possible or confirmed human carcinogen by IARC.ACGIH: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by ACGIH.NTP: No component of this product present at a level equal to or greater than 0.1% is identified as a anticipated or confirmed carcinogen by NTP.OSHA: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by OSHA.Reproductive toxicityClassified based on available data. For more details, see section 2Specific target organ toxicity - single exposureClassified based on available data. For more details, see section 2Specific target organ toxicity - repeated exposureClassified based on available data. For more details, see section 2Aspiration hazardClassified based on available data. For more details, see section 212. ECOLOGICAL INFORMATION12.1 ToxicityNo data available.12.2 Persistence and degradabilityNo data available.12.3 Bioaccumlative potentialNo data available.12.4 Mobility in soilNo data available.12.5 Results of PBT and vPvB assessmentPBT/vPvB assessment unavailable as chemical safety assessment not required or not conducted.12.6 Other adverse effectsNo data available.13. DISPOSAL CONSIDERATIONS13.1 Waste treatment methodsProductDispose substance in accordance with prevailing country, federal, state and local regulations.Contaminated packagingConduct recycling or disposal in accordance with prevailing country, federal, state and local regulations.14. TRANSPORT INFORMATIONDOT (US)This substance is considered to be non-hazardous for transport.IMDGThis substance is considered to be non-hazardous for transport.IATAThis substance is considered to be non-hazardous for transport.15. REGULATORY INFORMATIONSARA 302 Components:No chemicals in this material are subject to the reporting requirements of SARA Title III, Section 302.SARA 313 Components:This material does not contain any chemical components with known CAS numbers that exceed the threshold (De Minimis) reporting levels established by SARA Title III, Section 313.SARA 311/312 Hazards:No SARA Hazards.Massachusetts Right To Know Components:No components are subject to the Massachusetts Right to Know Act.Pennsylvania Right To Know Components:No components are subject to the Pennsylvania Right to Know Act.New Jersey Right To Know Components:No components are subject to the New Jersey Right to Know Act.California Prop. 65 Components:This product does not contain any chemicals known to State of California to cause cancer, birth defects, or anyother reproductive harm.16. OTHER INFORMATIONCopyright 2017 MedChemExpress. The above information is correct to the best of our present knowledge but does not purport to be all inclusive and should be used only as a guide. The product is for research use only and for experienced personnel. It must only be handled by suitably qualified experienced scientists in appropriately equipped and authorized facilities. The burden of safe use of this material rests entirely with the user. MedChemExpress disclaims all liability for any damage resulting from handling or from contact with this product.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
760-78-1_DL-正缬氨酸_MED11074技术资料_上海_Medbio脉铂

1g
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11127
Fmoc-L-丝氨酸
Fmoc-Ser-OH
73724-45-5
100g
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
CAS
包装
纯度
MedBio
MED11025
N-羟基琥珀酰亚胺
HOSu
6066-82-6
100g
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11040
丝氨酸苄酯盐酸盐
H-Ser-OBzl.HCl
1738-72-3
100g
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11086
5g
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11059
N-Fmoc-N'-Boc-L-2,3-二氨基丙酸
Fmoc-Dap(Boc)-OH
162558-25-0
1g
≥98%
纯度
MedBio
MED11049
Fmoc-N-三苯甲基-L-天冬酰胺
Fmoc-Asn(Trt)-OH
132388-59-1
100g
≥98%
品牌
货号
中文名称
法倔唑产品说明书

产品说明书Beijing Jin Ming Biotechnology Co.,Ltd. Tel/Fax*************邮箱:*******************网址:基本信息产品编号:F10536产品名称:FadrozoleCAS:102676-47-1储存条件粉末-20℃四年分子式:C14H13N3溶于液体-80℃六个月分子量223.11-20℃一个月化学名:法倔唑CGS16949A free base;(Rac)-FAD286Solubility(25°C):体外:DMSO100mg/mL(448.21mM) Ethanol100mg/mL(448.21mM) Water Insoluble<1mg/ml表示微溶或不溶。
普西唐提供的所有化合物浓度为内部测试所得,实际溶液度可能与公布值有所偏差,属于正常的批间细微差异现象。
请根据产品在不同溶剂中的溶解度选择合适的溶剂配制储备液;⼀旦配成溶液,请分装保存,避免反复冻融造成的产品失效。
生物活性产品描述Fadrozole(CGS16949A)是一种有效的、选择性aromatase抑制剂,IC50为4.5nM,对aromatase比对其他细胞色素P450酶更有选择性。
靶点Aromatase4.5nM体外研究Fadrozole hydrochloride is a very potent inhibitor of both human placental and rat ovarian aromatase.In hamster ovarian slices,fadrozole hydrochloride inhibits the production of estrogen with an IC50of0.03μM.The productionof progesterone is inhibited with an IC50of120μM.Synthesis of other cytochrome P-450dependent steroids canbe suppressed to various degrees with higher doses of fadrozole hydrochloride.体内研究Fadrozole hydrochloride is able to inhibit the aromatase-mediated androstenedione-induced uterine hypertrophy in immature female rats with an ED50of0.03mg/kg when given orally.In the same model,aminoglutethimide elicitsthe same effect with an ED50of30mg/kg when given orally.Fadrozole hydrochloride prevents the development ofboth benign and malignant spontaneus mammary neoplasns in female Sprague-Dawley rats.It also slows the spontaneousdevelopment of ptuitary pars dta mas in female rats,and reduces the of spontaneous hcu ar tumours in male and femalerats.Administration of fadrozole in male and female mice suppresses the production of17b-estradiol,accompaniedwith a70%reduction in parasite burden.This protective effect is associated in male mice with a recovery of thespecific cellular immune response.Interleukin-6(IL-6)serum levels,and its production by splenocytes,is augmentedby80%,together with a10-fold increase in its expression in testes of infected male mice.Fadrozole treatment returnsthese levels to baseline values.。
药用辅料中英文对照

药用辅料中英文对照药用辅料中英文对照1 阿拉伯胶(Acacia)2 乙酰舒泛钾(Acesulfame Potassium)3 冰醋酸(Acetic Acid,Glacial)4 乙酰枸橼酸三丁酯(AcetyltributylCitrate)5 乙酰枸橼酸三乙酯(AcetyltriethylCitrate)6 人血白蛋白(Albumin)7 乙醇(Alcohol)8 海藻酸(Alginic Acid)9 脂肪族聚酯(Aliphatic Polyesters)10 阿力糖(Alitame)11 杏仁油(Almond Oil)12 维生素E(Alpha Tocopherol)13 氨溶液(Ammonia Solution)14 维生素C(Ascorbic Acid)15 棕榈酸维生素C酯(Ascorbyl Palmitate)16 阿司帕坦(Aspartame)17 绿坡缕石(Attapulgite)18 皂土(Bentonite)19 苯扎氯铵(Benzalkonium Chloride)20 苄索氯铵(Benzethonium Chloride)21 苯甲酸(Benzoic Acid)22 苯甲醇(Benzyl Alcohol)23 苯甲酸苄酯(Benzyl Benzoate)24 溴硝丙二醇(Bronopol)25 丁羟茴醚(Butylated Hydroxyanisole)26 丁羟甲苯(Butylated Hydroxytoluene)27 羟苯丁酯(Butylparaben)28 碳酸钙(Calcium Carbonate)29 无水磷酸氢钙(Calcium Phosphate,Dibasic Anhydrous)30 磷酸氢钙二水合物(Calcium Phosphate,Dibasic Dihydrate)31 磷酸钙(Calcium Phosphate,Tribasic)32 硬脂酸钙(Calcium Stearate)33 硫酸钙(Calcium Sulfate)34 低芥酸菜籽油(Canola Oil)35 卡波姆(Carbomer)36 二氧化碳(Carbon Dioxide)37 羧甲纤维素钙(Carboxymethylcellulose Calcium)38 羧甲纤维素钠(Carboxymethylcellulose Sodium)39 角叉菜胶(Carrageenan)40 蓖麻油(Castor Oil)41 氢化蓖麻油(Castor Oil,Hydro-genated)42 微晶纤维素(Cellulose,Microcr ystalline)43 粉状纤维素(Cellulose,Powdered)44 微粉硅胶微晶纤维素(Cellulose, Silicified Microcrystalline)45 醋酸纤维素(Cellulose Acetate)46 纤维醋法酯(Cellulose Acetate Phthalate)47 角豆胶(Ceratonia)48 十八十六醇(Cetostearyl Alcohol)49 西曲溴铵(Cetrimide)50 十六醇(Cetyl Alcohol)51 壳聚糖(Chitosan)52 氯己定(Chlorhexidine)53 三氯叔丁醇(Chlorobutanol)54 氯甲酚(Chlorocresol)55 一氯二氟乙烷(Chlorodifluoroe-thane)56 氟里昂(Chlorofluorocabons)57 对氯间二甲酚(Chloroxylenol)58 胆固醇(Cholesterol)59 枸橼酸(Citric Acid Monohydrate)60 胶态二氧化硅(微粉硅胶)(Colloidal Silicon Dioxide)61 着色剂(Coloring Agents)62 玉米油(Corn Oil)63 棉籽油(Cottonseed Oil)64 甲酚(Cresol)65 交联羧甲纤维素钠(Croscarmellose Sodium)66 交联聚维酮(Crospovidone)67 环糊精(Cyclodextrins)68 环甲基硅酮(Cyclomethicone)69 苯甲地那铵(Denatonium Benzoate)70 葡萄糖结合剂(Dextrates)71 糊精(Dextrin)72 葡萄糖(Dextrose)73 邻苯二甲酸二丁酯(Dibutyl Phthalate)74 癸二酸二丁酯(Dibutyl Sebacate)75 二乙醇胺(Diethanolamine)76 邻苯二甲酸二乙酯(Diethyl Phthalate)77 二氟乙烷(Difluoroethane)78 二甲硅油(Dimethicone)79 二甲醚(Dimethyl Ether)80 邻苯二甲酸二甲酯(Dimethyl Phthalate)81 二甲亚砜(Dimethyl Sulfoxide)82 多库酯钠(Docusate Sodium)83 依地酸(乙二胺四乙酸)(Edetic Acid)84 乙酸乙酯(Ethyl Acetate)85 乙基麦芽酚(Ethyl Maltol)86 油酸乙酯(Ethyl Oleate)87 乙基香草醛(Ethyl Vanillin)88 乙基纤维素(Ethylcellulose)89 硬脂酸棕榈酸乙二醇酯(Ethylene Glycol Palmitostearate)90 羟苯乙酯(Ethylparaben)91 果糖(Fructose)92 富马酸(Fumaric Acid)93 明胶(Gelatin)94 液体葡萄糖(Glucose,Liquid)95 甘油(Glycerin)96 山萮酸甘油酯(Glyceryl Behenate)97 单油酸甘油酯(Glyceryl Monooleate)98 单硬脂酸甘油酯(Glyceryl Monostearate)99 硬脂酸棕榈酸甘油酯(Glyceryl Palmitostearate)100 四氢呋喃聚乙二醇醚(Glycofurol)101 瓜耳胶(Guar Gum)102 七氟丙烷(HFC)(Heptafluoro-propane)103 海克西定(Hexetidine)104 烷烃类(HC) (Hydrocarbons)105 盐酸(Hydrochloric Acid)106 羟乙纤维素(Hydroxyethyl Cellulose)107 羟乙甲纤维素(Hydroxyethylmethyl Cellulose)108 羟丙纤维素(Hydroxypropyl Cellulose)109 低取代羟丙纤维素(Hydroxypropyl Cellulose,Low-substituted) 110 羟丙甲纤维素(Hypromellose)111 羟丙甲纤维素酞酸酯(Hypromellose Phthalate)112 咪唑烷脲(Imidurea)113 异丙醇(Isopropyl Alcohol)114 肉豆蔻酸异丙酯(Isopropyl Myristate)115 棕榈酸异丙酯(Isopropyl Palmitate)116 白陶土(Kaolin)117 乳酸(Lactic Acid)118 拉克替醇(Lactitol)119 乳糖(Lactose)120 羊毛脂(Lanolin)121 含水羊毛脂(Lanolin,Hydrous)122 羊毛醇(Lanolin Alcohols)123 卵磷脂(Lecithin)124 硅酸镁铝(Magnesium Aluminum Silicate)125 碳酸镁(Magnesium Carbonate)126 氧化镁(Magnesium Oxide)127 硅酸镁(Magnesium Silicate)128 硬脂酸镁(Magnesium Stearate)129 三硅酸镁(Magnesium Trisilicate)130 苹果酸(Malic Acid)131 麦芽糖醇(Maltitol)132 麦芽糖醇溶液(Maltitol Solution)133 麦芽糖糊精(Maltodextrin)134 麦芽酚(Maltol)135 麦芽糖(Maltose)136 甘露醇(Mannitol)137 中链脂肪酸甘油三酯(Medium-chain Triglycerides) 138 葡甲胺(Meglumine)139 薄荷脑(Menthol)140 甲基纤维素(Methylcellulose)141 羟苯甲酯(Methylparaben)142 液体石蜡(Mineral Oil)143 轻质液体石蜡(Mineral Oil,Light)144 液体石蜡羊毛醇(Mineral Oil and Lanolin Alcohols) 145 单乙醇胺(Monoethanolamine)146 谷氨酸一钠(Monosodium Glutamate)147 硫代甘油(Monothioglycerol)148 氮(Nitrogen)149 一氧化二氮(Nitrous Oxide)150 油酸(Oleic Acid)151 橄榄油(Olive Oil)152 石蜡(Paraffin)153 花生油(Peanut Oil)154 凡士林(Petrolatum)155 凡士林羊毛醇(Petrolatum and Lanolin Alcohols)156 苯酚(Phenol)157 苯氧乙醇(Phenoxyethanol)158 苯乙醇(Phenylethyl Alcohol)159 醋酸苯汞(Phenylmercuric Acetate)160 硼酸苯汞(Phenylmercuric Borate)161 硝酸苯汞(Phenylmercuric Nitrate)162 磷酸(Phosphoric Acid)163 波拉克林钾(Polacrilin Potassium)164 泊洛沙姆(Poloxamer)165 葡聚糖(Polydextrose)166 聚乙二醇(Polyethylene Glycol)167 聚氧乙烯(Polyethylene Oxide)168 聚(甲基)丙烯酸树脂(Polymethacr-ylates)169 聚氧乙烯烷基醚(Polyoxyethylene Alkyl Ethers)170 聚氧乙烯蓖麻油衍生物(Polyoxyeth-ylene Castor Oil Derivatives) 171 聚山梨酯(Polyoxyethylene Sorbitan Fatty Acid Esters)172 硬脂酸聚氧乙烯酯(Polyoxyethylene Stearates)173 聚醋酸乙烯酞酸酯(Polyvinyl Acetate Phthalate)174 聚乙烯醇(Polyvinyl Alcohol)175 苯甲酸钾(Potassium Benzoate)176 碳酸氢钾(Potassium Bicarbonate)177 氯化钾(Potassium Chloride)178 枸橼酸钾(Potassium Citrate)179 氢氧化钾(Potassium Hydroxide)180 焦亚硫酸钾(Potassium Metabisulfite)181 山梨酸钾(Potassium Sorbate)182 聚维酮(Povidone)183 丙酸(Propionic Acid)184 没食子酸丙酯(Propyl Gallate)185 碳酸丙烯酯(Propylene Carbonate)186 丙二醇(Propylene Glycol)187 海藻酸丙二醇酯(Propylene Glycol Alginate)188 羟苯丙酯(Propylparaben)189 糖精(Saccharin)190 糖精钠(Saccharin Sodium)191 芝麻油(Sesame Oil)192 虫胶(Shellac)193 二氧化硅二甲硅油(Simethicone)194 海藻酸钠(Sodium Alginate)195 抗坏血酸钠(Sodium Ascorbate)196 苯甲酸钠(Sodium Benzoate)197 碳酸氢钠(Sodium Bicarbonate)198 氯化钠(Sodium Chloride)199 枸橼酸钠二水合物(Sodium Citrate Dihydrate)200 环拉酸钠(Sodium Cyclamate)201 氢氧化钠(Sodium Hydroxide)202 月桂硫酸钠(十二烷基硫酸钠)(Sodium Lauryl Sulfate) 203 焦亚硫酸钠(偏亚硫酸钠)(Sodium Metabisulfite) 204 磷酸氢二钠(Sodium Phosphate,Dibasic)205 磷酸二氢钠(Sodium Phosphate ,Monobasic)206 丙酸钠(Sodium Propionate)207 羧甲淀粉钠(Sodium Starch Glycolate)208 硬脂富马酸钠(Sodium Stearyl Fumarate)209 山梨酸(Sorbic Acid)210 山梨坦酯Sorbitan Esters(Sorbitan Fatty Acid Esters)211 山梨醇(Sorbitol)212 大豆油(Soybean Oil)213 淀粉(Starch)214 预胶化淀粉(Starch,Pregelatinized)215 灭菌玉米淀粉(Starch,Sterilizable Maize)216 硬脂酸(Stearic Acid)217 硬脂醇(Stearyl Alcohol)218 羟糖氯(Sucralose)219 蔗糖(Sucrose)220 可压性蔗糖(Sugar,Compressible)221 蔗糖粉(Sugar,Confectioner’s)222 蔗糖球形颗粒(Sugar Spheres)223 硫酸(Sulfuric Acid)224 葵花籽油(Sunflower Oil)225 氢化植物油(硬脂)栓剂基质(Sup-pository Bases,Hard Fat) 226 滑石粉(Talc)227 酒石酸(Tartaric Acid)228 四氟乙烷(HFC)(Tetrafluoroe-thane)229 硫柳汞(Thimerosal)230 二氧化钛(Titanium Dioxide)231 西黄蓍胶(Tragacanth)232 海藻糖(Trehalose)233 三醋汀(Triacetin)234 枸橼酸三丁酯(Tributyl Citrate)235 三乙醇胺(Triethanolamine)236 枸橼酸三乙酯(Triethyl Citrate)237 香草醛(Vanillin)238 氢化植物油(Vegetable Oil,Hydrogenated)239 水(Water)240 阴离子乳化蜡(Wax,Anionic Emulsifying)241 巴西棕榈蜡(Wax,Carnauba)242 十六醇酯蜡(Wax,Cetyl Esters)243 微晶蜡(Wax,Microcrystalline)244 非离子乳化蜡(聚西托醇乳化蜡)(Wax,Nonionic Emulsifying) 245 白蜡(Wax,White)246 黄蜡(Wax,Yellow)247 黄原酸胶(Xanthan Gum)248 木糖醇(Xylitol)796249 玉米朊(玉米蛋白)(Zein)250 硬脂酸锌(Zinc Stearate)。
EMEA:生物等效性指南

European Medicines AgencyPre-Authorisation Evaluation of Medicines for Human Use7 Westferry Circus, Canary Wharf, London, E14 4HB, UKTel. (44-20) 74 18 84 00 Fax (44-20) 74 18 86 13E-mail: mail@emea.europa.eu http://www.emea.europa.euLondon, 24 July 2008 Doc. Ref. CPMP/EWP/QWP/1401/98 Rev. 1COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE(CHMP)DRAFT GUIDELINE ON THE INVESTIGATION OF BIOEQUIVALENCEDRAFT AGREED BY THE EFFICACY WORKING PARTYJuly 2008 ADOPTION BY CHMP FOR RELEASE FOR CONSULTATION24 July 2008 END OF CONSULTATION (DEADLINE FOR COMMENTS) 31 January 2009This guideline will replace the “Note for guidance on the investigation of bioavailability and bioequivalence" CPMP/EWP/QWP/1401/98 and the related questions in the Q&A document (EMEA/CHMP/EWP/40326/2006). This guideline includes recommendations on BCS-based biowaivers.Comments should be provided to EWPSecretariat@emea.europa.eu using this templateKEYWORDS Bioequivalence, pharmacokinetics, biowaiver, in vitro dissolution, genericsGUIDELINE ON THE INVESTIGATION OF BIOEQUIVALENCETABLE OF CONTENTSEXECUTIVE SUMMARY (3)11.INTRODUCTION (BACKGROUND) (3)232.SCOPE (4)43.LEGAL BASIS (4)54.MAIN GUIDELINE TEXT (5)4.1D ESIGN, CONDUCT AND EVALUATION OF BIOEQUIVALENCE STUDIES (5)674.1.1Study design (5)84.1.2Reference and test product (6)94.1.3Subjects (7)104.1.4Study conduct (7)114.1.5Characteristics to be investigated (9)124.1.6Strength and dose to be investigated (10)134.1.7Chemical analysis (12)144.1.8Evaluation (12)154.1.9Narrow therapeutic index drugs (15)164.1.10Highly variable drugs or drug products (16)174.2I N-VITRO DISSOLUTION TESTS (16)184.2.1In-vitro dissolution tests complementary to bioequivalence studies (16)4.2.2In-vitro dissolution tests in support of biowaiver of strengths (16)19204.3V ARIATIONS (16)4.4S TUDY REPORT (17)2122DEFINITIONS (17)23APPENDIX I (19)24D ISSOLUTION TESTING (19)25APPENDIX II (21)B IOEQUIVALENCE STUDY REQUIREMENTS FOR DIFFERENT DOSAGE FORMS (21)2627APPENDIX III (24)BCS-BASED B IOWAIVER (24)2829APPENDIX IV (28)D ECISION TREE ON MEASUREMENT OF PARENT COMPOUND OR METABOLITE (28)3031APPENDIX V (29)D ECISION TREE ON SELECTION OF DOSE AND STRENGTH IN BIOEQUIVALENCE STUDIES (29)3233EXECUTIVE SUMMARY3334This guideline defines when bioequivalence studies are necessary and formulates requirements for 35their design, conduct, and evaluation. The guideline focuses primarily on bioequivalence forimmediate release dosage forms with systemic action.36(background)371. INTRODUCTION38Two medicinal products containing the same active substance are considered bioequivalent if their 39bioavailabilities (rate and extent) after administration in the same molar dose lie within acceptable 40predefined limits. These limits are set to ensure comparable in vivo performance, i.e. similarity interms of safety and efficacy.4142In bioequivalence studies, the plasma concentration time curve is used to assess the rate and extent of43absorption. Meaningful pharmacokinetic parameters and preset acceptance limits allow the final 44decision on bioequivalence of the tested products. AUC, the area under the concentration time curve, 45reflects the extent of exposure. C max, the maximum plasma concentration or peak exposure, and the46time to maximum plasma concentration, t max, are parameters that are influenced by absorption rate.It is the objective of this guideline to define when bioequivalence studies are necessary and to4748formulate requirements for their design, conduct, and evaluation. The possibility of using in vitro 49instead of in vivo studies is also addressed.The concept of bioequivalence forms the basis for approval of generic application, but it may also be5051applicable to hybrid application, extensions and variations applications, and to different formulations 52used during the development of a new medicinal product containing a new chemical entity.For generic applications, the purpose of establishing bioequivalence is to demonstrate equivalence in5354biopharmaceutic quality between the generic product and a reference medicinal product in order to 55allow bridging of clinical data associated with the reference medicinal product. The current definition56for generic products is found in Directive 2001/83/EC, Article 10(2)(b). In general, a generic product 57is a product which has the same qualitative and quantitative composition in active substances as the58reference medicinal product, the same pharmaceutical form as the reference medicinal product, and whose bioequivalence with the reference medicinal product has been demonstrated by appropriate 5960bioavailability studies. By definition it is considered that different salts, esters, ethers, isomers, 61mixtures of isomers, complexes or derivatives of an active substance are considered to be the same active substance, unless they differ significantly in properties with regard to safety and/or efficacy.6263Furthermore, various immediate-release oral pharmaceutical forms are considered to be one and the 64same pharmaceutical form. It is also stated in the Directive that bioavailability studies need not be65required if it can be demonstrated that the generic medicinal product meets the relevant criteria for a 66biowaiver.67Hybrid applications rely on the results of preclinical tests and clinical trials of an approved referencemedicinal product and include new data. These new data may include bioequivalence or comparative6869bioavailability data.70Also applications for extensions such as additional dosage forms, new strengths, new routes of 71administration often need support of bioequivalence in order to bridge data from the authorised 72reference medicinal product.73Variations for a change in composition or for significant manufacturing changes which may affect drug bioavailability may also require support of bioequivalence studies.7475During development of a new chemical entity, the principles of bioequivalence may be applied in 76order to bridge data between different formulations e.g. between a formulation used in the pivotal clinical studies and the to-be-marketed formulation. In such situations however, wider acceptance 7778limits may be acceptable if these are justified based on data provided with a complete application,adequately addressing the clinical relevance of the widening from both a safety and efficacy 79perspective. 802. SCOPE 81This guideline focuses on recommendations for bioequivalence studies for immediate release 82formulations with systemic action. 83Specific recommendations regarding bioequivalence studies for modified release products, 84transdermal products and orally inhaled products are given in other guidelines (see section 3). 85Recommendation for the comparison of biologicals to reference medicinal products can be found in 86guidelines on biosimilar products. Recommendations for pharmacokinetics of therapeutic proteins are 87also described in a specific guideline (CPMP/EWP/89249/04). 88In case bioequivalence cannot be demonstrated using drug plasma concentrations, in exceptional 89circumstances pharmacodynamic or clinical endpoints may be needed. This situation is outside the 90scope of this guideline and the reader is referred to therapeutic area specific guidelines. 91Furthermore, this guideline does not cover aspects related to generic substitution as this is subject to 92national legislation. 933. LEGAL BASIS 94This guideline applies to Marketing Authorisation Applications for human medicinal products 95submitted in accordance with the Directive 2001/83/EC as amended, under Art. 8(3) (full 96applications), Art 10b (fixed combination), Art. 10 (1) (generic applications), Art 10(3) (hybrid 97applications), and also for line extension and variation applications in accordance with Commission 98Regulations (EC) No 1084/2003 and 1085/2003. 99This guideline should be read in conjunction with the Annex I of Directive 2001/83/EC as amended, 100as well as European and ICH guidelines for conducting clinical trials, including those on: 101− General Considerations for Clinical Trials (ICH topic E8, CPMP/ICH/291/95) 102 − Guideline for Good Clinical Practice (ICH E6 (R1), CPMP/ICH/135/95) 103 − Structure and Content of Clinical Study Reports (ICH E3, CPMP/ICH/137/95) 104 − CHMP guidance for users of the centralised procedure for generics/hybrid applications 105 (EMEA/CHMP/225411/2006) 106− Modified Release Oral and Transdermal Dosage Forms: Section II (CPMP/EWP/280/96) 107 − Requirements for clinical documentation for orally inhaled products (OIP) including the 108 requirements for demonstration of therapeutic equivalence between two inhaled products for 109use in the treatment of Asthma and Chronic Obstructive Pulmonary Disease (COPD) 110(CPMP/EWP/4151/00 rev 1). 111− Fixed Combination Medicinal Products (CPMP/EWP/240/95) 112 − Clinical Requirements for Locally Applied, Locally Acting Products containing Known 113 Constituents (CPMP/EWP/239/95) 114− Good manufacturing practice (Eudralex volume 4). 115The guideline should also be read in conjunction with relevant guidelines on pharmaceutical quality. 116The test products used in the bioequivalence study must be prepared in accordance with GMP-117regulations. 118Bioequivalence trials should be conducted in accordance to Directive 2001/20/EC of the European 119parliament and of the Council. 120Companies may also apply for CHMP Scientific Advice, via the EMEA, for specific queries not 121covered by existing guidelines.1224. MAIN GUIDELINE TEXT1231244.1 Design, conduct and evaluation of bioequivalence studies125In the following sections, requirements for the design, conduct and evaluation of bioequivalencestudies investigating immediate release formulations with systemic action are described.126127The formulation and the characteristics of the active substance can affect the requirements for128bioequivalence studies. When the test product contains a different salt, ester, ether, isomer, mixture of 129isomers, complex or derivative of an active substance than the reference product, bioequivalence 130should be demonstrated in appropriate bioavailability studies. However, when the active substance in131test and reference products are identical or contain comparable salts, in vivo bioequivalence studies may, in some situations, not be required as described in APPENDIX II (bioequivalence study 132133requirements) and III (biowaiver).134The pharmacokinetic and physico-chemical properties of the substance affect the number of studies needed and the design of the studies. The choice of number of studies and study design should be 135136thoroughly justified based on the physico-chemical characteristics of the substance and its 137pharmacokinetic properties, discussing especially linearity in pharmacokinetics, activity of138metabolites, contribution of metabolites to the effect, the need for enantioselective analysis, and 139solubility of the active substance. In the context of this guideline, high solubility and low solubility is 140defined according to the Biopharmaceutics Classification System (BCS) definition of high and lowsolubility, as defined in APPENDIX III.141142The clinical overview of an application for marketing authorisation should list all studies carried out143with the product applied for. All bioequivalence studies comparing the product applied for with the reference product of interest must be submitted.144145design4.1.1 Study146The study should be designed in such a way that the formulation effect can be distinguished from 147other effects.148Standard design149If two formulations are going to be compared, a two-period, two-sequence single dose crossover150design is the design of choice. The treatment periods should be separated by an adequate wash out 151period.152Alternative designs153In general, single dose studies will suffice. However, in case of dose or time-dependent pharmacokinetics, resulting in markedly higher concentrations at steady state than expected from 154155single dose data, a potential difference in AUC between formulations may be larger at steady state 156than after single dose. Hence, a multiple dose study may be required in addition to the single dose 157study to ensure that the products are bioequivalent regarding AUC also at steady state. However, if the158single dose study indicates very similar PK profile for test and reference (the 90% confidence interval 159for AUC is within 90-111), the requirement for steady-state data may be waived.In certain cases when a single dose study cannot be conducted in healthy volunteers due to tolerability 160161reasons, and a single dose study is not feasible in patients, conduct of a multiple dose study in patients 162may be acceptable (see also section 4.1.6 Strength and Dose).A multiple dose study as an alternative to a single dose study may also be acceptable if problems of 163164sensitivity of the analytical method preclude sufficiently precise plasma concentration measurements165after single dose administration. As C max at steady state may be less sensitive to differences in the absorption rate than C max after single dose, bioequivalence should, if possible, be determined for C max 166167after the single dose administration (i.e. after the first dose of the multiple dose study) as a measure ofpeak exposure while extent of exposure can be based on demonstration of bioequivalence of AUC at 168169steady state.170In steady-state studies the administration scheme should preferably follow the highest usual dosagerecommendation (see also section 4.1.6 Strength and dose).171172Under certain circumstances, provided the study design and the statistical analyses are scientifically173sound, alternative well-established designs could be considered such as parallel design for substances 174with very long half-life and replicate designs e.g. for substances with highly variable pharmacokinetic 175characteristics (see section 4.1.10).1764.1.2 Reference and test productFor Article 10(1) and 10(3) applications the chosen reference medicinal product must be a medicinal 177178product authorised in the Community, on the basis of a complete dossier in accordance with the 179provisions of Article 8 of Directive 2001/83/EC, as amended. The product used as reference product in 180the bioequivalence study should be part of the global marketing authorisation of the reference medicinal 181product (as defined in Article 6(1) second subparagraph of Directive 2001/83/EC). The choice of the 182reference medicinal product should be justified by the applicant in Module 1.2, and Module 1, section 1831.5.2.184Test products in an application for a generic product are normally compared with the corresponding 185dosage form of a reference medicinal product.In an application for extension of a concerned medicinal product and when there are several dosage 186187forms of this medicinal on the market, the dosage form used for the initial approval of the concerned 188medicinal product (and which was used in clinical efficacy and safety studies) should be used ascomparative product, unless otherwise justified.189190For variations of a concerned medicinal product, the comparative medicinal product for use in191bioequivalence and dissolution studies is usually that authorised under the currently registered 192formulation, manufacturing process, packaging etc.193When variations to a generic product are made, the comparative medicinal product for the 194bioequivalence study should be the reference medicinal product.195The reference and test products should be packed in an individual way for each subject and period.196Packaging, which is a manufacturing operation, should be performed and documented in accordance 197with good manufacturing practice, including Annex 13 to the EU guide to GMP. It should be possible198to identify unequivocally the identity of the product administered to each subject at each trial period.Packaging and administration of the products to the subjects should therefore be documented in detail. 199200This documentation should include all precautions taken to avoid and identify potential dosing201mistakes.202Batch control results of the test and reference products should be reported. The assayed content of the203batch used as test product should not differ more than 5% from that of the batch used as reference 204product determined with the test procedure proposed for routine quality testing of the test product. In order to demonstrate that a representative batch of the reference product with regards to dissolution 205206and assay content has been selected, the applicant should present dissolution profiles and content 207analysis of at least 3 batches of the reference product, unless otherwise justified.The test product used in the study should be representative of the product to be marketed and this 208209should be justified by the applicant. In the case of oral solid forms for systemic action the test product 210should usually originate from a batch of at least 1/10 of production scale or 100,000 units, whicheveris greater, unless otherwise justified. The production of batches used should provide a high level of 211212assurance that the product and process will be feasible on an industrial scale. In case of a productionbatch smaller than 100,000 units, a full production batch will be required. If the product is subjected to 213214further scale-up, this should be properly validated.215Samples of the product from full production batches should be compared with those of the test batch, and should show similar in vitro dissolution profiles when employing suitable dissolution test 216217conditions (see Appendix I).218The study sponsor will have to retain a sufficient number of all investigational product samples in the 219study for one year in excess of the accepted shelf life or two years after completion of the trial or until 220approval whichever is longer to allow re-testing, if it is requested by the authorities.2214.1.3 SubjectsNumber of subjects222223The number of subjects to be included in the study should be based on an appropriate sample size 224calculation. The minimum number of subjects in a cross-over study should be 12.Selection of subjects225226The subject population for bioequivalence studies should be selected with the aim to permit detection 227of differences between pharmaceutical products. In order to reduce variability not related to 228differences between products, the studies should normally be performed in healthy volunteers unless 229the drug carries safety concerns that make this unethical. This model, in vivo healthy volunteers, is 230regarded adequate in most instances to detect formulation differences and the results will allow extrapolation to populations in which the reference product is approved (the elderly, children, patients 231232with renal or liver impairment, etc.).233The inclusion/exclusion criteria should be clearly stated in the protocol. In general, subjects should preferably be between 18 - 55 years old and of weight within the normal range according to accepted 234235normal values for the Body Mass Index. The subjects should be screened for suitability by means of 236clinical laboratory tests, an extensive review of medical history, and a comprehensive medical 237examination. Depending on the drug’s therapeutic class and safety profile, special medical 238investigations and precautions may have to be carried out before, during and after the completion of 239the study. Subjects could belong to either sex; however, the risk to women of childbearing potential 240should be considered on an individual basis. Subjects should preferably be non-smokers and without a 241history of alcohol or drug abuse. If moderate smokers are included (less than 10 cigarettes per day) 242they should be identified as such and the consequences for the results should be discussed. 243Phenotyping and/or genotyping of subjects may be considered for safety or pharmacokinetic reasons.In parallel design studies, the treatment groups should be comparable in all known prognostic 244245variables that affect the pharmacokinetics of the active substance (e.g. ethnic origin, smoking status, 246extensive/poor metabolic status). This is an essential pre-requisite to give validity to the study results. 247If the investigated active substance is known to have adverse effects and the pharmacological effects 248or risks are considered unacceptable for healthy volunteers, it may be necessary to use patients, under 249suitable precautions and supervision, instead. In such case the applicant should justify the alternative. 250conduct4.1.4 Study251StandardisationThe test conditions should be standardised in order to minimise the variability of all factors involved 252253except that of the products being tested. Therefore, it is recommended to standardise diet, fluid intake 254and exercise.The time of day for ingestion should be specified. As fluid intake may influence gastric passage for 255256oral administration forms, the test and reference products should be administered with a standardisedvolume of fluid (at least 150 ml). All meals and fluids taken after the treatment should also be 257258standardised in regard to composition and time of administration during the sampling period. As the 259bioavailability of an active moiety from a dosage form could be dependent upon gastrointestinaltransit times and regional blood flows, posture and physical activity may need to be standardised.260261The subjects should abstain from food and drinks, which may interact with circulatory,262gastrointestinal, hepatic or renal function (e.g. alcoholic or xanthine-containing beverages or 263grapefruit juice) during a suitable period before and during the study.264Subjects should not take any other concomitant medication (including herbal remedies) for an265appropriate interval before as well as during the study. In case concomitant medication is unavoidable and a subject is administered other drugs, for instance to treat adverse events like headache, the use 266267must be reported (dose and time of administration) and possible effects on the study outcome must be 268addressed.In case the study is to be performed under fasting conditions, subjects should fast during the night 269270prior to administration of the products, unless otherwise justified.271Sampling times272A sufficient number of samples to adequately describe the complete plasma concentration-time profile 273should be collected. The sampling schedule should include frequent sampling around C max to provide a 274reliable estimate of peak exposure. The sampling schedule should be planned to avoid C max being thefirst point of a concentration time curve. When partial AUC is to be determined, frequent early 275276sampling is recommended with preferably at least two quantifiable samples before expected t max. The277sampling schedule should also cover the plasma concentration time curve long enough to provide a reliable estimate of the extent of exposure which is achieved if AUC t is at least 80% of AUC∞. At least 278279three to four samples are needed during the terminal log-linear phase in order to reliably estimate the280terminal rate constant (which is needed for a reliable estimate of AUC∞).281A sampling period longer than 72 h is not considered necessary for any immediate release282formulation. Hence, for drugs with a long half-life, comparison of extent of exposure using truncated 283AUCs at 72 h is acceptable.Fasting or fed conditions284285The study should be conducted during fasting conditions unless the SPC recommends intake of the 286originator product only in the fed state. If the recommendation of food intake in the SPC is based onpharmacokinetic properties such as higher bioavailability, the bioequivalence study should be 287288conducted in the fed state. Also if the recommendation of food intake is intended to decrease adverse289events or to improve tolerability, it is recommended to conduct the bioequivalence study in fed state, although a bioequivalence study under fasting conditions could be acceptable if this has been 290291adequately justified.292For products with enhanced release characteristics differing from conventional immediate release 293formulations (e.g. microemulsions or solid dispersions), bioequivalence studies performed under both294fasted and fed conditions are required.295In cases where information is required in both the fed and fasted states, it is preferable to conduct a four-period single dose crossover design study (both products fed and fasted) rather than conducting 296297two separate bioequivalence studies in fed and fasted state, respectively. In a four-period crossover 298design study, the food effect on test and reference product can be evaluated which is not the case when299conducting two separate two-period, two-sequence single dose crossover design studies under fasting 300and fed conditions, respectively. In addition to the bioequivalence evaluation of test/reference in 301fasting and in fed state, the food effect can be presented for test and reference, i.e. the ratio302food/fasting and 90% confidence interval for test and reference, respectively.303In studies performed under fed conditions, the composition of the meal should be according to304recommendations in the SPC of the reference product. If no recommendation on the composition of305the meal is given in the reference product SPC, the meal should be a "standardized non high-fat meal" 306(about 650 kcal with about 30% of calories derived from fat). The composition of the meal should be 307described with regard to protein, carbohydrate and fat content (specified in grams, calories and relative 308caloric content (%)).3094.1.5 Characteristics to be investigated310Pharmacokinetic parameters311In studies to determine bioequivalence after a single dose, AUC t, AUC∞, C max and t max should be 312determined. Additional parameters that may be reported include the terminal rate constant, λz, and t1/2.For products where rapid absorption is of importance, partial AUCs can be used as a measure of early 313314exposure. The partial area can in most cases be truncated at the population median of t max values for 315the reference formulation. However, an alternative time point for truncating the partial AUC can be 316used when clinically relevant. The time point for truncating the partial AUC should be pre-specified 317and justified in the study protocol.318In studies to determine bioequivalence at steady state, AUCτ, C max,ss,C min,ss,t max,ss and fluctuation should be determined.319320Definitions of the pharmacokinetic parameters are given in section 6.321Additional parameters may be presented. The methods of estimating parameters should be specified.The use of compartmental methods for the estimation of parameters is not acceptable.322323Parent compound or metabolites324Recommendations for measuring parent compound and metabolite(s) depend on the contribution of parent compound and metabolite(s), respectively, to activity as detailed below and in Appendix IV. 325326In principle, evaluation of bioequivalence should be based upon measured concentrations of the parent compound. The reason for this is that C max of a parent compound is usually more sensitive to detect 327328differences between formulations in absorption rate than C max of a metabolite.329Also for inactive prodrugs, demonstration of bioequivalence for parent compound is the preferred 330option when the pharmacokinetics of pro-drug and active metabolite(s) is linear. In this situation, the 331active metabolite does not need to be measured. However, in case the pro-drug or active metabolites 332display non-linear pharmacokinetics (or it is difficult to conclude linear pharmacokinetics from 333available data), it is recommended to demonstrate bioequivalence for the main active metabolite. In 334such case, the parent compound does not need to be measured provided that it is inactive from efficacy 335and safety perspectives. Moreover, some pro-drugs may have low plasma concentrations, be quickly eliminated and have high variability, resulting in difficulties in demonstrating bioequivalence for 336337parent compound in a reasonably sized bioequivalence study. In this situation it is acceptable to 338demonstrate bioequivalence for the main active metabolite without measurement of parent compound. 339Furthermore, in situations where the pro-drug exposure is low and exposure to active metabolite is 340very much higher, it is acceptable to demonstrate bioequivalence for the main active metabolite 341without measurement of parent compound.The use of a metabolite as a surrogate for an active parent compound can only be considered if the 342343applicant presents convincing arguments demonstrating that it is not possible to reliably measure the 344parent compound after single dose administration or at steady state. However, as C max of the metabolite 345is usually less sensitive to differences in the absorption rate than C max of the parent drug, 346bioequivalence should, if possible, be determined for C max of the parent compound as a measure of 347peak exposure while extent of exposure can be based on demonstration of bioequivalence of AUC of metabolite. Furthermore, when using metabolite data as a substitute for parent drug concentrations, the 348349applicant should present any available data supporting the view that the parent drug exposure will be。
CAS号755038-65-4_Volasertib_MedBio技术介绍

Plk1,Plk2和Plk3的酶活性测定在连续稀释的抑制剂存在下进行,使用20ng重组激酶和10μg来自牛乳的酪蛋白作为底物。激酶反应最终体积为60μL,在30°C下进行45分钟[15 mM MgCl2,25 mM MOPS(pH 7.0),1 mM DTT,1%DMSO,7.5μMATP,0.3μCiγ-32P-ATP ]。加入125μL冰冷的5%TCA终止反应。将沉淀物转移至MultiScreen混合酯纤维素滤板后,用1%TCA洗涤板并用放射性法定量。剂量-反应曲线用于计算IC 50值。为了建立激酶选择性谱,通过合同研究组织进行额外的激酶测定,或者从商业来源购买试剂,并且根据供应商的说明进行测定。在测定设计中包括适当的阳性和阴性对照。
Thrombin Receptor Activatorfor Peptide 5 (TRAP-5)
141685-53-2
5mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11579
Olomoucine
Olomoucine
101622-51-9
5mg
≥98%
品牌
货号
中文名称
英文名称
1357470-29-1
5mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11520
Griseofulvin
Griseofulvin
126-07-8
10mM (in 1mL DMSO)
≥98%
CAS
1、产品物理参数:
常用名
伏拉塞替
英文名
OECD Test No. 460 Fluorescein Leakage Test Method for Identifying Ocular Corrosives and Severe Irrit

OECD/OCDE460Adopted: 2 October 2012© OECD, (2012)You are free to use this material for personal, non-commercial purposes without seeking prior consent from the OECD, provided the source is duly mentioned. Any commercial use of this material is subject to written permission from the OECD.OECD GUIDELINE FOR THE TESTING OF CHEMICALS Fluorescein Leakage Test Method for Identifying Ocular Corrosives and Severe IrritantsINTRODUCTION1. The Fluorescein Leakage (FL) test method is an in vitro test method that can be used under certain circumstances and with specific limitations to classify chemicals (substances and mixtures) as ocular corrosives and severe irritants, as defined by the United Nations (UN) Globally Harmonized System of Classification and Labelling of Chemicals (GHS) (Category 1), the European Union (EU) Regulation on Classification, Labelling and Packaging of Substances and Mixtures (CLP) (Category 1), and the U.S. Environmental Protection Agency (EPA) (Category I) (1) (2) (3). For the purpose of this Test Guideline, severe irritants are defined as chemicals that cause tissue damage in the eye following test substance administration that is not reversible within 21 days or causes serious physical decay of vision, while ocular corrosives are chemicals that cause irreversible tissue damage to the eye. These chemicals are classified as UN GHS Category 1, EU CLP Category 1, or U.S. EPA Category I.2. While the FL test method is not considered valid as a complete replacement for the in vivo rabbit eye test, the FL is recommended for use as part of a tiered testing strategy for regulatory classification and labelling. Thus, the FL is recommended as an initial step within a Top-Down approach to identify ocular corrosives/severe irritants, specifically for limited types of chemicals (i.e. water soluble substances and mixtures) (4)(5).3. It is currently generally accepted that, in the foreseeable future, no single in vitro eye irritation test will be able to replace the in vivo eye test (TG 405 (6)) to predict across the full range of irritation for different chemical classes. However, strategic combinations of several alternative test methods within a (tiered) testing strategy may be able to replace the in vivo eye test (5). The Top-Down approach (5) is designed to be used when, based on existing information, a chemical is expected to have high irritancy potential.Based on the prediction model detailed in paragraph 35, the FL test method can identify Category 1; EU CLP Category 1; U.S. EPA Category I) without any further testing. The same is assumed for mixtures although mixtures were not used in the validation. Therefore, the FL test method may be used to determine the eye irritancy/corrosivity of chemicals, following the460OECD/OCDEsequential testing strategy of TG 405 (6). However, a chemical that is not predicted as ocular corrosive or severe irritant with the FL test method would need to be tested in one or more additional test methods (in vitro and/or in vivo) that are capable of accurately identifying i) chemicals that are in vitro false negative ocular corrosives/severe irritants in the FL (UN GHS Category 1; EU CLP Category 1; U.S. EPA Category I); ii) chemicals that are not classified for eye corrosion/irritation (UN GHS No Category; EU CLP No Category; U.S. EPA Category IV); and/or iii) chemicals that are moderate/mild eye irritants (UN GHS Categories 2A and 2B; EU CLP Category 2; U.S. EPA Categories II and III).5. The purpose of this Test Guideline is to describe the procedures used to evaluate the potential ocular corrosivity or severe irritancy of a test substance as measured by its ability to induce damage to an impermeable confluent epithelial monolayer. The integrity of trans-epithelial permeability is a major function of an epithelium such as that found in the conjunctiva and the cornea. Trans-epithelial permeability is controlled by various tight junctions. Increasing the permeability of the corneal epithelium in vivo has been shown to correlate with the level of inflammation and surface damage observed as eye irritation develops.6. In the FL test method, toxic effects after a short exposure time to the test substance are measured by an increase in permeability of sodium fluorescein through the epithelial monolayer of Madin-Darby Canine Kidney (MDCK) cells cultured on permeable inserts. The amount of fluorescein leakage that occurs is proportional to the chemical-induced damage to the tight junctions, desmosomal junctions and cell membranes, and can be used to estimate the ocular toxicity potential of a test substance. Annex I provides a diagram of MDCK cells grown on an insert membrane for the FL test method.7. Definitions are provided in Annex II.INITIAL CONSIDERATIONS AND LIMITATIONS8. This Test Guideline is based on the INVITTOX protocol No. 71 (7) that has been evaluated in an international validation study by the European Centre for the Validation of Alternative Methods (ECVAM) (8), in collaboration with the US Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the Japanese Center for the Validation of Alternative Methods (JaCVAM).9. The FL test method is not recommended for the identification of chemicals which should be classified as mild/moderate irritants or of chemicals which should not be classified for ocular irritation (substances and mixtures) (i.e. GHS Cat. 2A/2B, no category; EU CLP Cat. 2, no category; US EPA Cat. II/III/IV), as demonstrated by the validation study (4) (8).10. The test method is only applicable to water soluble chemicals (substances and mixtures). The ocular severe irritation potential of chemicals that are water soluble and/or where the toxic effect is not affected by dilution is generally predicted accurately using the FL test method (8). To categorise a chemical as water soluble, under experimental conditions, it should be soluble in sterile calcium-containing (at a concentration of 1.0-1.8 mM), phenol red-free, Hanks’ Buffered Salt Solution (HBSS) at a concentration ≥ 250 mg/mL (one dose above the cut-off of 100 mg/mL). However, if the test substance is soluble below the concentration 100 mg/mL,2© OECD, (2012)OECD/OCDE 460 but already induces a FL induction of 20 % at that concentration (meaning FL20 < 100 mg/mL), it can still be classified as GHS Cat. 1 or EPA Cat. 1.11. The identified limitations for this test method exclude strong acids and bases, cell fixatives and highly volatile chemicals from the applicability domain. These chemicals have mechanisms that are not measured by the FL test method, e.g. extensive coagulation, saponification or specific reactive chemistries. Other identified limitations for this method are based upon the results for the predictive capacity for coloured and viscous test substance (8). It is suggested that both types of chemicals are difficult to remove from the monolayer following the short exposure period and that predictivity of the test method could be improved if a higher number of washing steps was used. Solid chemicals suspended in liquid have the propensity to precipitate out and the final concentration to cells can be difficult to determine. When substances within these chemical and physical classes are excluded from the database, the accuracy of FL across the EU, EPA, and GHS classification systems is substantially improved (8).12. Based on the purpose of this test method (i.e. to identify ocular corrosives/severe irritants only), false negative rates (see Paragraph 13) are not critical since such substances would be subsequently tested with other adequately validated in vitro tests or in rabbits, depending on regulatory requirements, using a sequential testing strategy in a weight of evidence approach (6) (see also paragraphs 3 and 4).13. Other identified limitations of the FL test method are based on false negative and false positive rates. When used as an initial step within a Top-Down approach to identify water soluble ocular corrosive/severe irritant substances and mixtures (UN GHS Category 1; EU CLP Category 1; U.S. EPA Category I), the false positive rate for the FL test method ranged from 7% (7/103; UN GHS and EU CLP) to 9% (9/99; U.S. EPA) and the false negative rate ranged from 54% (15/28; U.S. EPA) to 56% (27/48; UN GHS and EU CLP) when compared to in vivo results. Chemical groups showing false positive and/or false negative results in the FL test method are not defined here.14. Certain technical limitations are specific to the MDCK cell culture. The tight junctions that block the passage of the sodium-fluorescein dye through the monolayer are increasingly compromised with increasing cell passage number. Incomplete formation of the tight junctions results in increased FL in the non-treated control. Therefore, a defined permissible maximal leakage in the non-treated controls is important (see paragraph 38: 0% leakage). As with all in vitro assays there is the potential for the cells to become transformed over time, thus it is vital that passage number ranges for the assays are stated.15. The current applicability domain might be increased in some cases, but only after analyzing an expanded data set of studied test substances, preferably acquired through testing (4). This Test Guideline will be updated accordingly as new information and data are considered.16. For any laboratory initially establishing this assay, the proficiency chemicals provided in Annex III should be used. Laboratories can use these chemicals to demonstrate their technical competence in performing the FL test method prior to submitting FL assay data for regulatory hazard classification purposes.PRINCIPLE OF THE TEST3© OECD, (2012)460OECD/OCDE17. The FL test method is a cytotoxicity and cell-function based in vitro assay that is performed on a confluent monolayer of MDCK CB997 tubular epithelial cells that are grown on semi-permeable inserts and model the non-proliferating state of the in vivo corneal epithelium. The MDCK cell line is well established and forms tight junctions and desmosomal junctions similar to those found on the apical side of conjunctival and corneal epithelia. Tight and desmosomal junctions in vivo prevent solutes and foreign materials penetrating the corneal epithelium. Loss of trans-epithelial impermeability, due to damaged tight junctions and desmosomal junctions, is one of the early events in chemical-induced ocular irritation.18. The test substance is applied to the confluent layer of cells grown on the apical side of the insert. A short 1 min exposure is routinely used to reflect the normal clearance rate in human exposures. An advantage of the short exposure period is that water-based substances and mixtures can be tested neat, if they can be easily removed after the exposure period. This allows more direct comparisons of the results with the chemical effects in humans. The test substance is then removed and the non-toxic, highly fluorescent sodium-fluorescein dye is added to the apical side of the monolayer for 30 minutes. The damage caused by the test substance to the tight junctions is determined by the amount of fluorescein which leaks through the cell layer within a defined period of time.19. The amount of sodium-fluorescein dye that passes through the monolayer and the insert membrane into a set volume of solution present in the well (to which the sodium-fluorescein dye leaks in) is determined by measuring spectrofluorometrically the fluorescein concentration in the well. The amount of fluorescein leakage (FL) is calculated with reference to fluoresence intensity (FI) readings from two controls: a blank control, and a maximum leakage control. The percentage of leakage and therefore amount of damage to the tight junctions is expressed, relative to these controls, for each of the set concentrations of the test substance. Then the FL20 (i.e. concentration that causes 20% FL relative to the value recorded for the untreated confluent monolayer and inserts without cells), is calculated. The FL20 (mg/mL) value is used in the prediction model for identification of ocular corrosives and severe irritants (see paragraph 35).20. Recovery is an important part of a test substance’s toxicity profile that is also assessed by the in vivo ocular irritation test. Preliminary analyses indicated that recovery data (up to 72 h following the chemical exposure) could potentially increase the predictive capacity of INVITTOX Protocol 71 but further evaluation is needed and would benefit from additional data, preferably acquired by further testing (7). This Test Guideline will be updated accordingly as new information and data are considered.PROCEDUREPreparation of the cellular monolayer21. The monolayer of MDCK CB997 cells is prepared using sub-confluent cells growing in cell culture flasks in DMEM/Nutrient Mix F12 (1x concentrate with L-glutamine, 15 mM HEPES, calcium (at a concentration of 1.0-1.8 mM) and 10% heat-inactivated FCS/FBS). Importantly, all media/solutions used throughout the FL assay should contain calcium at a concentration between 1.8 mM (200 mg/L) and 1.0 mM (111 mg/L) to ensure tight junction formation and integrity. Cell passage number range should be controlled to ensure even and4© OECD, (2012)OECD/OCDE 460 reproducible tight junctions formation. Preferably, the cells should be within the passage range 3-30 from thawing because cells within this passage range have similar functionality, which aids assay results to be reproducible.22. Prior to performing the FL test method, the cells are detached from the flask by trypsinisation, centrifuged and an appropriate amount of cells is seeded into the inserts placed in 24-well plates (see Annex I). Twelve mm diameter inserts with membrane of mixed cellulose esters, a thickness of 80-150 µm and a pore size of 0.45 µm, should be used to seed the cells. In the validation study, Millicell-HA 12 mm inserts were used. The properties of the insert and membrane type are important as these may affect cell growth and chemical binding. Certain types of chemicals may bind to the Millicell-HA insert membrane, which could affect the interpretation of results. Proficiency chemicals (see Annex III) should be used to demonstrate equivalency if other membranes are used.23. Chemical binding to the insert membrane is more common for cationic chemicals, such as benzalkonium chloride, which are attracted to the positively charged membrane (8). Chemical binding to the insert membrane may increase the chemical exposure period, leading to an over-estimation of the toxic potential of the chemical, but can also physically reduce the leakage of fluorescein through the insert by binding of the dye to the cationic chemical bound to the insert membrane, leading to an under-estimation of the toxic potential of the chemical. This can be readily monitored by exposing the membrane alone to the top concentration of the chemical tested and then adding sodium-fluorescein dye at the normal concentration for the standard time (no cell control). If binding of the sodium-fluorescein dye occurs, the insert membrane appears yellow after the test material has been washed-off. Thus, it is essential to know the binding properties of the test substance in order to be able to interpret the effect of the chemical on the cells.24. Cell seeding on inserts should produce a confluent monolayer at the time of chemical exposure. 1.6 x 105 cells should be added per insert (400 µL of a cell suspension with a density of 4 x 105 cells / mL). Under these conditions, a confluent monolayer is usually obtained after 96 hours in culture. Inserts should be examined visually prior to seeding, so as to ensure that any damages recorded at the visual control described at paragraph 30 is due to handling.25. The MDCK cell cultures should be kept in incubators in a humidified atmosphere, at 5% ± 1% CO2and 37 ± 1 ºC. The cells should be free of contamination by bacteria, viruses, mycoplasma and fungi.Application of the Test and Control Chemicals26. A fresh stock solution of test substance should be prepared for each experimental run and used within 30 minutes of preparation. Test substances should be prepared in calcium-containing (at a concentration of 1.0-1.8 mM), phenol red-free, HBSS to avoid serum protein binding. Solubility of the chemical at 250 mg/mL in HBSS should be assessed prior to testing. If at this concentration the chemical forms a stable suspension or emulsion (i.e.maintains uniformity and does not settle or separate into more than one phase) over 30 minutes, HBSS can still be used as solvent. However, if the chemical is found to be insoluble in HBSS at this concentration, the use of other test methods instead of FL should be considered. The use of light mineral oil as a solvent, in cases where the chemical is found to be insoluble in HBSS, should be5© OECD, (2012)460OECD/OCDEconsidered with caution as there is not enough data available to conclude on the performance of the FL assay under such conditions.27. All chemicals to be tested are prepared in sterile calcium-containing (at a concentration of 1.0-1.8 mM), phenol red-free, HBSS from the stock solution, at five fixed concentrations diluted on a weight per volume basis: 1, 25, 100, 250 mg/mL and a neat or a saturated solution. When testing a solid chemical, a very high concentration of 750 mg/mL should be included. This concentration of chemical may have to be applied on the cells using a positive displacement pipette. If the toxicity is found to be between 25 and 100 mg/mL, the following additional concentrations should be tested twice: 1, 25, 50, 75, 100 mg/mL. The FL20value should be derived from these concentrations provided the acceptance criteria were met.28. The test substances are applied to the confluent cell monolayers after removal of the cell culture medium and washing twice with sterile, warm (37ºC), calcium-containing (at a concentration of 1.0-1.8 mM), phenol red-free, HBSS. Previously, the filters have been visually checked for any pre-existing damages that could be falsely attributed to potential incompatibilities with test chemicals. At least three replicates should be used for each concentration of the test substance and for the controls in each run. After 1 min of exposure at room temperature, the test substance should be carefully removed by aspiration, the monolayer should be washed twice with sterile, warm (37ºC), calcium-containing (at a concentration of 1.0-1.8 mM), phenol red-free, HBSS, and the fluorescein leakage should be immediately measured. 29. Concurrent negative (NC) and positive controls (PC) should be used in each run to demonstrate that monolayer integrity (NC) and sensitivity of the cells (PC) are within a defined historical acceptance range. The suggested PC chemical is Brij 35 (CAS No. 9002-92-0) at 100 mg/mL. This concentration should give approximately 30% fluorescein leakage (acceptable range 20-40% fluorescein leakage, i.e. damage to cell layer). The suggested NC chemical is calcium-containing (at a concentration of 1.0-1.8 mM), phenol red-free, HBSS (untreated, blank control).A maximum leakage control should also be included in each run to allow for the calculation of FL20 values. Maximum leakage is determined using a control insert without cells.Determination of fluorescein permeability30. Immediately after removal of the test and control substances, 400μL of 0.1 mg/mL sodium-fluorescein solution (0.01% (w/v) in calcium-containing [at a concentration of 1.0-1.8 mM], phenol red-free, HBSS) is added to the Millicell-HA inserts. The cultures are kept for 30 minutes at room temperature. At the end of the incubation with fluorescein, the inserts are carefully removed from each well. Visual check is performed on each filter and any damage which may have occurred during handling is recorded.31. The amount of fluorescein that leaked through the monolayer and the insert is quantified in the solution which remained in the wells after removal of the inserts. Measurements are done in a spectrofluorometer at excitation and emission wavelengths of 485 nm and 530 nm, respectively. The sensitivity of the spectrofluorometer should be set so that there is the highest numerical difference between the maximum FL (insert with no cells) and the minimum FL (insert with confluent monolayer treated with NC). Because of the differences in the used spectrofluorometer, it is suggested that a sensitivity is used which will give fluorescence intensity > 4000 at the maximum fluorescein leakage control. The maximum FL value should not be6© OECD, (2012)OECD/OCDE 460 greater than 9999. The maximum fluorescence leakage intensity should fall within the linear range of the spectrofluorometer used.Interpretation of results and Prediction model32. The amount of FL is proportional to the chemical-induced damage to the tight junctions. The percentage of FL for each tested concentration of chemical is calculated from the FL values obtained for the test substance with reference to FL values from the NC (reading from the confluent monolayer of cells treated with the NC) and a maximum leakage control (reading for the amount of FL through an insert without cells).The mean maximum leakage fluorescence intensity = xThe mean 0% leakage fluorescence intensity (NC) = yThe mean 100% leakage is obtained by subtracting the mean 0% leakage from the mean maximum leakage,i.e. x - y = z33. The percentage leakage for each fixed dose is obtained by subtracting the 0% leakage to the mean fluorescence intensity of the three replicate readings (m), and dividing this value by the 100% leakage, i.e. %FL = [(m-y) / z] x 100%, where:m = the mean fluorescence intensity of the three replicate measurements for the concentration involved% FL = the percent of the fluorescein which leaks through the cell layer34. The following equation for the calculation of the chemical concentration causing 20% FL should be applied:FL D = [(A-B) / (C-B)] x (M C –M B) + M BWhere:D = % of inhibitionA = % damage (20% fluorescein leakage)B = % fluorescein leakage < AC = % fluorescein leakage > AM C = Concentration (mg/mL) of CM B = Concentration (mg/mL) of B35. The cut-off value of FL20 for predicting chemicals as ocular corrosives/severe irritants is given below:7© OECD, (2012)460OECD/OCDE36. The FL test method is recommended only for the identification of water soluble ocular corrosives and severe irritants (UN GHS Category 1, EU CLP Category 1, U.S. EPA Category I) (see paragraphs 1 and 10).37. In order to identify water soluble chemicals (substances and mixtures) (4) (7) (8) as "inducing serious eye damage" (UN GHS/EU CLP Category 1) or as an "ocular corrosive or severe irritant" (U.S. EPA Category I), the test substance should induce an FL20 value of ≤ 100 mg/mL.Acceptance of results38. The mean maximum fluorescein leakage value (x) should be higher than 4000 (see paragraph 31), the mean 0% leakage (y) should be equal or lower than 300, and the mean 100% leakage (z) should fall between 3700 and 6000.39. A test is considered acceptable if the positive control produced 20% to 40% damage to the cell layer (measure as % fluorescein leakage).DATA AND REPORTINGData40. For each run, data from individual replicate wells (e.g. fluorescence intensity values and calculated percentage FL data for each test substance, including classification) should be reported in tabular form. In addition, means ± SD of individual replicate measurements in each run should be reported.Test Report41. The test report should include the following information:Test and Control Substances-Chemical name(s) such as the structural name used by the Chemical Abstracts Service (CAS), followed by other names, if known;-Chemical CAS number, if known;-Purity and composition of the substance or mixture (in percentage(s) by weight), to the extent this information is available;-Physical-chemical properties relevant to the conduct of the study (e.g. physical state, volatility, pH, stability, water solubility, chemical class);-Treatment of the test/control substance prior to testing, if applicable (e.g. warming, grinding);-Storage conditions;Justification of the Test Method and Protocol Used-Should include considerations regarding applicability domain and limitations of the test method;Test Conditions8© OECD, (2012)OECD/OCDE 460 -Description of cell system used, including certificate of authenticity and the mycoplasma status of the cell line;-Details of test procedure used;-Test substance concentration(s) used;-Duration of exposure to the test substance;-Duration of incubation with fluorescein;-Description of any modifications of the test procedure;-Description of evaluation criteria used;-Reference to historical data of the model (e.g. negative and positive controls, benchmark chemicals, if applicable);-Information on the technical proficiency demonstrated by the laboratory;Results-Tabulation of data from individual test substances and controls for each run and each replicate measurement (including individual results, means and SDs);-The derived classification(s) with reference to the prediction model and/or decision criteria used;-Description of other effects observed;Discussion of the Results-Should include considerations regarding a non-conclusive outcome (paragraph 35: FL20 > 100 mg/mL) and further testing;Conclusions9© OECD, (2012)460OECD/OCDELITERATURE1.UN (2009), United Nations Globally Harmonized System of Classification and Labelling ofChemicals (GHS), Third revised edition, New York & Geneva: United Nations Publications.ISBN: 978-92-1-117006-1. Available at:[/trans/danger/publi/ghs/ghs_rev03/03files_e.html]2.EC (2008), Regulation (EC) No 1272/2008 of the European Parliament and of the Council of16 December 2008 on classification, labelling and packaging of substances and mixtures,amending and repealing Directives 67/548/EEC and 1999/45/EC, and amending Regulation (EC) No 1907/2006, Official Journal of the European Union L353, 1-1355.3.U.S. EPA (1996), Label Review Manual: 2nd Edition, EPA737-B-96-001, Washington DC:U.S. Environmental Protection Agency.4.EC-ECVAM (2009), Statement on the scientific validity of cytotoxicity/cell-function based invitro assays for eye irritation testing. Available under Publications at: [http://ecvam.jrc.it/index.htm]5.Scott, L. et al. (2010), A proposed eye irritation testing strategy to reduce and replace in vivostudies using Bottom-Up and Top-Down approaches, Toxicol. In Vitro 24, 1-9.6.OECD (2002), Test No. 405: Acute Eye Irritation/Corrosion, OECD Guidelines for theTesting of Chemicals, Section 4, OECD Publishing. doi: 10.1787/9789264070646-en7.EC-ECVAM (1999), INVITOX Protocol 71: Fluorescein Leakage Test, Ispra, Italy:European Centre for the Validation of Alternative Methods (ECVAM). Available at: [http://ecvam-dbalm.jrc.ec.europa.eu]8.EC-ECVAM (2008), Fluorescein Leakage Assay Background Review Document as anAlternative Method for Eye Irritation Testing. Available under Validation Study Documents, Section Eye Irritation at: [http://ecvam.jrc.it/index.htm]9.OECD (2005), Guidance Document on the Validation and International Acceptance of Newor Updated Test Methods for Hazard Assessment, OECD Series on Testing and Assessment No. 34. OECD, Paris. Available at: [/env/testguidelines]10© OECD, (2012)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Inhibitors, Agonists, Screening Libraries
Data Sheet
BIOLOGICAL ACTIVITY:
VO–Ohpic trihydrate is an extremely potent inhibitor of PTEN with IC 50 of 46±10 nM.
IC50 & Target: IC50: 46±10 nM (PTEN)[1]
In Vitro: VO–OHpic with two OHpic ligands and an oxo ligand is a sterically demanding molecule, and one will therefore expect that binds substrate will affect the subsequent binding of the inhibitor due to steric hindrance. VO–OHpic significantly inhibits PTEN activity in low nanomolar concentrations (IC 50, 46±10 nM), which is in agreement with the previously determined potency (IC 50,35±2 nM) in a PIP 3–based assay. The inhibition constants K ic and K iu are determined to be 27±6 and 45±11 nM, respectively [1].VO–OHpic is an encouragingly specific and potent PTEN inhibitor. VO–OHpic is the most potent inhibitor (IC 50=35 nM) of the PTEN lipid phosphatase activity [2].In Vivo: PTEN is inhibited in mice by intra–peritoneal injection of VO–OHpic (10 μg/kg) 30 min before ischemia and then exposed them to 30 min of ischemia and 120 min of reperfusion. At the end of the experiment, myocardial infarct size is measured by triphenyltetrazolium chloride (TTC). Myocardial infarct size is significantly decreased in VO–treated mice (25±6 vs. 56±5 %, n=7,P<0.01). There is no difference in the area at risk between these two groups (46±3 vs. 57±3 %, n=7, P>0.05)[3].
PROTOCOL (Extracted from published papers and Only for reference)
Kinase Assay:[1]VO–OHpic is dissolved in DMSO (100 μM) and diluted further to the required concentration with 1% DMSO. For inhibition studies, PTEN is preincubated with VO–OHpic at RT for 10 min before substrate is added to initialise the reaction.
Background absorbance (malachite green assay) and fluorescence (OMFP assay) are determined with VO–OHpic in assay buffer and corrected in the data analysis [1].
Animal Administration: VO–OHpic (VO) is prepared in saline (Mice)[3].[3]Mice [3]
The experiment is performed with male C57BL6 mice. Briefly, mice are anesthetized with pentobarbital (70 mg/kg). The left coronary artery is occluded about 1–2 mm below the left auricle. Reperfusion is accomplished by loosening the ligature. The PTEN inhibitor VO–OHpic is administered by intra–peritoneal injection at the dosage of 10 μg/kg once 30 min before ischemia. Saline is used as control. At the end of the experiment, the animals are euthanized by transecting the aorta and removing the heart for infarct size determination.
References:
[1]. Mak LH, et al. Characterisation of the PTEN inhibitor VO–OHpic. J Chem Biol. 2010 Oct;3(4):157–63.
[2]. Rosivatz, E, et al. A small molecule inhibitor for phosphatase and tensin homologue deleted on chromosome 10 (PTEN). ACS Chem Biol. 2006 Dec 15;1(12):780–90.
[3]. Zu L, et al. PTEN inhibitors cause a negative inotropic and chronotropic effect in mice. Eur J Pharmacol. 2011 Jan 10;650(1):298–302.
Product Name:
VO–Ohpic (trihydrate)Cat. No.:
HY-13074CAS No.:
476310-60-8Molecular Formula:
C 12H 16N 2O 11V+Molecular Weight:
415.20Target:
PTEN Pathway:
PI3K/Akt/mTOR Solubility:10 mM in DMSO; H 2
O: < 0.1 mg/mL
Caution: Product has not been fully validated for medical applications. For research use only.
Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@
Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。